

Stress‐induced childhood‐onset neurodegeneration with ataxia and seizures (CONDSIAS) is an extremely rare, autosomal recessive, neuro‐degenerative disorder caused by inactivating mutations in Adenosine di‐phosphate ribosylhydrolase 2 (ADPRHL2) gene. This results in defective post‐translational modification of Adenosine di‐phosphate (ADP) ribosylated proteins, causing dysregulated cellular excitotoxicity and death, and progressive neurodegeneration. Patients usually present in the first decade with developmental delay, regression and seizures. Later disease onset manifests with gait ataxia, weakness, speech difficulty and cognitive impairment. Other manifestations include hearing impairment, polyneuropathy, easy fatiguability and dystonia. Exacerbation occurs in response to physical stress or febrile illness. We seek to highlight the occurrence of longitudinally extensive myelopathy as a novel manifestation of CONDSIAS, while also reporting the first case from India.
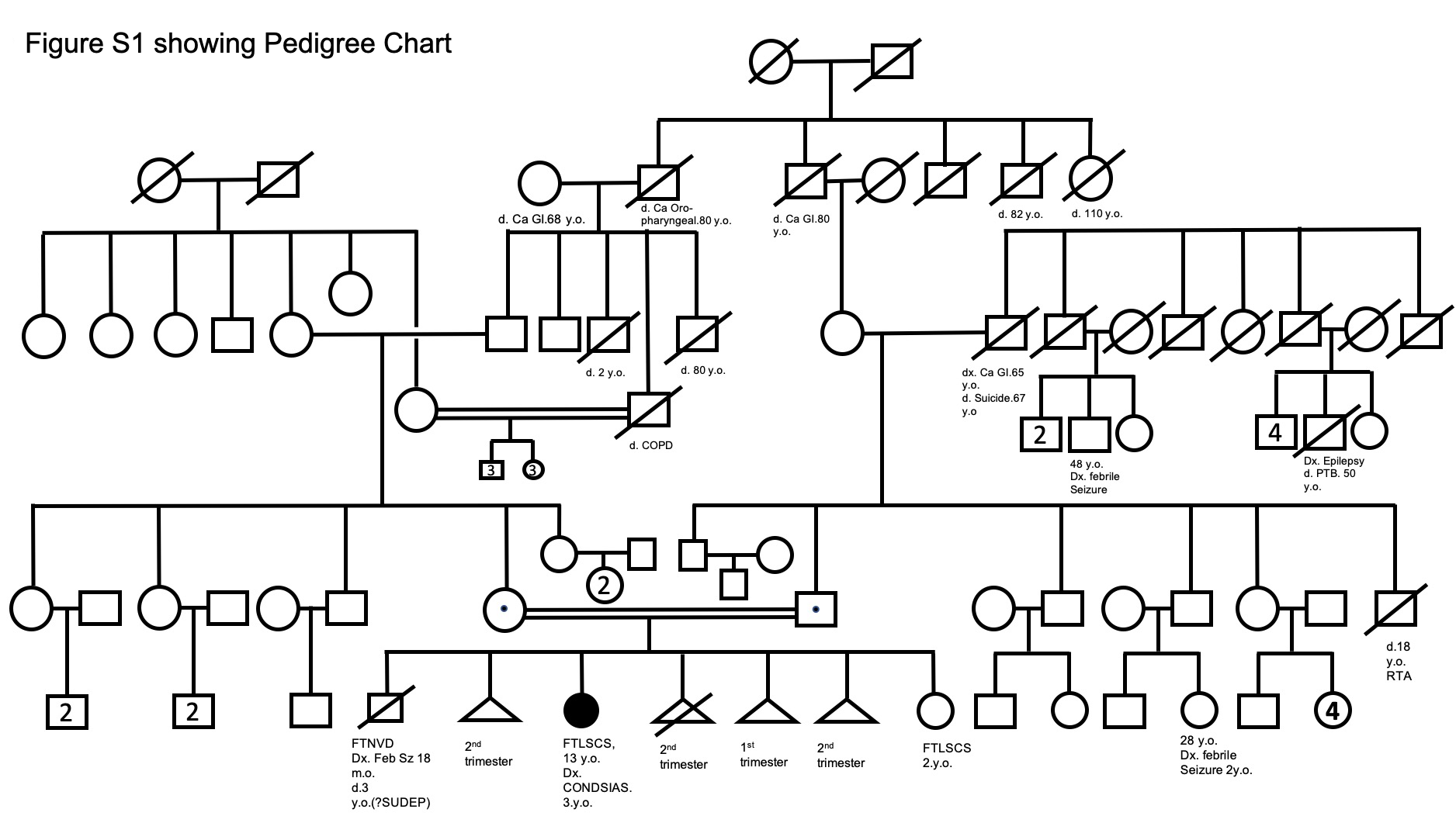
A 12‐year‐old girl, born of fourth‐degree consanguinity (Fig. S1), with normal developmental milestones till 3 years of age, developed transient, self‐resolving episodes of gait unsteadiness and abnormal posturing of the trunk and extremities, without loss of consciousness. Initially, the posturing was predominantly proximal and involved both upper limbs which progressed to distal upper and lower limb involvement in 6 years, and was associated with progressively increasing tremulousness, which was more on reaching out for objects. She was noticed to over/under‐shoot her intended targets, including her footwear. These recovered spontaneously, were more frequent during summer and were precipitated by febrile illnesses. Initially these episodes used to occur once or twice per month, but increased to once or twice every week in the next 3–4 years and have been occurring on a daily basis for the last 2 years. Initially, these episodes were transient and resolved completely in 3–5 minutes but have become persistent since the last 2 years.
At 9 years of age, she also developed behavioral abnormalities in the form of irritability, social awkwardness and delusion of persecution with suicidal ideations. She was prescribed anti‐psychotics without any relief.
She presented to our institute at the age of 12, when her scholastic performance dipped, had more frequent and prolonged episodes of unsteady gait and abnormal posturing (Video S1), hearing loss, visual hallucinations and excessive crying spells. Her family history was significant for bad obstetric history in the mother, along with history of epilepsy and sudden death of elder brother following a seizure at 3 years of age and febrile seizures in paternal cousin. Examination revealed a thin built with bilateral gaze‐evoked nystagmus and sensorineural hearing loss. Tone, power, reflexes and sensory examination was normal. Both axial and appendicular ataxia was present along with intermittent dystonic posturing of limbs.
She was being evaluated for childhood onset episodic ataxia and generalized dystonia syndromes when during her hospital stay, she developed acute onset symmetric quadriparesis with bulbar dysfunction, bladder and bowel involvement (onset to peak: 5 days). Her routine investigations were unremarkable (Table S1). Magnetic Resonance Imaging (MRI) brain and spine revealed cerebellar atrophy and longitudinally extensive holo‐cord transverse myelopathy (without contrast enhancement) respectively (Fig. 1). Cerebrospinal Fluid (CSF) analysis including pan‐viral Polymerase Chain Reaction (PCR) and Oligoclonal Bands (OCBs) were unremarkable. Serum aquaporin and anti‐ myelin oligodendrocyte glycoprotein (MOG) antibody were also negative. Electroencephalogram (EEG) showed generalized background slowing without epileptiform discharges. Nerve conduction study revealed sensory‐motor axonal polyneuropathy of lower limbs and muscle biopsy from quadriceps showed neurogenic changes. She was given intravenous methylprednisolone pulse 1000 mg for 5 days followed by intravenous immunoglobulin (IVIG) for 5 days (2 g/kg body weight) without significant clinical improvement. Her hospital course was complicated by sudden cardiac arrest and was revived by cardio‐pulmonary resuscitation. She subsequently developed regular rhythmic jaw tremors and upper limb dyskinesias (Video S2). Repeat neuroimaging after 4 weeks revealed additional hyperintensities in bilateral middle cerebellar peduncles with resolution of cord expansion.
FIG 1.
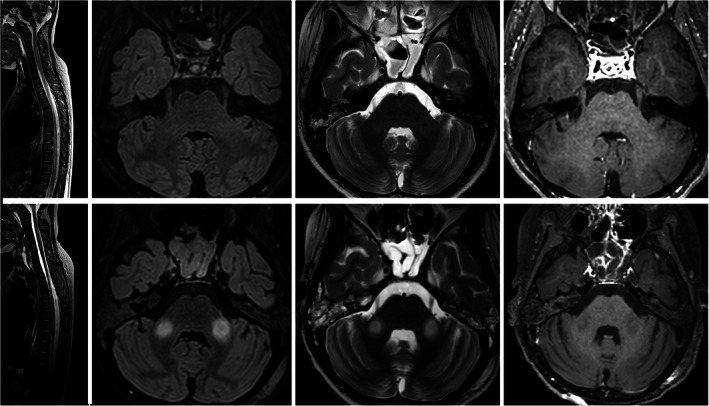
MRI brain and spine. Sagittal T2‐WI (A) of cervicodorsal spine shows long segment cord hyper intensity with cord expansion extending from C2 to conus. Axial Flair (B) and T2‐WI (C) at level of pons show no signal abnormality or enhancement in axial post‐gad T1‐WI (D). However, mild cerebellar atrophy was apparent. Follow‐up MRI: Sagittal T2‐WI (E) of cervicodorsal spine shows resolution of cord hyper intensity and expansion. Axial Flair (F) and T2‐WI (G) at level of pons show bilateral symmetrical hyper intensity in middle cerebellar peduncles. No enhancement is seen in n axial post‐gad T1‐WI (H).
A rare homozygous c.100 G > A (p.Asp34Asn) mutation in exon 1 of ADPRHL2 (+) gene (which results in CONDSIAS; Online Mendelian inheritence in Men [OMIM]#618170) was identified by targeted gene sequencing performed on the DNA extracted from her blood. Sentieon haplotype caller was used to identify clinically relevant variants, which were annotated according to published variants in literature and diseases databases like ClinVar, 1 OMIM, 2 and GWAS. 3 Our observed variation has been reported in a patient with CONDSIAS and it lies in the critical α‐helical loop within the ADP‐ribosylhydrolase domain, probably resulting in impairment of protein structure and activity. 4 The variant is classified as pathogenic in the ClinVar database, 5 defined as per American College of Medical Genetics Protocol. 6 Sanger sequencing confirmed the same. Genetic testing in parents revealed heterozygosity for the same mutation.
She had no definitive clinical improvement and was discharged on supportive treatment.
Our patient presented with episodic ataxic‐dystonic posturing from 3 years of age, which progressed with additional neurological deficits, finally compounded by quadriparesis due to holo‐cord myelopathy. Due to multi‐axial involvement in a young woman with positive family history, a mitochondrial cytopathy was suspected but genetic analysis diagnosed CONDSIAS. Danhauser et al.7 and Ghosh et al 4 have reported 12 and 16 patients of the same respectively (Table S2).
Our patient had prototypical features of CONDSIAS. However, the finding of acute longitudinally extensive myelopathy has not yet been described for CONDSIAS (spinal cord atrophy is described in some cases). 4 , 7 This could be associated or incidental. We tried to rule out the latter through exhaustive work up, which was unyielding. Although it is invariably fatal, increased reporting may provide for newer emerging therapies with improved outcomes.
The disease spectrum will constantly evolve with increasingly diagnosed cases. The diagnosis should be suspected in all children presenting with episodic ataxia, dystonia, severe seizures and peri‐oral tremors with/ without punctuation by sudden life threatening events, since a delay results in increasing number of attacks, leading to more morbidity and mortality. Although, no definitive treatment exists, prevention of precipitating events could help the afflicted individual lead a less disabled life.
Author Roles
1. Research project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript: A. Writing of the first draft, B. Review and Critique.
B.M.: 1A,B,C, 2B, 3A
S.F.: 1B, 2C, 3B
A.A.: 1A,B,C, 2A,B, 3A
D.M.R.: 1B, 2A,C, 3B
A.G.: 1B, 2C, 3B
A.K.S.: 1B, 2C, 3B
Disclosures
Ethical Compliance Statement: Written informed consent was taken from the patient's primary caregivers. The authors confirm that the approval of the institutional review board was not required for this work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: No funding was received for this work and the authors have no conflicts to report.
Financial Disclosures for Previous 12 Months: The authors have no disclosures to report.
Supporting information
Figure S1. Pedigree chart. 1st child: Male; born at full term by normal vaginal delivery; normal milestones till 18 months of age, when he developed febrile seizures. These progressively increased in frequency and he died in his sleep at 3 years of age (Sudden unexpected death in epilepsy [SUDEP]) second pregnancy: Intra‐uterine death (IUD) in the second trimester third pregnancy: Female; full term by Caesarean section (our proband case) fourth pregnancy: medically terminated in the early second trimester fifth and sixth pregnancies: IUDs at late first trimester and late second trimester respectively. Seventh pregnancy: female; Caesarean section; normal developmental milestones (presently 2 years old).
{kind=link}
Table S1. Laboratory Investigation parameters of the case.
Table S2. Comparison of clinical, laboratory, MRI and genetic features of the cases reported and our case.
Video S1. Video of the patient depicting a broad‐based ataxic gait with mild forward stoop due to dystonic ataxia (before admission with us). [patient in red top and blue pants].
Video S2. Video of the patient showing regular rhythmic jaw tremor which she developed during the course of hospitalization.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44(D1):D862–D868. 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McKusick VA. Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet 2007;80(4):588–604. 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS catalog, a curated resource of SNP‐trait associations. Nucleic Acids Res 2014;42(Database issue):D1001–D1006. 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ghosh SG, Becker K, Huang H, et al. Biallelic mutations in ADPRHL2, encoding ADP‐Ribosylhydrolase 3, lead to a degenerative pediatric stress‐induced epileptic ataxia syndrome. Am J Hum Genet 2018;103(3):431–439. 10.1016/j.ajhg.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. VCV000590303.1 ‐ ClinVar ‐ NCBI. https://www.ncbi.nlm.nih.gov/clinvar/variation/590303/. Accessed September 12, 2020.
- 6. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–423. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Danhauser K, Alhaddad B, Makowski C. Bi‐allelic ADPRHL2 mutations cause neurodegeneration with developmental delay, ataxia, and axonal neuropathy. Am J Hum Genet 2018;103(5):817–825. 10.1016/j.ajhg.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pedigree chart. 1st child: Male; born at full term by normal vaginal delivery; normal milestones till 18 months of age, when he developed febrile seizures. These progressively increased in frequency and he died in his sleep at 3 years of age (Sudden unexpected death in epilepsy [SUDEP]) second pregnancy: Intra‐uterine death (IUD) in the second trimester third pregnancy: Female; full term by Caesarean section (our proband case) fourth pregnancy: medically terminated in the early second trimester fifth and sixth pregnancies: IUDs at late first trimester and late second trimester respectively. Seventh pregnancy: female; Caesarean section; normal developmental milestones (presently 2 years old).
Table S1. Laboratory Investigation parameters of the case.
Table S2. Comparison of clinical, laboratory, MRI and genetic features of the cases reported and our case.
Video S1. Video of the patient depicting a broad‐based ataxic gait with mild forward stoop due to dystonic ataxia (before admission with us). [patient in red top and blue pants].
Video S2. Video of the patient showing regular rhythmic jaw tremor which she developed during the course of hospitalization.