

Abstract
A history of chronic cigarette smoking is known to increase risk for acute respiratory distress syndrome (ARDS), but the corresponding risks associated with chronic e-cigarette use are largely unknown. The chromosomal fragile site gene, WWOX, is highly susceptible to genotoxic stress from environmental exposures and thus an interesting candidate gene for the study of exposure-related lung disease. Lungs harvested from current versus former/never-smokers exhibited a 47% decrease in WWOX mRNA levels. Exposure to nicotine-containing e-cigarette vapor resulted in an average 57% decrease in WWOX mRNA levels relative to vehicle-treated controls. In separate studies, endothelial (EC)-specific WWOX knockout (KO) versus WWOX flox control mice were examined under ARDS-producing conditions. EC WWOX KO mice exhibited significantly greater levels of vascular leak and histologic lung injury. ECs were isolated from digested lungs of untreated EC WWOX KO mice using sorting by flow cytometry for CD31+ CD45−cells. These were grown in culture, confirmed to be WWOX deficient by RT-PCR and Western blotting, and analyzed by electric cell impedance sensing as well as an FITC dextran transwell assay for their barrier properties during methicillin-resistant Staphylococcus aureus or LPS exposure. WWOX KO ECs demonstrated significantly greater declines in barrier function relative to cells from WWOX flox controls during either methicillin-resistant S. aureus or LPS treatment as measured by both electric cell impedance sensing and the transwell assay. The increased risk for ARDS observed in chronic smokers may be mechanistically linked, at least in part, to lung WWOX downregulation, and this phenomenon may also manifest in the near future in chronic users of e-cigarettes.
Keywords: ARDS, e-cigarettes, WWOX, endothelium, cigarette smoke
Tobacco smoke exposure is the leading cause of preventable death worldwide owing to its contribution to the pathogenesis of chronic cardiovascular and pulmonary diseases (1). However, it has recently come to light that cigarette smoking likely also contributes to the development of an acute critical illness, acute respiratory distress syndrome (ARDS) (2–6). ARDS afflicts an estimated 200,000 patients/yr in the United States alone, kills ∼75,000, and is seriously debilitating for many survivors (7). It occurs in select individuals following an inciting illness or event such as acute aspiration, pneumonia, sepsis, and blunt force trauma (8). The cardinal, morbidity-producing feature of ARDS is noncardiogenic pulmonary edema resulting from pulmonary vascular barrier disruption with consequent alveolar flooding, and respiratory failure (9, 10). An increased incidence of ARDS has been observed in cigarette smokers versus nonsmokers following blunt force trauma, cardiac surgery, transfusion of blood products, or nonpulmonary sepsis (2–4, 6). The mechanisms linking cigarette smoke (CS) exposure and ARDS development are largely unknown. It is hoped that research in this area may yield novel therapeutic targets for ARDS altogether, if not for a subset of patients in whom environmental exposures produce a mechanistic predisposition.
Recently, the link between CS exposure and ARDS risk has been highlighted further by the emergence of e-cigarette or vaping product use–associated lung injury (EVALI), which has been occurring in epidemic proportions in the United States since the summer of 2019 (11). A significant proportion of EVALI seen thus far appears to be associated with specific chemical additives that have been vaped (12, 13). The overall rise in use of e-cigarettes has been fueled, in part, by the unsubstantiated notion that they are safer than traditional cigarettes because of the absence of combustion products in e-cigarette vapor versus CS. Thus, they are being used in many cases as a strategy for patients to quit smoking traditional cigarettes (14). However, notwithstanding the specific circumstances underlying the current EVALI epidemic, very little, if anything, is known about the long-term consequences of e-cigarette vapor exposure in terms of ARDS risk, as well as risks for the development of other CS-associated lung diseases.
A multitude of epigenetic modifications caused by CS, any combination of which may precipitate the CS-ARDS connection, include methylation at various CpG-containing sequences (15) as well as more dramatic changes such as rearrangements and deletions that occur with some frequency at chromosomal fragile sites (16, 17). Among both of these categories of CS-induced gene changes is the tumor suppressor WWOX, which resides at the second most active chromosomal fragile site in the human genome (18, 19). Apart from its heightened susceptibility to loss-of-function mutations and methylation-induced downregulation via environmental exposures including CS, its potential role in the pathogenesis of lung disease was elucidated by gene knockdown studies in mice and cultured pulmonary epithelial cells (20). Intratracheal instillation of WWOX-targeting siRNA in mice (vs. scrambled siRNA) was observed to result in widespread pulmonary neutrophilic inflammation, a finding that may be relevant to the pathogenesis of chronic lung diseases such as chronic obstructive pulmonary disease (COPD) (20). Because the intratracheal route of instillation was thought to primarily involve epithelial cells, this observed in vivo phenomenon was linked to in vitro observations of increased IL-8 secretion by human lung epithelial cells during WWOX knockdown (20).
Interestingly, during in vivo experiments, it was observed that vascular leak during LPS stimulation was greater in WWOX knockdown animals to a degree that was out of proportion to increases in inflammation (20). This raised the question of whether an inflammation-independent mechanism of endothelial barrier vulnerability was a part of the WWOX-deficient phenotype. It is also well known that although epithelial cells are on the “front lines” of inhalational toxins such as CS in the lung, endothelial cells are also susceptible to toxic effects (21).
For these reasons, in the current study, specific endothelial silencing of WWOX expression in cultured cells and mice was achieved to assess WWOX as a candidate gene for CS-ARDS.
Some of the results of these studies have been previously reported in the form of a preprint (https://doi.org/10.1101/2020.07.13.200832).
Methods
Reagents
Fluorescein isothiocyanate (FITC)-Dextran, SP100625, PP2, LPS, RBC Lysis Buffer, Dulbecco’s modified Eagle medium, BSA, deoxyribonuclease, Dispase II, and RIPA Buffer were purchased from Sigma-Aldrich. BCA Protein Assay Kit, Shandon Kwik-Diff kit, siPORT Amine transfection reagent, Lipofectamine 2000, High Capacity cDNA Reverse Transcription kit, Taqman Gene Expression Master Mix, and Taqman Gene Expression Assays for GAPDH and WWOX were from Thermo Fisher. Mini-PROTEAN TGX Precast Gels were from BioRad. QIAShredder and RNeasy Mini kits were from Qiagen. Anti-WWOX antibody was from Abcam. Human lung microvascular endothelial cells (ECs) and endothelial cell growth media were purchased from Lonza. Heparin was purchased from Hospira. Collagenase was from Worthington. ELISA kits for mouse cytokines (keratinocyte-derived chemokine [KC], MIP-2, MCP-1, IL-6, IL-1β, TNF-α), anti-mouse CD16/32, anti-mouse CD31-PE/Cy7, and anti-mouse CD45-Alexa700 antibodies were from BioLegend and R&D Systems. siGENOME WWOX-targeting and scrambled control siRNA was from Dharmacon/GE. pCMV entry vector containing the ORF corresponding to (Myc-DDK-tagged)-human WW domain-containing oxidoreductase ((WWOX), transcript variant 1, as well as a (Myc-DDK-tagged)-empty vector were purchased from OriGene. Propylene glycol, vegetable glycerin, and nicotine were from Sigma-Aldrich.
Cell Culture
ECs obtained from Lonza (primary cells) were grown in cell culture using endothelial growth media containing supplemental growth factors and 2% FBS according to the manufacturer’s instructions. Cells underwent a maximum of three passages for use in experiments.
For separate studies, ECs were isolated from the lungs of 8-week-old C57BL/6-WWOXflox/flox and Cdh5-CreERT2/WWOXflox/flox mice as in Kawasaki and colleagues (22). Following ketamine/xylazine administration, the thoracic cavity was surgically opened. Twenty milliliters of sterile PBS containing 10 U/ml heparin was instilled into the pulmonary vasculature via injection through the right ventricle until the lungs were blood-free. Mouse lungs were then harvested, minced, and digested in an enzyme complex as previously described. Washed cell pellets were resuspended and serially incubated with anti-mouse CD16/32 (to block Fc receptors), anti-mouse CD31-PE/Cy7, and anti-mouse CD45-Alexa700 antibodies. After washing with PBS/BSA, cells were sorted by flow cytometry (MoFlo Astrios Cell Sorter; Beckman Coulter) to isolate CD31+ CD45− ECs, which were then placed in EC growth media on gelatin-coated plates.
USA 300 MRSA
Wild-type USA 300 methicillin-resistant Staphylococcus aureus (MRSA) was prepared as in Chen and colleagues (23). Briefly, 3 ml of tryptic soy broth was inoculated with a single colony of bacteria and incubated overnight. One milliliter of this overnight culture was then used to inoculate 100 ml of tryptic soy broth for incubation until an optical density of 0.5 at 660 nm was reached. This was then centrifuged and washed for resuspension in sterile PBS at a concentration of 1 × 108 cfu per 30 μl. MRSA was heat-killed by incubation at 65°C for 1 hour before use in cell-based experiments. Live MRSA was used in mice.
Animals
All experiments and animal care procedures were approved by the University of Illinois at Chicago Animal Care and Use Committee. C57BL/6-WWOXflox/flox mice were donated generously by Dr. C. Marcelo Aldaz, M.D./Ph.D., of MD Anderson Cancer Center at the University of Texas. C57BL/6-Cdh5-CreERT2 mice were purchased from Jackson Laboratories. The two strains were crossed to produce Cdh5-CreERT2/WWOXflox/flox mice as in Ludes-Meyers and colleagues (24). They were housed in cages in a temperature-controlled room with a 12-hour dark/light cycle and with free access to food and water.
E-cigarette exposure in mice
E-cigarette liquid was generated in the lab using a 50:50 solution of propylene glycol and vegetable glycerin as the vehicle for 24 mg/ml nicotine, which corresponds to the concentration and composition of the popular vape pen liquid. Six- to 8-week-old male C57Bl/6 mice were exposed to vehicle versus nicotine-containing e-cigarette vapor via inExpose inhalation system (Scireq) for 1 hour a day, 5 days a week, and for 3 months as described previously (25). Thirty to 60 minutes after the final exposure, mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg), and lungs were harvested as described below. All e-cigarette–related animal protocols were approved by the University of California, San Diego Institutional Animal Care and Use Committee.
Murine models of ARDS
Six-week old male Cdh5-CreERT2/WWOXflox/flox mice and control animals received intraperitoneal injections of tamoxifen (75 mg/kg) for 5 consecutive days. Seven days later, they were anesthetized with isoflurane and underwent nasal instillation of USA 300 MRSA (0.75 × 108 cfu) versus an equivalent volume of sterile PBS. Mice were then housed in cages as described above for 18 hours, after which they underwent testing and lung harvesting as described below. In separate experiments, tamoxifen- versus vehicle-treated mice were anesthetized with isoflurane and underwent intratracheal instillation of 1 mg/kg LPS versus an equivalent volume of sterile saline. Eighteen hours later, they underwent testing and lung harvesting as described below.
FITC-dextran permeability assay
FITC-Dextran (70 kilodaltons) was used to measure pulmonary vascular leak as described previously (26). Briefly, FITC-Dextran was mixed to a concentration of 30 mg/ml in sterile PBS and sterile filtered. Thirty minutes before euthanasia, 100 μl of FITC-Dextran solution was injected into the retro-orbital venous plexus. BAL fluid (BALF) was collected as described below. Blood was collected during lung harvest and centrifuged to isolate serum. Fluorescence of equal volumes of BALF supernatant and serum were measured and relative changes in permeability were expressed as a ratio of FluoBALF/FluoSerum normalized to the control condition.
BALF collection and analysis
Mice were anesthetized with ketamine/xylazine. The trachea was exposed surgically and a small incision was made on the anterior surface. An 18-gauge blunt-end cannula was inserted into this opening and secured with a suture tied around the trachea. One milliliter of sterile saline was infused through this cannula and slowly withdrawn. This BALF was centrifuged to pellet cells and remove supernatant. RBCs were removed from the cell pellet using RBC lysis buffer according to the manufacturer’s instructions. The remaining cells (leukocytes) were pelleted and resuspended in a fixed volume of saline, and a small aliquot was used to measure cell concentration with an automated cell counter. The remaining cell suspension was used to make a cytospin prep on a glass slide for staining with the Shandon Kwik-Diff kit. A manual differential cell count was performed on 10 high-powered fields to determine relative percentages of neutrophils in the leukocyte population. Protein concentration was measured using the BCA Protein Assay kit, and the corresponding ELISA kits were used to measure concentrations of KC, MIP-2, MCP-1, IL-6, IL-1β, and TNF-α.
Lung harvesting for RT-PCR, Western blotting, and histology
Following BALF collection, the right ventricle was infused with sterile saline until the lungs were free of intravascular blood. The right middle lobe was excised and snap frozen in liquid nitrogen for RT-PCR and Western blotting. The remaining lobes were excised and placed in 10% formalin for 24 hours followed by transfer to 70% ethanol for storage until paraffin embedding and sectioning for hematoxylin and eosin (H&E) staining.
For RT-PCR, lungs were thawed, lysed, and homogenized, and RNA was isolated using the QIAShredder and RNeasy Mini kits per manufacturer’s instructions. cDNA was produced for each sample using the high-capacity cDNA Reverse Transcription kit from Thermo Fisher. Real-time PCR was performed using the Taqman Gene Expression Master Mix and Assays per manufacturer’s instructions. Relative quantification between controls and experimental samples of WWOX gene expression normalized for GAPDH was determined using the ΔΔCt method.
For Western blotting, snap-frozen lungs were thawed and homogenized in RIPA buffer. Homogenized samples were centrifuged to pellet debris and isolate protein lysates. Protein concentration of lysates was measured using the BCA Protein Assay kit, and samples were diluted in RIPA buffer until all had the same protein concentration. After dilution with SDS buffer, samples were loaded into 4–20% Mini-PROTEAN TGX Precast Gels and subjected to SDS-PAGE. Gels were transferred to nitrocellulose membranes that were then subjected to Western blotting using antibodies targeting proteins of interest in accordance with the manufacturer’s instructions. Densitometric analysis was performed using ImageJ software.
Electric Cell Impedance Sensing Permeability Assay
Endothelial cells were grown to confluence in polycarbonate wells containing evaporated gold microelectrodes purchased from Applied BioPhysics, and measurements of transendothelial electrical resistance (TER) were performed using an Electric Cell Impedance Sensing (ECIS) system (Applied BioPhysics) as described previously (27–29). Briefly, a 4,000-Hz alternating current was applied across the electrodes with a fixed amplitude and external resistance to establish a constant current of ∼1 μA. The in-phase and out-of-phase voltages between the electrodes were measured in real time and were used to calculate TER. Increased cell adherence and confluence correlates with higher TER, whereas cell retraction, rounding, or loss of adhesion leads to a decrease in TER (30). TER was monitored for stability before initiating MRSA/LPS treatment in all experiments, and a threshold of 2.5 × 103Ω was required for ECs to be considered confluent on the electrodes and suitable for study. TER from each microelectrode was measured at discrete time intervals and recorded for analysis.
FITC-Dextran Transwell Permeability Assay
ECs were grown to confluency on transwell inserts (EMD Millipore) containing 3-μm pores. Ten micrograms per milliliter of FITC-labeled dextran (70 kD) was added to the media over the cells along with vehicle, heat-killed MRSA (2 × 108 cfu/ml), or LPS (1 μg/ml). After 2 hours, 100 μl of media was extracted from below the transwell insert for measurement of FITC concentration by fluorometry (excitation/emission spectrum peak wavelengths of 495 nm and 519 nm) to determine the degree of EC monolayer permeability as described previously (31).
In vitro silencing of WWOX in human pulmonary endothelial cells
In vitro silencing of WWOX in human ECs (Lonza) was achieved using transfection of siRNA as described in Wolfson and colleagues (32). WWOX-targeting (CCAAGGACGGCUGGGUUUA) versus scrambled control siRNA (UAGCGACUAAACACAUCAA) was complexed with siPORT Amine transfection reagent. Serum-free media was used to bring the concentration of siRNA to 100 nM. Transfection of ECs was then performed as described previously (32). Seventy-two hours after transfection, ECs were analyzed by Western blotting for WWOX expression, and by ECIS for barrier function during LPS exposure.
Human lung tissue
The Vanderbilt Institutional Review Board approved this work as nonhuman subjects research (i.e., no institutional review board approval required). Lung tissue specimens were procured from organ donors whose lungs were declined for transplantation as part of the Beta-agonist for Oxygenation in Lung Donors study (33). At the time of procurement, lungs were resected without perfusion and were transported on ice to the investigators’ laboratory. Portions of each lobe were sampled and immediately frozen at −80°C in RNALater (Qiagen) until RNA extraction. Clinical history including history of lung disease and cigarette smoking was obtained from the donor medical record.
Statistical analysis
Either a Student’s t test or an ANOVA were performed where applicable on all data using GraphPad Prism software. *P < 0.05.
Results
Lung tissue from current human smokers versus former/never-smokers exhibits decreased WWOX expression. RNA was extracted from human lung tissue samples taken from lung donors with a history of regular cigarette smoking and analyzed by RT-PCR for levels of WWOX mRNA expression compared with that of former or never-smokers. As shown in Figure 1A, lung WWOX expression was 47% lower in current versus former or never-smokers (P < 0.05).
Figure 1.
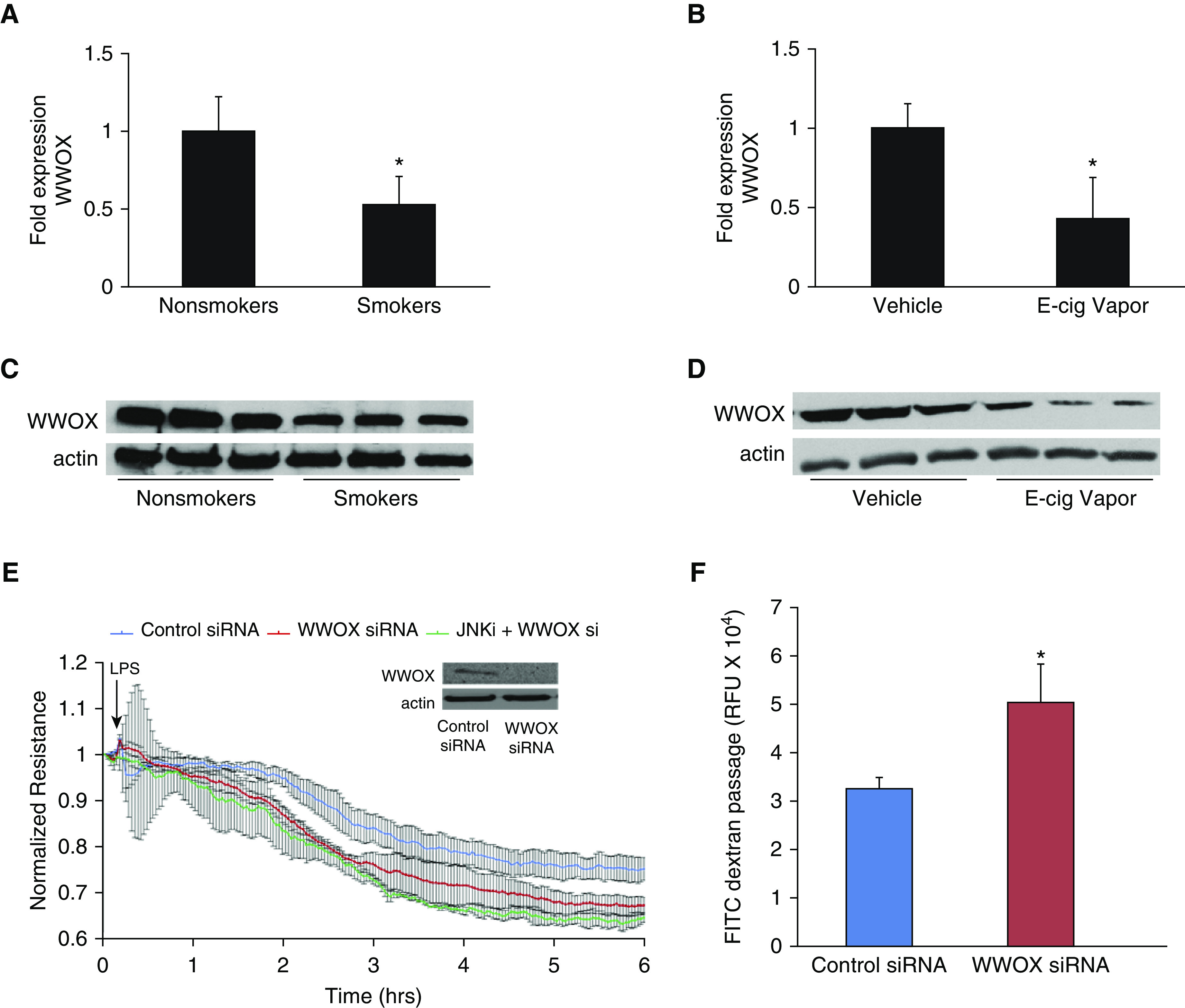
WWOX expression in human smokers/e-cigarette–exposed mice and the associated lung endothelial barrier consequences. (A) RNA was extracted from lung tissue harvested from current versus never-/former human smokers. RT-PCR for WWOX (Taqman Assay; Life Technologies) was performed. The bar graph depicts fold-changes in WWOX mRNA expression calculated by the ΔΔCt method with GAPDH as the internal control. Results are shown as means ± SD in n = 3 independent experiments. (B) RNA was extracted from lung tissue harvested from mice exposed to e-cigarette vapor with or without nicotine (control). RT-PCR for WWOX results are shown here. Results are shown as means ± SD in n = 3 independent experiments. The same lung tissues were analyzed by Western blotting for WWOX:actin expression. (C and D) Representative blots are shown. (E) Pulmonary endothelial cells (ECs) were transfected with either a WWOX-targeted or control scrambled siRNA (Dharmacon). A Western blot for WWOX confirms silencing. Transfected ECs were grown to confluency on gold electrodes for ECIS analysis. They were pretreated with a pharmacologic inhibitor of JNK (SP600125) versus vehicle followed by LPS. (F) In separate experiments, ECs were grown in a monolayer on transwell inserts containing 3-micron pores. Then, 10 μg/ml FITC-labeled dextran (3 kD) was added over the cells along with LPS. After 1 hour, media from below the transwell was analyzed by fluorometry for the presence of FITC. Results are shown as means ± SD in n = 3 independent experiments. *P < 0.05, compared with control by Student’s t test. E-cig = electronic-cigarette; ECIS = electrical cell impedance sensing; FITC = fluorescein isothiocyanate; RFU = relative fluorescent unit; si = silencing.
Lung tissue samples from the same donors were homogenized for analysis of protein expression by Western blotting. A representative blot is shown in Figure 1C and demonstrates that downregulation of mRNA translates to decreased protein expression.
Nicotine-Containing E-Cigarette Vapor Exposure in Mice Causes Knockdown of Mouse Lung WWOX Expression
C57Bl/6 mice were exposed to vehicle versus nicotine-containing e-cigarette vapor for 3 months. Lungs were harvested and examined by RT-PCR for levels of WWOX expression. Lungs from mice exposed to nicotine-containing e-cigarette vapor exhibited an average 57% decrease in WWOX expression compared with controls (P < 0.05) (Figure 1B). A potential concern regarding the validity of these results is that the RT-PCR method relies on relatively stable amounts of housekeeping gene expression between samples. Therefore, it is possible that in both the human smoker and the chronic e-cigarette exposure model, inflammatory cell accumulation might increase the relative amount of housekeeping gene expression in cell types that may not express similar levels of WWOX as the endogenous lung. To this end, mRNA levels of the endothelial-specific gene ACE-2 were measured in the same e-cigarette–exposed mouse lung samples and found to be stably expressed with no evidence of artificial downregulation (data not shown). Furthermore, WWOX expression has been noted in inflammatory cell types in addition to the major cell types of the lung (34), reducing the possibility that an influx of WWOX-deficient cell types artificially resulted in decreased WWOX expression as measured by RT-PCR.
Lung tissue samples from the same experimental groups were homogenized for analysis of protein expression by Western blotting. A representative blot is shown in Figure 1D and demonstrates that downregulation of mRNA translates to decreased protein expression.
WWOX-silenced Human Pulmonary Endothelial Cells Sustain Greater Barrier Dysfunction during LPS Exposure
Human lung microvascular endothelial cells (hLMVECs) were treated with scrambled, control versus WWOX-targeting siRNA, which resulted in efficient knockdown of WWOX expression (inset, Figure 1C). Cells were then grown to confluency on gold electrodes for ECIS assessments of TER in response to LPS. As shown in Figure 1C, treatment of WWOX-silenced hLMVECs with LPS resulted in larger decreases in TER compared with wild-type cells. In an earlier study (20), WWOX knockdown was associated with JNK-dependent proinflammatory signaling in alveolar epithelial cells. Therefore, WWOX-silenced hLMVECs were pretreated with the JNK inhibitor SP600125 before LPS exposure to determine whether a JNK-related pathway was involved in heightened barrier dysfunction related to WWOX knockdown in endothelial cells. JNK inhibition before LPS resulted in no significant rescue of barrier function toward that of wild-type cells (Figure 1C).
To confirm that the observed decreases in TER in control versus WWOX-silenced hLMVECs during LPS exposure correlated with increased permeability of the EC monolayer, transmonolayer FITC-dextran flux was measured. Control versus WWOX-silenced hLMVECs were grown to confluency on transwell inserts containing 3-μm pores and then treated with 10 μg/ml FITC-dextran (3 kD) added to the media overlying the monolayer. After exposure to 100 ng/ml LPS for 2 hours, media from the chamber underlying the transwell inserts was extracted for fluorometry. Knockdown of WWOX expression resulted in increased LPS-induced transmonolayer flux of FITC-dextran relative to wild-type monolayers (Figure 1D).
Generation of Endothelial-Specific WWOX KO Mice
C57BL/6-WWOXflox/flox mice were bred with C57BL/6-Cdh5-CreERT2 to generate tamoxifen-inducible EC-specific WWOX KO mice. Cre+ flox homozygotes were confirmed by PCR as in Ludes-Meyers and colleagues (24) (Figure 2A). Following tamoxifen treatment in mature animals to induce endothelial-specific WWOX knockdown, lungs were harvested for flow cytometric isolation of CD31+ CD45− endothelial cells (Figure 2B). Cells were propagated in culture and analyzed by RT-PCR (Figure 2C) and Western blotting (Figure 2D) to confirm WWOX knockdown.
Figure 2.
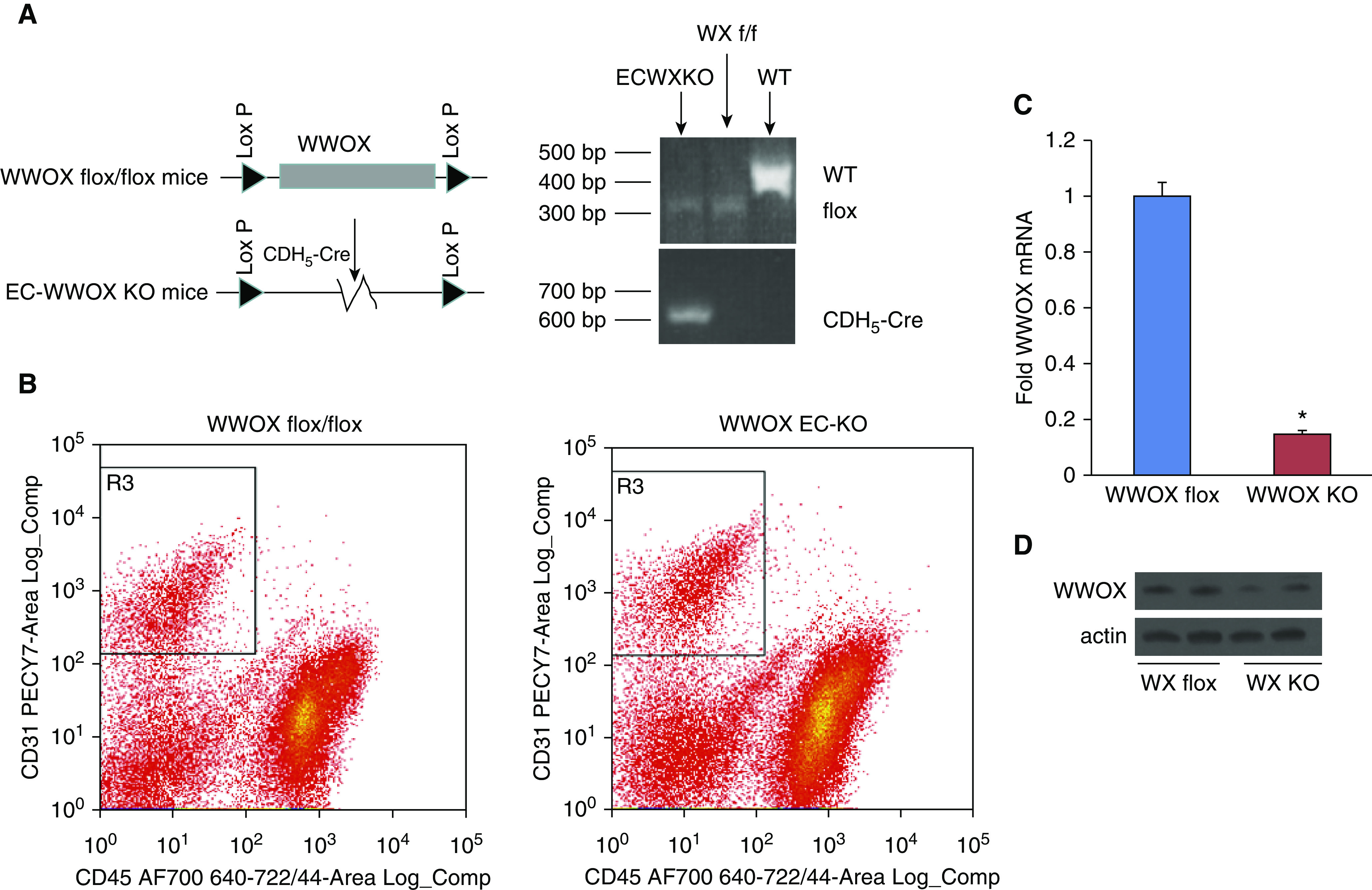
Generation of inducible endothelial-specific WWOX knockout (KO) mice. WWOX flox/flox mice were obtained from the laboratory of Dr. C. Marcelo Aldaz, M.D., Ph.D., of MD Anderson Cancer Center at the University of Texas. These were bred with mice containing a tamoxifen-inducible cre deleter under the control of the CDH5 promoter to obtain an inducible endothelial-specific WWOX KO. (A) Genotyping was performed as described in Ludes-Meyers and colleagues (24). (B) Lung endothelial cells were isolated from cell suspensions obtained from WWOX flox and KO mice using FACS to segregate CD45− CD31+ cells. These were grown in culture and analyzed for WWOX expression by RT-PCR (C) and Western blotting (D). Results are shown as means ± SD in n = 3 independent experiments. *P < 0.05, compared with control by Student’s t test. f/f = flox/flox; WX = WWOX; WT = wild-type.
EC-Specific WWOX KO Mice Exhibit Greater Amounts of Pulmonary Vascular Leak following Intratracheal LPS Instillation
A direct injury model of ARDS via intratracheal instillation of LPS was used to phenotype differences in vascular leak between EC WWOX KO mice and WWOX flox controls. Initially, C57BL/6-Cdh5-CreERT2 mice were compared with C57BL/6-WWOXflox/flox mice to determine whether endothelial expression of cre had any influence on endothelial cell permeability (35). No significant differences were noted, and thereafter, tamoxifen-treated C57BL/6-WWOXflox/flox mice were used as controls. As shown in Figure 3, BALF obtained from EC WWOX KO mice treated with LPS exhibited greater amounts of leukocyte numbers, protein concentration, and FITC-dextran flux from the vascular space compared with LPS-treated WWOX flox mice. Unlike the previously reported observations in whole-lung WWOX knockdown mice (20), there were no significant differences between untreated EC WWOX KO mice and controls. Increased vascular leak of cells and protein in LPS-treated EC WWOX KO mice was confirmed by histologic examination of H&E-stained lung sections (Figure 3D).
Figure 3.

EC-specific WWOX KO mice exhibit greater vascular leak during LPS-induced ARDS. Six WWOX flox (control) and six WWOX KO mice were treated with intratracheal instillation of either PBS or 1 mg/kg LPS (n = 3 per group). Eighteen hours later, all mice underwent BAL using 1 ml of PBS. Lungs were then harvested for Western blotting and histologic examination. (A, B, and D) Bar graphs depict BAL leukocyte counts, protein concentration, and FITC dextran flux as a measure of permeability. Results are shown as means ± SD in n = 3 independent experiments. *P < 0.05, compared with LPS-treated control by Student’s t test. (C) A representative hematoxylin and eosin–stained lung histologic section from each of the four experimental conditions is shown here.
Interestingly, although there was a trend toward increased concentrations of inflammatory cytokines in the BALF of EC WWOX KO mice treated with LPS compared with WWOX flox mice, none were statistically significant (Figure 4). This, along with the ECIS observations in Figure 1, which showed no rescue effect of JNK inhibition in the enhanced vascular leak noted in WWOX-silenced ECs, suggests that an inflammation-independent EC-specific mechanism underlies these phenomena.
Figure 4.
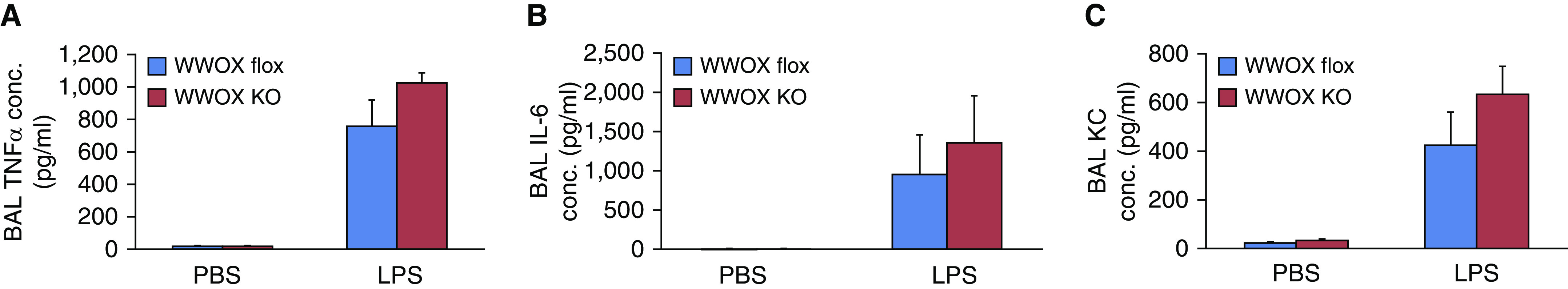
EC-specific WWOX KO mice do not exhibit greater BAL cytokines during LPS-induced ARDS. (A–C) Bar graphs depict BAL TNFα (A), IL-6 (B), and KC (C) concentrations. None of the increases in LPS-treated WWOX KO animals were statistically significantly different than those of LPS-treated controls. Results are shown as means ± SD in n = 3 independent experiments. KC = keratinocyte-derived chemokine. conc. = concentrations; KC = keratinocyte-derived chemokine.
EC-Specific WWOX KO Mice Injured in a Whole Bacterial MRSA Model of ARDS Exhibit Greater Amounts of Vascular Leak without Significant Increases in Inflammation Compared with WWOX Flox Mice
To further test the notion that enhanced vascular leak observed in EC WWOX KO mice is an inflammation-independent phenomenon, and to extend these observations to a more common clinical scenario, a whole bacterial model of ARDS induced by intratracheal instillation of heat-killed USA 300 MRSA was used to further phenotype these animals. As shown in Figures 5A and 5B, BALF from EC WWOX KO animals treated with MRSA exhibited greater amounts of leukocyte numbers and protein concentration compared with corresponding MRSA-treated WWOX flox mice. Histologic examination of H&E-stained lung sections confirmed increased vascular leak in EC WWOX KO mice (Figure 5C).
Figure 5.

EC-specific WWOX KO mice exhibit greater vascular leak during methicillin-resistant Staphylococcus aureus (MRSA)-induced ARDS. Six WWOX flox (control) and six WWOX KO mice were treated with intratracheal instillation of either PBS or 1 × 108 cfu of USA 300 MRSA (n = 3 per group). Eighteen hours later, all mice underwent BAL using 1 ml of PBS. Lungs were then harvested for Western blotting and histologic examination. (A and B) Bar graphs depict BAL leukocyte counts, and protein conc. as a measure of permeability. Results are shown as means ± SD in n = 3 independent experiments. *P < 0.05, compared with LPS-treated control by Student’s t test. (C) A representative hematoxylin and eosin–stained lung histologic section from each of the four experimental conditions is shown here. Scale bars, 200 μm. cfu = colony forming unit.
Interestingly, once again, although there was a trend toward increased proinflammatory cytokine (TNFα, IL-6, KC) secretion in BALF from MRSA-treated EC WWOX KO animals versus WWOX flox counterparts, these differences were not statistically significant (Figure 6).
Figure 6.
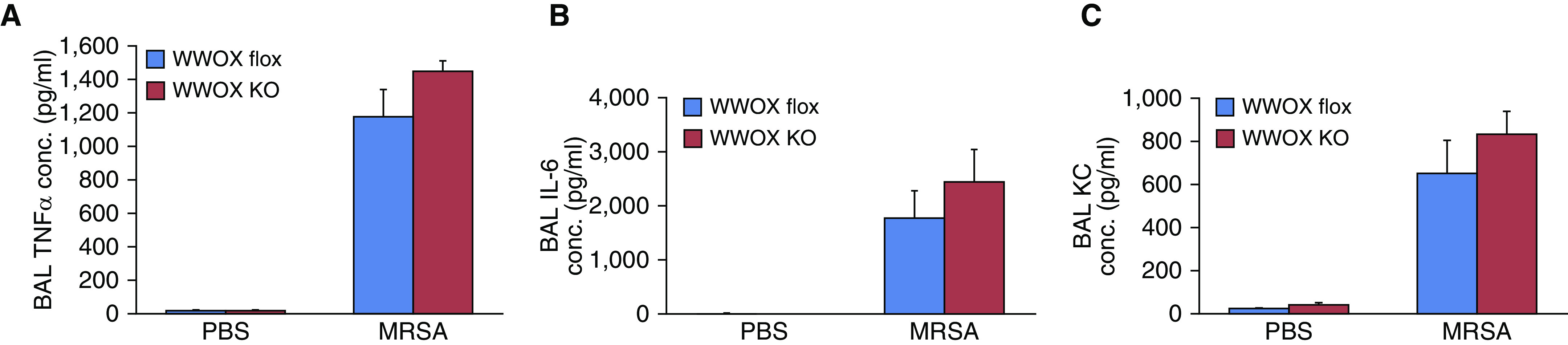
EC-specific WWOX KO mice do not exhibit greater BAL cytokines during MRSA-induced ARDS. (A–C) Bar graphs depict BAL TNFα (Α), IL-6 (B), and KC (C) concentrations. None of the increases in LPS-treated WWOX KO animals were statistically significantly different than those of LPS-treated controls. Results are shown as means ± SD in n = 3 independent experiments.
ECs Isolated from EC WWOX KO Mice Are More Susceptible to MRSA-induced Monolayer Barrier Disruption
ECs from WWOX KO and WWOX flox controls were isolated by flow cytometric sorting for CD31+ CD45− cells and grown in culture. They were then examined by ECIS measurement of TER and transwell FITC-dextran passage for their monolayer barrier properties during exposure to MRSA. WWOX KO ECs exhibited a greater decrease in TER during MRSA exposure compared with ECs from WWOX flox animals (Figure 7A). The correspondence of this observation to barrier disruption was confirmed by measurement of FITC-dextran passage across monolayers of ECs from WWOX KO mice versus controls in a transwell assay (Figure 7B).
Figure 7.
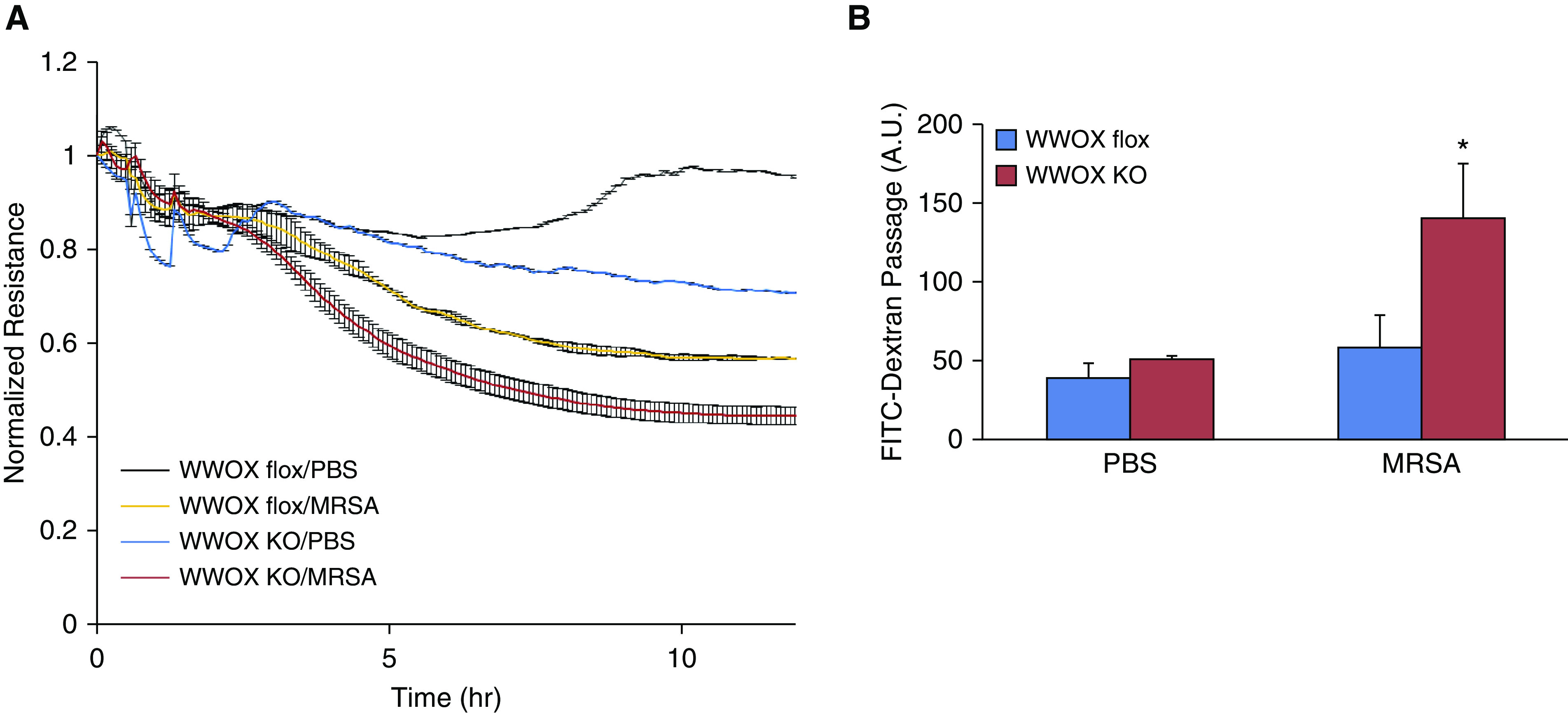
ECs isolated from EC WWOX KO mice are more susceptible to MRSA-induced monolayer barrier disruption. ECs from WWOX KO and WWOX flox controls were isolated by flow cytometric sorting for CD31+ CD45− cells and grown in culture. They were then examined by ECIS measurement of transendothelial resistance (TER) during exposure to heat-killed MRSA. (A) The scatter plot depicts changes in TER over the course of 10 hours following introduction of MRSA to the cell culture media. (B) In separate experiments, ECs from WWOX KO and WWOX flox controls were grown in a monolayer on transwell inserts containing 3-micron pores. Then, 10 μg/ml FITC-labeled dextran (3 kD) was added over the cells along with MRSA. After 3 hours, media from below the transwell was analyzed by fluorometry for the presence of FITC. Results are shown as means ± SD in n = 3 independent experiments. *P < 0.05, compared with MRSA-treated control by Student’s t test. A.U. = arbitrary unit.
Cell media supernatants from MRSA-stimulated ECs were examined for concentrations of secreted IL-6, KC, and MCP-1. These were all increased in the supernatants of WWOX KO cells compared with those from WWOX flox cells (Figure 8).
Figure 8.
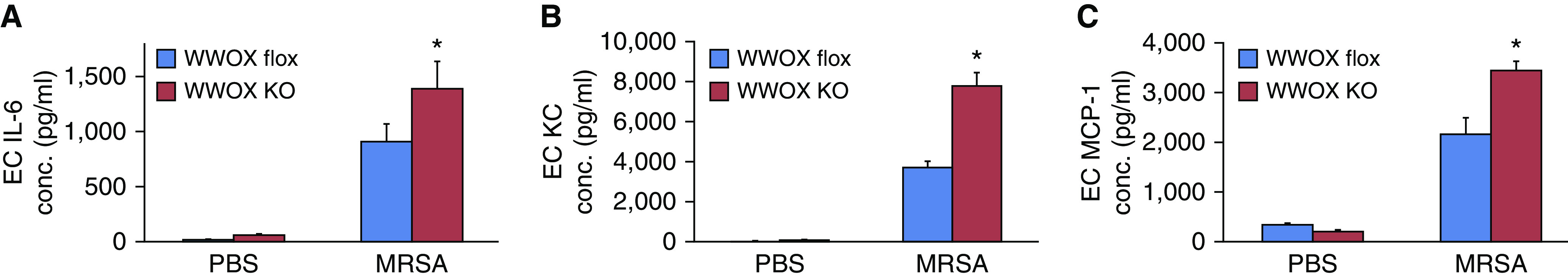
ECs isolated from EC WWOX KO mice produce more cytokines during MRSA exposure. (A–C) Cell media supernatants from MRSA-stimulated ECs were examined for concentrations of secreted IL-6 (A), KC (B), and MCP-1 (C). Bar graphs depict these results here. Results are shown as means ± SD in n = 3 independent experiments. *P < 0.05, compared with MRSA-treated control by Student’s t test. MPC-1 = monocyte chemoattractant protein-1.
Discussion
In the current study, it has been discovered that loss of lung WWOX expression has additional effects on lung disease susceptibility that were difficult to detect in prior global knockdown experiments that were associated with massive lung neutrophil influx (20). The current set of experiments focuses on lung vascular endothelium and suggests that CS/e-cigarette-related loss of WWOX expression in these lung cells may underlie the observed increase in susceptibility for severe ARDS seen epidemiologically in chronic smokers (2–4, 6).
Questions remain regarding both CS- and e-cigarette–mediated WWOX downregulation. The human lung samples used in this study were retained as part of the Beta agonist for Oxygenation in Lung Donors study published in 2014 (33). The clinical data obtained for that study included current versus former/never-smokers. Unfortunately, there are no additional data regarding the intensity or duration of current/prior smoking habits. Future work by our laboratory, focusing on the upcoming epidemic of e-cigarette–related lung disease, aims to define the minimum intensity and duration of chronic e-cigarette vapor exposure that would downregulate lung WWOX expression and therefore increase risk and severity of ARDS during conducive circumstances. The current study also does not pursue the mechanisms of CS-induced WWOX downregulation.
The mechanistic link between endothelial WWOX downregulation and enhanced LPS- or MRSA-induced vascular leak remains unknown. In the current study, inhibition of JNK did not appear to affect barrier susceptibility during WWOX knockdown (Figure 1), suggesting that c-Jun–related pathways are not a significant component of the underlying mechanism. The cell supernatant levels of inflammatory cytokines were elevated in MRSA-stimulated WWOX-deficient ECs compared with WWOX flox. Some of these may contribute to enhanced EC barrier disruption seen in WWOX-deficient ECs (36) via autocrine effects. However, interestingly, there were no significant differences in the BAL levels of permeability-promoting cytokines between WWOX flox and EC WWOX KO mice treated with MRSA. This observation suggests that additional inflammation-independent mechanisms exist by which EC WWOX deficiency may lead to increased vascular leak. It is possible that with greater numbers of mice and therefore greater statistical power, the increase in cytokines in the LPS/MRSA-treated EC WWOX KO mice relative to that of LPS/MRSA-treated controls will be found to be modestly greater with statistical significance. However, the key point is that at a power where BALF leukocytes are increased to a greater degree in LPS/MRSA-treated EC WWOX KO mice relative to the increase in LPS/MRSA-treated controls, the corresponding increase in inflammatory cytokines does not appear to be much different between these two equally stimulated groups. The key mechanism thought to account for this is augmented vascular leak via an EC-specific mechanism related to EC-specific WWOX knockdown.
The association between CS exposure and genotoxic stress at the chromosomal fragile site in which WWOX resides in the genome highlights the potential for chromosomal fragile sites to be examined in the context of lung disease. Nonrandom chromosomal aberrations are now known to play a significant role in the development of various human malignancies (37). Specific chromosomal locations in the human genome that are particularly susceptible to such structural alteration are called fragile sites (37). These have been further classified broadly into two categories. Those that are thought to be altered relatively rarely, and that have been observed in association with heritable disease, are termed rare chromosomal fragile sites, whereas those that are altered more frequently in conjunction with toxic environmental exposures are known as common chromosomal fragile sites (37). The mechanism of chromosomal fragility is thought to be due to structural factors that increase the tendency for gaps or constrictions to form at particular locations that may break during partial replication stress (37). Rare fragile sites occur only in select individuals and confer susceptibility to a particular heritable disease (37). The classic example of a rare fragile site is Fragile X syndrome, a heritable form of intellectual disability (37). Common chromosomal fragile sites, on the other hand, occur in nearly all individuals and are thought to accrue mutations somatically during genotoxic stress related to environmental exposures (37). Virtually all common fragile genes have been studied in the context of cancer, where a particular mutation at one of these sites would be perpetuated by additional oncogenic factors. The rate of accumulation in normal tissue is largely unknown. However, interestingly, in at least one study of WWOX expression in cancer, reduced WWOX expression was observed not only in the majority of cancer cells examined but also in about one-third of adjacent normal tissue (38).
CS has long been known to be one such genotoxic stress that increases the frequency of aberrations observed at common fragile sites (17). This association has been studied extensively in bone marrow and peripheral blood cells, where it is linked to the development of leukemias and lymphomas (17). However, the accumulation of aberrations at common fragile sites has not yet been studied in the lung, where environmental genotoxic stress may theoretically result in the greatest amount of accumulated aberrations over the life of an individual smoker.
Although the frequency of fragile site events or the rate of accumulation of chromosomal aberrations in the lungs of smokers is therefore largely unknown, it is interesting to note that WWOX, which resides at the second most active chromosomal fragile site in humans (39), is linked to signaling events relevant for a number of smoking-related lung diseases. WWOX is already known to interact with several molecules linked to the pathogenesis of lung disease including those in which pulmonary vascular leak is a pathophysiologic component. At least eight of the known binding partners of WWOX (39, 40) may connect it to non–cancer-related lung disease. These include NF-κB activating protein (41), ErbB4 (42), c-Jun (43–46), ezrin (47), Dvl2 (48), p53 (49, 50), and the R-SMADs (51, 52). Many of these molecules are already connected to the pathogenesis of lung diseases such as ARDS, asthma, COPD, pulmonary hypertension, and pulmonary fibrosis (53–63). Furthermore, WWOX polymorphisms have been associated with human COPD susceptibility and pulmonary function traits such as forced vital capacity (64–66).
The most critical inference that may be drawn from this study is with regard to the possible long-term effects of e-cigarette use. The mouse model uses a dosage and duration that is comparable to human exposures of up to 20 years. Lung WWOX expression was observed to decrease only in mice exposed to vapor containing nicotine. Therefore, this study potentially predicts that the consequences for chronic users of nicotine-containing e-cigarettes would include an increased risk of ARDS during lung infection as well as increased risk for the development of chronic lung diseases that are associated with neutrophilic inflammation. The development of a dual-hit e-cigarette exposure and MRSA-induced ARDS animal model is needed to confirm this association and to define the relative importance of WWOX downregulation versus other e-cigarette–induced gene expression changes in mediating these potential risks.
Footnotes
Supported by the services of the Cardiovascular Research Core at the University of Illinois in Chicago. Funding sources include the Natural National Science Foundation of China (81760351 and 81870011) (Z.Z) and the U.S. National Institutes of Health (HL126176 and HL103836) (L.B.W.); R01-HL-127342, R01-HL-133951, and 2R01 HL111656-06 (R.F.M.); and 1K08 HL140222-01A1 (S.S.).
Author Contributions: Conception and design: L.E.C.A., L.B.W., C.M.A., J.R.J., S.M.D., V.N., R.F.M., and S.S. Data acquisition: Z.Z., W.C., A.M., Z.S., and R.K. Analysis and interpretation: Z.Z., W.C., R.F.M., and S.S. Manuscript preparation: Z.Z., L.E.C.A., C.M.A., S.M.D., R.F.M., and S.S.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0145OC on October 15, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Jha P, Ramasundarahettige C, Landsman V, Rostron B, Thun M, Anderson RN, et al. 21st-century hazards of smoking and benefits of cessation in the United States. N Engl J Med. 2013;368:341–350. doi: 10.1056/NEJMsa1211128. [DOI] [PubMed] [Google Scholar]
- 2.Calfee CS, Matthay MA, Eisner MD, Benowitz N, Call M, Pittet JF, et al. Active and passive cigarette smoking and acute lung injury after severe blunt trauma. Am J Respir Crit Care Med. 2011;183:1660–1665. doi: 10.1164/rccm.201011-1802OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calfee CS, Matthay MA, Kangelaris KN, Siew ED, Janz DR, Bernard GR, et al. Cigarette smoke exposure and the acute respiratory distress syndrome. Crit Care Med. 2015;43:1790–1797. doi: 10.1097/CCM.0000000000001089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christenson JT, Aeberhard JM, Badel P, Pepcak F, Maurice J, Simonet F, et al. Adult respiratory distress syndrome after cardiac surgery. Cardiovasc Surg. 1996;4:15–21. doi: 10.1016/0967-2109(96)83778-1. [DOI] [PubMed] [Google Scholar]
- 5.Diamond JM, Lee JC, Kawut SM, Shah RJ, Localio AR, Bellamy SL, et al. Lung Transplant Outcomes Group. Clinical risk factors for primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med. 2013;187:527–534. doi: 10.1164/rccm.201210-1865OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toy P, Gajic O, Bacchetti P, Looney MR, Gropper MA, Hubmayr R, et al. TRALI Study Group. Transfusion-related acute lung injury: incidence and risk factors. Blood. 2012;119:1757–1767. doi: 10.1182/blood-2011-08-370932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 8.Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. ARDS Definition Task Force. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 9.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–327. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zimmerman JJ, Akhtar SR, Caldwell E, Rubenfeld GD. Incidence and outcomes of pediatric acute lung injury. Pediatrics. 2009;124:87–95. doi: 10.1542/peds.2007-2462. [DOI] [PubMed] [Google Scholar]
- 11.Layden JE, Ghinai I, Pray I, Kimball A, Layer M, Tenforde M, et al. Pulmonary illness related to e-cigarette use in Illinois and Wisconsin - preliminary report. N Engl J Med. 2020;382:903–916. doi: 10.1056/NEJMoa1911614. [DOI] [PubMed] [Google Scholar]
- 12.Blount BC, Karwowski MP, Morel-Espinosa M, Rees J, Sosnoff C, Cowan E, et al. Evaluation of bronchoalveolar lavage fluid from patients in an outbreak of e-cigarette, or vaping, product use-associated lung injury - 10 states, August-October 2019. MMWR Morb Mortal Wkly Rep. 2019;68:1040–1041. doi: 10.15585/mmwr.mm6845e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butt YM, Smith ML, Tazelaar HD, Vaszar LT, Swanson KL, Cecchini MJ, et al. Pathology of vaping-associated lung injury. N Engl J Med. 2019;381:1780–1781. doi: 10.1056/NEJMc1913069. [DOI] [PubMed] [Google Scholar]
- 14.Hajek P, Phillips-Waller A, Przulj D, Pesola F, Myers Smith K, Bisal N, et al. A randomized trial of e-cigarettes versus nicotine-replacement therapy. N Engl J Med. 2019;380:629–637. doi: 10.1056/NEJMoa1808779. [DOI] [PubMed] [Google Scholar]
- 15.Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–447. doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ban S, Cologne JB, Neriishi K. Effect of radiation and cigarette smoking on expression of FUdR-inducible common fragile sites in human peripheral lymphocytes. Mutat Res. 1995;334:197–203. doi: 10.1016/0165-1161(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 17.Kao-Shan CS, Fine RL, Whang-Peng J, Lee EC, Chabner BA. Increased fragile sites and sister chromatid exchanges in bone marrow and peripheral blood of young cigarette smokers. Cancer Res. 1987;47:6278–6282. [PubMed] [Google Scholar]
- 18.Thavathiru E, Ludes-Meyers JH, MacLeod MC, Aldaz CM. Expression of common chromosomal fragile site genes, WWOX/FRA16D and FHIT/FRA3B is downregulated by exposure to environmental carcinogens, UV, and BPDE but not by IR. Mol Carcinog. 2005;44:174–182. doi: 10.1002/mc.20122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang W, Cui S, Ma J, Lu Q, Kong C, Liu T, et al. Cigarette smoking extract causes hypermethylation and inactivation of WWOX gene in T-24 human bladder cancer cells. Neoplasma. 2012;59:216–223. doi: 10.4149/neo_2012_028. [DOI] [PubMed] [Google Scholar]
- 20.Singla S, Chen J, Sethuraman S, Sysol JR, Gampa A, Zhao S, et al. Loss of lung WWOX expression causes neutrophilic inflammation. Am J Physiol Lung Cell Mol Physiol. 2017;312:L903–L911. doi: 10.1152/ajplung.00034.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Q, Gottlieb E, Rounds S. Effects of cigarette smoke on pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2018;314:L743–L756. doi: 10.1152/ajplung.00373.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawasaki T, Chen W, Htwe YM, Tatsumi K, Dudek SM. DPP4 inhibition by sitagliptin attenuates LPS-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2018;315:L834–L845. doi: 10.1152/ajplung.00031.2018. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Feng G, Guo Q, Wardenburg JB, Lin S, Inoshima I, et al. Transcriptional events during the recovery from MRSA lung infection: a mouse pneumonia model. PLoS One. 2013;8:e70176. doi: 10.1371/journal.pone.0070176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ludes-Meyers JH, Kil H, Parker-Thornburg J, Kusewitt DF, Bedford MT, Aldaz CM. Generation and characterization of mice carrying a conditional allele of the Wwox tumor suppressor gene. PLoS One. 2009;4:e7775. doi: 10.1371/journal.pone.0007775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crotty Alexander LE, Drummond CA, Hepokoski M, Mathew D, Moshensky A, Willeford A, et al. Chronic inhalation of e-cigarette vapor containing nicotine disrupts airway barrier function and induces systemic inflammation and multiorgan fibrosis in mice. Am J Physiol Regul Integr Comp Physiol. 2018;314:R834–R847. doi: 10.1152/ajpregu.00270.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grommes J, Vijayan S, Drechsler M, Hartwig H, Mörgelin M, Dembinski R, et al. Simvastatin reduces endotoxin-induced acute lung injury by decreasing neutrophil recruitment and radical formation. PLoS One. 2012;7:e38917. doi: 10.1371/journal.pone.0038917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia JG, Schaphorst KL, Shi S, Verin AD, Hart CM, Callahan KS, et al. Mechanisms of ionomycin-induced endothelial cell barrier dysfunction. Am J Physiol. 1997;273:L172–L184. doi: 10.1152/ajplung.1997.273.1.L172. [DOI] [PubMed] [Google Scholar]
- 28.Giaever I, Keese CR. A morphological biosensor for mammalian cells. Nature. 1993;366:591–592. doi: 10.1038/366591a0. [DOI] [PubMed] [Google Scholar]
- 29.Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci USA. 1992;89:7919–7923. doi: 10.1073/pnas.89.17.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia JG, Schaphorst KL, Verin AD, Vepa S, Patterson CE, Natarajan V. Diperoxovanadate alters endothelial cell focal contacts and barrier function: role of tyrosine phosphorylation. J Appl Physiol (1985) 2000;89:2333–2343. doi: 10.1152/jappl.2000.89.6.2333. [DOI] [PubMed] [Google Scholar]
- 31.Martins-Green M, Petreaca M, Yao M. An assay system for in vitro detection of permeability in human “endothelium”. Methods Enzymol. 2008;443:137–153. doi: 10.1016/S0076-6879(08)02008-9. [DOI] [PubMed] [Google Scholar]
- 32.Wolfson RK, Chiang ET, Garcia JG. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res. 2011;81:189–197. doi: 10.1016/j.mvr.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ware LB, Landeck M, Koyama T, Zhao Z, Singer J, Kern R, et al. California Transplant Donor Network. A randomized trial of the effects of nebulized albuterol on pulmonary edema in brain-dead organ donors. Am J Transplant. 2014;14:621–628. doi: 10.1111/ajt.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sbrana I, Veroni F, Nieri M, Puliti A, Barale R. Chromosomal fragile sites FRA3B and FRA16D show correlated expression and association with failure of apoptosis in lymphocytes from patients with thyroid cancer. Genes Chromosomes Cancer. 2006;45:429–436. doi: 10.1002/gcc.20305. [DOI] [PubMed] [Google Scholar]
- 35.Perl AK, Zhang L, Whitsett JA. Conditional expression of genes in the respiratory epithelium in transgenic mice: cautionary notes and toward building a better mouse trap. Am J Respir Cell Mol Biol. 2009;40:1–3. doi: 10.1165/rcmb.2008-0011ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sedgwick JB, Menon I, Gern JE, Busse WW. Effects of inflammatory cytokines on the permeability of human lung microvascular endothelial cell monolayers and differential eosinophil transmigration. J Allergy Clin Immunol. 2002;110:752–756. doi: 10.1067/mai.2002.128581. [DOI] [PubMed] [Google Scholar]
- 37.Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 38.Guler G, Uner A, Guler N, Han SY, Iliopoulos D, Hauck WW, et al. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer. 2004;100:1605–1614. doi: 10.1002/cncr.20137. [DOI] [PubMed] [Google Scholar]
- 39.Del Mare S, Salah Z, Aqeilan RI. WWOX: its genomics, partners, and functions. J Cell Biochem. 2009;108:737–745. doi: 10.1002/jcb.22298. [DOI] [PubMed] [Google Scholar]
- 40.Aldaz CM, Ferguson BW, Abba MC. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim Biophys Acta. 2014;1846:188–200. doi: 10.1016/j.bbcan.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Everhart MB, Han W, Sherrill TP, Arutiunov M, Polosukhin VV, Burke JR, et al. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury. J Immunol. 2006;176:4995–5005. doi: 10.4049/jimmunol.176.8.4995. [DOI] [PubMed] [Google Scholar]
- 42.Schmiedl A, Behrens J, Zscheppang K, Purevdorj E, von Mayersbach D, Liese A, et al. Lipopolysaccharide-induced injury is more pronounced in fetal transgenic ErbB4-deleted lungs. Am J Physiol Lung Cell Mol Physiol. 2011;301:L490–L499. doi: 10.1152/ajplung.00131.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arndt PG, Young SK, Lieber JG, Fessler MB, Nick JA, Worthen GS. Inhibition of c-Jun N-terminal kinase limits lipopolysaccharide-induced pulmonary neutrophil influx. Am J Respir Crit Care Med. 2005;171:978–986. doi: 10.1164/rccm.200406-712OC. [DOI] [PubMed] [Google Scholar]
- 44.Guo RF, Lentsch AB, Sarma JV, Sun L, Riedemann NC, McClintock SD, et al. Activator protein-1 activation in acute lung injury. Am J Pathol. 2002;161:275–282. doi: 10.1016/S0002-9440(10)64179-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee HS, Kim HJ, Moon CS, Chong YH, Kang JL. Inhibition of c-Jun NH2-terminal kinase or extracellular signal-regulated kinase improves lung injury. Respir Res. 2004;5:23. doi: 10.1186/1465-9921-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu M, Shi L, Chen M, Chen S, Zou X. Effects of c-Jun N-terminal kinase signaling pathway on severe acute pancreatitis-associated lung injury. Pancreas. 2012;41:358–366. doi: 10.1097/MPA.0b013e3182297f09. [DOI] [PubMed] [Google Scholar]
- 47.Adyshev DM, Dudek SM, Moldobaeva N, Kim KM, Ma SF, Kasa A, et al. Ezrin/radixin/moesin proteins differentially regulate endothelial hyperpermeability after thrombin. Am J Physiol Lung Cell Mol Physiol. 2013;305:L240–L255. doi: 10.1152/ajplung.00355.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pongracz JE, Stockley RA. Wnt signalling in lung development and diseases. Respir Res. 2006;7:15. doi: 10.1186/1465-9921-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barabutis N, Dimitropoulou C, Birmpas C, Joshi A, Thangjam G, Catravas JD. p53 protects against LPS-induced lung endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2015;308:L776–L787. doi: 10.1152/ajplung.00334.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu G, Park YJ, Tsuruta Y, Lorne E, Abraham E. p53 attenuates lipopolysaccharide-induced NF-kappaB activation and acute lung injury. J Immunol. 2009;182:5063–5071. doi: 10.4049/jimmunol.0803526. [DOI] [PubMed] [Google Scholar]
- 51.Akbarshahi H, Sam A, Chen C, Rosendahl AH, Andersson R. Early activation of pulmonary TGF-β1/Smad2 signaling in mice with acute pancreatitis-associated acute lung injury. Mediators Inflamm. 2014;2014:148029. doi: 10.1155/2014/148029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li LF, Lee CS, Liu YY, Chang CH, Lin CW, Chiu LC, et al. Activation of Src-dependent Smad3 signaling mediates the neutrophilic inflammation and oxidative stress in hyperoxia-augmented ventilator-induced lung injury. Respir Res. 2015;16:112. doi: 10.1186/s12931-015-0275-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett BL. c-Jun N-terminal kinase-dependent mechanisms in respiratory disease. Eur Respir J. 2006;28:651–661. doi: 10.1183/09031936.06.00012106. [DOI] [PubMed] [Google Scholar]
- 54.Hosokawa S, Haraguchi G, Sasaki A, Arai H, Muto S, Itai A, et al. Pathophysiological roles of nuclear factor kappaB (NF-kB) in pulmonary arterial hypertension: effects of synthetic selective NF-kB inhibitor IMD-0354. Cardiovasc Res. 2013;99:35–43. doi: 10.1093/cvr/cvt105. [DOI] [PubMed] [Google Scholar]
- 55.Li L, Wei C, Kim IK, Janssen-Heininger Y, Gupta S. Inhibition of nuclear factor-κB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension. 2014;63:1260–1269. doi: 10.1161/HYPERTENSIONAHA.114.03220. [DOI] [PubMed] [Google Scholar]
- 56.Malenfant S, Neyron AS, Paulin R, Potus F, Meloche J, Provencher S, et al. Signal transduction in the development of pulmonary arterial hypertension. Pulm Circ. 2013;3:278–293. doi: 10.4103/2045-8932.114752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Jesus Perez V, Yuan K, Alastalo TP, Spiekerkoetter E, Rabinovitch M. Targeting the Wnt signaling pathways in pulmonary arterial hypertension. Drug Discov Today. 2014;19:1270–1276. doi: 10.1016/j.drudis.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1) Suppl:S20–S31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonner JC. Mesenchymal cell survival in airway and interstitial pulmonary fibrosis. Fibrogenesis Tissue Repair. 2010;3:15. doi: 10.1186/1755-1536-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fujimoto H, D’Alessandro-Gabazza CN, Palanki MS, Erdman PE, Takagi T, Gabazza EC, et al. Inhibition of nuclear factor-kappaB in T cells suppresses lung fibrosis. Am J Respir Crit Care Med. 2007;176:1251–1260. doi: 10.1164/rccm.200609-1288OC. [DOI] [PubMed] [Google Scholar]
- 61.Morrisey EE. Wnt signaling and pulmonary fibrosis. Am J Pathol. 2003;162:1393–1397. doi: 10.1016/S0002-9440(10)64271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhandary YP, Shetty SK, Marudamuthu AS, Ji HL, Neuenschwander PF, Boggaram V, et al. Regulation of lung injury and fibrosis by p53-mediated changes in urokinase and plasminogen activator inhibitor-1. Am J Pathol. 2013;183:131–143. doi: 10.1016/j.ajpath.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gauldie J, Kolb M, Ask K, Martin G, Bonniaud P, Warburton D. Smad3 signaling involved in pulmonary fibrosis and emphysema. Proc Am Thorac Soc. 2006;3:696–702. doi: 10.1513/pats.200605-125SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loth DW, Soler Artigas M, Gharib SA, Wain LV, Franceschini N, Koch B, et al. Genome-wide association analysis identifies six new loci associated with forced vital capacity. Nat Genet. 2014;46:669–677. doi: 10.1038/ng.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xie C, Chen X, Qiu F, Zhang L, Wu D, Chen J, et al. The role of WWOX polymorphisms on COPD susceptibility and pulmonary function traits in Chinese: a case-control study and family-based analysis. Sci Rep. 2016;6:21716. doi: 10.1038/srep21716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang L, Qiu F, Fang W, Zhang L, Xie C, Lu X, et al. The functional copy number variation-67048 in WWOX contributes to increased risk of COPD in southern and eastern Chinese. COPD. 2015;12:494–501. doi: 10.3109/15412555.2014.948993. [DOI] [PubMed] [Google Scholar]