

Abstract
Augmented glycolysis due to metabolic reprogramming in lung myofibroblasts is critical to their profibrotic phenotype. The primary glycolysis byproduct, lactate, is also secreted into the extracellular milieu, together with which myofibroblasts and macrophages form a spatially restricted site usually described as fibrotic niche. Therefore, we hypothesized that myofibroblast glycolysis might have a non–cell autonomous effect through lactate regulating the pathogenic phenotype of alveolar macrophages. Here, we demonstrated that there was a markedly increased lactate in the conditioned media of TGF-β1 (transforming growth factor-β1)–induced lung myofibroblasts and in the BAL fluids (BALFs) from mice with TGF-β1– or bleomycin-induced lung fibrosis. Importantly, the media and BALFs promoted profibrotic mediator expression in macrophages. Mechanistically, lactate induced histone lactylation in the promoters of the profibrotic genes in macrophages, consistent with the upregulation of this epigenetic modification in these cells in the fibrotic lungs. The lactate inductions of the histone lactylation and profibrotic gene expression were mediated by p300, as evidenced by their diminished concentrations in p300-knockdown macrophages. Collectively, our study establishes that in addition to protein, lipid, and nucleic acid molecules, a metabolite can also mediate intercellular regulations in the setting of lung fibrosis. Our findings shed new light on the mechanism underlying the key contribution of myofibroblast glycolysis to the pathogenesis of lung fibrosis.
Keywords: pulmonary fibrosis, myofibroblast, macrophage, lactate, histone lactylation
Lung fibrosis is a typical pathological feature of a large group of diseases known as interstitial lung disease (ILD) (1, 2). This pathology is characterized by a progressive and nonrelenting injury repair process, which leads to normal lung architecture destruction, lung scarring, and ultimate organ failure (1, 2). Idiopathic pulmonary fibrosis (IPF) is the most severe form of ILD and has an average life expectancy of 3–5 years after diagnosis (1). Currently, there are only two U.S. Food and Drug Administration–approved drugs to treat IPF (nintedanib and pirfenidone). However, the two medications do not cure the disease but just modestly slow down the decline of lung functions (1, 3, 4).
Myofibroblasts are widely known as one of the most important cells that are critically involved in the pathogenesis of IPF (5, 6). These cells are the primary effectors that produce excessive collagens and other extracellular matrix proteins in the fibrotic lungs (5, 6). Myofibroblasts have also been shown to secrete or release a variety of protein, lipid, and nucleic acid molecules that contribute to the pathological phenotype of other types of cells in the fibrotic lungs (2, 5, 6). Not surprisingly, lung myofibroblasts are the most targeted cells for developing novel therapeutics to treat this disease (5, 6). In addition to myofibroblasts, alveolar macrophages have also been found to play a critical role in the progression of lung fibrosis (7–11). These cells demonstrate a profibrotic phenotype in the diseased lungs largely by producing a number of soluble factors, such as TGF-β (transforming growth factor-β), PDGF (platelet-derived growth factor), VEGF (vascular endothelial growth factor,) and THBS1 (thrombospondin 1), which can differentiate resident fibroblasts into myofibroblasts (10–13). Downregulating the profibrotic activity of alveolar macrophages or depleting this group of cells is effective in treating experimental lung fibrosis (13–15).
Recently, the fibrotic niches have been gaining much attention as a primary site of active pathogenesis in the lungs (16–18). A niche is described as a spatially restricted multicellular structure, which different types of cells are in contact with or in close proximity to (16–18). These cells have been found to conduct intercellular communications via cell contacts and/or paracrine actions. The fibrotic niches in the lung consist of at least myofibroblasts, macrophages, and alveolar epithelial cells (16–18), suggesting that each of these cells is subject to multilateral effects.
Metabolic dysregulation has been increasingly recognized to play an important role in the pathogenesis of many pulmonary disorders, such as lung fibrosis, pulmonary hypertension, and chronic obstructive pulmonary disease (19–22). Previously, we and others demonstrated that lung myofibroblasts displayed augmented aerobic glycolysis (23–27). We showed that the increased glycolysis promoted myofibroblast differentiation (23). The delineation of the cell autonomous effects of glycolysis is critical to the understanding of the therapeutic efficacy of glycolytic inhibition in treating experimental lung fibrosis (23, 25). However, we speculated that the glycolytic escalation in the lung myofibroblasts might have some pathological effects that were beyond the cell boundaries because, first, the byproduct of glycolysis, lactate, can be secreted into the extracellular environment (19). Second, lung myofibroblasts are frequently localized at fibrotic niches with alveolar epithelial cells and macrophages (16–18). In this study, we strived to answer the question of whether the augmented glycolysis in lung myofibroblasts has a non–cell autonomous role that may contribute to the pathogenesis of pulmonary fibrosis.
Methods
Reagents
L-(+)-lactic acid was from Sigma-Aldrich. Human recombinant TGF-β1 was from Peprotech. HiPerFect Transfection Reagent and RNA isolation kit RNeasy Mini were from Qiagen. Bleomycin was from Hospira.
Experimental Pulmonary Fibrosis Model
Two-month old male C57BL/6 mice were purchased from the Jackson Laboratory. The mouse lung fibrosis model was established by intratracheal instillation of bleomycin or adenovirus expressing active TGF-β1, as previously detailed (28, 29). All animal experiment protocols were approved by the University of Alabama at Birmingham institutional animal care and use committee.
Human Lung Tissues
IPF and failed donor lung tissues were obtained from the University of Alabama Birmingham Tissue Procurement and Cell Culture Core. The protocol was approved by the University of Alabama at Birmingham institutional review board.
Establishment of Mouse Bone Marrow–derived Macrophages
Mouse bone marrow–derived macrophages (BMDMs) were derived from bone marrow cells of C57BL/6 mice as previously described (30). Briefly, after erythrocyte lysis, bone marrow cells were cultured in Dulbecco’s modified Eagle medium containing 10% FBS and 50 ng/ml murine M-CSF (macrophage colony-stimulating factor) (R&D Systems) for 5–6 days. The differentiated cells were then split and plated for the following experiments.
Isolation of Primary Mouse Alveolar Macrophages
Mouse alveolar macrophages were purified as previously described (31). Briefly, cells were recovered from BAL fluid (BALF). After red blood cell lysis, BAL cells were plated for 1 hour, which was followed by an extensive wash to remove unattached cells. The attached cells were used as alveolar macrophages.
Fibroblast- and Myofibroblast-conditioned Media Preparation
Human fibroblast line MRC5 cells (American Type Culture Collection) were treated with or without 2 ng/ml TGF-β1 for 48 hours. The cells were washed three times and cultured in serum-free Opti-MEM for another 2 days. The cell culture media were centrifuged, filtered through a 0.2-μm pore size syringe filter, and stored for the following experiments.
Fluorescent Immunohistochemistry and Immunocytochemistry
Immunofluorescence stainings for paraffin sections of human or mouse lungs or cells cultured on coverslips were performed as previously described (32). Rabbit anti–lactyl-lysine antibody (PTM-1401) was from PTM Biolabs. Rat anti-F4/80 antibody (clone BM8) was from eBioscience. Mouse anti-CD68 antibody (66231–2-Ig) was from ProteinTech. Alexa Fluor 488– and Alexa Fluor 594–conjugated secondary antibodies were from Thermo Fisher Scientific.
Real-Time PCR
mRNA concentrations were determined by real-time PCR using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). Primer sequences are listed in Table E1 in the data supplement. To calculate the fold change in the expression of these genes, ΔCt (cycle threshold) = Ct of tubulin − Ct of individual genes was first obtained. ΔΔCt = ΔCt of treated groups − ΔCt of untreated control groups was then obtained. Fold change was calculated as 2ΔΔCt, with control groups as one fold.
Western Blotting
Western blotting was performed as previously described (29). Rabbit anti–lactyl-lysine antibody (PTM-1401) was from PTM Biolabs. Rabbit anti–histone H3 (4499) and rabbit anti-p300 (70088) antibodies were from Cell Signaling Technology. Mouse anti–α-tubulin (T5168) and anti–β-actin (A2228) antibodies were from Sigma-Aldrich.
siRNA Transfection
siRNA transfection was performed using HiPerFect reagents (Qiagen) according to the manufacturer’s instructions. ON-TARGETplus negative control siRNA and specific mouse p300 siRNA pool were from Dharmacon.
Lactate Assay
The intracellular and extracellular concentrations of lactate were determined using the Lactate Colorimetric/Fluorometric Assay Kit (K607–100; BioVision) according to the manufacturer’s instructions.
Chromatin IP Assay
Chromatin IP assays were performed as previously described (30). Briefly, BMDMs were fixed with 1% formaldehyde in PBS for 10 minutes and collected in radioimmunoprecipitation assay (RIPA) buffer. Genomic DNA was then sheared by sonication to lengths of approximately 200–500 bp. The IP was performed with anti–lactyl-lysine antibody (PTM-1401; PTM Biolabs) and Dynabeads Protein G (Invitrogen). Genomic DNA in the immunocomplexes was purified using Qiagen miniprep columns (Qiagen). Primer sequences to amplify the proximal promoter regions of mouse genes are listed in Table E2.
Statistics
One-way ANOVA followed by the Bonferroni test was used for multiple group comparisons. The two-tailed Student’s t test was used for comparison between two groups. P < 0.05 was considered statistically significant.
Results
Increased Glycolysis Is Characteristic of Lung Fibrosis
In accordance with the previous finding that there was aberrant glycolysis in the fibrotic lungs (20, 23), here, we observed a marked increase in lactate concentrations in the BALFs from mice with bleomycin-induced lung fibrosis compared with those from mice that were treated with saline (Figure 1A). The lactate buildup in the BALFs was likely due to enhanced cellular glycolysis because the lactate concentration in the lung extracts from bleomycin-treated mice was significantly higher than that from the control animals (Figure 1B). The elevated lactate was also observed in the BALFs and lung extracts from mice with TGF-β1–induced pulmonary fibrosis (Figures 1C and 1D). Congruent with the augmented glycolysis in the fibrotic lungs, there was considerably increased lactate in the supernatants of TGF-β1–induced lung myofibroblasts (Figure 1E). Taken together, these data suggest that the enhanced glycolysis in the fibrotic lungs is attributable, at least partially, to the glycolytic myofibroblasts. The above findings also indicate that the extracellular milieu in the fibrotic lung is enriched with the key byproduct of glycolysis (i.e., lactate).
Figure 1.

Increased glycolysis is characteristic of lung fibrosis. (A and B) Eight-week old C57BL/6 mice were i.t. instilled with saline or bleomycin (1.5 U/kg in 50 μl saline). Three weeks after treatment, mice were killed, and their BAL fluids (BALFs) and lungs were harvested. Lactate concentrations in the BALFs and lungs were determined (n = 5 mice for each group; mean ± SEM). **P < 0.01 and ***P < 0.001 by two-tailed Student’s t test. (C and D) C57BL/6 mice were i.t. instilled with control adenovirus or adenovirus expressing active TGF-β1 (transforming growth factor-β1) (1 × 1011 pfu/kg body weight). Two weeks after treatment, mice were killed, and lactate concentrations in the BALFs and lungs were determined (n = 3–5 mice for each group; mean ± SEM). **P < 0.01 and ***P < 0.001 by two-tailed Student’s t test. (E) Human lung fibroblast MRC5 cells were treated with or without TGF-β1 (2 ng/ml) for 48 hours. Lactate concentrations in the culture media were determined (n = 3; mean ± SD). ***P < 0.001 by two-tailed Student’s t test. Ad-con = control adenovirus; Ad-TGF-β1 = adenovirus expressing active TGF-β1; BLM = bleomycin; pfu = plaque-forming unit.
Alveolar Macrophages in Fibrotic Lungs Demonstrate A Profibrotic Phenotype
In the search for the non–cell autonomous pathological effect of glycolysis in fibrotic lungs, we set our sight first on alveolar macrophages because these cells are in close proximity to the glycolytic myofibroblasts in the fibrotic niches (16, 17, 33). Furthermore, alveolar macrophages have been well recognized to play a crucial role in the pathogenesis of lung fibrosis, mostly through their profibrotic activity in the diseased lungs (10–13). We and others previously confirmed the profibrotic phenotype of these cells (30, 31, 34, 35). We show here that a number of genes identified by our earlier RNA-sequencing analysis (GSE98468) (31) and known to have important contributions to this pathology, such as ARG1, OPN, PDGFA, THBS1, and VEGFA (34, 36, 37), demonstrated significantly greater expression in the alveolar macrophages from the lungs of mice with both bleomycin- and TGF-β1–induced pulmonary fibrosis compared with those from control animals (Figures 2A and 2B).
Figure 2.

Alveolar macrophages in fibrotic lungs demonstrate profibrotic phenotype. (A) C57BL/6 mice were i.t. instilled with saline or BLM. Three weeks after treatment, BALFs were harvested, and alveolar macrophages were isolated. Total RNAs were purified, and concentrations of the indicated genes were determined by real-time PCR (n = 3 mice for each group; mean ± SEM). *P < 0.05 and **P < 0.01 by two-tailed Student’s t test. (B) C57BL/6 mice were i.t. instilled with Ad-con or Ad-TGF-β1. Two weeks after treatment, alveolar macrophages were isolated, and total RNAs were purified. Concentrations of the indicated genes were determined by real-time PCR (n = 4 mice for each group; mean ± SEM). *P < 0.05 and **P < 0.01 by two-tailed Student’s t test. ARG1 = arginase 1; OPN = osteopontin; PDGFA = platelet-derived growth factor A; THBS1 = thrombospondin 1; VEGFA = vascular endothelial growth factor A.
Lung Myofibroblast–conditioned Media and BALFs from Mice with Lung Fibrosis Promote Macrophage Profibrotic Phenotype
Despite the overwhelming evidence revealing the profibrotic activities of alveolar macrophages in lung fibrosis (13, 34, 35), it is less understood how these cells acquire the profibrotic phenotype. As there has been an increasing appreciation that the phenotype and function of macrophages are largely dictated by the distinct tissue environment that they inhabit (13), we reasoned that the local buildup of lactate that resulted from the augmented glycolysis in fibrotic lungs might contribute to the profibrotic activity of these cells. To test this concept, we first treated macrophages in vitro with conditioned media from TGF-β1–induced myofibroblasts. As shown in Figure 3A, cells treated with myofibroblast-conditioned media demonstrated markedly increased expression of a selected group of the profibrotic mediators that were identified previously by us and others (30, 31, 34, 35). We then treated macrophages with BALFs from mice with or without bleomycin-induced lung fibrosis and found that BALFs from fibrotic lungs also induced the expression of these mediators, although to a lesser extent than the lung myofibroblast–conditioned media (Figure 3B).
Figure 3.

Lung myofibroblast–conditioned media, BALFs from mice with lung fibrosis, or lactate promote macrophage profibrotic phenotype. (A) Bone marrow–derived macrophages (BMDMs) were treated with conditioned media from normal fibroblasts or TGF-β1–induced myofibroblasts. Twenty-four hours after treatment, cells were harvested, and concentrations of the indicated genes were determined by real-time PCR (n = 4; mean ± SD). **P < 0.01 and ***P < 0.001 by two-tailed Student’s t test. (B) BMDMs were treated with BALFs from mice that were treated i.t. with saline or BLM for 3 weeks. Twenty-four hours after treatment, concentrations of the indicated genes were determined by real-time PCR. n = 3; mean ± SD. **P < 0.01 by two-tailed Student’s t test. (C) BMDMs were treated with or without 20 mM lactate for 24 hours. Cells were harvested, and concentrations of the indicated genes were determined by real-time PCR (n = 4; mean ± SD). ***P < 0.001 by two-tailed Student’s t test. (D) Primary alveolar macrophages) were isolated from BALFs of C57BL/6 mice and treated with 20 mM lactate for 24 hours. Concentrations of the indicated genes were determined by real-time PCR (n = 3; mean ± SD). *P < 0.05 and ***P < 0.001 by two-tailed Student’s t test. AM = alveolar macrophage; con = control; Fbs C.M. = fibroblast-conditioned media; MyoFbs C.M. = myofibroblast-conditioned media.
Lactate Induces Profibrotic Phenotype in Macrophage
Given that there was increased lactate in the conditioned media from lung myofibroblasts and BALFs from the fibrotic mouse lungs, we speculated that it might be lactate that induced the profibrotic activation of macrophages. To test this hypothesis, we directly treated BMDMs with lactate at a physiologically relevant concentration and found that lactate was potent in stimulating the macrophage profibrotic phenotype (Figure 3C). Lactate also promoted the expression of profibrotic mediators in naive alveolar macrophages (Figure 3D). Collectively, these findings suggest that lactate derived from the augmented glycolysis in fibrotic lungs plays an important role in the regulation of the profibrotic activity of alveolar macrophages in the fibrotic lungs.
Lactate Induces Histone Lactylation in Macrophages
Epigenetic modification is well known to be one of the most important regulatory mechanisms that control macrophage phenotype. Lactylation, which occurs at histone lysine residues, is a recently discovered epigenetic modification (38, 39). Histone lactylation has also been found to be associated with transcriptional activation (38, 39). Given that lactate promoted the expression of the profibrotic mediators in macrophages, we hypothesized that this was achieved by lactate-induced histone lactylation at the promoter regions of these profibrotic genes. To test this hypothesis, we first confirmed that there was an increased concentration of intracellular lactate in the macrophages treated with the conditioned media from lung myofibroblasts or treated directly with lactate (Figures 4A and 4D). Concordantly, histone lactylation was also increased in the treated cells (Figures 4B, 4C, and 4E). Interestingly, although the pan anti–lactyl-lysine antibody used in this study theoretically detects all proteins with this modification, the proteins with augmented lysine lactylation in lactate-treated macrophages were indeed predominantly histones, as reflected by the absence of an evident alteration of lactylation in other proteins of all sizes (Figure 4C). These data clearly indicate that lysine residues of histones are more accessible to lactylation than those of other proteins. Furthermore, we found that there was enhanced histone lactylation in the lungs of mice with bleomycin-induced pulmonary fibrosis as well as in human IPF lungs compared with that in the normal control animals (Figures 4F and 4G). The heightened epigenetic modification was partially localized to macrophages in the lungs (Figures 4F and 4G). To assess further histone lactylation in these cells, we performed the immunofluorescent staining with purified alveolar macrophages and found that cells from fibrotic mouse lungs demonstrated greater amounts of this epigenetic modification (Figures 4H and 4I). Taken together, these data suggest that lactate induces histone lactylation in the macrophages of fibrotic lungs, thus boosting the profibrotic phenotype of these cells.
Figure 4.

Lactate induces histone lactylation in macrophages. (A) BMDMs were treated with or without 20 mM lactate for 24 hours. The cells were lysed, and lactate contents in the cellular lysates were determined (n = 3; mean ± SD). ***P < 0.001 by two-tailed Student’s t test. (B) BMDMs were treated with or without 20 mM lactate as in A. Concentrations of the indicated proteins in whole cell lysates or HCl-extracted histones were determined by Western blotting. (C) BMDMs were treated with or without lactate. The entire blot was developed for lactyl-lysine. (D) BMDMs were treated with conditioned media from normal fibroblasts or TGF-β1–induced myofibroblasts. Twenty-four hours after treatment, cells were lysed, and lactate contents in the cellular lysates were determined (n = 3; mean ± SD). *P < 0.05 by two-tailed Student’s t test. (E) BMDMs were treated with conditioned media from normal fibroblasts or TGF-β1–induced myofibroblasts as in (D). Concentrations of the indicated proteins were determined by Western blotting. (F) C57BL/6 mice were i.t. instilled with saline or BLM. Three weeks after treatment, mice were killed, and lung slices were prepared. The expression of F4/80 and lactyl-histone were evaluated by immunofluorescence staining and fluorescence microscopy. (G) Slices of normal con and idiopathic pulmonary fibrosis lungs were prepared. The expression of CD68 and lactyl-histone were evaluated by immunofluorescence staining and fluorescence microscopy. (H and I) C57BL/6 mice were i.t. instilled with saline or BLM. Three weeks after treatment, BALFs were harvested, and alveolar macrophages were isolated and cultured on coverslips. The expression of F4/80 and lactyl-histone were evaluated by immunofluorescence staining and fluorescence microscopy (H). Single-cell fluorescence intensity was measured by ImageJ for ∼100 randomly selected cells from each group, and the average was calculated. ***P < 0.001 by two-tailed Student’s t test. The box-and-whisker plots depict the 25th and 75th percentiles and the median, minimum, and maximum values (I). (F–H) Nuclei were counterstained with DAPI. Scale bars, 100 μm. AE = HCl-extracted; IPF = idiopathic pulmonary fibrosis; MFI = mean fluorescence intensity; WCE = whole cell lysates.
Lactate Increases Histone Lactylation Amounts at the Promoters of the Profibrotic Genes in Macrophages
To demonstrate that histone lactylation was indeed involved in the lactate induction of profibrotic mediators in macrophages, we performed a chromatin IP assay to assess histone lactylation amounts at the promoters of these genes. As shown in Figure 5A, we found that histone lactylation concentrations at the proximal promote regions of ARG1, PDGFA, THBS1, and VEGFA were significantly increased in lactate-treated macrophages. In contrast, there was no alteration in the histone lactylation at the promoter of the ACTB gene that was not induced in lactate-treated cells (Figure 5B). Taken together, the above findings suggest that lactate induces the expression of these profibrotic mediators by inducing histone lactylation at their promoters.
Figure 5.

Lactate increases histone lactylation amounts at the promoters of the profibrotic genes in macrophages. BMDMs were treated with or without 20 mM lactate. Twenty-four hours after treatment, the cells were fixed with 1% formaldehyde and collected in radioimmunoprecipitation assay (RIPA) buffer for sonication. A chromatin IP assay was performed using anti–lactyl-lysine antibody. (A and B) The histone lactylation amounts within the proximal promoter regions of the indicated genes (A) and reference ACTB gene (B) were determined by real-time PCR (n = 6; mean ± SD). *P < 0.05, **P < 0.01, and ***P < 0.001 by two-tailed Student’s t test. ACTB = actin β.
p300 Mediates Lactate-induced Histone Lactylation and Profibrotic Mediator Expression in Macrophages
The acetyltransferase p300 has been implicated in the histone lactylation (38, 39). To determine whether p300 mediated lactate-induced histone lactylation and the profibrotic gene expression in macrophages, we knocked down p300 and found that lactate-induced histone lactylation was considerably diminished in p300-knockdown macrophages (Figures 6A and 6B). Furthermore, we found that lactate induction of the profibrotic gene expression was dampened when p300 was knocked down in these cells, which was accompanied by reduced histone lactylation at the promoters of these genes (Figures 6C and 6D). Together, these data suggest that the lactate promotion of the expression of profibrotic genes is dependent on p300-mediated histone lactylation at their promoters.
Figure 6.
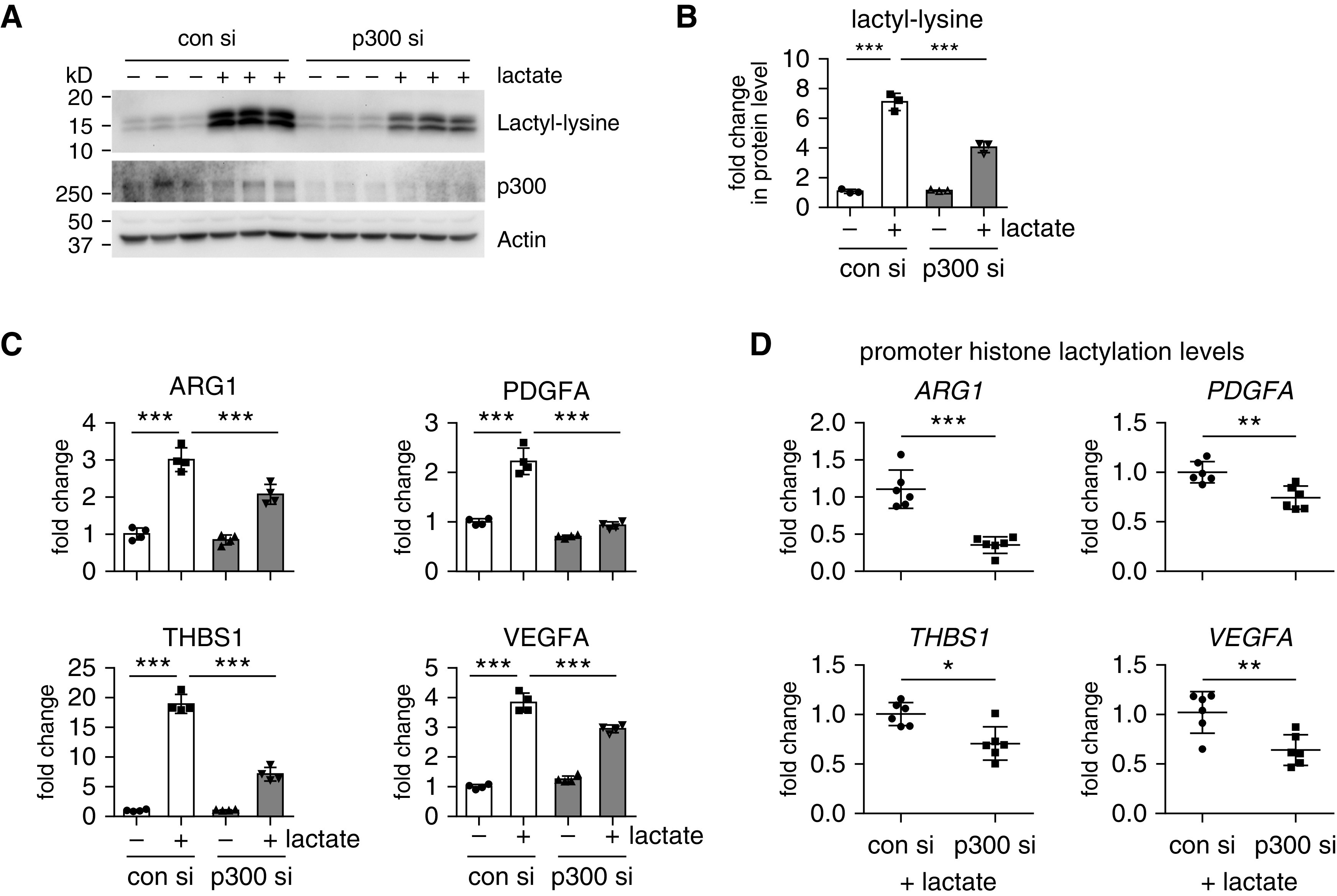
p300 mediates lactate-induced histone lactylation and profibrotic mediator expression in macrophages. (A and B) BMDMs were transfected with 20 nM con siRNAs or p300 siRNAs. Twenty-four hours after transfection, the cells were treated with or without 20 nM lactate for another 24 hours. The histone lactylation amounts were determined by Western blotting (A) and densitometric analyses performed using ImageJ (B) (n = 3; mean ± SD). ***P < 0.001 by one-way ANOVA with Bonferroni’s post hoc test. (C) BMDMs were transfected and treated as in A and B. The concentrations of the indicated genes were determined by real-time PCR. ***P < 0.001 by one-way ANOVA with Bonferroni’s post hoc test. (D) BMDMs were transfected and treated as in A–C. The histone lactylation amounts at the promoters of the indicated genes were determined by chromatin IP assay as in Figure 5 (n = 6; mean ± SD). *P < 0.05, **P < 0.01, and ***P < 0.001 by two-tailed Student’s t test. si = siRNAs.
Knockdown p300 Attenuates the Profibrotic Phenotype Induced by Lung Myofibroblast–conditioned Media in Macrophages
To determine whether p300 mediated histone lactylation was also involved in the macrophage profibrotic phenotype induced by the lung myofibroblast–conditioned media, we performed an experiment similar to the one with lactate treatment. As shown in Figures 7A and 7B, lung myofibroblast–conditioned media–induced histone lactylation in macrophages was diminished when p300 was knocked down. Concordant with this finding, lung myofibroblast–conditioned media–induced profibrotic gene expression was also attenuated in p300-knockdown cells (Figure 7C). Altogether, these findings suggest that lactate in the lung myofibroblast–conditioned media induces p300-mediated histone lactylation, thereby enhancing the profibrotic phenotype in macrophages.
Figure 7.

Knockdown p300 attenuates the profibrotic phenotype induced by lung myofibroblast–conditioned media in macrophages. (A and B) BMDMs were transfected with 20 nM con si or p300 si. Twenty-four hours after transfection, the cells were treated with conditioned media from normal fibroblasts or TGF-β1–induced myofibroblasts for another 24 hours. The histone lactylation amounts were determined by Western blotting (A) and densitometric analyses performed using ImageJ (B). (C) BMDMs were transfected and treated as in A and B. The concentrations of the indicated genes were determined by real-time PCR. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA with Bonferroni’s post hoc test.
Discussion
Dysregulation of cellular metabolism has been increasingly recognized to play an important role in the pathogeneses of various pulmonary disorders (19–22). Specifically, there has been convincing evidence showing that glycolytic escalation in lung fibroblasts, smooth muscle cells, and endothelial cells makes a key contribution to the progressions of lung fibrosis and pulmonary hypertension (23, 40, 41). The augmented glycolysis leads to disruptions in the tricarboxylic acid cycle, energy production, and various signaling events, thereby promoting the pathological phenotype of these cells (23, 40, 41). However, these underlying pathogenic mechanisms were invariably envisioned from a cell-autonomous point of view. A partially non–cell autonomous role of lactate, namely, activated lung fibroblast–secreted lactate-promoting myofibroblast differentiation, in the pathogenesis of this disorder has also been previously elegantly demonstrated (42, 43). Here, we extend those studies by presenting novel evidence suggesting that the dysregulated glycolysis does not only play a cell-autonomous role but also a non–cell autonomous role in lung fibrogenesis. We have shown that in addition to protein, nucleic acid, and lipid molecules, a metabolite may also function as a paracrine mediator to promote this pathology. Our study has thus advanced the understanding of the interregulations among different types of pulmonary cells during pathogenic lung repair. However, despite the promising implication of the lactate-augmented macrophage profibrotic activity, we also need to recognize the limitation of this finding in that we have not had direct evidence demonstrating the participation of the lactate-induced macrophage phenotypic alterations in the regulation of pulmonary fibrosis. Therefore, the pathological significance of the effect of myofibroblast-derived lactate on lung macrophages in the initiation and progression of this disease remains an open and interesting question.
Although we showed that there was increased histone lactylation in the macrophages of the fibrotic lungs, we need to acknowledge that the elevated epigenetic modification was not only restricted to these cells. This should not have come as a surprise because all the neighboring cells in a fibrotic niche are exposed to the same lactate buildup and subjected to similar lactate-induced histone lactylation. In addition, although we have shown that histone lactylation may promote the expression of many profibrotic genes by macrophages in the lung, it remains unclear what role this epigenetic modification played in the phenotypic alterations of other types of cells that are involved in the pathogenesis of lung fibrosis, such as alveolar epithelial cells and mesenchymal progenitors. Addressing these remaining questions will be crucial to the understanding of how extracellular lactate, and perhaps other metabolites, contributes to the initiation and progression of this disease.
We have confirmed that p300 mediates lactate-induced histone lactylation in macrophages in vitro. However, we do realize that whether the augmentation of this epigenetic modification in macrophages is mediated by p300 in vivo in the fibrotic lung is still an unanswered question. In addition, we do not believe that p300 represents an ideal therapeutic target for those pulmonary diseases that are featured by an escalated glycolysis. One of the reasons for this is that p300 is also one of the most significant acetyltransferases and has a myriad of other cellular functions (44). Targeting p300 is likely to have drastic effects that are far beyond what histone lactylation is supposed to produce, thereby causing unwanted side effects.
Our study indicates an unconventional therapeutic approach for lung fibrosis in that a metabolite, but not a protein, a lipid, or a nucleic acid, is a potential target. We speculate that lactate manipulation could be achieved through several approaches, including by depleting extracellular lactate and thereby reducing lactate entry into the lung macrophages and other types of cells or by directly blocking lactate intake by lung macrophages. However, these strategies are not without caveats. First, there is no precedent concerning how to deplete a single metabolite from an extracellular setting. Second, membrane transporters for lactate are capable of mediating both the import and export of this molecule (45). Blocking lactate uptake from the extracellular milieu may concurrently hamper the export of lactate from macrophages, which also leads to an intracellular lactate buildup. Therefore, it seems only logical to target a nontraditional mediator with some unorthodox strategies that remain to be identified. Regardless, our study does present a novel therapeutic opportunity for treating pulmonary fibrosis.
Our data suggest that lactate from glycolytic lung myofibroblasts in the fibrotic lung is critically involved in promoting the pathological phenotype of macrophages in the lung, which contributes to the pathogenesis of this disorder. It is also known that there is augmented glycolysis in different types of cells in other pulmonary disorders, including pulmonary hypertension and chronic obstructive pulmonary disease (40, 41, 46, 47). Thus, our finding that lactate has a non–cell autonomous role suggests that there is an additional mechanism by which the increased glycolysis participates in the pathogeneses of these disorders.
Intercellular communications through metabolites have been previously described between cancer epithelial and stromal cells (48). However, a role of metabolites as intercellular messengers in pulmonary disorders has not been investigated. Our study on lactate sets an example of basic strategies for future explorations of other metabolites in lung fibrosis. To uncover significant extracellular metabolic messengers, a targeted or nontargeted metabolic profiling on BALFs from lungs of animals with experimental pulmonary fibrosis and human patients with IPF seems necessary. Identifying distinct extracellular metabolites, tracing their fate, and defining their functional impact in relevant cells will undoubtedly expand the understanding of intercellular regulations during lung injury and repair. Those future studies will be also vital to the discovery of novel therapeutic targets and strategies for the treatment of pulmonary fibrosis, which remains a formidable health challenge.
Supplementary Material
Footnotes
Supported by U.S. National Institutes of Health grant HL135830 and Department of Defense grant W81XWH-20-1-0226.
Author Contributions: H.C. and G.L. designed the study. H.C., N.X., S.B., J.G., D.J., R.-M.L., and G.L. performed the experiments and/or analyzed the data. Q.L.M. contributed critical materials. H.C., T.D., R.-M.L., and G.L. wrote the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0360OC on October 19, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. 2012;380:680–688. doi: 10.1016/S0140-6736(12)61144-1. [DOI] [PubMed] [Google Scholar]
- 3.Mora AL, Rojas M, Pardo A, Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Discov. 2017;16:755–772. doi: 10.1038/nrd.2017.170. [DOI] [PubMed] [Google Scholar]
- 4.Brownell R, Kaminski N, Woodruff PG, Bradford WZ, Richeldi L, Martinez FJ, et al. Precision medicine: the new frontier in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193:1213–1218. doi: 10.1164/rccm.201601-0169CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu B, Phan SH. Myofibroblasts. Curr Opin Rheumatol. 2013;25:71–77. doi: 10.1097/BOR.0b013e32835b1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philip K, Mills TW, Davies J, Chen NY, Karmouty-Quintana H, Luo F, et al. HIF1A up-regulates the ADORA2B receptor on alternatively activated macrophages and contributes to pulmonary fibrosis. FASEB J. 2017;31:4745–4758. doi: 10.1096/fj.201700219R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tao B, Jin W, Xu J, Liang Z, Yao J, Zhang Y, et al. Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J Immunol. 2014;193:2801–2811. doi: 10.4049/jimmunol.1303463. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. 2019;129:2619–2628. doi: 10.1172/JCI124615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol. 2016;17:9–17. doi: 10.1038/ni.3320. [DOI] [PubMed] [Google Scholar]
- 12.Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol. 2017;79:593–617. doi: 10.1146/annurev-physiol-022516-034356. [DOI] [PubMed] [Google Scholar]
- 13.Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol Med. 2016;22:303–316. doi: 10.1016/j.molmed.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 14.McCubbrey AL, Barthel L, Mohning MP, Redente EF, Mould KJ, Thomas SM, et al. Deletion of c-flip from cd11b hi macrophages prevents development of bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol. 2018;58:66–78. doi: 10.1165/rcmb.2017-0154OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ucero AC, Bakiri L, Roediger B, Suzuki M, Jimenez M, Mandal P, et al. Fra-2-expressing macrophages promote lung fibrosis in mice. J Clin Invest. 2019;129:3293–3309. doi: 10.1172/JCI125366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joshi N, Watanabe S, Verma R, Jablonski RP, Chen CI, Cheresh P, et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur Respir J. 2020;55:1900646. doi: 10.1183/13993003.00646-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu G, Summer R. Cellular metabolism in lung health and disease. Annu Rev Physiol. 2019;81:403–428. doi: 10.1146/annurev-physiol-020518-114640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang YP, Lee SB, Lee JM, Kim HM, Hong JY, Lee WJ, et al. Metabolic profiling regarding pathogenesis of idiopathic pulmonary fibrosis. J Proteome Res. 2016;15:1717–1724. doi: 10.1021/acs.jproteome.6b00156. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Kumar A, Carmeliet P. Metabolic pathways fueling the endothelial cell drive. Annu Rev Physiol. 2019;81:483–503. doi: 10.1146/annurev-physiol-020518-114731. [DOI] [PubMed] [Google Scholar]
- 22.Michaeloudes C, Bhavsar PK, Mumby S, Xu B, Hui CKM, Chung KF, et al. Role of metabolic reprogramming in pulmonary innate immunity and its impact on lung diseases. J Innate Immun. 2020;12:31–46. doi: 10.1159/000504344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM, et al. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med. 2015;192:1462–1474. doi: 10.1164/rccm.201504-0780OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernard K, Logsdon NJ, Ravi S, Xie N, Persons BP, Rangarajan S, et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J Biol Chem. 2015;290:25427–25438. doi: 10.1074/jbc.M115.646984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodwin J, Choi H, Hsieh MH, Neugent ML, Ahn JM, Hayenga HN, et al. Targeting hypoxia-inducible factor-1α/pyruvate dehydrogenase kinase 1 axis by dichloroacetate suppresses bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2018;58:216–231. doi: 10.1165/rcmb.2016-0186OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin X, Choudhury M, Kang JH, Schaefbauer KJ, Jung MY, Andrianifahanana M, et al. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-β. Sci Signal. 2019;12:eaax4067. doi: 10.1126/scisignal.aax4067. [DOI] [PubMed] [Google Scholar]
- 27.Hu X, Xu Q, Wan H, Hu Y, Xing S, Yang H, et al. PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab Invest. 2020;100:801–811. doi: 10.1038/s41374-020-0404-9. [DOI] [PubMed] [Google Scholar]
- 28.Cui H, Ge J, Xie N, Banerjee S, Zhou Y, Antony VB, et al. Mir-34a inhibits lung fibrosis by inducing lung fibroblast senescence. Am J Respir Cell Mol Biol. 2017;56:168–178. doi: 10.1165/rcmb.2016-0163OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui H, Xie N, Jiang D, Banerjee S, Ge J, Sanders YY, et al. Inhibition of glutaminase 1 attenuates experimental pulmonary fibrosis. Am J Respir Cell Mol Biol. 2019;61:492–500. doi: 10.1165/rcmb.2019-0051OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui H, Banerjee S, Guo S, Xie N, Ge J, Jiang D, et al. Long noncoding RNA Malat1 regulates differential activation of macrophages and response to lung injury. JCI Insight. 2019;4:e124522. doi: 10.1172/jci.insight.124522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie N, Cui H, Ge J, Banerjee S, Guo S, Dubey S, et al. Metabolic characterization and RNA profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2017;313:L834–L844. doi: 10.1152/ajplung.00235.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui H, Jiang D, Banerjee S, Xie N, Kulkarni T, Liu RM, et al. Monocyte-derived alveolar macrophage apolipoprotein E participates in pulmonary fibrosis resolution. JCI Insight. 2020;5:e134539. doi: 10.1172/jci.insight.134539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobie R, Henderson NC. Unravelling fibrosis using single-cell transcriptomics. Curr Opin Pharmacol. 2019;49:71–75. doi: 10.1016/j.coph.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 34.Liu SS, Lv XX, Liu C, Qi J, Li YX, Wei XP, et al. Targeting degradation of the transcription factor C/EBPβ reduces lung fibrosis by restoring activity of the ubiquitin-editing enzyme a20 in macrophages. Immunity. 2019;51:522–534, e7. doi: 10.1016/j.immuni.2019.06.014. [DOI] [PubMed] [Google Scholar]
- 35.Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity. 2016;44:582–596. doi: 10.1016/j.immuni.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laddha AP, Kulkarni YA. VEGF and FGF-2: promising targets for the treatment of respiratory disorders. Respir Med. 2019;156:33–46. doi: 10.1016/j.rmed.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Pardo A, Gibson K, Cisneros J, Richards TJ, Yang Y, Becerril C, et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005;2:e251. doi: 10.1371/journal.pmed.0020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–580. doi: 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Notarangelo G, Haigis MC. Sweet temptation: from sugar metabolism to gene regulation. Immunity. 2019;51:980–981. doi: 10.1016/j.immuni.2019.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Cao Y, Zhang X, Wang L, Yang Q, Ma Q, Xu J, et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc Natl Acad Sci USA. 2019;116:13394–13403. doi: 10.1073/pnas.1821401116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kovacs L, Cao Y, Han W, Meadows L, Kovacs-Kasa A, Kondrikov D, et al. Pfkfb3 in smooth muscle promotes vascular remodeling in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2019;200:617–627. doi: 10.1164/rccm.201812-2290OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med. 2012;186:740–751. doi: 10.1164/rccm.201201-0084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kottmann RM, Trawick E, Judge JL, Wahl LA, Epa AP, Owens KM, et al. Pharmacologic inhibition of lactate production prevents myofibroblast differentiation. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1305–L1312. doi: 10.1152/ajplung.00058.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Attar N, Kurdistani SK. Exploitation of ep300 and crebbp lysine acetyltransferases by cancer. Cold Spring Harb Perspect Med. 2017;7:a026534. doi: 10.1101/cshperspect.a026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Payen VL, Mina E, Van Hée VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Mol Metab. 2020;33:48–66. doi: 10.1016/j.molmet.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li M, Riddle S, Zhang H, D’Alessandro A, Flockton A, Serkova NJ, et al. Metabolic reprogramming regulates the proliferative and inflammatory phenotype of adventitial fibroblasts in pulmonary hypertension through the transcriptional corepressor c-terminal binding protein-1 Circulation 20161341105–1121.[Published erratum appears in Circulation 139:e1.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michaeloudes C, Kuo CH, Haji G, Finch DK, Halayko AJ, Kirkham P, et al. COPDMAP. Metabolic re-patterning in copd airway smooth muscle cells. Eur Respir J. 2017;50:1700202. doi: 10.1183/13993003.00202-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, et al. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and malignancy. Cell Metab. 2019;29:124–140, e10. doi: 10.1016/j.cmet.2018.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.