

Abstract
Sjögren’s syndrome (SS) is a chronic rheumatic autoimmune disorder affecting multiple organ systems. The clinical findings in SS patients show considerable heterogeneity and overlap with other autoimmune diseases. In addition, the autoimmune response in SS initiates several years before the appearance of clinical symptoms. Thus, understanding the pathogenic mechanisms involved in the disease process have been a challenge. Several animal model systems of SS-like disease have been developed to overcome these issues. The New Zealand Black (NZB) × New Zealand White (NZW) F1 (NZB/W F1) mouse represents the first spontaneous mouse model of SS. In this review, we provide a historical perspective and detailed description of this mouse model focusing on exocrine gland histopathology, autoantibody populations, and glandular dysfunction. Considering that NZB/W F1 mice also develop a systemic lupus erythematosus (SLE)-like disease, this mouse model mimics the clinical presentation of polyautoimmunity seen in a sizable subset of SS patients. It is plausible that such patients will require distinct therapeutic interventions necessary to treat both SLE and SS. Therefore, the NZB/W F1 mouse is a powerful tool to decipher pathogenic mechanisms involved in SS related polyautoimmunity and develop appropriate therapeutic strategies.
Keywords: Sjögren’s syndrome, Lupus, Mouse, NZB/W F1, Salivary Gland, Polyautoimmunity
1. Introduction
Sjögren’s syndrome (SS) is one of the three most common systemic rheumatic autoimmune disorders affecting a large number of people worldwide [1,2]. The disease is typically diagnosed in older adults during the fourth or fifth decade of life and shows a strong female to male bias with a ratio of 10:1 [3]. Circulating antibodies targeting intracellular proteins, and immune cells infiltrating different tissues gives SS its autoimmune character [4]. The prominent clinical feature of SS is dry mouth and dry eye, caused by the exocrine salivary and lacrimal gland dysfunction [5]. Fatigue, arthralgia, arthritis, and peripheral neuropathy are some of the common extraglandular manifestations of SS. Other organ systems like the lungs, kidneys, skin, and gastrointestinal tract are affected to varying degrees. Most patients also present with hematological abnormalities, including hypergammaglobulinemia, monoclonal gammopathy, hypocomplementemia, and cytopenias [6]. There is considerable heterogeneity in the clinical presentation of SS, and this has been a significant obstacle for accurate diagnosis and therapy [7].
SS has a strong genetic component and many of the genetic loci associated with SS map to different innate and adaptive immune pathways [8]. The appearance of circulating autoantibodies several years to decades before the clinical presentation further supports an autoimmune etiology for SS [9,10]. There is a consensus that interactions between a genetically dysregulated immune system and varied environmental stimuli manifest as autoimmune responses in SS. The triggers responsible for initiating autoimmunity in SS remain unclear, but viral infections are considered as the primary candidates [11,12]. Whether a systemic infection, or an exocrine gland infection, or both, are responsible for driving early events in SS is difficult to decipher in patients. Another challenge is to identify how the disease progresses from benign autoimmunity to a fulminant clinical presentation. Thus, to address these issues, several animal model systems have been developed in the past five decades [13–15]. However, considering the heterogeneity in immunological abnormalities and clinical manifestations of SS, it is not surprising that no single animal model can claim to recapitulate the full spectrum of SS. Each model has its strengths and limitations in investigating human disease. Several reviews in the literature provide a glimpse into multiple mouse models that develop some aspect of SS [13–15]. In this review, we describe the New Zealand Black (NZB) × New Zealand White (NZW) F1 (NZB/W F1) mouse, the first spontaneous mouse model system of SS, and its relevance for investigating pathogenic mechanisms of SS.
2. Classification criteria for SS in patients and their application to mice
The signs and symptoms of SS are not unique to the disease and show notable overlaps with other rheumatic autoimmune disorders, thereby making the diagnosis of SS a complex task [1]. Over the past three decades, various classification schemes were developed to address this issue [16–18]. These schemes have helped to formulate guidelines for recruiting patients into research protocols and clinical trials. The latest American College of Rheumatology – European League Against Rheumatism (ACR- EULAR) 2016 classification criteria for SS [18] are based on an objective assessment of glandular inflammation, autoantibodies to SSA/Ro antigens, and glandular dysfunction (Table 1). For an individual to be classified as a primary SS patient, the cumulative score for five items shown in Table 1 has to be ≥ 4.0. Also, the individual has to respond positively to at least one of five questions dealing with subjective oral and ocular dryness measures. Alternatively, based on the EULAR SS Disease Activity Index (ESSDAI) questionnaire, the individual is required to have one positive response in any of the 12 domains representing different organ systems. Thus, a complex evaluation of both symptoms and signs involving multiple organs determine the diagnosis of SS.
Table 1.
ACR-EULAR 2016 Classification Criteria for SS [18] and their application to NZB/W F1 mouse model for SS.
| Item | Weight / Score | NZB/W F1 |
|---|---|---|
| Labial salivary gland with focal lymphocytic sialadenitis and focus-score ≥ 1. | 3 | ✓a |
| Anti-SSA (Ro) + | 3 | ? |
| Ocular staining score ≥ 5 (or van Bijsterfeld score ≥ 4) on at least one eye | 1 | X |
| Schirmer ≤ 5 mm/5 min on at least one eye | 1 | ✓b |
| Unstimulated whole saliva flow rate ≤ 0.1 mL/min | 1 | ✓c |
Inclusion criteria: The patient should respond positively to at least one of the following symptoms of ocular or oral dryness. 1)Have you had daily, persistent, troublesome dry eyes for more than 3 months? 2)Do you have a recurrent sensation of sand or gravel in the eyes? 3)Do you use tear substitutes more than 3 times a day? 4)Have you had a daily feeling of dry mouth for more than 3 months? 5)Do you frequently drink liquids to aid in swallowing dry food?
Alternatively, there should be a suspicion of SS from ESSDAI questionnaire (at least one domain with positive item)
Histopathology data from submandibular and parotid glands.
Modified Schirmer test data shows lower tear volumes than non-autoimmune mouse strains.
Agonist induced saliva shows age dependent drop in volume.
While this scheme has proven valuable as a guideline for human clinical studies, the verbatim extension of these parameters to determine whether a mouse model represents SS is untenable. A reconfiguration of the three primary objective criteria (glandular inflammation, autoantibodies, and glandular dysfunction) is required to best model the human disease in mice. Rather than minor labial gland biopsy, histopathological investigation in mice can be done in the 3 major salivary glands (parotid, submandibular, and sublingual).
Almost 25–30% of SS patients do not harbor anti-SSA/Ro antibodies, but have anti-nuclear antibodies, rheumatoid factor, and other specificities indicating a loss of tolerance to self and the establishment of auto-reactivity (Table 2) [6]. Thus, mouse strains that are negative for anti-SSA/Ro, but develop other autoantibodies and glandular inflammation represent a specific subset of SS patients.
Table 2.
The prevalence of major autoantibody specificities in Sjögren’s Syndrome patientsa and their presence in NZB/W F1 mice.
| Autoantibody | Prevalence (% positive) | NZB/W F1 |
|---|---|---|
| Anti-nuclear antibody (ANA) | 7749/9784 (79.2) | ✓ |
| Anti-Ro/SSA | 7617/10417 (73.1) | ? |
| Anti-La/SSB | 4662/10362 (45) | ? |
| Rheumatoid Factor (RF) | 4245/8758 (48.5) | ✓ |
| Cryoglobulins | 342/4732 (7.2) | ✓ |
Summary of data published by Brito-Zeron et al., [6] in 10,500 patients from 22 countries.
Number positive/Number tested.
Evaluating unstimulated saliva flow in mice is technically challenging, and necessitates measuring agonist-induced saliva production. Currently, there are no established standards for normal levels of tear and saliva production in mice. Our study measuring saliva volumes in age-matched female mice showed a wide variation between different strains [19]. Therefore, the best approach to evaluating glandular dysfunction is using longitudinal studies to monitor a reduction in saliva or tear volumes over time. In this review we demonstrate that the NZB/W F1 mouse fulfils 3 out of the 4 objective classification criteria established for SS.
3. NZB/W F1 mouse as a model for SS
Almost 61 years ago, Bielschowsky et al. described the development of autoantibodies and hemolytic anemia in the NZB mouse strain [20]. To determine the genetic transmission of autoimmune hemolytic anemia, Helyer and Howie crossed the NZB mouse with different mouse strains to generate F1 hybrids [21]. In 1963, they described the NZB/W F1 hybrid mouse as a strain that developed lupus-like nephritis with a high penetrance [22]. In an extensive phenotypic analysis (500 females and 157 males), Howie et al. [23] meticulously described the development of anti-nuclear antibodies, renal immune-complex deposition, proteinuria, azotemia, and survival of NZB/W F1 mice. Anti-nuclear antibodies began to appear in these mice by 2–4 months of age, followed approximately two months later (4–6 months of age) by immune-complex deposition in the kidneys and the establishment of glomerulonephritis. By 8–10 months of age, more than 50% of mice developed severe proteinuria, which preceded death. The mean survival reported in this original investigation for females was 279.8 days, and for males was 438.8 days.
The first description of NZB/W F1 mice as a model for SS was published by Harry Kessler [24]. In a comparison of parental NZB (n=18), NZW (n=29), and the F1 hybrids (n=29), it was noted that by 4 months of age, the NZB and the NZB/W F1 mice showed mononuclear cell infiltrates within their salivary and lacrimal glands. These lesions became progressively severe with age. The NZW mice did not show any pathological changes. Salivary gland dysfunction in NZB/W F1 mice was evident by the reduction in salivary amylase concentration by almost 50% compared to NZW mice. Since this historical description of salivary and lacrimal gland disease, several studies in the literature have reported the development of different features characteristic of SS in NZB/W F1 mice. Herein, we provide an overview of NZB/W F1 mouse as a model for SS with reference to exocrine gland histopathology, autoantibody responses, and glandular dysfunction. For an overview of genetics and immune cell defects in NZB/W F1 mice, the readers are referred to excellent articles in the literature [25–27].
4. Histopathology
Inflammation of the exocrine glands is a preeminent feature of SS. In some patients, this is accompanied by pain and swelling in the glands. The establishment of tertiary lymphoid structures associated with the formation of malignant lymphomas is a cause of increased mortality in SS [28]. Thus, minor labial salivary gland biopsies offer a valuable diagnostic and prognostic tool in SS patients. In NZB/W F1 mice, extensive evaluation of salivary gland pathology has been performed over the past five decades. In this review, we first provide a historical perspective on histopathological analysis of salivary glands in SS patients and its application to studying salivary gland pathology in the NZB/W F1 mice.
4.1. A historical perspective of glandular inflammation in SS
The initial description of SS included keratoconjunctivitis sicca, xerostomia, and polyarthritis, as a triad of manifestations occurring in a sicca complex [29]. In 1963, Waterhouse evaluated focal inflammation (adenitis) in submandibular and parotid salivary glands and lacrimal glands from 239 subjects undergoing autopsy and reported that salivary and lacrimal gland involvement might occur independently of arthritis [30]. In the gland parenchyma, aggregates of >50 cells, predominantly lymphocytes and histiocytes, were considered a focus of inflammation. Plasma cells were seen at the periphery of some large foci. Glands with at least two such foci were considered to be positive for inflammation. Focal adenitis was more prevalent in women (70%) than men (30%), especially in women over 45 years. The foci were most frequent in the submandibular glands compared to the parotid or lacrimal glands. In 1966, Calman and Reifman reported focal adenitis in the minor salivary glands from the buccal mucosa in a patient of xerostomia and keratoconjunctivitis, thereby offering an avenue for early diagnosis of SS [31]. Subsequently, an expanded study of 40 patients and 60 controls established the value of labial salivary gland biopsies in the diagnosis of SS [32]. The labial salivary glands from patients showed focal and diffuse infiltration of lymphocytes, histiocytes, and plasma cells in between healthy acinar tissue.
Over five decades after the description of focal adenitis, the primary criteria for diagnosis and quantification of disease severity in SS continues to be the identification of areas in labial salivary glands occupied by lymphocytic aggregates with >50 cells per focus [33]. The phenotyping of infiltrating immune cells for T and B cell surface markers organized to form germinal centers also allows determination of the risk for lymphoma development [34]. However, it should be noted that distinct histopathological features such as hyperplasia of the ductal epithelium and formation of epithelial-myoepithelial cell islands, has been noted in parotid gland biopsies obtained from Mikulicz’s disease patients [35], and about 40% of SS cases [36]. These reports suggest that although minor labial gland pathology allows for diagnosis of SS, the study of major glands will provide an in-depth understanding into disease pathogenesis. In this context, the NZB/W F1 mice are an invaluable tool for investigating the mechanisms of inflammatory cell-mediated pathology in major salivary glands.
4.2. Focal adenitis in salivary and lacrimal glands of NZB/W F1 mice
The salivary and lacrimal glands from NZB/W F1 male and female mice have been studied at different ages and provide insights into the initiation and evolution of the cellular lesions. Similar to the human disease, glandular inflammation appears earlier and more frequently in females compared to males. The lymphocytic infiltration is seen first in the submandibular glands and lacrimal glands by 4 months of age and later appears in the parotid glands [37–39]. Sublingual glands, which consist predominantly of mucous acini in mice, are rarely involved. Despite the differences in kinetics of inflammation in submandibular and parotid glands, the characteristics of cellular infiltrates in both glands are similar. Lymphocytes appear around blood vessels and medium-sized intra-lobular striated ducts of the submandibular glands [24,40].
The lumen of lymphatic vessels shows the accumulation of lymphocytes and a gradual expansion of the infiltrates into the inter-lobar interstitial regions (Figure 1A). The inflammatory foci consist predominantly of CD4 T cells and macrophages close to the acinar and ductal epithelium. Larger foci show significant numbers of B cells, organizing into germinal center-like structures (Figure 1B). Plasma cells are present extending along the periphery of larger foci. There is a progressive degeneration of the vascular wall and basement membrane lining the ducts. The immune cells invade into the acini through the discontinuity in the basement membrane by 6–8 months of age. There is a loss of polarity in the ductal and acinar epithelial cells, followed by a destruction of these cells resulting in acinar atrophy. TUNEL staining shows apoptosis of the glandular epithelium and of infiltrating lymphocytes.
Figure 1. Salivary gland inflammation in NZB/W F1 female mice at 6–7 months of age.
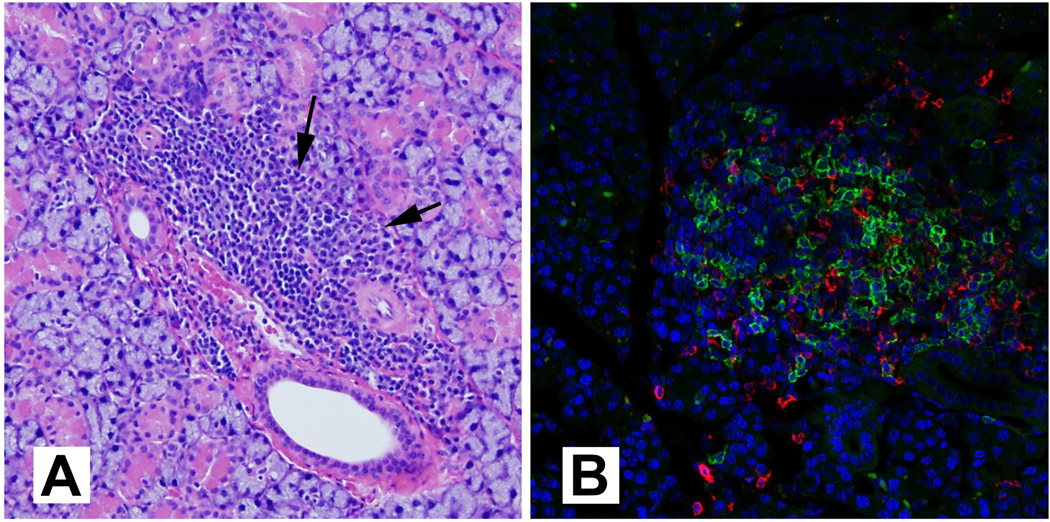
(A) Representative image of a hematoxylin and eosin-stained submandibular salivary gland showing a large peri-ductal and peri-vascular lymphocytic focus, extending into the gland parenchyma (arrows). Magnification 10×. (B) Immunostaining of the submandibular salivary gland showing the presence of B cells (anti-B220, green) and T cells (anti-CD4, red) within a lymphocytic focus. Magnification 20×.
IgG deposits are seen in basement membranes around ducts, acini, and capillaries in areas of inflammation, but significantly, also at sites distant from the inflammatory foci [39]. Autoantibodies to acinar and ductal antigens are found in circulation [41,42]. It should be noted that immunoglobulin deposits were also reported in salivary glands of SS patients [43]. These antibody deposits are of particular interest since recent studies show a significant role for autoantibodies in driving glandular dysfunction, independent of local inflammation [44,45]. Overall the light microscopy studies demonstrate that critical characteristics of focal adenitis in NZB/W F1 mice are remarkably similar to those seen in SS patients.
4.3. Ultrastructural studies provide mechanistic insights into disease pathogenesis
The failure of tolerance mechanisms and inflammatory cells forming the lymphocytic foci in exocrine glands have been under the spotlight for investigations in SS, and have yielded a vast body of literature on disease pathogenesis. Nonetheless, how this inflammation causes dryness remains unresolved [46,47]. A critical observation is that the severity of lymphocytic infiltration does not necessarily correlate with the extent of glandular dysfunction in SS patients or mice [48,49].
In patients, the major salivary glands show hyperplasia of the ductal epithelium and formation of epithelial-myoepithelial cell islands. These cells are either remnants of the ductal epithelium, myoepithelial cells, or both [50]. Since such features are not found in NZB/W F1 mice, some investigators have opined that the NZB/W F1 lesion was more a “vasculitis” that progresses to fibrosis and not the “epithelitis” seen in patients [51,52]. However, electron micrographs at five months of age show that myoepithelial cells, adjacent to the infiltrating lymphocytes, have increased disorganization of myofilaments, pyknotic nuclei, and degenerating organelles [39]. These changes are associated with detachment of the cells from the basement membrane, destruction of the myoepithelial cells, and the subsequent degeneration of the ductal epithelium. Similar changes are also seen in the secretory acini, subsequently leading to acinar atrophy. A recent report on SS patients showed reduced staining for alpha-smooth muscle actin in myoepithelial cells around acini and intercalated ducts, implying a loss of contractile function contributing to reduced saliva output [53]. These results from mouse and patient glands suggest that further investigations into the role of myoepithelial cells in SS pathogenesis are needed. Such studies will provide clues on pathways and mechanisms of glandular dysfunction.
Fibrosis and fatty replacement of the glandular tissue are prominent features found in labial biopsies from SS patients [54]. These glandular changes are most significantly associated with the patients’ age, and the influence on disease severity or functional loss is debated [55]. Although NZB/W F1 mice fail to show extensive fibrosis or fatty replacement in the glandular tissue, electron micrographs of salivary glands with lymphocytic foci display an increase in peri-arteriolar collagen fibers [52], and early deposition of peri-ductal laminin [39]. The integrin molecule, VLA-6, on NK cells and T cells recognizes laminin, suggesting that the laminin deposits may facilitate the recruitment of immune cells into the glands [38]. In addition, the increased ICAM expression on the endothelium and elevated cytokines in the systemic circulation [56] are all potential mechanisms for salivary gland inflammation in NZB/W F1 mice. In summary, the histopathological studies in NZB/W F1 mice have given insights into the evolution of the inflammatory focus and have helped to develop a dynamic picture of the disease process in the salivary glands. Although logistically difficult, longitudinal histopathological studies in SS patients are needed to understand how salivary gland inflammation contributes to glandular dysfunction.
5. Autoantibodies
The presence of circulating antibodies to multiple self-proteins is a hallmark of SS and confers the disorder with its autoimmune characteristic. Studies in different patient cohorts worldwide have established that autoantibodies target multiple self- proteins in SS. The Sjögren Big Data Consortium recently published data on immunological tests obtained from 10,500 SS patients from 22 countries [6]. Table 2 summarizes the prevalence of the dominant autoantibody specificities in SS patients and their presence in NZB/W F1 mice. Many of the autoantibody specificities are associated with specific clinical features and these findings have been elegantly reviewed by Kyriakidis et al. [57] and recently by Scofield et al. [58]. In this review, we have critically evaluated the presence of SS-associated autoantibody specificities in the NZB/W F1 mouse.
5.1. Anti-nuclear antibody (ANA)
ANA is the most prevalent autoantibody detected in SS patients (Table 2) and is the earliest specificity appearing well before the onset of clinical symptoms [9, 10]. In an exhaustive review on the geoepidemiology of SS, ANA positivity was reported in 75 to 90% of the patients [2]. The highest prevalence of ANA was in Asia, (89.13%, 1485/1666), followed by Europe (81.62%, 10283/12598), North America (76.92%, 950/1235), and Latin America (75.58%, 1003/1327). Although a high titer of ANA is an indication of ongoing autoimmunity, it is not the most robust predictor specific to SS. Hence, it has been removed from the 2016 ACR/EULAR classification criteria [18].
However, the importance of ANA in SS is exemplified by the observations that ANA- positive patients are young, and they have higher disease activity scores (ESSDAI) with greater involvement in the lymphadenopathy, cutaneous, hematological and biological ESSDAI domains [6].
In NZB/W F1 mice, ANA were first detected by indirect immunofluorescence or by latex nucleoprotein flocculation techniques, in approximately 20% of mice by the age of 2–4 months [23]. By 12 months of age, the incidence became 100%. In other reports using indirect immunofluorescence assays on human leukocyte substrates, NZB/W F1 mice showed a low titer of ANA (1:2 serum dilution) as early as 5wks of age that steadily increased as the mice got old. By 5–6 months, the ANA was more pronounced, with the titer doubling further in mice surviving to 9 months of age [59].
A recent report on the clinical relevance of indirect immunofluorescence staining by immune sera on human cells of epithelial origin (HEp-2) provides guidelines for interpreting the ANA patterns [60]. Sera from SS patients show nuclear (fine speckled, punctate nucleolar, and centromere-associated protein-F like), cytoplasmic (reticular/anti-mitochondrial), and mitotic (nuclear mitotic apparatus) staining patterns on HEp-2 cells. The presence of heterogeneous antibody populations targeting different antigens is responsible for these numerous ANA staining patterns in SS patients. In NZB/W F1 mice, the nuclear staining shows either a homogenous or a speckled pattern, while other ANA stained along the nuclear rim. Whether ANA from SS patients and NZB/W F1 mice target similar or different cellular antigens is not known.
Proteomic identification of these antigens might shed more light on the evolution of autoimmune reactivity and the role of ANA in SS pathogenesis.
5.2. Anti-SSA and Anti-SSB
Ro60 and Ro52 proteins encoded by the TROVE2 and TRIM21 genes, represent the Sjögren’s syndrome antigen A (SSA). The SSB gene encodes the Sjögren’s syndrome antigen B (SSB) or the La autoantigen. Ro60 and La proteins are a part of the Ro-RNP complex via their binding to the small cytoplasmic hY-RNAs [61]. The Ro52 protein belongs to a large family of the tripartite motif (TRIM) containing proteins and is an E3 ubiquitin ligase [62]. In contrast to Ro60 and La, only a tiny proportion of Ro52 is found associated with the RNP complex [63]. Autoantibodies targeting the Ro and La proteins are detected in 70% and 45% of SS patients, respectively; [6] and anti-Ro are included in the classification criteria for SS [18].
The presence of anti-Ro and anti-La antibodies in NZB/W F1 mice has been demonstrated mainly in assays employing purified recombinant proteins. In one of the earliest reports, sera obtained from female and male NZB/W F1 mice at 30 and 33 weeks of age, respectively, failed to show any Ro and La reactivity in an Ouchterlony double diffusion assay [64]. This assay detects antibodies reactive with the native conformation of the protein, and the sensitivity can be substantially lower than other immunoassays. Thus, many later studies have employed purified recombinant Ro and La proteins for antibody detection. In our studies, 5-month-old female and male mice showed weak reactivity to denatured recombinant Ro60, Ro52, and La/SSB [65]. These antibodies appeared to be poly-reactive, as incubation of sera with lupus-associated autoantigen, SmD, significantly abrogated reactivity to the Ro and La proteins.
Interestingly, following treatment of NZB/W F1 mice with Freund’s incomplete adjuvant (IFA), the levels of these anti-Ro60 antibodies were amplified [49]. Reactivity to recombinant Ro protein has also been reported by other groups [39,42]. In sera analyzed by using a commercial assay kit, mice were deemed anti-Ro/SSA positive from 18–22 weeks of age. However, very low OD405 readings (0.01 to 0.12) and lack of information on the number of positive mice indicates that these data need to be interpreted with caution. Similarly, reactivity to denatured Ro60 and recombinant La/SSB was demonstrated in 34-week old mice, and these antibodies declined following treatment of mice with antagonists targeting TLR7, TLR8, and TLR9 [66].
Considered together, a critical analysis of published reports suggests that NZB/W F1 mice do not develop immunoprecipitating, high affinity, and high titer anti-Ro and anti-La antibody responses that are comparable to those seen in SS patients. Thus, the NZB/W F1 mice represent a subset of SS patients that are anti-Ro/La negative.
5.3. Rheumatoid Factor (RF)
RF of IgM, IgG, and IgA class are detected in SS patients, with a prevalence ranging from 36 to 74% [57]. Although the diagnostic value of RF to distinguish SS from other autoimmune disorders is debatable, they have a considerable prognostic value [67, 68]. The presence of RF is associated with higher disease severity and extraglandular manifestations. In NZB/W F1 mice, although IgM RF (reactive with mouse IgG) was detected, their levels were not significantly elevated compared to healthy age-matched non-autoimmune C57BL/6 and CBA/St mice [69]. In a study by Singh et al. [70], IgG RF was detected in about 30% of mice by 6 months of age. Genetic deletion of β2 microglobulin in NZB/W F1 mice resulted in a dramatic increase in RF positivity to 80%, compared to littermate WT mice at 35% positivity. In another report, RF of the IgG1 isotype was not readily detected in sera of young NZB/W F1 mice but could be induced following the injection of mice with protein-G binding material from human RA synovial fluid. Overall, these data collectively demonstrate that NZB/W F1 mice retain RF reactive B cells that can be activated into producing RF.
5.4. Cryoglobulins
Immunoglobulins that precipitate at temperatures below the normal body temperature of 37°C are considered as cryoglobulins [71]. The composition of cryoglobulins is heterogeneous, comprising of IgM and IgG antibodies, with some even showing RF activity. In the Sjögren Big data project, of the 4732 SS patients tested, 342 or 7.3% were positive for cryoglobulins [6]. The most striking observation from this study was that patients with cryoglobulinemia showed the highest mean ESSDAI score among all immunological subsets (anti-Ro, anti-La, anti-Ro+La, ANA, RF, C4, and C3). The mean ESSADI of patients with cryoglobulins was four-fold higher than patients negative for any immunological marker and almost three-fold higher for patients with positive ANA or anti-Ro. The presence of cryoglobulins in SS is also associated with an increased risk of lymphomagenesis [72].
Andrews et al. [69] showed that at six months of age, the average cryoglobulin level in NZB/W F1 female mice was 340 μg/ml, compared to (100–200 μg/ml) in non- autoimmune mice. The cryoglobulins were predominantly IgM and IgG1 antibodies, followed by IgG2a. In addition, C3 complement was found in 20% of cryoglobulins.
Despite these observations, the correlation between cryoglobulin levels and disease severity in NZB/W F1 mice is not known.
5.5. Other Autoantibodies
The presence of anti-dsDNA antibodies is a hallmark of systemic lupus erythematosus (SLE), and these antibodies are readily detected in NZB/W F1 mice. Anti-dsDNA antibodies start appearing by 4–5 months of age, and their titer increases almost three-fold by nine months [69]. The frequency of anti-dsDNA reported in SS patients is highly variable and appears to be assay dependent. Anti-DNA reactivity was seen in 63% (20/32) of SS patient sera when measured by ELISA using calf thymus DNA [73]. However, none of these sera were positive in an immunofluorescence assay employing Crithidia luciliae as a substrate. Other studies show that 4–6% of SS patients have antibodies to native DNA [74]. Interestingly, this report demonstrated that antibodies to ribonucleoproteins (RNP) were associated with a higher incidence of extraglandular manifestations [74].
Similarly, there was a strong association between anti-RNP positivity and hypergammaglobulinemia, myositis, and pulmonary involvement in SS patients with connective tissue disease [75]. Several other autoantibody specificities like anti- centromere, anti-mitochondrial, anti-smooth muscle, anti-carbonic anhydrase, anti- alpha fodrin, anti-muscarinic 3 receptor, and anti-TRIM38 have been reported in SS patients [57, 58]. Our literature search did not reveal that the presence of these antibodies was explicitly investigated in NZB/W F1 mice.
In summary, some of the autoantibody specificities in NZB/W F1 mice are shared with a subset of SS patients. Based on the current data in the literature, the presence of anti-Ro/La in NZB/W F1 mice is debatable. It needs resolution by immunoassays that use proteins in their native conformation. The identity of proteins recognized by ANA in SS patients will provide important information on antigens that might contribute towards disease pathogenesis. Regardless, the presence of circulating autoantibodies in SS patients and NZB/W F1 mice provides evidence for B cell hyperactivity and immune dysregulation.
6. Glandular Dysfunction
The defining clinical features of SS include reduced tear and saliva production leading to dry eye and dry mouth. It is now well accepted that both immune and non- immune mechanisms contribute to glandular dysfunction. In NZB/W F1 mice, reduced volume of agonist-induced salivation and altered saliva protein content are indicators of salivary gland dysfunction. Tear volume measurement has been the mainstay for the evaluation of lacrimal gland dysfunction.
6.1. Salivary gland dysfunction
The first evidence for salivary gland dysfunction in NZB/W F1 mice was provided in the original description of the model by Kessler [24]. Following the subcutaneous administration of the sialagogue methacholine hydrochloride (1 mg/kg body weight), saliva was aspirated from the floor of the mouth. Amylase concentration in NZB/W F1 saliva was almost twofold lower than that observed for NZW. No significant differences in sodium, potassium, or chloride levels were noted. However, no information on the total saliva volume or salivary flow rate was provided.
Since xerostomia is a predominant complaint in SS patients, the amount of fluid secretion is a crucial measure of salivary gland function. NZB/W F1 mice show an age- dependent decline in parotid saliva production [47]. Females at 6–6.5 months made significantly less saliva (11 μl/100 gm body weight/min) compared to younger 2.3–2.5 months old females (22 μl/100 gm body weight/min). In contrast, male NZB/W F1 failed to show a substantial drop in saliva production with age (15 μl/100 g body weight/min to 11 μl/100g body weight/min). A point of interest was that NZB/W F1 males had significantly lower saliva volume than age-matched non-autoimmune B6 male mice.
In our studies, we have also reported a significant drop in saliva production in female NZB/W F1 mice by six months of age [49]. This saliva drop was significantly accelerated (to 3.6 months) in mice injected with IFA. Interestingly, the glandular dysfunction in IFA treated mice did not correlate with the severity of inflammation. Instead, glandular dysfunction was associated with increased frequency of plasmacytoid dendritic cells within the submandibular glands of IFA treated mice. These findings suggest that activation of innate immunity contributed to salivary gland dysfunction in NZB/W F1 mice.
Further proof for the role of innate immunity, specifically type I interferon (IFN), in glandular dysfunction derives from our studies in NZB/W F1 mice treated with the TLR3 agonist, poly (I:C) [76–78]. A rapid but transient loss in saliva production occurred in poly (I:C) treated mice [76]. This functional loss was dependent on type I IFN as mice deficient in interferon receptor were partially protected from the effects of poly (I:C) [77]. Another consequence of poly (I:C) treatment was an accelerated onset of sialadenitis with greater severity [78]. In this context, it is of interest to note that well before the knowledge of TLRs or cytoplasmic RNA sensors, Steinberg, Baron, and Talal demonstrated that poly (I:C) injected NZB/W F1 mice made type I IFN, had an earlier onset of anti-DNA, and developed accelerated lupus-nephritis [79]. However, the effects on salivary gland disease were not investigated. Based on these observations in NZB/W F1 mice, the possibility that glandular dysfunction in SS patients might be strongly influenced by type I IFN should be considered. The minor labial salivary glands in SS patients harbor plasmacytoid dendritic cells (pDCs), which are considered to be the most prolific type I IFN producing cells. Whether the presence of pDCs in salivary glands of SS patients is associated with glandular dysfunction needs to be carefully evaluated.
6.2. Lacrimal gland dysfunction
The Schirmer test is used as an objective measure to evaluate dry eye in SS patients [80]. A patient is considered to have a dry eye if the wetting distance of the filter paper strip placed inside the lower eyelid in 5 min is less than 5 mm. A modified Schirmer test using a Whatman#2 filter paper strip (1.5 mm × 15 mm) showed that 6–7- month-old NZB/W F1 mice gave a lower wetting distance (mean= 3.1 mm; range 1.7–6.0 mm) compared to age-matched NZW mice (mean=4.8 mm; a range of 3.0–8.0 mm) [81]. This first description of tear measurement in NZB/W F1 mice provides evidence for lacrimal gland dysfunction in this mouse strain.
Direct measurement of tear production was performed by inserting a micro- capillary into a severed lacrimal duct. The secreted fluid was collected for 10 to 15 minutes, with or without carbachol stimulation [82]. Surprisingly, despite the presence of lacrimal gland inflammation in old NZB/W F1 mice, the mean fluid volumes from 8- month-old NZB/W F1 were higher than 2-month-old mice. These observations show a complete dissociation between the severity of lacrimal gland inflammation and tear production. In this study, the age-matched non-autoimmune Swiss Webster females showed significantly higher tear production at all ages compared to the NZB/W F1 mice.
The lack of correlation between inflammation and dysfunction has led to the hypothesis that inherent glandular defects lead to lower fluid secretion in SS. Support for this thesis is provided by patch-clamp studies that show a reduction in calcium flux in NZB/W F1 lacrimal gland acinar cells compared to those from Swiss Webster mice [83]. However, the acinar cells from older NZB/W F1 mice showed increased levels of basal calcium, thereby suggesting an inability to sequester the intracellular calcium stores. Recent studies in salivary glands of SS patients show that a poor response of acinar cells to carbachol stimulation was linked with reduced expression of inositol 1,4,5 triphosphate receptors (IP3R), specifically IP3R2 and IP3R3 that play a role in releasing Ca2+ from the lumen of the endoplasmic reticulum [84]. Whether similar mechanisms exist in lacrimal gland acinar cells from NZB/W F1 mice needs to be investigated.
A severe dry eye often contributes to corneal and conjunctival damage, which is evaluated by staining of the ocular surface with fluorescein and lissamine green [85]. Ocular staining score ≥ 5 (or van Bijsterfeld score ≥ 4) on at least one eye is one of the classification criteria for SS [18]. Despite lacrimal gland inflammation, NZB/W F1 females (n=43) failed to show significant differences in tear film osmolarity and ocular surface staining compared to age-matched non-autoimmune BALB/c and DBA mice [86]. In this study, NZB/W F1 males (n=31) had lower lacrimal gland inflammation compared to female mice and also failed to show corneal or conjunctival damage.
In summary, NZB/W F1 mice show ample evidence for reduced saliva and tear production. The glandular dysfunction does not correlate with the severity of inflammatory cell infiltration within the exocrine salivary and lacrimal gland. This observation echoes the findings from several SS patients showing discordance between fluid secretion and the magnitude of glandular inflammation.
7. SS and polyautoimmunity
The NZB/W F1 female mice recapitulate multiple features of SS, including exocrine gland histopathology, presence of autoantibodies, and glandular dysfunction. These mice also develop anti-dsDNA and severe lupus-like nephritis, and they have been studied extensively as an animal model for SLE. Historically, a patient with an autoimmune disease like SLE, rheumatoid arthritis, or systemic sclerosis who also fulfilled SS classification criteria was considered a secondary SS patient [17]. This nomenclature was based on the temporal appearance of SS, which is typically diagnosed in women at an older age compared to the other autoimmune diseases [87–89].
Qualifying SS as a “secondary” disease has unintentionally propagated an assumption that this codification may have an etiologic basis. An alternative concept is to describe the presence of more than one well-defined autoimmune disease in a single individual, as polyautoimmunity [90, 91]. In this regard, the NZB/W F1 mouse represents a model for polyautoimmunity in SS.
The coexistence of multiple autoimmune diseases is not uncommon in clinical practice. A cross-sectional study showed that one-third of SS patients (134/440; 32.6%) had signs of other diseases, including autoimmune thyroid disease, rheumatoid arthritis, SLE, and inflammatory bowel disease [92]. In different SLE patient cohorts, as many as 7–33% of the patients also fulfill the classification criteria for SS [37, 88, 93–95]. An extensive analysis of clinical and laboratory parameters showed that SLE patients with SS form a distinct subset from the SLE patients without SS [91] suggesting that distinct cellular and molecular pathways might be operating in these patients. The NZB/W F1 mouse is a valuable tool for elucidating pathways responsible for concomitantly driving SS and lupus. It is also a model for evaluating therapeutic strategies to treat both disorders.
The goals in developing new drugs for SS include improvement of subjective measures, increased salivary flow, and reduce xerostomia. NZB/W F1 mice with established SS showed improved salivary gland function and reduced inflammation after treatment with an antibody to CD40 [96]. An ongoing clinical trial with CFZ533, a new monoclonal anti-CD40, showed promising improvement in SS disease activity and patient-reported indices [97]. Whether CFZ5333 is useful in the treatment lupus nephritis is under investigation [98]. The mechanisms underlying the therapeutic effects are unclear, and the NZB/W F1 mice will provide some insights into the pathways modulated by anti-CD40 treatment. In this regard, other approaches, such as calorie- reduction, have yielded promising results in alleviating both the exocrine gland and kidney disease in NZB/W F1 mice [99]. Whether these or other strategies succeed in patients remains to be determined.
Highlights.
NZB/W F1 mouse is a model for polyautoimmunity in Sjögren’s syndrome.
NZB/W F1 mouse is a valuable tool for investigating pathogenic mechanisms and devise therapeutic strategies for Sjögren’s syndrome patients with lupus.
NZB/W F1 mouse recapitulates several features of the human disease.
Acknowledgements.
This work was supported by grants from the National Institute of Dental and Craniofacial Research (DE025030), National Institutes of Health, USA. The funding source had no involvement in study design; in the collection, analysis and interpretation of data. Expert assistance from the OMRF imaging core and the OMRF flow cytometry core is gratefully acknowledged.
Footnotes
Declarations of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- [1].Sarmiento-Monroy JC, Mantilla RD, Rojas-Villarraga A, et al. Sjögren’s syndrome In: Anaya JM, Shoenfeld Y, Rojas-Villarraga A, et al., editors. Autoimmunity: From Bench to Bedside [Internet]: El Rosario: University Press; 2013. Jul 18, Chapter 28. [PubMed] [Google Scholar]
- [2].Restrepo-Jimenez P, Molano-Gonzalez N, Anaya J-M. Geoepidemiology of Sjogren’s syndrome in Latin America. Jt Bone Spine 2019;86:620–6. doi: 10.1016/j.jbspin.2019.02.004. [DOI] [PubMed] [Google Scholar]
- [3].Ramos-Casals M, Brito-Zerón P, Kostov B, Sisó-Almirall A, Bosch X, Buss D, et al. Google-driven search for big data in autoimmune geoepidemiology: Analysis of 394,827 patients with systemic autoimmune diseases. Autoimmun Rev 2015;14:670–9. doi: 10.1016/j.autrev.2015.03.008. [DOI] [PubMed] [Google Scholar]
- [4].Psianou K, Panagoulias I, Papanastasiou AD, de Lastic A-L, Rodi M, Spantidea PI, et al. Clinical and immunological parameters of Sjögren’s syndrome. Autoimmun Rev 2018;17:1053–64. doi: 10.1016/j.autrev.2018.05.005. [DOI] [PubMed] [Google Scholar]
- [5].Vivino F, Bunya VY, Massaro-Giordano G, Johr CR, Giattino SL, Schorpion A, et al. Sjogren’s syndrome: An update on disease pathogenesis, clinical manifestations and treatment. Clin Immunol 2019;203:81–121. doi: 10.1016/j.clim.2019.04.009. [DOI] [PubMed] [Google Scholar]
- [6].Brito-Zeron P, Acar-Denizli N, Ng W-F, Zeher M, Rasmussen A, Mandl T, et al. How immunological profile drives clinical phenotype of primary Sjogren’s syndrome at diagnosis: analysis of 10,500 patients (Sjogren Big Data Project). Clin Exp Rheumatol 2018;36 Suppl 1:102–12. [PubMed] [Google Scholar]
- [7].James JA, Guthridge JM, Chen H, Lu R, Bourn RL, Bean K, et al. Unique Sjogren’s syndrome patient subsets defined by molecular features. Rheumatology 2020;59:860–8. doi: 10.1093/rheumatology/kez335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat Genet 2013;45:1284–94. doi: 10.1038/ng.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jonsson R, Theander E, Sjöström B, Brokstad K, Henriksson G. Autoantibodies present before symptom onset in primary Sjögren syndrome. JAMA - J Am Med Assoc 2013;310:1854–5. doi: 10.1001/jama.2013.278448. [DOI] [PubMed] [Google Scholar]
- [10].Theander E, Jonsson R, Sjöström B, Brokstad K, Olsson P, Henriksson G. Prediction of Sjögren’s syndrome years before diagnosis and identification of patients with early onset and severe disease course by autoantibody profiling. Arthritis Rheumatol 2015;67:2427–36. doi: 10.1002/art.39214. [DOI] [PubMed] [Google Scholar]
- [11].Bartoloni E, Alunno A, Gerli R. The dark side of Sjögren’s syndrome: the possible pathogenic role of infections. Curr Opin Rheumatol 2019;31:505–11. doi: 10.1097/BOR.0000000000000631. [DOI] [PubMed] [Google Scholar]
- [12].Maślińska M. The role of Epstein-Barr virus infection in primary Sjögren’s syndrome. Curr Opin Rheumatol 2019;31:475–83. doi: 10.1097/BOR.0000000000000622. [DOI] [PubMed] [Google Scholar]
- [13].van Blokland SCA, Versnel MA. Pathogenesis of Sjögren’s syndrome: characteristics of different mouse models for autoimmune exocrinopathy. Clin Immunol 2002;103:111–24. doi: 10.1006/clim.2002.5189. [DOI] [PubMed] [Google Scholar]
- [14].Park Y-S, Gauna A, Cha S. Mouse models of primary Sjogren’s syndrome. Curr Pharm Des 2015;21:2350–64. doi: 10.2174/1381612821666150316120024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xiao F, Han M, Wang X, Gong X, Huang E, Zhu Z, et al. Animal models of Sjögren’s syndrome: an update. Clin Exp Rheumatol 2019;37 Suppl 1:209–16. [PubMed] [Google Scholar]
- [16].Vitali C, Bombardieri S, Moutsopoulos HM, Balestrieri G, Bencivelli W, Bernstein RM, et al. Preliminary criteria for the classification of Sjögren’s syndrome. Results of a prospective concerted action supported by the European Community. Arthritis Rheum 1993;36:340–7. doi: 10.1002/art.1780360309. [DOI] [PubMed] [Google Scholar]
- [17].Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002;61:554–8. doi: 10.1136/ard.61.6.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data- driven methodology involving three international patient cohorts. Arthritis Rheumatol 2017;69:35–45. doi: 10.1002/art.39859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bagavant H, Trzeciak M, Papinska J, Biswas I, Dunkleberger ML, Sosnowska A, et al. A method for the measurement of salivary gland function in mice. J Vis Exp 2018; (131):57203. doi: 10.3791/57203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bielschowsky M, Helver BJ, Howie JB. Spontaneous anemia in mice of the NZB/BL strain. Proc Univ Otago Med Sch 1959;37:9–11. [Google Scholar]
- [21].Helyer BJ, Howie JB. Positive lupus erythematosis tests in a cross-bred strain of mice NZB/BL-NZY/BL. Proc Univ Otago Med Sch 1961;39:17–8. [Google Scholar]
- [22].Helyer BJ, Howie JB. Renal disease associated with positive lupus erythematosus tests in a cross-bred strain of mice. Nature 1963;197:197. doi: 10.1038/197197a0. [DOI] [PubMed] [Google Scholar]
- [23].Howie JB, Helyer BJ. The immunology and pathology of NZB mice. Adv Immunol 1968;9:215–66. doi: 10.1016/s0065-2776(08)60444-7. [DOI] [PubMed] [Google Scholar]
- [24].Kessler HS. A laboratory model for Sjogren’s syndrome. Am J Pathol 1968;52:671–85. [PMC free article] [PubMed] [Google Scholar]
- [25].Borchers A, Ansari AA, Hsu T, Kono DH, Gershwin ME. The pathogenesis of autoimmunity in New Zealand mice. Semin Arthritis Rheum 2000;29:385–99. doi: 10.1053/sarh.2000.7173. [DOI] [PubMed] [Google Scholar]
- [26].Kono DH, Burlingame RW, Owens DG, Kuramochi A, Balderas RS, Balomenos D, et al. Lupus susceptibility loci in New Zealand mice. Proc Natl Acad Sci USA 1994;91:10168–72. doi: 10.1073/pnas.91.21.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Drake CG, Rozzo SJ, Vyse TJ, Palmer E, Kotzin BL. Genetic contributions to lupus-like disease in (NZB × NZW)F1 mice. Immunol Rev 1995;144:51–74. doi: 10.1111/j.1600-065x.1995.tb00065.x. [DOI] [PubMed] [Google Scholar]
- [28].Flores-Chávez A, Kostov B, Solans R, Fraile G, Maure B, Feijoo-Massó C, et al. Severe, life-threatening phenotype of primary Sjögren’s syndrome: clinical characterisation and outcomes in 1580 patients (GEAS-SS Registry). Clin Exp Rheumatol 2018;36 Suppl 1:121–9. [PubMed] [Google Scholar]
- [29].Sjögren H Zur kenntnis der Keratoconjunctivitis Sicca. Keratitis filiformis bei hypofunction der Tränendrüsen. (A new conception of keratoconjunctivitis sicca (keratitis filiformis in hypofunction of the lacrimal glands). Acta Ophthalmol 1933:1–151. [Google Scholar]
- [30].Waterhouse JP. Focal adenitis in salivary and lacrimal glands. Proc R Soc Med 1963;56:911–8. [PMC free article] [PubMed] [Google Scholar]
- [31].Calman HI, Reifman S. Sjogren’s syndrome. Report of a case. Oral Surg Oral Med Oral Pathol 1966;21:158–62. doi: 10.1016/0030-4220(66)90236-2. [DOI] [PubMed] [Google Scholar]
- [32].Chisholm DM, Mason DK. Labial salivary gland biopsy in Sjogren’s disease. J Clin Pathol 1968;21:656–60. doi: 10.1136/jcp.21.5.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fisher BA, Jonsson R, Daniels T, Bombardieri M, Brown RM, Morgan P, et al. Standardisation of labial salivary gland histopathology in clinical trials in primary Sjögren’s syndrome. Ann Rheum Dis 2017;76:1161–8. doi: 10.1136/annrheumdis-2016-210448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Carubbi F, Alunno A, Cipriani P, Coletti G, Bigerna B, Manetti M, et al. Different operators and histologic techniques in the assessment of germinal center-like structures in primary Sjögren’s syndrome minor salivary glands. PLoS One 2019;14:e0211142. doi: 10.1371/journal.pone.0211142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Morgan WS, Castleman B. A clinicopathologic study of Mikulicz’s disease. Am J Pathol 1953;29:471–503. [PMC free article] [PubMed] [Google Scholar]
- [36].Bloch KJ, Buchanan WW, Wohl MJ, Bunim JJ. Sjögren’s Syndrome. A clinical, pathological, and serological study of sixty-two cases. Medicine 1965;44:187–231. [PubMed] [Google Scholar]
- [37].Jonsson R, Tarkowski A, Backman K, Klareskog L. Immunohistochemical characterization of sialadenitis in NZB X NZW F1 mice. Clin Immunol Immunopathol 1987;42:93–101. doi: 10.1016/0090-1229(87)90176-0. [DOI] [PubMed] [Google Scholar]
- [38].Hayashi T, Shirachi T, Hasegawa K. Relationship between sialoadenitis and periductal laminin expression in the submandibular salivary gland of NZBxNZWF(1) mice. J Comp Pathol 2001;125:110–6. doi: 10.1053/jcpa.2001.0493. [DOI] [PubMed] [Google Scholar]
- [39].Hayashi T, Hayashi H, Fujii T, Adachi C, Hasegawa K. Ultrastructure of myoepithelial cells as a target cell in sialoadenitis of submandibular glands of lupus-prone female NZB×NZWF1 mice. Virchows Arch 2008;453:177–88. doi: 10.1007/s00428-008-0627-4. [DOI] [PubMed] [Google Scholar]
- [40].Greenspan JS, Daniels TE, Talal N, Sylvester RA. The histopathology of Sjögren’s syndrome in labial salivary gland biopsies. Oral Surg Oral Med Oral Pathol 1974;37:217–29. doi: 10.1016/0030-4220(74)90417-4. [DOI] [PubMed] [Google Scholar]
- [41].Ohashi Y, So SK, Minasi PN, Tabbara KF. The presence of cytotoxic autoantibody to lacrimal gland cells in NZB/W mice. Invest Ophthalmol Vis Sci 1985;26:214–9. [PubMed] [Google Scholar]
- [42].Hayashi T, Adachi C, Hasegawa K. Systemic treatment of anti-CD4CD25 T cell monoclonal antibody exacerbates sialoadenitis in submandibular glands during the early life in lupus-prone female NZB × NZWF mice. J Oral Pathol Med 2009;38:234–40. doi: 10.1111/j.1600-0714.2008.00730.x. [DOI] [PubMed] [Google Scholar]
- [43].Fernandes JD, Nico MMS, Aoki V, Bologna S, Romiti R, Levy-Neto M, et al. Xerostomia in Sjogren’s syndrome and lupus erythematosus: a comparative histological and immunofluorescence study of minor salivary glands alterations. J Cutan Pathol 2010;37:432–8. doi: 10.1111/j.1600-0560.2009.01368.x. [DOI] [PubMed] [Google Scholar]
- [44].Gao J, Cha S, Jonsson R, Opalko J, Peck AB. Detection of anti-type 3 muscarinic acetylcholine receptor autoantibodies in the sera of Sjögren’s syndrome patients by use of a transfected cell line assay. Arthritis Rheum 2004;50:2615–21. doi: 10.1002/art.20371. [DOI] [PubMed] [Google Scholar]
- [45].Szczerba BM, Kaplonek P, Wolska N, Podsiadlowska A, Rybakowska PD, Dey P, et al. Interaction between innate immunity and Ro52-induced antibody causes Sjogren’s syndrome-like disorder in mice. Ann Rheum Dis 2016;75:617–22. doi: 10.1136/annrheumdis-2014-206297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nikolov NP, Illei GG. Pathogenesis of Sjogren’s syndrome. Curr Opin Rheumatol 2009;21:465–70. doi: 10.1097/BOR.0b013e32832eba21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wolff A, Scott J, Woods K, Fox PC. An investigation of parotid gland function and histopathology in autoimmune disease-prone mice of different age groups. J Oral Pathol Med 1991;20:486–9. doi: 10.1111/j.1600-0714.1991.tb00409.x. [DOI] [PubMed] [Google Scholar]
- [48].Wolska N, Rybakowska P, Rasmussen A, Brown M, Montgomery C, Klopocki A, et al. Brief report: patients with primary Sjögren’s syndrome who are positive for autoantibodies to tripartite motif-containing protein 38 show greater disease severity. Arthritis Rheumatol 2016;68:724–9. doi: 10.1002/art.39497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Deshmukh US, Ohyama Y, Bagavant H, Guo X, Gaskin F, Fu SM. Inflammatory stimuli accelerate Sjogren’s syndrome-like disease in (NZB × NZW)F1 mice. Arthritis Rheum 2008;58:1318–23. doi: 10.1002/art.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chaudhry AP, Cutler LS, Yamane GM, Satchidanand S, Labay G, Sunderraj M. Light and ultrastructural features of lymphoepithelial lesions of the salivary glands in Mikulicz’s disease. J Pathol 1986;148:239–50. doi: 10.1002/path.1711480308. [DOI] [PubMed] [Google Scholar]
- [51].Carlsoo B, Ostberg Y. Ultrastructural observations on the parotitis autoimmunica in the NZB/NZW hybrid mice. Acta Otolaryngol 1978;85:298–306. doi: 10.3109/00016487809111939. [DOI] [PubMed] [Google Scholar]
- [52].Carlsoo B, Ostberg Y. The autoimmune submandibular sialoadenitis of the NZB/NZW hybrid mice. A light and electron microscopical investigation. Arch Otorhinolaryngol 1979;225:57–65. doi: 10.1007/BF00455876. [DOI] [PubMed] [Google Scholar]
- [53].Sisto M, Lorusso L, Ingravallo G, Tamma R, Nico B, Ribatti D, et al. Reduced myofilament component in primary Sjogren’s syndrome salivary gland myoepithelial cells. J Mol Histol 2018;49:111–21. doi: 10.1007/s10735-017-9751-2. [DOI] [PubMed] [Google Scholar]
- [54].Aqrawi LA, Jensen JL, Øijordsbakken G, Ruus A-K, Nygård S, Holden M, et al. Signalling pathways identified in salivary glands from primary Sjögren’s syndrome patients reveal enhanced adipose tissue development. Autoimmunity 2018;51:135–46. doi: 10.1080/08916934.2018.1446525. [DOI] [PubMed] [Google Scholar]
- [55].Leehan KM, Pezant NP, Rasmussen A, Grundahl K, Moore JS, Radfar L, et al. Minor salivary gland fibrosis in Sjogren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin Exp Rheumatol 2018;36 Suppl 1:80–8. [PMC free article] [PubMed] [Google Scholar]
- [56].Hayashi T, Hasegawa K, Ichinohe N. ICAM-1 expression on endothelium and systemic cytokine production in cutaneous neutrophilic leukocytoclastic vasculitis in NZB×NZWF1 mice. Histol Histopathol 2005;20:45–52. doi: 10.14670/HH-20.45. [DOI] [PubMed] [Google Scholar]
- [57].Kyriakidis NC, Kapsogeorgou EK, Tzioufas AG. A comprehensive review of autoantibodies in primary Sjögren’s syndrome: clinical phenotypes and regulatory mechanisms. J Autoimmun 2014;51:67–74. doi: 10.1016/j.jaut.2013.11.001. [DOI] [PubMed] [Google Scholar]
- [58].Scofield RH, Fayyaz A, Kurien BT, Koelsch KA. Prognostic value of Sjögren’s syndrome autoantibodies. J Lab Precis Med 2018;3. doi: 10.21037/jlpm.2018.08.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Walker SE, Bole GGJ. Selective suppression of autoantibody responses in NZB/NZW mice treated with long-term cyclophosphamide. Arthritis Rheum 1975;18:265–72. doi: 10.1002/art.1780180312. [DOI] [PubMed] [Google Scholar]
- [60].Damoiseaux J, Andrade LEC, Carballo OG, Conrad K, Francescantonio PLC, Fritzler MJ, et al. Clinical relevance of HEp-2 indirect immunofluorescent patterns: the International Consensus on ANA patterns (ICAP) perspective. Ann Rheum Dis 2019;78:879–89. doi: 10.1136/annrheumdis-2018-214436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pruijn GJ, Slobbe RL, Van Venrooij WJ. Structure and function of La and Ro RNPs. Mol Biol Rep 1990;14:43–8. doi: 10.1007/BF00360410. [DOI] [PubMed] [Google Scholar]
- [62].Wada K, Kamitani T. Autoantigen Ro52 is an E3 ubiquitin ligase. Biochem Biophys Res Commun 2006;339:415–21. doi: 10.1016/j.bbrc.2005.11.029. [DOI] [PubMed] [Google Scholar]
- [63].Fabini G, Rutjes SA, Zimmermann C, Pruijn GJ, Steiner G. Analysis of the molecular composition of Ro ribonucleoprotein complexes. Identification of novel Y RNA-binding proteins. Eur J Biochem 2000;267:2778–89. doi: 10.1046/j.1432-1327.2000.01298.x. [DOI] [PubMed] [Google Scholar]
- [64].Hoffman RW, Alspaugh MA, Waggie KS, Durham JB, Walker SE. Sjögren’s syndrome in MRL/l and MRL/n mice. Arthritis Rheum 1984;27:157–65. doi: 10.1002/art.1780270206. [DOI] [PubMed] [Google Scholar]
- [65].Deshmukh US, Lewis JE, Gaskin F, Dhakephalkar PK, Kannapell CC, Waters ST, et al. Ro60 peptides induce antibodies to similar epitopes shared among lupus- related autoantigens. J Immunol 2000;164:6655–61. doi: 10.4049/jimmunol.164.12.6655. [DOI] [PubMed] [Google Scholar]
- [66].Zhu F-G, Jiang W, Bhagat L, Wang D, Yu D, Tang JX, et al. A novel antagonist of Toll-like receptors 7, 8 and 9 suppresses lupus disease-associated parameters in NZBW/F1 mice. Autoimmunity 2013;46:419–28. doi: 10.3109/08916934.2013.798651. [DOI] [PubMed] [Google Scholar]
- [67].Lee K-A, Kim K-W, Kim B-M, Won J-Y, Kim H-A, Moon H-W, et al. Clinical and diagnostic significance of serum immunoglobulin A rheumatoid factor in primary Sjogren’s syndrome. Clin Oral Investig 2019;23:1415–23. doi: 10.1007/s00784-018-2545-4. [DOI] [PubMed] [Google Scholar]
- [68].Maślińska M, Mańczak M, Kwiatkowska B. Usefulness of rheumatoid factor as an immunological and prognostic marker in PSS patients. Clin Rheumatol 2019;38:1301–7. doi: 10.1007/s10067-019-04438-z. [DOI] [PubMed] [Google Scholar]
- [69].Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med 1978;148:1198–215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Singh RR, Yang JQ, Kim PJ, Halder RC. Germline deletion of β2 microglobulin or CD1d reduces anti-phospholipid antibody, but increases autoantibodies against non-phospholipid antigens in the NZB/W F1 model of lupus. Arthritis Res Ther 2013;15. doi: 10.1186/ar4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet 2012;379:348–60. doi: 10.1016/S0140-6736(11)60242-0. [DOI] [PubMed] [Google Scholar]
- [72].Kapsogeorgou EK, Voulgarelis M, Tzioufas AG. Predictive markers of lymphomagenesis in Sjögren’s syndrome: From clinical data to molecular stratification. J Autoimmun 2019;104:102316. doi: 10.1016/j.jaut.2019.102316. [DOI] [PubMed] [Google Scholar]
- [73].Salonen EM, Miettinen A, Walle TK, Koskenmies S, Kere J, Julkunen H. Anti- telomere antibodies in systemic lupus erythematosus (SLE): a comparison with five antinuclear antibody assays in 430 patients with SLE and other rheumatic diseases. Ann Rheum Dis 2004;63:1250–4. doi: 10.1136/ard.2003.011890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Fauchais AL, Martel C, Gondran G, Lambert M, Launay D, Jauberteau MO, et al. Immunological profile in primary Sjogren syndrome: clinical significance, prognosis and long-term evolution to other auto-immune disease. Autoimmun Rev 2010;9:595–9. doi: 10.1016/j.autrev.2010.05.004. [DOI] [PubMed] [Google Scholar]
- [75].Abbara S, Seror R, Henry J, Chretien P, Gleizes A, Hacein-Bey-Abina S, et al. Anti- RNP positivity in primary Sjogren’s syndrome is associated with a more active disease and a more frequent muscular and pulmonary involvement. RMD Open 2019;5:e001033. doi: 10.1136/rmdopen-2019-001033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Deshmukh US, Nandula SR, Thimmalapura P-R, Scindia YM, Bagavant H. Activation of innate immune responses through Toll-like receptor 3 causes a rapid loss of salivary gland function. J Oral Pathol Med 2009;38:42–7. doi: 10.1111/j.1600-0714.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Nandula S-R, Dey P, Corbin KL, Nunemaker CS, Bagavant H, Deshmukh US. Salivary gland hypofunction induced by activation of innate immunity is dependent on type I interferon signaling. J Oral Pathol Med 2013;42:66–72. doi: 10.1111/j.1600-0714.2012.01181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Nandula S-R, Scindia YM, Dey P, Bagavant H, Deshmukh US. Activation of innate immunity accelerates sialoadenitis in a mouse model for Sjögren’s syndrome-like disease. Oral Dis 2011;17:801–7. doi: 10.1111/j.1601-0825.2011.01839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Steinberg AD, Baron S, Talal N. The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinic- polycytidylic acid. Proc Natl Acad Sci U S A 1969;63:1102–7. doi: 10.1073/pnas.63.4.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Beckman KA, Luchs J, Milner MS. Making the diagnosis of Sjögren’s syndrome in patients with dry eye. Clin Ophthalmol 2016;10:43–53. doi: 10.2147/OPTH.S80043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kessler HS, Cubberly M, Manski W. Eye changes in autoimmune NZB and NZB × NZW mice. Comparison with Sjogren’s syndrome. Arch Ophthalmol 1971;85:211–9. doi: 10.1001/archopht.1971.00990050213016. [DOI] [PubMed] [Google Scholar]
- [82].Paranyuk Y, Claros N, Birzgalis A, Moore LC, Brink PR, Walcott B. Lacrimal gland fluid secretion and lymphocytic infiltration in the NZB/W mouse model of Sjogren’s syndrome. Curr Eye Res 2001;23:199–205. doi: 10.1076/ceyr.23.3.199.5468. [DOI] [PubMed] [Google Scholar]
- [83].Walcott B, Matthews G, Brink P. Differences in stimulus induced calcium increases in lacrimal gland acinar cells from normal and NZB/NZW F1 female mice. Curr Eye Res 2002;25:253–60. doi: 10.1076/ceyr.25.4.253.13489. [DOI] [PubMed] [Google Scholar]
- [84].Teos LY, Zhang Y, Cotrim AP, Swaim W, Won JH, Ambrus J, et al. IP3R deficit underlies loss of salivary fluid secretion in Sjögren’s Syndrome. Sci Rep 2015;5:13953. doi: 10.1038/srep13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rasmussen A, Stone DU, Kaufman CE, Hefner KS, Fram NR, Siatkowski RL, et al. Reproducibility of ocular surface staining in the assessment of Sjögren syndrome- related Keratoconjunctivitis Sicca: implications on disease classification. ACR Open Rheumatol 2019;1:292–302. doi: 10.1002/acr2.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Gilbard JP, Hanninen LA, Rothman RC, Kenyon KR. Lacrimal gland, cornea, and tear film in the NZB/NZW F1 hybrid mouse. Curr Eye Res 1987;6:1237–48. doi: 10.3109/02713688709025234. [DOI] [PubMed] [Google Scholar]
- [87].Moutsopoulos HM, Webber BL, Vlagopoulos TP, Chused TM, Decker JL. Differences in the clinical manifestations of sicca syndrome in the presence and absence of rheumatoid arthritis. Am J Med 1979;66:733–6. doi: 10.1016/0002-9343(79)91110-0. [DOI] [PubMed] [Google Scholar]
- [88].Manoussakis MN, Georgopoulou C, Zintzaras E, Spyropoulou M, Stavropoulou A, Skopouli FN, et al. Sjögren’s syndrome associated with systemic lupus erythematosus: clinical and laboratory profiles and comparison with primary Sjögren’s syndrome. Arthritis Rheum 2004;50:882–91. doi: 10.1002/art.20093. [DOI] [PubMed] [Google Scholar]
- [89].Baer AN, Maynard JW, Shaikh F, Magder LS, Petri M. Secondary Sjogren’s syndrome in systemic lupus erythematosus defines a distinct disease subset. J Rheumatol 2010;37:1143–9. doi: 10.3899/jrheum.090804. [DOI] [PubMed] [Google Scholar]
- [90].Kollert F, Fisher BA. Equal rights in autoimmunity: is Sjögren’s syndrome ever “secondary”? Rheumatology 2020;59:1218–25. doi: 10.1093/rheumatology/keaa009. [DOI] [PubMed] [Google Scholar]
- [91].Anaya J-M, Rojas-Villarraga A, Mantilla RD, Arcos-Burgos M, Sarmiento-Monroy JC. Polyautoimmunity in Sjögren syndrome. Rheum Dis Clin North Am 2016;42:457–72. doi: 10.1016/j.rdc.2016.03.005. [DOI] [PubMed] [Google Scholar]
- [92].Amador-Patarroyo MJ, Arbelaez JG, Mantilla RD, Rodriguez-Rodriguez A, Cárdenas-Roldán J, Pineda-Tamayo R, et al. Sjögren’s syndrome at the crossroad of polyautoimmunity. J Autoimmun 2012;39:199–205. doi: 10.1016/j.jaut.2012.05.008. [DOI] [PubMed] [Google Scholar]
- [93].Jonsson H, Nived O, Sturfelt G, Norberg R. Symptomatic secondary Sjögren’s syndrome in patients with systemic lupus erythematosus (SLE). Relation to anti- SS-A and anti-SS-B autoantibodies. Scand J Rheumatol Suppl 1986;61:166–9. [PubMed] [Google Scholar]
- [94].Pan HF, Ye DQ, Wang Q, Li WX, Zhang N, Li XP, et al. Clinical and laboratory profiles of systemic lupus erythematosus associated with Sjögren syndrome in China: a study of 542 patients. Clin Rheumatol 2008;27:339–43. doi: 10.1007/s10067-007-0720-0. [DOI] [PubMed] [Google Scholar]
- [95].Ruacho G, Kvarnström M, Zickert A, Oke V, Rönnelid J, Eketjäll S, et al. Sjögren syndrome in systemic lupus erythematosus: a subset characterized by a systemic inflammatory state. J Rheumatol 2020;47:865–75. doi: 10.3899/jrheum.190250. [DOI] [PubMed] [Google Scholar]
- [96].Perper SJ, Westmoreland S V, Karman J, Twomey R, Seagal J, Wang R, et al. Treatment with a CD40 antagonist antibody reverses severe proteinuria and loss of saliva production and restores glomerular morphology in murine systemic lupus erythematosus. J Immunol 2019;203:58–75. doi: 10.4049/jimmunol.1900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Fisher B, Zeher M, Ng WF, Bombardieri M, Posch M, Papas AS,et al. The novel Anti-CD40 monoclonal antibody CFZ533 shows beneficial effects in patients with primary Sjögren’s syndrome: a phase IIA double-blind, placebo-controlled randomized trial [abstract]. Arthritis Rheumatol 2017;69 (suppl.10). [Google Scholar]
- [98].ClinicalTrials.gov [Internet]. National Library of Medicine (US). August 1, 2018. Identifier NCT03610516. Safety, pharmacokinetics and preliminary efficacy study of CFZ533 in patients with lupus nephritis. Available from: www.clinicaltrials.gov/ct2/show/N n.d. Accessed June 8, 2020
- [99].Chandrasekar B, McGuff HS, Aufdermorte TB, Troyer DA, Talal N, Fernandes G. Effects of calorie restriction on transforming growth factor beta 1 and proinflammatory cytokines in murine Sjogren’s syndrome. Clin Immunol Immunopathol 1995;76:291–6. doi: 10.1006/clin.1995.1128. [DOI] [PubMed] [Google Scholar]