

Abstract
Autism spectrum disorders (ASDs) represent a spectrum of neurodevelopmental disorders characterized by impaired social interaction, repetitive or restrictive behaviors, and problems with speech. According to a recent report by the Centers for Disease Control and Prevention, one in 68 children in the US is diagnosed with ASDs. Although ASD-related diagnostics and the knowledge of ASD-associated genetic abnormalities have improved in recent years, our understanding of the cellular and molecular pathways disrupted in ASD remains very limited. As a result, no specific therapies or medications are available for individuals with ASDs. In this review, we describe the neurodevelopmental processes that are likely affected in the brains of individuals with ASDs and discuss how patient-specific stem cell-derived neurons and organoids can be used for investigating these processes at the cellular and molecular levels. Finally, we propose a discovery pipeline to be used in the future for identifying the cellular and molecular deficits and developing novel personalized therapies for individuals with idiopathic ASDs.
1 |. INTRODUCTION
Autism spectrum disorders(ASD) is defined as a category of neurodevelopmental disorders characterized by social and communicative deficits and restricted or repetitive patterns of behavior (the diagnostic and statistical manual of mental disorders [DSM-5]). Historically, it has been difficult to diagnose and study ASD because each of the ASD-related deficits is widely present in the general population, and ASD symptoms are often presented in conjunction with other neurological and psychiatric conditions, including intellectual disability (ID), epilepsy, and attention-deficit hyperactivity disorder (ADHD).1 Correspondingly, ASD is a highly heterogeneous group of disorders with a substantial degree of patient-to-patient variability, and many questions remain related to the etiology, heterogeneity, and epidemiology of ASD. For example, it is still unclear why ASD is four times more prevalent in boys than in girls,2 and to what extent different genetic and environmental risk factors contribute to its pathogenesis. Nevertheless, increased public awareness of this condition and improved early diagnostics have allowed us to find substantially more ASD-affected individuals (ASD prevalence is estimated to be 1%−1.5%), and to initiate well-powered studies for identifying the genetic basis3 and brain level functional connectivity4 associated with ASD.
Early twin studies have estimated that ASD is one of the most heritable disorders, with genetic risk factors accounting for approximately 90% of ASD liability.5 This view, however, has been challenged in more recent studies that estimated genetics to be responsible for 21%−38% and nongenetic in-utero (environmental) factors for 58%−78% of the liability.6,7 The most current view is that ASD is a predominantly genetic disorder with an estimated heritability of 74%−84%,8,9 the pathogenesis of which can be influenced by both pre- and postnatal environmental factors, including maternal smoking, infections, and pesticide exposure.10,11
Both candidate-gene- and whole-genome-based approaches have been employed for the identification of genetic variants associated with ASDs. Common variants have been estimated to account for 12%−52% of the liability, while rare genomic variants, primarily de novo mutations, account for 2.6%−15%.9,12 However, the identification of specific common variants has been difficult due to an insufficient number of cases, and only recently, a study of 18 381 patients and 27 969 controls has found 12 common variants that implicate around 30 neighboring genes in ASD pathogenesis.13 Although this is an important step forward in understanding the complex ASD genetics, it will take some time to disentangle the potential functional impact of such variants. In genome-wide association studies (GWAS), a variant per se may not be associated with a disease, but rather report on an association with the region defined by the linkage disequilibrium.14
On the other hand, studies aimed at the identification of rare variants have been very fruitful and provided us with a list of about 100 high-confidence risk genes significantly associated with ASD.15–18 This knowledge has allowed us to begin the identification of potentially affected neurobiological processes and developmental stages in the human brain.19–21 In addition, it has enabled the generation of mutant animals for studying the cellular and molecular mechanisms disrupted by ASD mutations. There are many excellent reviews devoted to the discussion of ASD genetics and the value of animal models for studying ASDs.19,22–27
In this review, we aim to discuss the main genetic and environmental risk factors and the key biological processes occurring during the ASD-related neurodevelopmental time window (Figure 1), as well as the evidence implicating the disruption of these processes in ASDs. We focus on how induced pluripotent stem cell (iPSC)-based approaches are being used and could be used further for studying ASDs. Finally, we propose a novel diagnostics and drug-discovery pipeline for the development of novel personalized therapies for individuals with ASDs.
FIGURE 1.
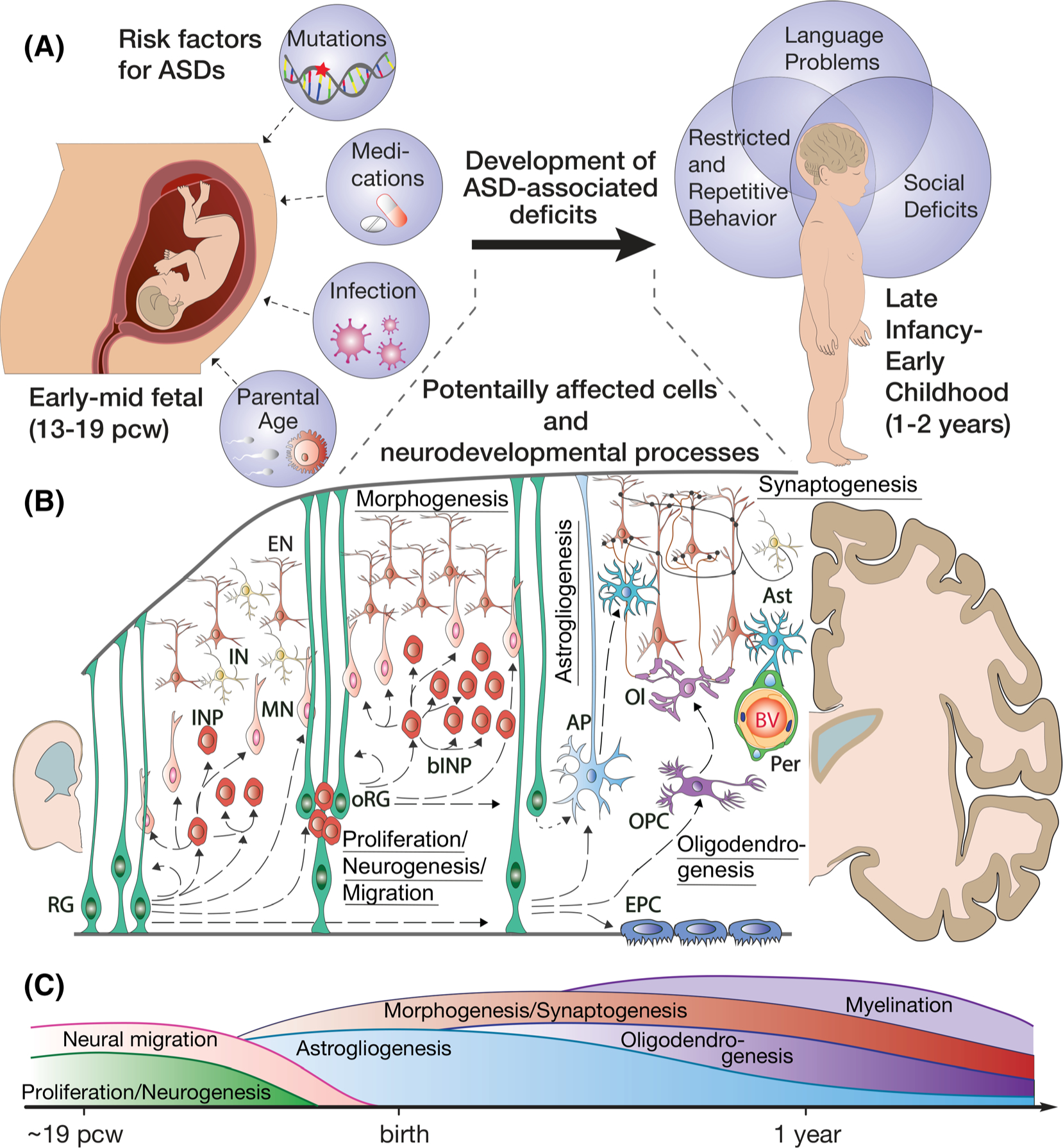
Potentially disrupted neurodevelopmental processes in ASDs. A, Key risk factors and behavioral manifestations associated with ASDs. B, Neurodevelopmental processes and cells that could be potentially affected in the developing cortex of ASD patients. Processes: proliferation, neurogenesis, migration, gliogenesis (astrogliogenesis and oligodendrogenesis, morphogenesis, and synaptogenesis). Cells: RG, ventricular radial glia; INP, intermediate neural progenitors; oRG, outer radial glia; bINP, basal intermediate neural progenitors; MN, migrating neurons; EN, excitatory neurons; IN, inhibitory neurons; AP, astrocyte progenitors; Ast, astrocytes; OPC, oligodendrocyte progenitors; Ol, oligodendrocytes; Per, pericytes; BV, blood vessels; EPC, ependymal cells. C, Time course for key neurodevelopmental processes in the human cortex
2 |. MAJOR RISK FACTORS FOR ASD
Disruption of key pre- and early postnatal neurodevelopmental processes can cause severe problems in the offspring, including early termination, birth defects, developmental delays, and neuropsychiatric disorders. A wide array of studies in the past few decades have identified a specific set of factors associated with an increased risk for the development of ASDs. These include genetic mutations and environmental factors that can work independently or together to disrupt or perturb normal neurodevelopmental programs. In this section, we discuss the major genetic and environmental risk factors that have been shown to contribute to ASD risk (Figure 1).
2.1 |. Genetic risk factors for ASD
It is estimated that the large bulk of genetic heritability for ASDs is due to common variants. Early GWAS were not very fruitful in the identification of high-confidence common variants in ASDs, due to insufficient statistical power.19 However, recent large-scale meta-analysis studies combining both previously published and unpublished data sets have begun to accumulate sufficient sample sizes to identify high-confidence common ASD risk variants, implicating ~60 nearby genes13,28 (Table 1). In addition, more than 100 rare and de novo variants, including those known to cause syndromic ASDs such as Fragile X Syndrome, Timothy Syndrome, Tuberous Sclerosis, PTEN Syndrome, and Phelan McDermid Syndrome, have been reproducibly identified in association with ASDs.17,30 Many of these high-confidence genes are co-expressed in the cortex during mid-fetal developmental stages,19–21 implicating disruptions in the associated neurodevelopmental processes in ASD development (Figure 1).
TABLE 1.
Summary of key risk factors and potentially affected cells and disrupted neurodevelopmental processes
| ASD classification | Risk factor | Potentially affected cells in the cortex29 | Potentially disrupted neurodevelopmental processes | |
|---|---|---|---|---|
| Syndromic ASD | RS | MECP2 | Ubiquitous | Proliferation, neurogenesis, migration, morphogenesis, synaptogenesis, gliogenesis, BBB formation |
| FXS | FMR1 | |||
| TSCS | TSC1, TSC2 | |||
| PTENS | PTEN | |||
| TS | CACNA1C | Neurons, mural and endothelial cells | Migration, morphogenesis, synaptogenesis, BBB formation | |
| PMS | SHANK3 | |||
| Idiopathic ASD | Common variants13,28 | APOPT1, BAG4, BAG5, CUEDC2, DDHD2, FGFR1, FOXP1, KLC1, KMT2E, LSM1, MARK3, POU3F2, PPDPF, PTBP2, SEC13, SLC30A9, SRM, SRPK2, TMEM33, WHSC1L1, XRN2, ZNF318, ZNF441 | Ubiquitous | Proliferation, neurogenesis, migration, morphogenesis, synaptogenesis, gliogenesis, BBB formation |
| BEND4, CKB, MMS22L, NUDT1, XRCC3 | Progenitors | Proliferation, neurogenesis, migration | ||
| CADPS, EEF1A2, MACROD2, NEGRI | Neurons | Migration, morphogenesis, synaptogenesis, | ||
| GHRLOS, KIZ, FHIT, NKX2–2 | Glia | Gliogenesis, synaptogenesis | ||
| PRKG1, SOX7 | Mural and endothelial cells | BBB formation | ||
| ABCC10, ATP2B2, ATDN2, C8orp4, CRIP3, DCAF4L1, GHRL, LETM2, TLINC00852, LOC102723661, MIR1284, MIR378B, MIR6780B, MIR885, MROH5, NKX2–4, PINX1, PLPP5, PTK6, SLC22A7, TRMT61A, TTBK1, ZNF440, ZNF491, ZNF823 | Very few or undetected | |||
| Rare variants30,31 | ADNP, AKAP9, ANK2, ANKRD11, AP2S1, ARID IB, ASH1L, BCL11A, CHD2, CREBBP, CTNNB1, CTTNBP2, CUL3, DIP2A, DNMT3A, DPYSL2, DYNC1H1, DYRK1A, ERBB2IP, ETFB, FOXP1, GIGYF1, GNAI1, GRIA2, HDLBP, HECTD4, ILF2, INTS6, IRF2BPL, KDM5B, KDM6B, KIAA0232, KMT2C, KMT2E, LDB1, MAPI A, MBD5, MED13L, MIB1, NAA15, NCKAP1, NCOA1, NINL, NLGN3, NRXN1, NSD1, NUP155, PHF12, PHF21A, POGZ, PPP2R5D, PTEN, RANBP17, RFX3, RORB, SATB1, SETD5, SIN3A, SKI, SLC6A1, SMARCC2, SPAST, STXBP1, SUV420H, TAOK1, TBL1XR1, TCF20, TCF4, TLK2, TM9SF4, TNRC6B, TRIM23, TRIO, TRIP 12, UBR1, VEZF1, WAC, WDFY3, ZMYND8, ZNF559 | Ubiquitous | Proliferation, neurogenesis, migration, morphogenesis, synaptogenesis, gliogenesis, BBB formation | |
| ASXL3, CACNA1E, CACNA2D3, CELF4, CDH8, DEAF1, DSCAM, ELAVL3, FOXP2, GABRB2, GABRB3, GRIN2B, KCNMA1, KCNQ3, MYT1L, NR3C2, SCN1A, SCN2A, SHANK2, SHANK3, TBR1, | Neurons | Migration, morphogenesis, synaptogenesis, | ||
| COROIA, GFAP, KAT2B, LRRC4C, SCN1A, SRPR, TCF7L2 | Glia | Gliogenesis, synaptogenesis | ||
| MKX, PHF2, SHANK3, TEK | Mural and endothelial cells | BBB formation | ||
| ACHE, APH1A, CAPN12, KATNAL2, KMT5B, MFRP, NACC1, OR52M1, P2RX5, PAX5, PPP1R9B, PPP5C, PRR12, PTK7, SYNGAP1, TRAF7, USP45 | Very few or undetected | |||
| Medications | Valproic Acid, nAchRα antagonists | Progenitors, neurons | Proliferation, neurogenesis, migration, synaptogenesis | |
| Maternal infection | Cytomegalovirus, Herpes Simplex Virus, Rubella, Measles, Mumps, Varicella | Progenitors, neurons | Proliferation, neurogenesis, migration, synaptogenesis |
Note: ASDs are broadly classified into syndromic and idiopathic. Syndromic ASD have been detected in patients with neurodevelopmental disorders, including Rett Syndrome (RS), Fragile X Syndrome (FXS), Tuberous Sclerosis Complex Syndrome (TSCS), PTEN Syndrome (PTENS), Timothy Syndrome (TS), and Phelan-McDermid Syndrome (PMS). For most of these syndromes, we know potentially affected cells and neurodevelopmental processes, based on the gene expression profiles and results of studies in rodent and human iPSC-based models. Idiopathic ASD has been associated with both common and rare genetic abnormalities, as well as certain medications and maternal infections. We extracted genes that have been found to carry common and rare genetic variants detected in the most recent studies and analyzed the expression pattern of these genes in different cells in the developing human cortex using publicly available database (http://cortex-dev.cells.ucsc.edu/). Using this analysis, all genes were subjectively classified into six groups: ubiquitously expressed in most cells; Primarily expressed in neural progenitors; neurons, including both excitatory and inhibitory neurons; glial cells, including astrocytes, oligodendrocyte progenitors, and choroid plexus cells; mural cells and/or endothelial cells; and those expressed in only few cells and/or at very low to undetectable levels.
Additional insights regarding affected cells and cellular processes can be gained from recent single-cell mRNA sequencing studies29,32 performed on single cells obtained from the human fetal cortex at different developmental stages. We investigated the expression profiles from both GWAS and rare genes in the Nowakowski database (http://cortex-dev.cells.ucsc.edu/). Interestingly, we found that the majority of ASD genes (80 of 127 associated with rare variants and 32 of 63 associated with common variants) are ubiquitously expressed among different cells in the developing human cortex, including neural progenitors, immature and maturing neurons, and glial, mural, and endothelial cells (Table 1). This suggests that pathogenic mutations in a large proportion of ASD genes may induce global alterations in neurodevelopment that could lead to diverse clinical outcomes and manifestations. Indeed, some of these genes have been extensively investigated already, including PTEN and CHD8, and have been associated with an array of developmental abnormalities, including ASDs, macrocephaly, global developmental delay, and intellectual disability.3,33 This indicates that other ubiquitously expressed ASD genes, if disrupted, may contribute to similar neurodevelopmental deficits.
Many ASD genes appear to be specifically expressed only in certain cells. For example, GRIN2B, TBR1, CACNA1E, MYT1L, and SCN2A are relatively neuron-specific and are expressed in excitatory and/or inhibitory neurons. Pathogenic mutations in these genes may specifically disrupt neuronal properties, such as neurogenesis, migration, morphogenesis, and synaptogenesis. On the other hand, expression of GFAP, KAT2B, LRRC4C, SCN1A, TCF7L2, KIZ, FHIT, and CORO1A seems to be enriched in glial cells, including astrocytes, oligodendrocytes progenitor cells (OPCs), and microglia. Thus, it is conceivable that mutations in these genes may disrupt homeostasis, synaptic connectivity, and immune responses in the brain.
Surprisingly, a few genes, such as SOX7, TEK, and SHANK3, show preferential expression in endothelial cells, suggesting that their mutation may cause brain-blood barrier dysfunction and a broad spectrum of associated deficits. The inclusion of SHANK3 among these genes was unexpected. SHANK3 has been extensively investigated with respect to excitatory synaptic transmission in neurons, but never in endothelial cells. Additional research is needed to elucidate the role of SHANK3 in endothelial cells and brain-blood barrier formation and function.
Finally, a number of ASD genes show little or no detectable expression among various cells in the developing human cortex (Table 1). This could be related to the lack of single-cell transcriptomic data collected from cells at later gestational and postnatal timepoints and/or from cells obtained from other brain regions. Indeed, Nowakowski and colleagues investigated the properties of cells obtained from the developing cortex and ganglionic eminence at 5–37 PCW.29 Thus, a larger number of sampled brain regions, as well as a greater number of timepoints, may provide us with a more complete view of the most vulnerable developmental time points and brain regions for therapeutic interventions.
2.2 |. Environmental risk factors for ASD
There are several lines of evidence implicating environmental factors in the development of ASD. (For review, see Reference 34). Here, we review the major ones, such as parental age, medication, and maternal infection, and discuss their potential influences on different cells and neurodevelopmental processes.
It is well documented that parental age is associated with incidence of adverse health outcomes in the offspring, including early fetal deaths,35 birth defects,36 and neuropsychiatric disorders.37,38 A recent meta-analysis of 27 studies on the association of parental age with ASD showed a dose-dependent relationship between both paternal and maternal age with ASD risk, with an increase of 10 years in parental age associated with a ~20% higher risk for autism in the offspring.39 The effect of paternal age is likely related to the increased prevalence of germline harmful de novo mutations.40 Increased maternal age during pregnancy can lead to greater fetal exposure of accumulated toxins, such as air pollutants and pesticides, and may contribute to ASD risk in the offspring.41
Epidemiological studies have identified a wide array of teratogens that increase autism risk when exposed during first trimester.42 Recently, it has been demonstrated that two medications taken during pregnancy, valproate, and neuronal nicotinic acetylcholine receptor antagonists, are associated with higher ASD risk in the offspring.37 Of these, valproic acid (VPA) is the most extensively studied. VPA is an anticonvulsant that has been shown to disrupt neural tube closure.43 The first case report of autism following valproate exposure was published in 1994,44 and since then, it has been shown that maternal use of valproate during pregnancy increases absolute risk of ASD of children by two to threefold.45 Over the past decade, the VPA animal model of ASD has risen to become one of the most widely used animal models for ASD, producing autistic-like behaviors in animals akin to those observed in patients.46,47 In rats, the critical period of VPA-induced social impairments was found to be E12,48 which marks the end of neural tube closure and the beginning of neurogenesis. Additionally, animal models show that VPA exposure disrupts neural proliferation49,50 and neuronal activity and connectivity.51,52 Taken together, VPA may disrupt multiple processes in the developing human brain, including neurogenesis and connectivity.
Another environmental factor that contributes to the risk of ASD is prenatal exposure to viral infections. Indeed, maternal exposure to numerous viruses during pregnancy, including cytomegalovirus, herpes simplex virus, and rubella virus, have been reported in association with ASD.53 Although some of these viruses may infect the fetus prenatally or perinatally, emerging evidence suggest that maternal immune response to pathogens particularly during the first and second trimesters may be the main risk factor for ASD in the offspring.54,55 Maternal IgG antibodies can cross the placenta and provide the developing fetus with protection against infections; however, autoantibodies can also cross the placenta and trigger harmful reactions in the fetus, leading to severe developmental abnormalities. It has also been demonstrated that simply triggering maternal immune response in pregnant rodents at mid-pregnancy can lead to ASD-related behavioral deficits in the offspring.56 Together, these studies point to the early neurodevelopmental stages and processes that when disrupted by immune responses to viral infections could lead to ASD-related deficits.
3 |. STEM CELL-BASED PLATFORMS FOR MODELING ASD-RELATED PHENOTYPES
Human stem cells are a new tool in our arsenal for modeling human development and disorders at the cellular and molecular levels. Stem cell-based platforms hold several key advantages over traditional animal models: (a) they enable the study and characterization of human-specific disease-relevant cell types, (b) they reflect the genetic background of patient cohorts, and (c) they can be used for high-throughput screening for candidate drugs.57 There are three main methods for producing human neural cells from stem cells: directed differentiation, reprogramming, and organoid formation (Figure 2). Many excellent reviews have provided in-depth discussions of the directed differentiation,59,60 reprogramming,61–63 and organoid techniques,64–68 as well as the potential uses of these techniques for disease modeling and drug discovery.69–71 Here, we only summarize the techniques and primarily focus on their key advantages and disadvantages for studying human cortical development and ASDs.
FIGURE 2.
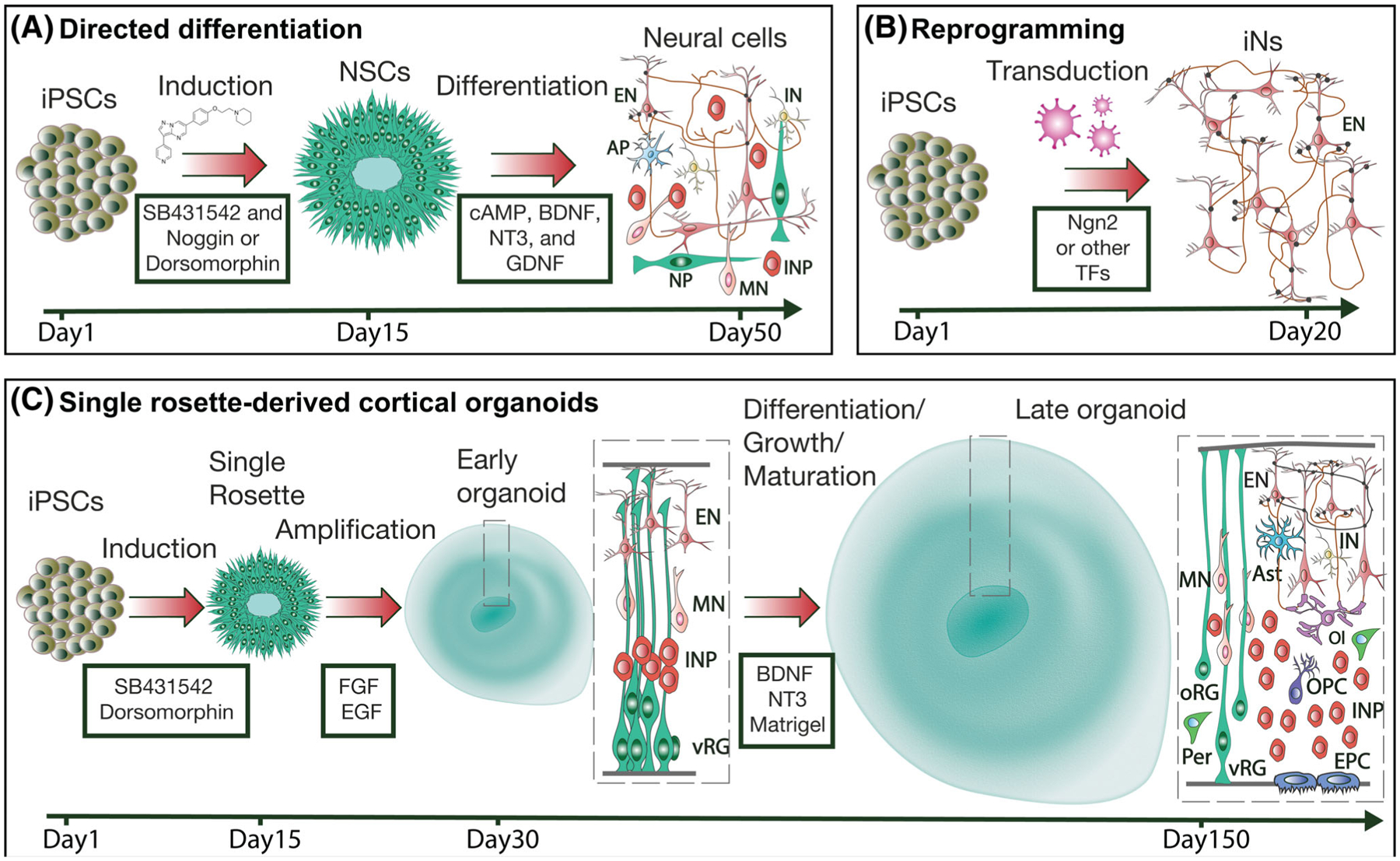
Stem cell-based tool box for modeling human cortical development and disorders. A, Typical protocol for directed neural differentiation of human pluripotent stem cells. During the neural induction step, stem cells are converted into neuroepithelial cells/neural stem cells. A dual SMAD inhibition protocol is often used for this step.58 These cells are typically grown in clusters of neural rosettes. During differentiation, neural stem cells are exposed to trophic factors, such as cAMP, BDNF, NT3, and GDNF, which promote neuronal differentiation, maturation, and survival. After 50 days of differentiation, the cultures typically consist of a mixed population of cells, including neural progenitors (NP), intermediate neural progenitors (INP), migrating neurons (MN), astrocyte progenitors (AP), excitatory neurons (EN), and inhibitory neurons (IN). B, Typical protocol for reprogramming of human pluripotent stem cells into induced neurons (iN). Stem cells are transduced with lentiviruses carrying specific transcription factor(s) and an antibiotic selection cassette required for efficient conversion and selection. A TET-ON/OFF system is typically used to control the level and timing of expression. For example, a relatively uniform population of functionally mature excitatory neurons can be produced via Ngn2-mediated reprogramming. C, Protocol for generating cortical organoids. We propose that cortical organoids can be generated from single neural rosettes to improve organoid-to-organoid reproducibility and facilitate reliable identification of cellular and molecular deficits in ASDs. The protocol will consist of a neural induction step for the production of neural rosettes that can be isolated and propagated in suspension culture. Initially, isolated single rosettes will be grown in the presence of growth factors to promote neural stem cell proliferation. Later, single rosette-derived organoids will be imbedded in Matrigel (or another scaffolding material) and cultured in the presence of trophic factors, such as BDNF and NT3, to promote differentiation, growth, and maturation. In contrast to 2D protocols, a diversity of properly organized neural cells can be generated in organoids at different developmental stages, including ventricular and outer radial glia (vRG and oRG, respectively); intermediate progenitors (INP); migrating, neurons (MN); excitatory and inhibitory neurons (EN and IN, respectively); astrocytes (Ast); oligodendrocytes and oligodendrocyte progenitors (Ol and OPC, respectively); pericytes (Per); and ependymal cells (EPC)
3.1 |. Directed differentiation
The potential of human stem cells for disease modeling has been appreciated since the development of techniques for human embryonic stem cell isolation and in vitro propagation.72 Using lessons learned from studying embryonic development in different model organisms, we know the key signaling cascades required for efficient differentiation of human stem cells into neuroepithelial cells of different positional identities. It has been demonstrated that dual SMAD inhibition using bone morphogenetic protein (BMP) and transforming growth factor (TGF)-beta pathway inhibitors is important for quick and efficient conversion of stem cells into neuroepithelial cells, which can then further be specifically directed to become neural progenitors of different regional identities.58 For example, anterior fate is a default neurodevelopmental program that can be achieved without any morphogens73,74 or with exposure to WNT inhibitors to suppress posteriorization.75–77 To produce more posterior neural progenitors, WNT/β-catenin pathway activators, fibroblast growth factors (FGFs), and retinoic acid (RA) have been used in a concentration- and time-dependent fashion.78–85 Although the role of WNT signaling in the posteriorization of neural tissue is well established,81,86 the roles of the FGF and RA signaling cascades are less clear. For example, it has also been demonstrated that activation of the FGF signaling cascades is essential for the specification of telencephalic tissue87,88 and RA is important for the specification of basal cortical intermediate progenitors and upper layer cortical neurons.89 These questions need to be resolved in future studies, and the timing and concentration of the morphogens required for the production of different tissues may vary. On the other hand, it is well-documented that activation of the Sonic Hedgehog (SHH) pathway is required for efficient ventralization of neural tissue.90 Correspondingly, co-application of a SHH agonist together with a WNT agonist or antagonist is important for the production of anterio-ventral or posterior-ventral neural progenitors, respectively.76,91,92 Thus, a typical protocol for directed differentiation consists of at least three steps (Figure 2A): (a) neural induction using BMP/TGF-beta inhibitors; (b) proliferation/specification of neural progenitors via exposure to different growth factors and morphogens delivered at different concentrations during different developmental stages; and (c) differentiation/maturation by withdrawal of growth factors and application of trophic factors, such as brain-derived neurotrophic factor (BDNF) and cyclic adenosine monophosphate (cAMP) with or without glial-derived neurotrophic factor (GDNF), nerve growth factor (NGF), and neurotrophin 3 (NT3).
3.1.1 |. Advantages
Directed differentiation is a relatively time-effective and simple approach to produce different subtypes of neural progenitors and neurons for modeling early aspects of human brain development. Neurons produced using this method demonstrate predictable acquisition of neuronal identities and expected functional intrinsic and synaptic maturation. This system also allows controlled delivery of morphogens and growth factors to fine-tune the specification and differentiation processes. It is also amenable for virus-based manipulations of gene expression profiles in loss- and gain-of-function experiments designed to elucidate molecular mechanisms.93–95 Correspondingly, this has been the most widely used human stem cell-based system for modeling molecular deficits in genetic neurodevelopmental disorders.
3.1.2 |. Disadvantages
The key disadvantages associated with the directed differentiation approach are the heterogeneity of differentiated cells, unpredictable yield of desired cells, delayed functional synaptic maturation and spinogenesis, and reduced production or absence of certain later-born cells, including superficial cortical neurons, astrocytes, and oligodendrocytes. The heterogeneity is primarily associated with variable proportions of neural progenitors and immature and mature neurons present in the cultures at different time points. One way to overcome this problem is by using cell-type specific reporters, such as lenti- or adeno-associated viruses carrying fluorescent proteins under cell-type specific promoters.94–96 Functional synaptic maturation of differentiated human stem cell-derived neurons could be improved by co-culturing them with primary rodent astrocytes and/or by treating them with trophic factors such as BDNF or insulin-like growth factor 1 (IGF1).93,94,97 This approach, however, has not been effective for improving spinogenesis, and directly differentiated neurons rarely develop mushroom-like looking dendritic spines. This problem is likely related to the substantially prolonged time-course for human neuronal development maturation, as differentiated human neurons do not survive that long in 2D culture (in general no longer than 2–3 months) to become fully mature. Improved spinogenesis has been observed in human neurons transplanted to the mouse brain by 6 to 9 months posttransplantation.74
3.2 |. Reprogramming
To circumvent some of the challenges related to the directed differentiation approach, particularly slow functional maturation and reduced production of glial cells, methods have been developed to allow for direct transcription factor (TF)-induced reprogramming of human stem cells or skin fibroblasts into the cells of interest.98–100 TF-induced reprogramming has been used for producing neural progenitors, synaptically mature cortical excitatory and inhibitory neurons, astrocytes, and oligodendrocytes from both fibroblasts and stem cells.69,101,102 In contrast to the reprogramming of human stem cells into neurons, the reprogramming of adult human skin fibroblasts requires miRNAs miR-9/9* and miR-124 to alter the epigenetic landscape and make it accessible for TFs.100,103,104 Importantly, neurons produced from fibroblasts maintain age-related epigenetic marks, which makes them particularly suited for modeling later-onset neurodegenerative disorders.104,105 In general, TF-based reprogramming of stem cells involves two simple steps: first, infection of stem cells with a combination of TFs under the control of a conditional promoter to induce the reprogramming (Figure 2B), and second, coculture of reprogrammed cells with other cells, including astrocytes for neurons or neurons for astrocytes, to improve their functional and structural maturation.
3.2.1 |. Advantages
TF-induced reprogramming provides a quick and efficient way to produce a relatively homogeneous population of functionally mature cells. For example, human neurons produced from stem cells through induced expression of neurogenic factor neurogenin 2 (Ngn2) uniformly express cell type-specific markers consistent with their layer 2/3 cortical identities, including BRN2 and SATB2, and demonstrate robust action potentials and AMPA-mediated post-synaptic currents.106 Accordingly, this system has been used for investigating functional neuronal deficits in association with ASDs.107–109 This system is also amenable for virus-based manipulations to study molecular mechanisms,108 and it is particularly suitable for high-throughput screening.110
3.2.2 |. Disadvantages
There are several limitations that are important to remember when designing disease-modeling experiments using TF-induced cells. First, the majority of TFs used for reprogramming are helix-loop-helix TFs111 that regulate (both directly and indirectly) a myriad of important molecular pathways,112–115 including those found to be dysfunctional in association with ASDs. Correspondingly, disease-related phenotypes could be compensated, skewed, or exaggerated in TF-induced cells. Indeed, it has been demonstrated that ASD-associated morphological deficits detected in maturing cortical neurons obtained through directed differentiation are not detected in Ngn2-induced neurons.116 It was found that the genes responsible for the development of the phenotype are already expressed at very early developmental stages in neural stem cells, and that skipping these stages via induced Ngn2 expression results in rapid morphological maturation and compensation of ASD-related transcriptional and morphological deficits.116 On the other hand, both induced and differentiated neurons show similar intrinsic and AMPA-mediated synaptic deficits in association with SHANK3 deficiency.108 Further, somewhat aberrant gene expression profiles have been observed in Ngn2-induced neurons. For example, mature Ngn2-induced neurons express an unnatural combination of both neural stem cell and mature neuronal genes, such as NESTIN and NEUN117 or OCT4, SOX2, NANOG, and PSD95.106 In addition, even though Ngn2-induced neurons have the identities of layer 2/3 cortical excitatory neurons, they primarily express vGlut2,106 which is largely expressed by subcortical and peripheral primary neurons.118 Furthermore, Ngn2-induced neurons express relatively low levels of NMDA receptors and no functional synaptic NMDA receptors,106 which is a major limitation for modeling ASDs and other psychiatric disorders associated with genetic abnormalities in NMDA receptors.15 The expression levels of NMDA receptors, particularly the ASD-associated GRIN2B subunit, in Ngn2-induced neurons could be elevated by treatment with SMAD and WNT inhibitors during reprogramming117; however, the treatment does not solve the problem completely, as NMDA-mediated synaptic currents are still substantially smaller than AMPA-mediated currents and the GRIN2A subunit is expressed at very low levels in the majority of cells. In primary neurons, NMDA-mediated synaptic currents develop earlier than AMPA-mediated currents (silent synapses) and regulate post-synaptic expression of AMPA receptors.119
3.3 |. 3D cultures
3D self-organized cultures of neural cells (often referred as organoids or spheroids) are the most recent addition to our arsenal of stem cell-based models for studying human brain development and disorders.68,120 This approach is based on the ability of stem cells to self-organize and provide required self-patterning factors for morphogenesis and differentiation.121 It has been demonstrated that 3D self-organized neural cultures, produced from either pluripotent stem cells or multipotent neural stem cells, recapitulate many important aspects of early human cortical development, including sequential neurogenesis, migration, gliogenesis, and synaptogenesis.73,122–125 Importantly, these cultures also contain outer radial glia (oRG),122,125,126 cells that are difficult to produce in 2D cultures and are the key contributors to the dramatic expansion of the human cortex.127,128 3D self-organized neural cultures can also be maintained in vitro for as long as 2 years,129 which allows them to reach the later stages of neurodevelopment that are impossible to reach with 2D cultures and, correspondingly, to produce later-born neural cells, including astrocytes and oligodendrocytes.129–131 The protocols for generating 3D self-organized neural cultures usually consist of three steps: (a) aggregation of dissociated stem cells in suspension culture and maintenance in neuronal media to promote neural induction and the production of neuroepithelial cells; (b) after the formation of neuroepithelial cell layers, 3D cell aggregates are embedded in Matrigel (MG) or transferred into MG-containing media to further support tissue growth and morphogenesis. It remains unclear which MG components contribute to this process and how they do so. It has been demonstrated that for retinal organoids, MG in the media can be substituted with purified laminin/entactin proteins and recombinant nodal protein.132 Moreover, some protocols do not use MG at all124; and (c) for the production of region-specific organoids, the culture media can be supplemented with additional morphogens and/or inhibitors. For example, SHH activators have been used to produce ventral organoids,133,134 WNT activators for midbrain and hindbrain organoids,125,135 FGF2 for cerebellar organoids,136 and WNT activators and RA for spinal cord organoids.137 In addition, different region-specific organoids can be fused with each other in different combinations for studies of the development of more complex interregional brain processes, such as migration and connectivity.64 Indeed, a robust unidirectional migration of inhibitory neurons was detected upon fusion of ventral to dorsal telencephalic organoids.133,134 The range of applications for which organoids are used as a model is growing at a fast pace, and accordingly, organoids are expected to become the main model system for investigating human brain development and disorders.
3.3.1 |. Advantages
The main advantages of 3D cultures are the organized diversity of human neural cells and their prolonged survival in vitro. This system is uniquely suitable for modeling human-specific aspects of brain development and disorders. Accordingly, 3D cultures have been used for investigations of the developmental mechanisms responsible for the production of human oRG, astrocytes, and oligodendrocytes, and for modeling neurodevelopmental disorders, such as ASDs,138 Timothy syndrome,133 and microcephaly.123,125,139 3D cultures are also amenable to genetic manipulations for studying molecular mechanisms.123
3.3.2 |. Disadvantages
Organoid-to-organoid variability is a major issue associated with this approach140 that substantially limits the use of organoids for disease modeling and drug discovery.65,67 There are at least two major sources of variability: (a) the presence of non-neuroectodermal cells and (b) variation in the number of neural rosettes (neural tube-like structures) per organoid and, correspondingly, disorganized distribution and orientation of cells inside the organoids. Non-ectodermal cells, including mesodermal progenitors140 and microglia,141 have primarily been detected in organoids produced from pluripotent stem cells. The proportion of these cells varies from batch-to-batch and from organoid-to-organoid. Correspondingly, it is difficult to predict how the presence or absence of these cells influences the patterning and differentiation of neuroectodermal cells, especially considering that some of these cells can secrete important morphogens, such as BMP and WNT (epidermis/ectoderm), SHH (notochord/mesoderm), and RA (somite/mesoderm). However, it also important to note that the presence of different cell types in organoids may enable the production of brain-relevant cell types that are difficult to produce otherwise.68,121
Non-ectodermal cells, however, have not been detected in spheroids, because these 3D cultures are produced from neural stem cells, which have a differentiation potential restricted to neural lineages. It has also been demonstrated using bulk RNA sequencing and immunostaining that spheroid generation is consistent between different iPSC lines and different batches.142 In the future, single-cell mRNA sequencing using cells obtained from different spheroids will be required to verify spheroid-to-spheroid variability. The main source of variability in spheroids, however, is the variable number of neural rosettes per spheroid. Neural rosettes consist of neuroepithelial cells and neural progenitors that express and secrete different growth factors, including FGF and vascular endothelial growth factor (VEGF), which are known to influence the migration, development, and maturation of neurons.143 Accordingly, it is likely that morphological and functional maturation of neurons in multi-rosette containing organoids are different, and potentially delayed, as compared to the maturation of neurons in organoids with a single rosette or few rosettes. Indeed, cortical neurons with elaborate and radially oriented dendritic arbors and dendritic spines have yet to be observed in spheroids. The multi-rosette problem could be potentially solved by using organoids/spheroids produced from a single neural rosette. It has been reported that single neural rosettes can be isolated from 2D culture144 and propagated in suspension to generate excitatory and inhibitory neurons.145 Our lab has developed a new protocol for generating single rosette-derived cortical organoids (Figure 2C) that grow large in suspension culture (reaching 4 mm in diameter) and consist of functionally and structurally mature cortical excitatory and inhibitory neurons, astrocytes, oligodendrocytes, and mural cells (http://organoid.chpc.utah.edu).
In summary, iPSC-derived neurons and organoids are a highly promising approach for modeling the cellular and molecular deficits in ASDs and other neurodevelopmental disorders. The main advantages are patient-specific genetic background (which is essential for modeling complex genetic disorders), human-specific cells (such as oRG), and human-and patient-specific gene expression profiles. The major challenges are the absence of standardized and robust protocols for producing different neural cell types and organoids.
4 |. DEVELOPMENTAL PROCESSES AFFECTED IN ASD
Since the diagnosis of ASD can be made by the age of 2 to 3, and nonspecific associations can be detected as early as 12 months,146 the window for disrupted neurodevelopment is thought to occur between the start of CNS development during the early prenatal time period to the early postnatal months (Figure 1). Here, we categorize early neurodevelopmental processes into four major categories (proliferation and neurogenesis, migration, morphogenesis and synaptogenesis, and gliogenesis and myelination) and discuss how these processes, if disrupted, could contribute to ASD pathogenesis based on the results of clinical, genetic, and iPSC-based studies.
4.1 |. Proliferation and neurogenesis
Proliferation and neurogenesis are the earliest processes in the developing nervous system (Figure 1). During these stages, neuroepithelial cells lining the innermost portion of the neural tube, known as the ventricular zone, divide symmetrically to each produce two identical daughter cells to substantially expand the progenitor pool. After several rounds of division, neuroepithelial cells gradually transform into radial glia (RG), which divide both symmetrically and asymmetrically. During asymmetric divisions, RG produce one RG and one basal intermediate neural progenitor (INP, also known as basal progenitors) or two INPs.147 INPs also divide both symmetrically and asymmetrically to produce either an INP and a neuron, or two neurons. During the later stages of neurogenesis, INPs occupy the space above the ventricular zone, forming another mitotic zone in the cortex called subventricular zone. Importantly, the subventricular zone is vastly expanded in humans as compared to rodents, partly due to the enhanced proliferative capacity and morphological differences of human INPs.148,149 The INPs located in the outer subventricular zone contribute to the majority of cortical neurons in the human brain. There are many factors, both intrinsic and extrinsic, that regulate proliferation and neurogenesis in the mammalian and human brain.147,150,151 These include factors such as FGF, NGF, and NT3, which not surprisingly are also factors that aid in the generation of iPSC-derived neurons.
4.1.1 |. Clinical evidence
The deficits of proliferation and neurogenesis in ASD has been extensively investigated through clinical evaluation, structural imaging, and examination of postmortem brain tissue. Clinically, about 30% of ASD individuals can be subclassified as having complex autism, which comprises of patients exhibiting significant dysmorphology or microcephaly.152 ASD is associated with decreased head circumference early in infancy, but significantly increased head circumference after 6 months.153 Further, ASD is associated with early brain enlargement and increased extra-axial fluid.154 These clinical observations suggest that mechanisms that control brain size and morphology, such as neurogenesis and neural proliferation, could be disrupted in a subset of ASD patients with altered head circumferences. Correspondingly, evidence from structural imaging studies show that brain size of ASD children is reduced at birth, followed by rapid rate of brain growth, and early cessation of growth by 2 to 4 years of age.155 Postmortem brain tissue in children with ASD have significantly increased number of neurons, particularly in the prefrontal cortex,156 suggesting that regional proliferation could be impaired. Further, cytoarchitectural differences in the subventricular zone have been identified in adult ASD patients.157
In summary, evidence for the possibly disrupted neurogenesis and neural proliferation can be observed from clinical head size measurements, structural imaging studies on brain size, and examination of postmortem brain tissues. However, only a subset of ASD patients will present with these phenotypes, and the causative link to neurogenesis and proliferation is missing. Further, since the bulk of neurogenesis occurs prenatally,158 and the most striking observations in ASD brain size occurs after 6 months, it is possible that altered neurogenesis, gliogenesis, synaptic pruning, and cell morphology abnormalities could all contribute to the aforementioned findings.
4.1.2 |. Genetic evidence
Previously identified and well-known ASD genes with specific roles in neurogenesis and proliferation include PTEN, WDFY3, ANKRD11, and CHD2. PTEN inhibits the activity of PI3K/AKT signaling pathway and plays a critical role in regulation of NPC proliferation.159 WDFY3 encodes for an adaptor protein and plays a role in macroautophagy, and WDFY3 loss-of-function leads to disrupted NPC proliferation.160 ANKRD11, also known as Ankyrin repeat domain containing protein 11, is a transcriptional regulator that interacts with histone deacetylases.161 Knock-down of ANKRD11 was found to reduce NPC proliferation.162 CHD2 is important for modulation of chromatin remodeling, and suppression of CHD2 results in differentiation of RG into intermediate progenitors.163 In addition, the key neurogenic transcription factor genes, including ASH1L and MYT1L that have been used for direct reprogramming of iPSCs, are among the high-confidence ASD genes.17
Recent GWAS meta-analysis has identified a number of candidate ASD risk genes that may potentially play a role in neurogenesis.13 These include genes that modulate Wnt signaling (SLC30A9, SOX7), energy homeostasis (CKB, NUDT12), and cell cycle regulation (KIZ). Functional studies are necessary to determine their specific contributions to disrupted neurogenesis in ASD.
4.1.3 |. Evidence from iPSCs studies
Perhaps the most compelling evidence for the role of neurogenesis deficits in syndromic and nonsyndromic ASDs come from patient-derived iPSC studies. Marchetto and colleagues demonstrated that NPCs derived from eight ASD individuals with early brain overgrowth phenotypes showed rapid proliferation, as measured by NPC population doubling time, with shortening of G1 and S phases.93 This provides direct evidence that proliferation and neurogenesis are impaired in human cells derived from ASD patients. Intriguingly, Wnt/BRN2 signaling was found to be impaired in ASD NPCs, as well as reduced expression of glutamatergic and increased expression of inhibitory markers. Another group showed, using iPSC-derived cerebral organoids, that idiopathic autism is associated with accelerated cell cycle, and overproduction of GABAergic neurons.138 These findings suggest that disrupted neurogenesis may in part alter the ratio of distinct neuronal subtypes, with possible consequences on excitatory-inhibitory balance and network deficits.
In syndromic ASDs, deficits in proliferation have been observed in Fragile X Syndrome164,165 and Tuberous Sclerosis.166–168 Sheridan et al.164 were one of the first groups to characterize cellular phenotypes related to FXS using iPSC-based systems. In this early study, they generated iPSCs from three patients with FXS, characterized their differentiation into neurons and glia, and observed aberrant neuronal differentiation from FXS iPSCs correlated with reduced FMRP expression.164 In another study, Telias et al.165 performed directed differentiation of embryonic stem cells from three patients with FXS into neurons, and observed that FMR1 expression increased during neural differentiation in control but not FXS lines. FX neural progenitors showed abnormal expression of key NPC genes, including SOX1, NOTCH1, and PAX6, and reduced expression of neuronal genes TUBB3 and TAU. Further, FXS-neural rosettes developed significantly slower than controls, and showed decreased neurogenic potential and a striking bias towards the production of GFAP+ glial cells.165 These studies did not explore whether patient neurons showed altered ratio of glutamatergic to GABAergic neurons.
Li et al. (2017) generated iPSC-derived neural progenitor cells and neurons from a single TSC patient with well-characterized heterozygous mutation in TSC2 gene. The authors observed morphological perturbations in TSC neurons, including cell hypertrophy, and altered neurite branching and complexity, that may be related to hyperactivity of mTOR. Further, TSC progenitor cells exhibited higher proliferation rates, as measured through BrdU staining.169 Similarly, Zucco et al.168 generated iPSC-derived NPCs and neurons from two patients with TSC, and found dysregulation of PI3K/AKT/mTORC1 pathway. Characterization of NPC proliferation via Ki67 staining revealed normal NPC proliferative capacity, but delayed neuronal differentiation into mature neurons.168 Finally, Nadadhur et al.167 generated iPSC-derived neurons and oligodendrocytes from two TSC patients, and showed that TSC neurons in monoculture exhibited hyperexcitability, with markedly increased spontaneous calcium transients. The morphological perturbations as observed by Li et al. (2017) were not observed in neuronal monocultures from this study by Nadadhur et al.167; however, patient neurons cocultured with patient and control oligodendrocytes showed significant increases in soma size and axon density.167
4.1.4 |. Summary
iPSC-based approaches have identified deficits in patient-derived neural progenitor cells in nonsyndromic and syndromic ASDs, including Fragile X Syndrome and Tuberous Sclerosis, implicating disruptions in proliferation and neurogenesis in the etiology of ASD. However, many of these early iPSC-based studies had low sample sizes, and it remains unclear whether differences in genetic background, experimental setup, and other factors may affect patient phenotypes. It is also unknown whether deficits early in neurodevelopment may be compensated at later developmental stages. Indeed, although Mariani et al.138 observed disrupted cell cycle length at the early iPSC and progenitor stages in patients with idiopathic ASD, overall size of patient-derived organoids was not perturbed.138 Further, as neurogenesis and proliferation occur at earlier time points than other cellular phenotypes such as disrupted excitability and synaptic connectivity, it is unclear whether and how such deficits contribute to the development of these later-onset cellular phenotypes. In addition, the cell-subtype specificity of neurogenesis and proliferation-related deficits should be further explored, particularly in light of emerging evidence that the ratio of glutamatergic to GABAergic neurons could be affected.93,138 As Nadadhur et al.167 demonstrated using the neuron and oligodendrocyte coculture assay, a thorough and systematic investigation of cell-autonomous and cell-nonautonomous effects may be necessary to elucidate otherwise hidden phenotypes. This may be particularly important, as factors secreted by different progenitors, astrocytes, oligodendrocytes, and ependymal cells may regulate proliferation and neurogenesis.170 In this regard, 3D organoids can be particularly advantages, as they comprise a diversity of neural and glial cell types that enable the study of cell-autonomous and nonautonomous phenotypes.
4.2 |. Migration
Following the events of early neurogenesis, newly produced neurons migrate from the ventricular zone or the subventricular zone in an inside-out fashion to occupy space in the cortex (Figure 1), with late-born neurons residing more superficially than early-born neurons.171 As the brain further develops, and the distance of radial migration increases, neurons switch from relying on cellular locomotion to relying on scaffolding support from RG guides to reach their destination. Eventually, the radial migration of palial neural precursors results in the formation of a six-layered cortical structure, and the tangential migration of subpalial immature neural precursors into the cortex gives rise to inhibitory interneurons.172 Different guidance molecules, including Reelin, Ephrins, Semaphorins, Netrins, and Slits have been shown to be critical for proper migration and the establishment of distinct brain networks.173
4.2.1 |. Clinical evidence
Clinical evidence supporting cell migration deficits in ASD have been primarily obtained from the studies of postmortem brain tissue. Indeed, ectopic cell clustering and increased numbers of cells in layer 1 and subplate have been observed in the cortical tissue obtained from individuals with ASD.174 Further, heterotopias and disrupted cytoarchitecture in sub-cortical, periventricular, hippocampal and cerebellar regions as well as abnormal migration of granule cells within the dentate gyrus have been observed.175 Finally, patches of abnormal laminar cytoarchitecture and disorganization in the prefrontal and temporal cortexes, spanning multiple cortical layers, have been detected in postmortem brain from ASD patients.31 Although the mechanisms that give rise to these histopathologic findings in ASD brains are not understood, the finding of periventricular heterotopias and disrupted laminar cytoarchitecture strongly suggest that certain neurons fail to reach their target destinations due at least in part to the disruption of mechanisms that regulate cell migration to defined regions.176 Although histopathological findings were observed in nearly every individual in the aforementioned studies, there is heterogeneity in the abundance and severity of histopathology across individuals, which could reflect genetic or phenotypic differences. How ASD genes and patient-specific genetic backgrounds contribute to abhorrent patient-specific histopathology remains poorly understood.
4.2.2 |. Genetic evidence
Early genetic studies have identified mutations in RELN in association with ASD.177 RELN is located within autism susceptibility locus 1 (AUTS1) and encodes for a large secreted glycoprotein and regulates many steps of neuronal migration, including somal translocation, radial migration, and terminal translocation among other functions.178 RELN-related deficits have been observed in ASDs177 and other disorders, including lissencephaly, schizophrenia, bipolar disorder, epilepsy, and Alzheimer’s disease.179–182 Other ASD-associated genes include AUTS2, which modulates radial migration and neurite extension.183 A transcriptomic profiling study of iPSC-derived neurons from individuals with idiopathic ASD shows enriched differentially expressed genes in functional pathways related to axon guidance, neuronal migration, and synaptic function.184
Interestingly, POU3F2 has been identified as a candidate risk gene located within rs238834 through a recent ASD GWAS meta-analysis.13 POU3F2 encodes for member of the POU-III class of neural transcription factor that plays a role in cortical neuron migration and layering.185 This gene was previously implicated in bipolar disorder.186 Its contribution to the development of ASD-related phenotypes, however, has not been investigated.
4.2.3 |. Evidence from iPSC studies
Migration deficits have been reported in association with syndromic ASDs. Specifically, abnormal migration of neural progenitors has been observed in excitatory neural progenitors in association with Rett syndrome187 and inhibitory neural progenitors in association with Timothy syndrome.133 Zhang et al.187 developed a novel 3D based migration assay, in which fluorescently labeled neural progenitors were loaded onto the top layer of a two-layered 3D hydrogel matrix, and migration from the top to bottom layer containing either neurons or astrocytes was evaluated. The investigators observed that neural progenitors derived from Rett syndrome patients and isogenic MECP2 mutation lines showed significantly impaired migration compared to healthy controls.187 Other iPSC-based studies of Rett syndrome did not directly look for neuronal migration phenotypes.93,188–191 Birey et al.133 generated 3D cortical spheroids from three patients with Timothy Syndrome, and found impaired interneuron migration in patient spheroids that was rescued with suppression of L-type calcium channel activity.133 However, migration-related deficits were not detected in excitatory neural progenitors from Timothy syndrome patients using the neurosphere migration assay.192 Given that only few studies specifically examined migration-related deficits and that different techniques were utilized for these experiments, a systematic evaluation of migration deficits is needed to elucidate the contribution of neural migration in syndromic and idiopathic ASDs. Furthermore, it is possible that 3D-based methods that more closely mimic human brain microenvironments may be necessary to observe migration-related deficits compared to 2D methods.193
4.2.4 |. Summary
To date, numerous 2D systems have been developed to model migration deficits. These include the Boyden Chamber Assay, the Stripe Choice Assay,194 and the neurosphere migration assay.195 3D-based systems have also been developed to characterize migration, such as the 3D hydrogel-based migration assay187 and the interneuron migration assay.133,134,196 These assays have identified deficits related to migration from patients with syndromic ASDs. Although both types of systems are suitable for modeling migration deficits in association with ASDs, 3D organoid-based approaches have several key advantages: (a) Unlike iPSC-derived neurons grown in 2D, 3D organoids comprise of diverse cell types including astrocytes and oligodendrocytes, which are known to support neuronal migration197,198 (b) Fusion of organoids leads to the robust generation of migrating interneurons. Generation and fusion of region-specific organoids may be used to model region-specific migratory mechanisms that are disrupted in pathologic conditions such as ASD. These advantages make 3D organoids the ideal model for examining migration-related deficits in ASDs.
4.3 |. Morphogenesis and synaptogenesis
Following the events of neurogenesis, proliferation, and migration, neurons start establishing proper synaptic connections with each other (Figure 1). Presynaptic neurons should have differentiated axons ready for making connections, and post-synaptic cells should have developed dendrites and all the molecular machinery in place for accepting the connections and transferring electrical and biochemical signals into molecular events inside the cell. Neurite outgrowth (both axono- and dendritogenesis) involves actin remodeling.199 At the tip of the neurite is a specialized structure known as the growth cone, which responds to numerous chemotactic and chemotrophic cues to guide their morphology and trajectory.200 Synapse formation is a complex multi-step process that begins with cellular contact between neurons and is mediated by cell-adhesion molecules, synaptogenic factors, and pre and post-synaptic structures.201,202 Axonal and dendritic growth cones, guided by external guidance cues, extend and retract their filopodia and make transient contact with their targets. If the contact becomes stable, a nascent synapse is formed. This leads to the subsequent activation of cell adhesion molecules such cadherin, neurexin and neuroligin, and molecular changes in the presynaptic and postsynaptic cells. Pre and postsynaptic proteins are recruited to the nascent synapse, including presynaptic active zone proteins Bassoon, SNAP-25, and N-cadherin,203 synaptic vesicle protein synaptophysin,204 post-synaptic scaffolding proteins PSD95, GKAP, and SHANKs,205 and AMPA and NMDA receptor subunits.206 As the nascent synapse matures, morphologic changes to actin cytoskeleton leads to the formation of postsynaptic dendritic spines,207,208 which are the sites of the majority of excitatory synaptic inputs and are crucial regulators for synaptic strength and stability.209 Neuronal activity can subsequently modulate the strength and architecture of the maturing synapse, in the form of long-term potentiation and depression.
4.3.1 |. Clinical evidence
The formation of appropriate excitatory and inhibitory synapses is critical to normal brain functioning. It has been hypothesized that disrupted ratio of excitation and inhibition in the brain could contribute to ASD etiology,210 based on the observation that ASD is associated with perceptual processing abnormalities, such as hypersensitivity to auditory and tactile stimuli, and seizures. Indeed, it has been demonstrated that 20%−30% of patients with autism also develop epilepsy.211 Excitatory-inhibitory balance in the brain is largely thought to be controlled by the activity of glutamatergic and GABAergic neurons within neural circuits. Thus, deficits in critical components of excitatory and inhibitory synapse formation (eg, guidance cues, neurite outgrowth, cell adhesion molecules, dendritic spine formation, pre and post-synaptic machinery, and impaired synaptic transmission) may all contribute in some way to the disruption of excitatory-inhibitory balance in neuronal circuits, particularly in brain networks important for the sensory and executive processing, language, and social cognition, and thus contributing to ASD etiology. Indeed, a recent postmortem brain tissue study showed evidence for significantly decreased GABA receptor density in the anterior cingulate cortex of seven ASD individuals, four of whom previously had seizures.212 Several other studies of ASD postmortem tissue in the widely implicated cerebellum also reported abnormalities in GABA signaling,213–215 and nicotinic cholinergic receptor binding.216 Evidence for disrupted GABA signaling has also been identified in the hippocampus,217,218 cingulate cortex and fusiform gyrus,212,219 and parietal and frontal cortices.214 Additionally, there is evidence for morphologic deficits in neurites from ASD neurons,220–222 and dendritic spine densities,223 both of which can disrupt synapse formation.
Functional magnetic resonance imaging (fMRI) provides complementary insight into the neural circuitry disrupted in patients with ASD (For review of methods: see Reference 224). A recent meta-analysis of fMRI studies in ASD show that ASD patients exhibit hypoactivity of the prefrontal cortex during executive functioning tasks, as well as impaired cerebellar and superior temporal engagement during motor and language tasks, respectively.225 Studies examining neural circuitry of ASD patients at rest suggest a pattern of local hyperconnectivity and long-range hypoconnectivity in ASD.226 However, it is unclear whether these differences in network excitability are a result of disrupted early neurodevelopment, circuit assembly, synaptic transmission, or compensatory mechanisms. Indeed, emerging evidence suggests that disrupted excitatory-inhibitory ratio in autism may be merely compensatory.227 A full review of fMRI findings in ASD is found elsewhere.226,228
4.3.2 |. Genetic evidence
A wide array of rare and common variants in genes important for neurite outgrowth, synapse formation, and function have been identified in association with ASD. Common ASD risk genes implicated from recent GWAS meta-analysis,13 including CADPS, NEGR1, KCNN2, and CKB, are known to regulate dense-core vesicle exocytosis, synaptogenesis, and both excitatory and inhibitory synaptic transmission.229–232 A recent study of de novo mutations from 30 ASD families identified 37 genes with potentially pathogenic variants, and functional screening of a subset of candidate genes revealed that 8 of 14 genes regulated neurite outgrowth in mouse neuroblastoma Neuro2A cells,233 suggesting that neurite outgrowth may be a common process disrupted in association with ASD. Indeed, well-known ASD candidate genes associated with neurite outgrowth include TSC1/2, MET, KIDLIA, TAOK2, and AUTS2.234 TSC1/2 mutations are associated with Tuberous Sclerosis, and are part of a set of syndromic ASD genes including TSC1/2, PTEN, and FMR1 that disrupt the mTOR signaling pathway.235 Disruptions in these mTOR pathway genes are known to cause major disruption in the neurite outgrowth, dendritic spine density, and synaptic transmission.236–238 MET encodes for an AMP-activated kinase (AMPK), which promotes neural proliferation and neurite outgrowth.239,240 KIDLIA is a neural-specific X-linked gene that is important for normal neuronal migration and dendritic outgrowth potentially through inhibition of RhoA activity.234 TAOK2 encodes for a protein that participates in Sema3A/Neuropilin1 activity and helps regulate neurite outgrowth.241 AUTS2, previously discussed for its role in neuronal migration, has also been shown to regulate neurite outgrowth, through the same Rac1-mediated pathway.183
Genes associated with cell adhesion are among the most commonly implicated ASD genes detected using GWAS noise reduction approach.242 These genes include NLGN3/4, NRXN1, CDH9/10, CNTNAP2/4, and KIRREL3. NRXN1 and NLGN3/4 are well-known synaptic cell adhesion molecules. NLGN3/4 and NRXN1 encode for neuroligin-3/4 and neurexin-1, respectively. Neuroligins are binding partners for neurexins, and are important for the formation of neuronal synapses and presynaptic differentiation following cell-cell contact.243 NRXN1 and NLGN3/4 in particular may be critical for formation and function of both excitatory and inhibitory synapses in different brain regions,107,244–247 and thus disruptions in this pathway could lead to disrupted neural circuitry. CDH9/10 encode for cadherin-9 and cadherin-10, respectively, and are members of the cadherin family known to be involved in regulating many important neurodevelopmental processes, including neurogenesis, migration, neurite outgrowth, and synaptic specificity and plasticity.248–254 CNTNAP2 encodes for Caspr2, another member of the neurexin family of proteins that is expressed in neurons and oligodendrocytes.255 In neurons, Caspr2 acts as a scaffold and is necessary for K+ channel localization at the juxtaparanodal regions of myelinated axons.255,256 Further, CNTNAP2 plays a role in neuronal migration and GABAergic neuron development and morphology.257,258 CNTNAP4 encodes for Caspr4 and is highly enriched in GABAergic interneurons. Moreover, loss of Caspr4 is associated with decreased proliferation of neural progenitor cells and decreased GABA signaling.259,260 KIRREL3 is expressed by DG neurons and GABAergic interneurons within the hippocampus and functions to mediate Kirrel3-expressing DG and GABAergic neuron synapse formation.261
Major ASD genes required for proper synapse development and function include SHANK2, SHANK3, SYN1, and SYN2. Mutations in SHANK3 have been identified in 0.5%−2% of individuals with ASD262 and are known to cause Phelan-McDermid Syndrome, which is one of the most common genetic causes of ASD. SHANK3 encodes for a scaffolding protein located at the post-synaptic density (PSD) of excitatory synapses. SHANK3 plays key roles in excitatory synaptic transmission and dendritic spine formation.94,108,263 SYN1/2 encode for Synapsin-1 and Synapsin-2, which are part of the Syn family of presynaptic synaptic vesicle-associated proteins that play key roles in synaptic transmission.264 Synapsin-1/2 has been shown to regulate availability of synaptic vesicles,265–269 neurite outgrowth,266,268–271 synaptic density,269 and inhibitory synaptic transmission.272
Whole exome sequencing of 238 families from the Simons Simplex Collection revealed significant ASD association for SCN2A, an epilepsy-associated gene encoding a subunit of voltage-gated sodium channels.273–275 In addition to the “classic” synaptic ASD risk genes, whole exome sequencing of nearly 4000 autism individuals identified 33 high-confidence genes (FDR < 0.1), half of which were either novel genes or genes previously not classified as true risk genes.15 These include GABRB3, which encodes for a GABA-A receptor subunit,276 CACNA2D, which encodes for an auxiliary subunit of voltage-gated calcium channels,277 and CTTNBP2, a regulator of dendritic spine formation.278
4.3.3 |. Evidence from iPSC studies
iPSC-based models have identified deficits in morphogenesis and synaptogenesis in patients with Fragile X Syndrome, Tuberous Sclerosis, Timothy Syndrome, and Phelan-McDermid Syndrome. In an early study, Doers and colleagues generated iPSC-derived neurons from three patients with FXS, and performed a neurosphere outgrowth assay. FXS-derived forebrain neurospheres showed significantly reduced neurite outgrowth compared to controls.279 However, whether synaptogenesis or synaptic transmission are affected remains to be determined. In Timothy Syndrome, Krey et al.280 demonstrated that expression of Cav1.2 channels harboring TS mutations in neurons led to activity-dependent dendritic retraction in both rodent and human neurons.280
In Tuberous Sclerosis, three iPSC-based studies identified deficits related to morphogenesis and synapse formation. Costa and colleagues generated engineered isogenic iPSC lines with hetero- and homozygous TSC2 deletions. Interestingly, bilallelic TSC2 deletion led to mTORC1 hyperfunction in neural rosettes, impaired neural differentiation, and increased astroglial differentiation. Further, TSC2 null neurons showed increased soma size, dendritic outgrowth, disrupted synaptic transmission, and downregulation of genes related to synaptic transmission, including CNTNAP2, NLG3, and KCC2.281 In a later study, Li et al.166 generated iPSC-derived neurons from a single TSC patient harboring a mutation in TSC2.166 Characterization of the patient line revealed increased NPC proliferation and mTOR pathway activation, astrocyte proliferation, as well as increased neuron soma size. However, in contrast to the previous study, neurite length was found to be significantly reduced. More recently, Nadadhur and colleagues characterized iPSC-derived neurons from two TSC patients, one with a heterozygous TSC1 mutation, and another with mutation in TSC2. The team discovered that patient neurons were hyper-excitable, and that hyperexcitability can be mitigated with rapamycin treatment. Interestingly, patient neurons in the monoculture showed significant increase in dendritic branching, whereas patient neurons cocultured with oligodendrocytes showed significantly increased axon density and soma size. When the cocultured cells were transplanted into the mouse brain, it was observed that grafted patient cells showed significantly increased neurite lengths.167
Morphologic and synapse-related deficits represent the primary cellular phenotypes identified in engineered and patient-derived iPSC-based models of Phelan-McDermid Syndrome (PMDS). Shcheglovitov et al.94 generated iPSC-derived neurons from two patients with PMDS with hemizygous 22q13 deletions encompassing SHANK3. The group found that patient-derived neurons had disrupted excitatory-inhibitory balance, with defects in excitatory but not inhibitory synaptic transmission that can be compensated with IGF1.94 Since PMDS is thought to be primarily caused by the loss of a copy of SHANK3, Yi and colleagues generated engineered iPSC lines harboring heterozygous and homozygous SHANK3 deletions.108 SHANK3-deficient neurons harbored significantly decreased neurite outgrowth, increased input resistance, hyperexcitability, and disrupted excitatory synaptic transmission, which corresponded with findings from PMDS patients.94,108 They also demonstrated that excitability deficits in SHANK3-deficient neurons develop at least in part be due to reduced surface expression of HCN channels.108 iPSC-derived olfactory placodal neurons from two ASD patients with Shank3 microdeletions harbored synaptic deficits, as well as morphologic deficits in neuron soma size and significantly larger number of primary neurites and increased neurite length. These deficits were rescued with SHANK3 overexpression.282 Gouder et al.263 generated iPSC-derived neurons from four patients with ASD and SHANK3 mutations, and identified abnormal dendritic spine morphology and spine density which correlated with SHANK3 mRNA levels.263 Huang et al.283 performed shRNA-mediated SHANK3 knockdown in human iPSCs during neural differentiation, and discovered that SHANK3 knockdown led to significantly decreased dendritic lengths, neuron soma size, and excitatory and inhibitory synaptic transmission. Surprisingly, SHANK3 knockdown significantly altered neurite complexity in excitatory, but not inhibitory or dopamine neurons.283 Intriguingly, in contrast to SHANK3, SHANK2-deficient iPSC-derived neurons obtained from individuals with ASD showed increased excitatory synaptic transmission and more elaborated dendritic arbors as compared to control neurons.95 This suggests that down-and upregulated morphogenesis and synaptogenesis could be associated with SHANK-related abnormalities and ASD in human neurons.
4.3.4 |. Summary
In summary, synaptogenesis and synapse maturation is a complex multistep process, and variants in ASD genes involved with neurite outgrowth, cell-cell contact, synapse maturation, and synaptic transmission have been implicated in idiopathic and syndromic forms of ASD. The first synapses in the brain are observed around the 23rd week of gestation,284 and production of synapses continue to rise, peaking around the first year of life. As synaptogenesis occurs at a relatively later developmental time point, deficits in synaptogenesis may serve as a convergent phenotype for the development of ASD. Indeed, transcriptomic analysis of temporal, prefrontal, and cerebellar tissues from 19 ASD individuals showed an enrichment of differentially expressed genes in ontological processes related to synaptic function, vesicular transport, and neuronal projection.285 It is also important to remember, however, that deficits in other developmental processes, such as neurogenesis, migration, and gliogenesis, can all contribute to disrupted synapse formation and synaptic transmission. Therefore, cortical organoids may be advantages for assaying morphogenesis and synaptogenesis, as they contain different populations of CNS cell types and more closely recapitulate diseased brain environment.
4.4 |. Gliogenesis and myelination
Around mid to late-gestation, RG switches from neuro- to gliogenesis, and primarily contribute to the production of astrocytes and oligodendrocytes in the brain286,287 (Figure 1). A number of morphogens are involved in neurogenesis-to-gliogenesis switching, including Notch, Wnt, LIF, BMP, and Shh (For review see Reference 287). Transcriptionally, Ascl1 promotes neuronal differentiation, whereas Hes1 and Olig2 promotes astrocyte and oligodendrocyte cell fates, respectively.288
Astrocytes are predominantly produced during late-gestation and early postnatal phases. During late embryonic brain development, astrocytes are generated from RG and progenitors in the subventricular zone.289,290 During early postnatal phase, local glial proliferation contributes to the bulk of astrocyte production.291 Morphologically, immature astrocytes are observed to extend long, meandering filopodial processes. However, by the third postnatal week, astrocytes become more spongiform and show only few long filopodial processes.292 As astrocytes continue to mature, distinct nonoverlapping astrocytic domains become apparent.292 The formation of these astrocytic domains may be crucial for the maintenance of neuronal circuits.293 Astrocytes perform multiple important functions in the brain, including synaptogenesis, ion homeostasis, prevention of oxidative, stress neurotransmitter recycling, nutrient support, synaptic pruning, and synaptic transmission and plasticity.294
Oligodendrocytes are primarily differentiated from a subpallial domain of progenitors at later-prenatal to early-postnatal stages.295,296 In rodents, oligodendrocyte production peaks during postnatal week 2.297 Oligodendrocyte progenitors migrate into the cortex and differentiate into premyelinating oligodendrocytes upon reaching their destinations. Oligodendrocytes are produced in excessive abundance, and must compete for axonal survival factors or undergo apoptosis.298,299 The premyelinating oligodendrocytes rapidly extend growth-cone like structures, which are guided by extracellular and intracellular signals.300,301 Myelination begins around gestation week 28.302 During myelination, oligodendrocytes wrap around the axon with lateral extension of the myelin membrane layers towards the nodal regions, akin to the wrappings of a “croissant”.303,304 Cytoplasmic channels within the compacted myelin layers aid in the transport of membrane material, which is regulated through PI3K/ATK/mTOR pathway.303 In humans, myelination starts during the third trimester, peaks during the first year of life, and continues into adulthood.302
Unlike astrocytes and oligodendrocytes, microglia are derived from primitive myeloid progenitors in the yolk sac.305,306 Microglia precursors migrate into the CNS at around gestation week 4 to 5, and continues to proliferate, migrate and aid in neurodevelopment.286,307,308 Microglia density peaks around mid-gestation and begins to decrease after gestation week 31 to 35.308 Mature microglia are known to modulate immune response following infection or injury, clear apoptotic cells, and contribute to synaptic pruning.309,310 Resting microglia are highly dynamic cells that constantly extend and retracting their processes to sample the extracellular space. They establish direct contacts with neurons, astrocytes, blood vessels, and synapses.311,312
4.4.1 |. Clinical evidence
Given the wide-reaching roles of astrocytes, oligodendrocytes, and microglia in supporting neuronal and neural circuit functions, it is possible that disruptions in development of these cells or abnormal activation (reactivation) will impact neural development at multiple levels and contribute to ASD pathology.313 Indeed, recent studies show evidence of increased GFAP, a marker of reactive astrocytes, in postmortem ASD brains.314–316 Additionally, astrocyte-relevant markers aquaporin 4 and connexin 43 were also found to be significantly altered, with increased aquaporin 4 expression in the superior frontal cortex and significantly decreased expression of connexin 43 in the cerebellum of ASD brains.317 Elevated expression of astrocytic markers could be related to the neuroinflammation hypothesis in ASD, which suggests that neuroinflammation, such as those caused by maternal infections, may contribute to risk for ASD.318 Clinical evidence for this hypothesis comes from studies reporting increased levels of pro-inflammatory markers in postmortem ASD brains,319,320 as well as evidence of altered levels of peripheral pro-inflammatory cytokines in children with ASD.321–323 As microglia are the chief immune cells in the CNS, it is generally thought that microglia activation is critical for properly responding to CNS infections, and that disruption to microglia function can lead to chronic neuroinflammation and ASD.294 Supporting this notion is evidence that in 13 individuals with ASD, five cases demonstrated aberrant microglial morphology, with enlarged soma, reduced number of and shortening and thickening of processes.324 Additionally, Suzuki et al.325 recruited 20 individuals with ASD and used Positron emission tomography (PET) to examine the binding potential of [11C] (R)-PK11195, a representative measure of microglia activation, and subsequently found increased microglial activation in the brain of ASD patients, as compared to healthy controls.325 However, whether neuroinflammation is a consequence of or a precursor to ASD pathology is not fully understood.
The extent of myelination in the brain could be assessed using diffusion tensor imaging (DTI), specifically measuring the myelin water fraction (MWF) of white matter based on the idea that myelin acts as a barrier to the diffusion of water molecules.326 Studies using this approach show that young adults with ASD demonstrate significantly reduced MWF compared to matched controls,327 suggesting that myelin alterations may play a role in ASD pathology. Further, ASD patients were found to exhibit increased igG and igA autoantibodies against myelin basic protein, suggesting the possibility of autoimmune-mediated destruction of myelin sheath in ASD pathogenesis.328 Importantly, Smith-Lemli-Opitz syndrome (SLOS), an inborn error of cholesterol biosynthesis, is also highly associated with autism spectrum disorder, with ~50% of individuals meeting criteria for autism.329 As cholesterol is necessary for myelin synthesis, it is conceivable that deficits in myelination may lead to autistic symptoms in some patients with SLOS.
4.4.2 |. Genetic evidence
Genetic evidence for the role of astrocytes and oligodendrocytes are limited, partly because the function of ASD genes in nonneuronal cell-types is vastly understudied. In a recent study, Stogsdill et al.330 showed that neurexin-neuroligin interactions between neurons and astrocytes were critical for astrocyte morphogenesis and synaptogenesis.330 Further, the group demonstrated that contact-mediated mechanisms, as opposed to synaptic activity or extracellular factors, are important for astrocyte morphogenesis, which suggest that other ASD-associated cell adhesion genes may also be worthy of investigation. For oligodendrocytes, it has been shown that ASD risk genes CHD7 and CHD8 play key roles in regulating oligodendrocyte precursor cell proliferation, differentiation, and survival, and CHD7 loss-of-function leads to marked decreases in oligodendrocyte precursor cells in rodent brains.331 In Rett Syndrome, it has been demonstrated that MECP2 is expressed in both neurons and glial cells,332 and that loss of MECP2 function impacts not only neuronal function, but also astrocytes and microglia. Maezawa and colleagues demonstrated that MECP2-null microglia released toxic levels of glutamate, causing abnormal neuronal morphology and synaptic damage.333 Intriguingly, early introduction of wild-type microglia into MECP2-null mice significantly improved various facets of MECP2-null phenotypes.334 Astrocytes harboring MECP2 mutation cocultured with neurons failed to facilitate normal neuronal development, with impaired dendritic outgrowths.332 Further, preferential re-expression of MECP2 in astrocytes in global MECP2 null mice rescued behavioral deficits and aberrant neuronal morphology and synaptic transmission deficits.335 In summary, the role of well-known ASD risk genes on nonneuronal cell functions and their impact on neuronal development and wiring is underappreciated. Dysfunction in glial cells may serve as important drivers for ASD-related neuronal phenotypes. Systematic evaluation of the functional impact of ASD genes on astrocyte, oligodendrocyte, and microglial functions may uncover novel disease mechanisms and provide new methods for intervention.
Gene expression studies also support the role of glial cells in ASD pathology. In a major study, Voineagu and colleagues examined the transcriptomic profile of brain tissue from 19 individuals with ASD and 17 controls. Two gene coexpression modules were found to be associated with ASD: one module was enriched for genes related to synaptic function, neurotransmitter and vesicular transport, and neuronal projection; the second module was enriched for markers of astrocytes and activated microglia.285 Gene expression analysis of 104 ASD postmortem brain tissue samples showed enrichment of genes related to microglial activation and immune response.336 The most significant differentially expressed gene was MAL, which encodes for a transmembrane protein involved with membrane signaling in T cells and myelination.337,338 Overall, the limited genetic and transcriptomic evidence supports the notion that astrocytes, oligodendrocytes, and microglia may all contribute to ASD pathology and represent promising avenues for future investigation.
4.4.3 |. Evidence from iPSC studies
To date, very few studies generated and characterized glial cells from syndromic and nonsyndromic ASD patients. Russo and colleagues generated mixed neuronal cultures consisting of neurons and astrocytes from three individuals with nonsyndromic ASD. ASD cultures contained significantly reduced glutamate levels, expression of synaptic proteins, and synaptic transmission.339 Further, ASD-derived astrocytes generated increased levels of reactive oxygen species, showed impaired ability to support neuronal development, and produced significantly elevated levels of IL-6. Intriguingly, blocking IL-6 led to enhanced synaptogenesis in all three patient lines.339 Williams et al. generated iPSC-derived astrocytes and examined the effect of MECP2 mutations on astrocyte function. Patient-derived astrocytes showed impaired ability to support neurons, with wild-type neurons grown on mutant astrocytes exhibiting decreased soma size, neurite outgrowths and synaptic transmission.340 Intriguingly, IGF-1 and GPE were sufficient to partially rescue neuronal deficits.
4.4.4 |. Summary
Human iPSC-based studies on the roles of astrocytes, microglia and oligodendrocytes in ASD are severely lacking. As previous studies by Russo et al.339 and Williams et al.340 have shown, patient-derived glial cells can heavily modulate neuronal phenotypes and modulating their function may be a promising therapeutic approach. Although it is known that significant interactions and cross-talk exist between neurons, astrocytes, microglia, and oligodendrocytes,313,341 technical challenges limit the study of interactions between these cells in a 2D iPSC-based system. Thus, the use of cortical organoids to model these complex interactions may be beneficial. Indeed, cortical organoids can be grown to contain a mixture of neurons, astrocytes, oligodendrocytes, and even microglia.124,125,129–131,140,141 The use of CRISPR-based genome engineering can enable the selective rescue or knockdown of specific neuronal or glial populations to determine the causal effect of each cell type on neuronal or glial cells of interest, as well as the effect on global network activity342
5 |. FUTURE iPSC-BASED DIAGNOSTIC AND DRUG-DISCOVERY PIPELINE FOR ASD
In the past few decades, we have made significant strides in defining and understanding the clinical manifestations and biological underpinnings related to this group of disorders. Early studies into syndromic comorbidities of ASDs and gene expression studies in postmortem brain tissue of autistic patients have shed light on the genetic and biological pathways that are potentially related to ASDs. Animal studies have been performed to investigate the effect of candidate genes on autistic phenotypes and biological mechanisms. However, genetic heterogeneity, symptom variability, and our evolving understanding of autistic behaviors and what we clinically define as ASD have all hindered progress in understanding and treating these disorders.
It is now clear that ASDs are complex and multifaceted disorders that can be caused by disruptions in multiple biological pathways and, as such, are unlikely to be fully dissected using a “one size fits all” model.343 There is a need to understand whether and how disruption in various stages of neurodevelopment may lead to a common set of symptoms that are observed in specific subsets of ASD patients in light of patients’ genetic background (sub-phenotyping at the cellular level). Therefore, new approaches are needed to systematically identify and characterize the biological convergence points that can serve as biomarkers for specific autistic phenotypes. A previously proposed convergence point may be synaptic homeostasis23,344,345; however, a method is needed for the systematic examination of many other possible convergence points, including proliferation and neurogenesis, migration, and gliogenesis and myelination.
Although mouse models are routinely used to investigate ASD-related deficits and to test candidate drugs, the drugs identified to date have shown low efficacy in humans, limiting their translational potential.346–348 By using human-specific cells, iPSC-based approaches may be advantageous for screening cellular phenotypes and candidate drugs compared to rodent models.57,71 To gain insights into the cellular and molecular phenotypes associated with ASDs, syndromic ASDs, such as Phelan-McDermid Syndrome, Rett Syndrome, Timothy Syndrome, and Fragile X Syndrome are being studied using differentiated and induced iPSC-derived neurons. However, the vast majority of ASDs are idiopathic, with complex genetics or no clear causes. For these cases, it is difficult to generate a priori hypotheses as to which molecular and cellular pathways need to be investigated and which cellular and molecular assays should be utilized.
We propose an iPSC- and organoid-based precision medicine discovery pipeline for identifying the cellular etiology of idiopathic ASDs, determining etiology-specific drug candidates, and grouping patients for clinical trials based on cellular etiology (Figure 3). First, patients with ASDs are recruited, whole-genome sequencing is performed, and patient-specific iPSCs are generated using an efficient and standardized protocol. Then, patient-specific organoids are produced for investigating the cellular phenotypes that have previously been associated with ASDs, including abnormal proliferation and neurogenesis, migration, morphogenesis and synaptogenesis, and gliogenesis. The protocol for organoid production should also be standardized to minimize line-to-line, organoid-to-organoid, and batch-to-batch variability. Single-cell RNA sequencing could be used for an unbiased assessment of the cellular phenotypes (based on the proportions of different cells and enrichment in genes associated with certain pathways) as well as a quality control measure to confirm the consistency. In addition, a neurosphere migration assay in combination with immunocytochemistry and imaging could be used to assess the migration deficits and morphology of differentiated neurons, and multielectrode arrays could be used to test for functional and synaptic deficits.
FIGURE 3.
Precision medicine pipline for diagnostic and drug-discovery for ASDs. Historically, only limited information has been available regarding the cellular and molecular deficits associated with ASDs (Past). Therefore, most affected individuals have received no treatments or have been treated with non-specific mediations. We now know that many genetic abnormalities contribute to the development of ASDs, and we have already identified the cellular and molecular deficits associated with some of these abnormalities using animal models and 2D cultures of patient-specific and engineered iPSC-derived neurons (Present). In the future, we propose to use patient-specific organoids to screen for cellular and molecular deficits in individuals with idiopathic ASDs for use in diagnostic strategies. 2D cultures of stem cell-derived neurons would be used for phenotypic drug screening. Identified candidates would then be validated with patient-specific organoids and tested in animal models for safety, pharmacology, and toxicity. Finally, individuals with idiopathic autism would be classified into groups based on the cellular etiology and recruited into group-specific clinical trials
Once cellular deficits have been identified in organoids, patients and patient-specific iPSCs could be grouped based on the type of cellular deficits identified and further used for pharmacologic screening with an appropriate 2D model. For example, induced neurons could be used for screening drug candidates that can rescue functional or synaptic deficits and differentiated neurons for screening drug candidates that can rescue neurogenesis, migration, or morphogenesis. Challenges, however, may arise if cellular phenotypes present in organoids are not reproduced in 2D models. In this scenario, different differentiation methods and coculture systems can be utilized.
Once the data for a substantial number of patients have been analyzed via this pipeline, whole genome sequencing data could be utilized to identify genetic variants associated with a particular cellular phenotype and the corresponding molecular pathways. Using this pipeline, we should be able to stratify the patient population and categorize patients into treatment groups based on the cellular etiology. This pipeline could potentially be applicable for studies of other complex neuropsychiatric disorders with an unclear neurodevelopmental origin, such as schizophrenia and bipolar disorder.
Funding information
Alan B. Slifka Foundation, Grant/Award Number: Slifka/Ritvo award; Brain and Behavior Research Foundation, Grant/Award Number: NARSAD Young Investigator Grant; Brain Research Foundation, Grant/Award Number: Fay/Frank Seed Grant; National Institute of Mental Health, Grant/Award Number: 1R01MH113670 - 01A1; National Institute of Neurological Disorders and Stroke, Grant/Award Number: NS104963-01A1; Utah Genome Project; Utah Neuroscience Initiative; Whitehall Foundation, Grant/Award Number: 20150886
REFERENCES
- 1.Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 2013;12:406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baio J, Wiggins L, Christensen DL, et al. Prevalence of autism Spectrum disorder among children aged 8 years — autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill Summ. 2018;67:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buxbaum JD, Cai G, Chaste P, et al. Mutation screening of the PTEN gene in patients with autism Spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet. 2007; 144B:484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Martino A, Yan CG, Li Q, et al. The autism brain imaging data exchange: Towards a large-scale evaluation of the intrinsic brain architecture in autism. Molecular Psychiatry. 2014;19: 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey A, Le Couteur A, Gottesman I, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. [DOI] [PubMed] [Google Scholar]
- 6.Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frazier TW, Thompson L, Youngstrom EA, et al. A twin study of heritable and shared environmental contributions to autism. Journal of Autism and Developmental Disorders. 2014;44:2013–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. The heritability of autism Spectrum disorder. JAMA. 2017;318:1182–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan PF, Geschwind DH. Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell. 2019;177:162–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karimi P, Kamali E, Mousavi SM, Karahmadi M. Environmental Factors Influencing the Risk of Autism. J Res Med Sci. 2017; 22:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Gaitsch H, Poon H, Cox NJ, Rzhetsky A. Classification of common human diseases derived from shared genetic and environmental determinants. Nat Genet. 2017;49:1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaugler T, Klei L, Sanders SJ, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grove J, Ripke S, Als TD, et al. Common risk variants identified in autism spectrum disorder. bioRxiv. 2017;224774. [Google Scholar]
- 14.Bush WS, Moore JH. Chapter 11: genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Rubeis S, He X, Goldberg AP, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515: 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iossifov I, Ronemus M, Levy D, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanders SJ, He X, Willsey AJ, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satterstrom FK, Kosmicki JA, Wang J, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. bioRxiv. 2019;484113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geschwind DH, State MW. Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 2015;14: 1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parikshak NN, Luo R, Zhang A, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willsey AJ, Sanders SJ, Li M, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banerjee S, Riordan M, Bhat MA. Genetic aspects of autism spectrum disorders: insights from animal models. Front Cell Neurosci. 2014;8:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourgeron T From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16: 551–563. [DOI] [PubMed] [Google Scholar]
- 24.Jeste SS, Geschwind DH. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat Rev Neurol. 2014;10:74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Y-H, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78:8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaiser T, Feng G. Modeling psychiatric disorders for developing effective treatments. Nat Med. 2015;21:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahin M, Sur M. Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science. 2015; 350:aab3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anney RJL, Ripke S, Anttila V, et al. The Autism Spectrum Disorders Working Group of the Psychiatric Genomics Consortium, 2017. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Molecular Autism. 2017;8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nowakowski TJ, Bhaduri A, Pollen AA, et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science. 2017;358:1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satterstrom FK, Kosmicki JA, Wang J, et al. Novel genes for autism implicate both excitatory and inhibitory cell lineages in risk. bioRxiv. 2018;484113. [Google Scholar]
- 31.Stoner R, Chow ML, Boyle MP, et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med. 2014; 370:1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong S, Zhang S, Fan X, et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature. 2018;555:524–528. [DOI] [PubMed] [Google Scholar]
- 33.Bernier R, Golzio C, Xiong B, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158: 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dietert RR, Dietert JM, Dewitt JC, 2010. Environmental risk factors for autism. Emerg Health Threats J 4, 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nybo Andersen A-M, Hansen KD, Andersen PK, Davey Smith G. Advanced paternal age and risk of fetal death: a cohort study. Am J Epidemiol. 2004;160:1214–1222. [DOI] [PubMed] [Google Scholar]
- 36.McIntosh GC, Olshan AF, Baird PA. Paternal age and the risk of birth defects in offspring. Epidemiology. 1995;6:282–288. [DOI] [PubMed] [Google Scholar]
- 37.Janecka M, Kodesh A, Levine SZ, et al. Association of Autism Spectrum Disorder with Prenatal Exposure to medication affecting neurotransmitter systems. JAMA Psychiat. 2018;75:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salem Yaniv S, Levy A, Wiznitzer A, Holcberg G, Mazor M, Sheiner E. A significant linear association exists between advanced maternal age and adverse perinatal outcome. Arch Gynecol Obstet. 2011;283:755–759. [DOI] [PubMed] [Google Scholar]
- 39.Wu S, Wu F, Ding Y, Hou J, Bi J, Zhang Z. Advanced parental age and autism risk in children: a systematic review and meta-analysis. Acta Psychiatr Scand. 2017;135:29–41. [DOI] [PubMed] [Google Scholar]
- 40.Wong WSW, Solomon BD, Bodian DL, et al. New observations on maternal age effect on germline de novo mutations. Nat Commun. 2016;7:10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rossignol DA, Genuis SJ, Frye RE. Environmental toxicants and autism spectrum disorders: a systematic review. Transl Psychiatry. 2014;4:e360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arndt TL, Stodgell CJ, Rodier PM. The teratology of autism. Int J Dev Neurosci. 2005;23:189–199. [DOI] [PubMed] [Google Scholar]
- 43.Hughes A, Greene NDE, Copp AJ, Galea GL. Valproic acid disrupts the biomechanics of late spinal neural tube closure in mouse embryos. Mech Dev. 2018;149:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christianson AL, Chester N, Kromberg JGR. Fetal valproate syndrome: clinical and Neuro-developmental features in two sibling pairs. Dev Med Child Neurol. 1994;36:361–369. [DOI] [PubMed] [Google Scholar]
- 45.Christensen J, Grønborg TK, Sørensen MJ, et al. Prenatal valproate exposure and risk of autism Spectrum disorders and childhood autism. JAMA. 2013;309:1696–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mabunga DFN, Gonzales ELT, Kim J, Kim KC, Shin CY. Exploring the validity of Valproic acid animal model of autism. Exp Neurobiol. 2015;24:285–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruhela RK, Sarma P, Soni S, Prakash A, Medhi B. Congenital malformation and autism spectrum disorder: insight from a rat model of autism spectrum disorder. Indian J Pharm. 2017;49: 243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim KC, Kim P, Go HS, et al. The critical period of valproate exposure to induce autistic symptoms in Sprague–Dawley rats. Toxicol Lett. 2011a;201:137–142. [DOI] [PubMed] [Google Scholar]
- 49.Fujimura K, Mitsuhashi T, Shibata S, Shimozato S, Takahashi T. In utero exposure to Valproic acid induces neocortical Dysgenesis via Dysregulation of neural progenitor cell proliferation/differentiation. J Neurosci. 2016;36:10908–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jung G-A, Yoon J-Y, Moon B-S, et al. Valproic acid induces differentiation and inhibition of proliferation in neural progenitor cells via the beta-catenin-Ras-ERK-p21Cip/WAF1 pathway. BMC Cell Biol. 2008;9:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brumback AC, Ellwood IT, Kjaerby C, et al. Identifying specific prefrontal neurons that contribute to autism-associated abnormalities in physiology and social behavior. Mol Psychiatry. 2018;23: 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rinaldi T, Kulangara K, Antoniello K, Markram H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. PNAS. 2007;104:13501–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zerbo O, Qian Y, Yoshida C, Grether JK, Van de Water J, Croen LA. Maternal infection during pregnancy and autism Spectrum disorders. J Autism Dev Disord. 2015;45:4015–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beversdorf DQ, Manning SE, Hillier A, et al. Timing of prenatal stressors and autism. J Autism Dev Disord. 2005;35:471–478. [DOI] [PubMed] [Google Scholar]
- 55.Meyer U, Yee BK, Feldon J. The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse? Neuroscientist. 2007;13:241–256. [DOI] [PubMed] [Google Scholar]
- 56.Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dolmetsch R, Geschwind DH. The human brain in a dish: the promise of iPSC-derived neurons. Cell. 2011;145:831–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hansen DV, Rubenstein JLR, Kriegstein AR. Deriving excitatory neurons of the Neocortex from pluripotent stem cells. Neuron. 2011;70:645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suzuki IK, Vanderhaeghen P. Is this a brain which I see before me? Modeling human neural development with pluripotent stem cells. Development. 2015;142:3138–3150. [DOI] [PubMed] [Google Scholar]
- 61.Ho SM, Topol A, Brennand KJ. From “directed differentiation” to “neuronal induction”: Modeling neuropsychiatric disease. Bio-mark Insights. 2015;10:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu Y-L, Yoo AS. Mechanistic insights into MicroRNA-induced neuronal reprogramming of human adult fibroblasts. Front Neurosci. 2018;12:522–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang N, Ng YH, Pang ZP, Südhof TC, Wernig M. Induced neuronal cells: how to make and define a neuron. Cell Stem Cell. 2011;9:517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amin ND, Paşca SP. Building models of brain disorders with three-dimensional Organoids. Neuron. 2018;100:389–405. [DOI] [PubMed] [Google Scholar]
- 65.Di Lullo E, Kriegstein AR. The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci. 2017;18: 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kelava I, Lancaster MA. Stem cell models of human brain development. Cell Stem Cell. 2017;18:736–748. [DOI] [PubMed] [Google Scholar]
- 67.Quadrato G, Brown J, Arlotta P. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat Med. 2016;22:1220–1228. [DOI] [PubMed] [Google Scholar]
- 68.Sasai Y Cytosystems dynamics in self-organization of tissue architecture. Nature. 2013b;493:318–326. [DOI] [PubMed] [Google Scholar]
- 69.Ichida JK, Kiskinis E. Probing disorders of the nervous system using reprogramming approaches. EMBO J. 2015;34:1456–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paşca SP, Panagiotakos G, Dolmetsch RE. Generating human neurons in vitro and using them to understand neuropsychiatric disease. Annu Rev Neurosci. 2014;37:479–501. [DOI] [PubMed] [Google Scholar]
- 71.Sun Y, Dolmetsch RE. How induced pluripotent stem cells are informing drug discovery in psychiatry. Swiss Med Wkly. 2016; 146:w14241–w14241. [DOI] [PubMed] [Google Scholar]
- 72.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998; 282:1145–1147. [DOI] [PubMed] [Google Scholar]
- 73.Eiraku M, Watanabe K, Matsuo-Takasaki M, et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell. 2008;3: 519–532. [DOI] [PubMed] [Google Scholar]
- 74.Espuny-Camacho I, Michelsen KA, Gall D, et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron. 2013;77:440–456. [DOI] [PubMed] [Google Scholar]
- 75.Mariani J, Simonini MV, Palejev D, et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci. 2012;109:12770–12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maroof AMM, Keros S, Tyson JAA, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Watanabe K, Kamiya D, Nishiyama A, et al. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci. 2005;8:288–296. [DOI] [PubMed] [Google Scholar]
- 78.Amoroso MW, Croft GF, Williams DJ, et al. Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J Neurosci. 2013;33:574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boulting GL, Kiskinis E, Croft GF, et al. A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22: 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kirkeby A, Grealish S, Wolf DA, et al. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012;1: 703–714. [DOI] [PubMed] [Google Scholar]
- 82.Koch P, Opitz T, Steinbeck JA, Ladewig J, Brüstle O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci U S A. 2009;106:3225–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li XJ, Du ZW, Zarnowska ED, et al. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol. 2005;23: 215–221. [DOI] [PubMed] [Google Scholar]
- 84.Marklund M, Sjödal M, Beehler BC, Jessell TM, Edlund T, Gunhaga L. Retinoic acid signalling specifies intermediate character in the developing telencephalon. Development. 2004;131: 4323–4332. [DOI] [PubMed] [Google Scholar]
- 85.Maury Y, Côme J, Piskorowski RA, et al. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat Biotechnol. 2015; 33:89–96. [DOI] [PubMed] [Google Scholar]
- 86.Nordström U, Jessell TM, Edlund T. Progressive induction of caudal neural character by graded Wnt signaling. Nat Neurosci. 2002;5:525–532. [DOI] [PubMed] [Google Scholar]
- 87.Gutin G, Fernandes M, Palazzolo L, et al. FGF signalling generates ventral telencephalic cells independently of SHH. Development. 2006;133:2937–2946. [DOI] [PubMed] [Google Scholar]
- 88.Paek H, Gutin G, Hébert JM. FGF signaling is strictly required to maintain early telencephalic precursor cell survival. Development. 2009;136:2457–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shi Y, Kirwan P, Smith J, Robinson HPC, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 2012;15:477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ericson J, Morton S, Kawakami A, Roelink H, Jessell TM. Two critical periods of sonic hedgehog signaling required for the specification of motor neuron identity. Cell. 1996;87:661–673. [DOI] [PubMed] [Google Scholar]
- 91.Liu Y, Weick JP, Liu H, et al. Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat Biotechnol. 2013;31:440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nicholas CR, Chen J, Tang Y, et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended time-line and mimics human neural development. Cell Stem Cell. 2013;12:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marchetto MCN, Carromeu C, Acab A, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shcheglovitov A, Shcheglovitova O, Yazawa M, et al. Shank3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zaslavsky K, Zhang W-B, McCready FP, et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci. 2019;22:556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sun Y, Paşca SP, Portmann T, et al. A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet syndrome patients. elife. 2016;5:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Song H, Stevens CF, Gage FH. Neural stem cells from adult hippocampus develop essential properties of functional CNS neurons. Nat Neurosci. 2002;5:438–445. [DOI] [PubMed] [Google Scholar]
- 98.Pang ZP, Yang N, Vierbuchen T, et al. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yoo AS, Sun AX, Li L, et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature. 2011;476:228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Masserdotti G, Gascón S, Götz M. Direct neuronal reprogramming: learning from and for development. Development. 2016;143:2494–2510. [DOI] [PubMed] [Google Scholar]
- 102.Tsunemoto RK, Eade KT, Blanchard JW, Baldwin KK. Forward engineering neuronal diversity using direct reprogramming. EMBO J. 2015;34:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Abernathy DG, Kim WK, McCoy MJ, et al. MicroRNAs induce a permissive chromatin environment that enables neuronal sub-type-specific reprogramming of adult human fibroblasts. Cell Stem Cell. 2017;21:332–348.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Victor MB, Richner M, Olsen HE, et al. Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat Neurosci. 2018;21: 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huh CJ, Zhang B, Victor MB, et al. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. elife. 2016;5:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang Y, Pak C, Han Y, et al. Rapid Sngle-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pak C, Danko T, Zhang Y, et al. Human neuropsychiatric disease modeling using conditional deletion reveals synaptic transmission defects caused by heterozygous mutations in NRXN1. Cell Stem Cell. 2015;17:316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yi F, Danko T, Botelho SC, et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science. 2016;352:aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Benevento M, Iacono G, Selten M, et al. Histone Methylation by the Kleefstra Syndrome Protein EHMT1 Mediates Homeostatic Synaptic Scaling. Neuron. 2016;91:341–355. [DOI] [PubMed] [Google Scholar]
- 110.Liu Y, Yu C, Daley TP, et al. CRISPR activation screens systematically identify factors that Drive neuronal fate and reprogramming. Cell Stem Cell. 2018;23:758–771.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517–530. [DOI] [PubMed] [Google Scholar]
- 112.Battiste J, Helms AW, Kim EJ, et al. Ascl1 defines sequentially generated lineage-restricted neuronal and oligodendrocyte precursor cells in the spinal cord. Development. 2007;134:285–293. [DOI] [PubMed] [Google Scholar]
- 113.Busskamp V, Lewis NE, Guye P, et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol Syst Biol. 2014;10:760–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mall M, Kareta MS, Chanda S, et al. Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature. 2017;544:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Treutlein B, Lee QY, Camp JG, et al. Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature. 2016;534:391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schafer ST, Paquola ACM, Stern S, et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat Neurosci. 2019;22:243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nehme R, Zuccaro E, Ghosh SD, et al. Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep. 2018;23:2509–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.El Mestikawy S, Wallén-Mackenzie Å, Fortin GM, Descarries L, Trudeau LE. From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nat Rev Neurosci. 2011;12: 204–216. [DOI] [PubMed] [Google Scholar]
- 119.Kerchner GA, Nicoll R. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9: 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9:2329–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sasai Y Next-generation regenerative medicine: organogenesis from stem cells in 3D culture. Cell Stem Cell. 2013a;12:520–530. [DOI] [PubMed] [Google Scholar]
- 122.Kadoshimaa T, Sakaguchi H, Nakanoa T, et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci. 2013;110:20285–20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lancaster MA, Renner M, Martin C-A, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Paşca AM, Sloan SA, Clarke LE, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Qian X, Nguyen HN, Song MM, et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 2016;165:1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Watanabe M, Buth JE, Vishlaghi N, et al. Self-organized cerebral Organoids with human-specific features predict effective drugs to combat Zika virus infection. Cell Rep. 2017;21:517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lui JH, Hansen DV, Kriegstein AR. Development and evolution of the human neocortex. Cell. 2011;146:18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rakic P Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci. 2009;10:724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sloan SA, Darmanis S, Huber N, et al. Human astrocyte maturation captured in 3D cerebral cortical spheroids derived from pluripotent stem cells. Neuron. 2017;95:779–790.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Madhavan M, Nevin ZS, Shick HE, et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat Methods. 2018;15:700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Marton RM, Miura Y, Sloan SA, et al. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat Neurosci. 2019;22:484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Eiraku M, Sasai Y. Mouse embryonic stem cell culture for generation of three-dimensional retinal and cortical tissues. Nat Protoc. 2012;7:69–79. [DOI] [PubMed] [Google Scholar]
- 133.Birey F, Andersen J, Makinson CD, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545: 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xiang Y, Tanaka Y, Patterson B, et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell. 2017;21:383–398.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jo J, Xiao Y, Sun AX, et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell. 2016;19: 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015;10:537–550. [DOI] [PubMed] [Google Scholar]
- 137.Hor JH, Soh ES-Y, Tan LY, et al. Cell cycle inhibitors protect motor neurons in an organoid model of spinal muscular atrophy. Cell Death Dis. 2018;9:1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mariani J, Coppola G, Zhang P, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell. 2015;162:375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bershteyn M, Nowakowski TJ, Pollen AA, et al. Human iPSC-derived cerebral Organoids model cellular features of Lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell. 2017;20(4):435–449. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Quadrato G, Nguyen T, Macosko EZ, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ormel PR, de Sá RV, van Bodegraven EJ, et al. Microglia innately develop within cerebral organoids. Nat Commun. 2018; 9:4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yoon SJ, Elahi LS, Pașca AM, et al. Reliability of human cortical organoid generation. Nat Methods. 2019;16:75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ladewig J, Koch P, Brüstle O. Auto-attraction of neural precursors and their neuronal progeny impairs neuronal migration. Nat Neurosci. 2014;17:24–26. [DOI] [PubMed] [Google Scholar]
- 144.Knight GT, Lundin BF, Iyer N, et al. Engineering induction of singular neural rosette emergence within hPSC-derived tissues. elife. 2018;7:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee CT, Chen J, Kindberg AA, et al. CYP3A5 mediates effects of cocaine on human neocorticogenesis: studies using an in vitro 3D self-organized hPSC model with a single cortex-like unit. Neuropsychopharmacology. 2017;42:774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zwaigenbaum L, Bryson S, Rogers T, Roberts W, Brian J, Szatmari P. Behavioral manifestations of autism in the first year of life. Int J Dev Neurosci. 2005;23:143–152. [DOI] [PubMed] [Google Scholar]
- 147.Stiles J, Jernigan TL. The basics of brain development. Neuropsychol Rev. 2010;20:327–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kalebic N, Gilardi C, Stepien B, et al. Neocortical expansion due to increased proliferation of basal progenitors is linked to changes in their morphology. Cell Stem Cell. 2019;24:535–550.e9. [DOI] [PubMed] [Google Scholar]
- 149.Ortega JA, Memi F, Radonjic N, et al. The subventricular zone: a key player in human neocortical development. Neuroscientist. 2018;24:156–170. [DOI] [PubMed] [Google Scholar]
- 150.Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. [DOI] [PubMed] [Google Scholar]
- 151.Urbán N, Guillemot F. Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front Cell Neurosci. 2014; 8:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Miles JH, Takahashi TN, Bagby S, et al. Essential versus complex autism: definition of fundamental prognostic subtypes. Am J Med Genet A. 2005;135A:171–180. [DOI] [PubMed] [Google Scholar]
- 153.Courchesne E, Carper R, Akshoomoff N. Evidence of brain over-growth in the first year of life in autism. JAMA. 2003;290: 337–344. [DOI] [PubMed] [Google Scholar]
- 154.Shen MD, Nordahl CW, Young GS, et al. Early brain enlargement and elevated extra-axial fluid in infants who develop autism spectrum disorder. Brain. 2013;136:2825–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Redcay E, Courchesne E. When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biol Psychiatry. 2005; 58:1–9. [DOI] [PubMed] [Google Scholar]
- 156.Courchesne E, Campbell K, Solso S. Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Res. 2011;1380:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kotagiri P, Chance SA, Szele FG, Esiri MM. Subventricular zone cytoarchitecture changes in autism. Dev Neurobiol. 2014;74: 25–41. [DOI] [PubMed] [Google Scholar]
- 158.Rakic P Progress: neurogenesis in adult primate neocortex: an evaluation of the evidence. Nat Rev Neurosci. 2002;3:65–71. [DOI] [PubMed] [Google Scholar]
- 159.Groszer M, Erickson R, Scripture-Adams DD, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. [DOI] [PubMed] [Google Scholar]
- 160.Orosco LA, Ross AP, Cates SL, et al. Loss of Wdfy3 in mice alters cerebral cortical neurogenesis reflecting aspects of the autism pathology. Nat Commun. 2014;5:4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhang A, Yeung PL, Li C-W, et al. Identification of a novel family of Ankyrin repeats containing cofactors for p160 nuclear receptor Coactivators. J Biol Chem. 2004;279:33799–33805. [DOI] [PubMed] [Google Scholar]
- 162.Gallagher D, Voronova A, Zander MA, et al. Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev Cell. 2015;32:31–42. [DOI] [PubMed] [Google Scholar]
- 163.Shen T, Ji F, Yuan Z, Jiao J. CHD2 is required for embryonic neurogenesis in the developing cerebral cortex. Stem Cells. 2015; 33:1794–1806. [DOI] [PubMed] [Google Scholar]
- 164.Sheridan SD, Theriault KM, Reis SA, et al. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS ONE. 2011;6:e26203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Telias M, Segal M, Ben-Yosef D. Neural differentiation of fragile X human embryonic stem cells reveals abnormal patterns of development despite successful neurogenesis. Dev Biol. 2013; 374:32–45. [DOI] [PubMed] [Google Scholar]
- 166.Li Y, Barkovich MJ, Karch CM, et al. Regionally specific TSC1 and TSC2 gene expression in tuberous sclerosis complex. Sci Rep. 2018;8:13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nadadhur AG, Alsaqati M, Gasparotto L, et al. Neuron-glia interactions increase neuronal phenotypes in tuberous sclerosis complex patient iPSC-derived models. Stem Cell Reports. 2019;12: 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zucco AJ, Pozzo VD, Afinogenova A, Hart RP, Devinsky O, D’Arcangelo G. Neural progenitors derived from tuberous sclerosis complex patients exhibit attenuated PI3K/AKT signaling and delayed neuronal differentiation. Mol Cell Neurosci. 2018;92: 149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li Y, Cao J, Chen M, et al. Abnormal Neural Progenitor Cells Differentiated from Induced Pluripotent Stem Cells Partially Mimicked Development of TSC2 Neurological Abnormalities. Stem Cell Reports. 2017;8(4):883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Falk S, Götz M. Glial control of neurogenesis. Curr Opin Neurobiol. 2017;47:188–195. [DOI] [PubMed] [Google Scholar]
- 171.Nadarajah B, Alifragis P, Wong ROL, Parnavelas JG. Neuronal migration in the developing cerebral cortex: observations based on real-time imaging. Cereb Cortex. 2003;13:607–611. [DOI] [PubMed] [Google Scholar]
- 172.Corbin JG, Nery S, Fishell G. Telencephalic cells take a tangent: non-radial migration in the mammalian forebrain. Nat Neurosci. 2001;4(Suppl):1177–1182. [DOI] [PubMed] [Google Scholar]
- 173.Park HT, Wu J, Rao Y. Molecular control of neuronal migration. BioEssays. 2002;24:821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hutsler JJ, Love T, Zhang H. Histological and magnetic resonance imaging assessment of cortical layering and thickness in autism spectrum disorders. Biol Psychiatry. 2007;61:449–457. [DOI] [PubMed] [Google Scholar]
- 175.Wegiel J, Kuchna I, Nowicki K, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010;119:755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Watrin F, Manent J-B, Cardoso C, Represa A. Causes and consequences of Gray matter heterotopia. CNS Neurosci Ther. 2015; 21:112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lammert DB, Howell BW. RELN mutations in autism Spectrum disorder. Front Cell Neurosci. 2016;10:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hirota Y, Nakajima K. Control of neuronal migration and aggregation by Reelin Signaling in the developing cerebral cortex. Front Cell Dev Biol. 2017;5:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chang BS, Duzcan F, Kim S, et al. The role of RELN in lissencephaly and neuropsychiatric disease. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:58–63. [DOI] [PubMed] [Google Scholar]
- 180.Guidotti A, Auta J, Davis JM, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. [DOI] [PubMed] [Google Scholar]
- 181.Herring A, Donath A, Steiner KM, et al. Reelin depletion is an early phenomenon of Alzheimer’s pathology. J Alzheimers Dis. 2012;30:963–979. [DOI] [PubMed] [Google Scholar]
- 182.Persico AM, D’Agruma L, Maiorano N, et al. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol Psychiatry. 2001;6:150–159. [DOI] [PubMed] [Google Scholar]
- 183.Hori K, Hoshino M. Neuronal migration and AUTS2 syndrome. Brain Sci. 2017;7(5):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.DeRosa BA, Hokayem JE, Artimovich E, et al. Convergent pathways in idiopathic autism revealed by time course Transcriptomic analysis of patient-derived neurons. Sci Rep. 2018;8:8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. [DOI] [PubMed] [Google Scholar]
- 186.Mühleisen TW, Leber M, Schulze TG, et al. Genome-wide association study reveals two new risk loci for bipolar disorder. Nat Commun. 2014;5:3339. [DOI] [PubMed] [Google Scholar]
- 187.Zhang Z-N, Freitas BC, Qian H, et al. Layered hydrogels accelerate iPSC-derived neuronal maturation and reveal migration defects caused by MeCP2 dysfunction. Proc Natl Acad Sci U S A. 2016;113:3185–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Amenduni M, De Filippis R, Cheung AYL, et al. iPS cells to model CDKL5-related disorders. Eur J Hum Genet. 2011;19: 1246–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cheung AYL, Horvath LM, Grafodatskaya D, et al. Isolation of MECP2-null Rett syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011; 20:2103–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hotta A, Cheung AYL, Farra N, et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat Methods. 2009;6:370–376. [DOI] [PubMed] [Google Scholar]
- 191.Kim K-Y, Hysolli E, Park I-H. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc Natl Acad Sci U S A. 2011b;108:14169–14174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Paşca SP, Portmann T, Voineagu I, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17:1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Duval K, Grover H, Han L-H, et al. Modeling physiological events in 2D vs. 3D cell culture. Physiology. 2017;32:266–277. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 194.Azzarelli R, Oleari R, Lettieri A, Andre’ V, Cariboni A. In vitro, ex vivo and in vivo techniques to study neuronal migration in the developing cerebral cortex. Brain Sci. 2017;7(5):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Topol A, Tran NN, Brennand KJ. A guide to generating and using hiPSC derived NPCs for the study of neurological diseases. J Vis Exp. 2015;96:e52495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bagley JA, Reumann D, Bian S, Lévi-Strauss J, Knoblich JA. Fused dorsal-ventral cerebral organoids model complex interactions between diverse brain regions. Nat Methods. 2017;14: 743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Cardenas A, Kong M, Alvarez A, Leyton HM. Signaling pathways involved in neuron-astrocyte adhesion and migration. Curr Mol Med. 2014;14:275–290. [DOI] [PubMed] [Google Scholar]
- 198.Doretto S, Malerba M, Ramos M, et al. Oligodendrocytes as regulators of neuronal networks during early postnatal development. PLoS ONE. 2011;6(5):e19849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sainath R, Gallo G. Cytoskeletal and signaling mechanisms of neurite formation. Cell Tissue Res. 2015;359:267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lowery LA, Van Vactor D. The trip of the tip: understanding the growth cone machinery. Nat Rev Mol Cell Biol. 2009;10: 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Garner CC, Zhai RG, Gundelfinger ED, Ziv NE. Molecular mechanisms of CNS synaptogenesis. Trends Neurosci. 2002;25: 243–250. [DOI] [PubMed] [Google Scholar]
- 202.McAllister AK. Dynamic aspects of synapse formation. Annu Rev Neurosci. 2007;30:425–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, et al. Assembling the presynaptic active zone: a characterization of an active zone precursor vesicle. Neuron. 2001;29:131–143. [DOI] [PubMed] [Google Scholar]
- 204.Okabe S, Miwa A, Okado H. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci. 2001;21:6105–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gerrow K, Romorini S, Nabi SM, Colicos MA, Sala C, El-Husseini A. A preformed complex of postsynaptic proteins is involved in excitatory synapse development. Neuron. 2006;49: 547–562. [DOI] [PubMed] [Google Scholar]
- 206.Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. [DOI] [PubMed] [Google Scholar]
- 207.Lai K-O, Jordan BA, Ma X-M, Srivastava DP, Tolias KF. Molecular mechanisms of dendritic spine development and plasticity. Neural Plast. 2016;2016:2078121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. [DOI] [PubMed] [Google Scholar]
- 209.Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. [DOI] [PubMed] [Google Scholar]
- 210.Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bolton PF, Carcani-Rathwell I, Hutton J, Goode S, Howlin P, Rutter M. Epilepsy in autism: features and correlates. Br J Psychiatry. 2011;198:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Oblak AL, Gibbs TT, Blatt GJ. Reduced GABAA receptors and benzodiazepine binding sites in the posterior cingulate cortex and fusiform gyrus in autism. Brain Res. 2011;1380:218–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fatemi SH, Folsom TD, Reutiman TJ, Thuras PD. Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum. 2009a;8:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD. GABA(a) receptor downregulation in brains of subjects with autism. J Autism Dev Disord. 2009b;39:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yip J, Soghomonian JJ, Blatt GJ. Decreased GAD65 mRNA levels in select subpopulations in the cerebellar dentate nuclei in autism: an in situ hybridization study. Autism Res. 2009;2:50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lee M, Martin-Ruiz C, Graham A, et al. Nicotinic receptor abnormalities in the cerebellar cortex in autism. Brain. 2002;125: 1483–1495. [DOI] [PubMed] [Google Scholar]
- 217.Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord. 2001;31:537–543. [DOI] [PubMed] [Google Scholar]
- 218.Lawrence YA, Kemper TL, Bauman ML, Blatt GJ. Parvalbumin-, calbindin-, and calretinin-immunoreactive hippocampal interneuron density in autism. Acta Neurol Scand. 2010;121:99–108. [DOI] [PubMed] [Google Scholar]
- 219.Oblak A, Gibbs TT, Blatt GJ. Decreased GABAA receptors and benzodiazepine binding sites in the anterior cingulate cortex in autism. Autism Res. 2009;2:205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lepagnol-Bestel A-M, Maussion G, Boda B, et al. SLC25A12 expression is associated with neurite outgrowth and is upregulated in the prefrontal cortex of autistic subjects. Mol Psychiatry. 2008;13:385–397. [DOI] [PubMed] [Google Scholar]
- 221.Mukaetova-Ladinska EB, Arnold H, Jaros E, Perry R, Perry E. Depletion of MAP2 expression and laminar cytoarchitectonic changes in dorsolateral prefrontal cortex in adult autistic individuals. Neuropathol Appl Neurobiol. 2004;30:615–623. [DOI] [PubMed] [Google Scholar]
- 222.Raymond GV, Bauman ML, Kemper TL. Hippocampus in autism: a Golgi analysis. Acta Neuropathol. 1996;91:117–119. [DOI] [PubMed] [Google Scholar]
- 223.Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94. [DOI] [PubMed] [Google Scholar]
- 224.Zhu D, Zhang T, Jiang X, et al. Fusing DTI and FMRI data: a survey of methods and applications. NeuroImage. 2014;102(Pt 1):184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Philip RCM, Dauvermann MR, Whalley HC, Baynham K, Lawrie SM, Stanfield AC. A systematic review and meta-analysis of the fMRI investigation of autism spectrum disorders. Neurosci Biobehav Rev. 2012;36:901–942. [DOI] [PubMed] [Google Scholar]
- 226.Hull JV, Dokovna LB, Jacokes ZJ, Torgerson CM, Irimia A, Van Horn JD. Resting-State functional connectivity in autism Spectrum disorders: a review. Front Psych. 2017;7:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Antoine MW, Langberg T, Schnepel P, Feldman DE. Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron. 2019;101:648–661.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dichter GS. Functional magnetic resonance imaging of autism spectrum disorders. Dialogues Clin Neurosci. 2012;14:319–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Berwin B, Floor E, Martin TF. CAPS (mammalian UNC-31) protein localizes to membranes involved in dense-core vesicle exocytosis. Neuron. 1998;21:137–145. [DOI] [PubMed] [Google Scholar]
- 230.Mitra R, Ferguson D, Sapolsky R. SK2 potassium channel over-expression in basolateral amygdala reduces anxiety, stress-induced corticosterone and dendritic arborization. Mol Psychiatry. 2009;14:847–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+ −mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. [DOI] [PubMed] [Google Scholar]
- 232.Singh K, Loreth D, Pöttker B, et al. Neuronal growth and behavioral alterations in mice deficient for the psychiatric disease-associated Negr1 gene. Front Mol Neurosci. 2018;11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hashimoto R, Nakazawa T, Tsurusaki Y, et al. Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. J Hum Genet. 2016;61:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Gilbert J, Man H-Y, 2016. The X-linked autism protein KIAA2022/KIDLIA regulates Neurite outgrowth via N-cadherin and δ-catenin Signaling. eNeuro. 2016;3(5):ENEURO.0238–16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Garami A, Zwartkruis FJT, Nobukuni T, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–1466. [DOI] [PubMed] [Google Scholar]
- 236.Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience. 2008; 151:476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kwon C-H, Luikart BW, Powell CM, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50: 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Magdalon J, Sánchez-Sánchez SM, Griesi-Oliveira K, Sertié AL. Dysfunctional mTORC1 Signaling: a convergent mechanism between syndromic and nonsyndromic forms of autism Spectrum disorder? Int J Mol Sci. 2017;18(3):659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ahn M-J, Cho G-W. Metformin promotes neuronal differentiation and neurite outgrowth through AMPK activation in human bone marrow-mesenchymal stem cells. Biotechnol Appl Biochem. 2017;64:836–842. [DOI] [PubMed] [Google Scholar]
- 240.Gutierrez H, Dolcet X, Tolcos M, Davies A. HGF regulates the development of cortical pyramidal dendrites. Development. 2004; 131:3717–3726. [DOI] [PubMed] [Google Scholar]
- 241.de Anda FC, Rosario AL, Durak O, et al. An autism spectrum disorder susceptibility gene, TAO2, is important for basal dendrite formation in the neocortex. Nat Neurosci. 2012;15:1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Hussman JP, Chung R-H, Griswold AJ, et al. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol Autism. 2011;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. [DOI] [PubMed] [Google Scholar]
- 244.Graf ER, Zhang X, Jin S-X, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rothwell PE, Fuccillo MV, Maxeiner S, et al. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158:198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Tabuchi K, Blundell J, Etherton MR, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Basu R, Duan X, Taylor MR, et al. Heterophilic type II Cadherins are required for high-magnitude synaptic potentiation in the Hippocampus. Neuron. 2017;96:160–176.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Bekirov IH, Needleman LA, Zhang W, Benson DL. Identification and localization of multiple classic cadherins in developing rat limbic system. Neuroscience. 2002;115:213–227. [DOI] [PubMed] [Google Scholar]
- 250.Friedman LG, Riemslagh FW, Sullivan JM, et al. Cadherin-8 expression, synaptic localization and molecular control of neuronal form in prefrontal cortico-striatal circuits. J Comp Neurol. 2015;523:75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Gil OD, Needleman L, Huntley GW. Developmental patterns of cadherin expression and localization in relation to compartmentalized thalamocortical terminations in rat barrel cortex. J Comp Neurol. 2002;453:372–388. [DOI] [PubMed] [Google Scholar]
- 252.Smith KR, Jones KA, Kopeikina KJ, et al. Cadherin-10 maintains excitatory/inhibitory ratio through interactions with synaptic proteins. J Neurosci. 2017;37:11127–11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wang K, Zhang H, Ma D, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009; 459:528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Williams ME, Wilke SA, Daggett A, et al. Cadherin-9 regulates synapse-specific differentiation in the developing Hippocampus. Neuron. 2011;71:640–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Poliak S, Gollan L, Martinez R, et al. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24:1037–1047. [DOI] [PubMed] [Google Scholar]
- 256.Poliak S, Salomon D, Elhanany H, et al. Juxtaparanodal clustering of shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Gao R, Piguel NH, Melendez-Zaidi AE, et al. CNTNAP2 stabilizes interneuron dendritic arbors through CASK. Mol Psychiatry. 2018;23:1832–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Peñagarikano O, Abrahams BS, Herman EI, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Karayannis T, Au E, Patel JC, et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature. 2014;511:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Yin F-T, Futagawa T, Li D, et al. Caspr4 interaction with LNX2 modulates the proliferation and neuronal differentiation of mouse neural progenitor cells. Stem Cells Dev. 2015;24:640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Martin EA, Muralidhar S, Wang Z, et al. The intellectual disability gene Kirrel3 regulates target-specific mossy fiber synapse development in the hippocampus. elife. 2015;4:e09395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Leblond CS, Nava C, Polge A, et al. Meta-analysis of SHANK mutations in autism Spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 2014;10:e1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Gouder L, Vitrac A, Goubran-Botros H, et al. Altered spinogenesis in iPSC-derived cortical neurons from patients with autism carrying de novo SHANK3 mutations. Sci Rep. 2019; 9:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Giovedì S, Darchen F, Valtorta F, Greengard P, Benfenati F. Synapsin is a novel Rab3 effector protein on small synaptic vesicles. II. Functional effects of the Rab3A-synapsin I interaction. J Biol Chem. 2004;279:43769–43779. [DOI] [PubMed] [Google Scholar]
- 265.Ceccaldi PE, Grohovaz F, Benfenati F, Chieregatti E, Greengard P, Valtorta F. Dephosphorylated synapsin I anchors synaptic vesicles to Actin cytoskeleton: an analysis by videomicroscopy. J Cell Biol. 1995;128:905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Corradi A, Fadda M, Piton A, et al. SYN2 is an autism predisposing gene: loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum Mol Genet. 2014;23:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Fassio A, Patry L, Congia S, et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet. 2011;20:2297–2307. [DOI] [PubMed] [Google Scholar]
- 268.Ferreira A, Kosik KS, Greengard P, Han HQ. Aberrant neurites and synaptic vesicle protein deficiency in synapsin II-depleted neurons. Science. 1994;264:977–979. [DOI] [PubMed] [Google Scholar]
- 269.Perlini LE, Botti F, Fornasiero EF, et al. Effects of phosphorylation and neuronal activity on the control of synapse formation by synapsin I. J Cell Sci. 2011;124:3643–3653. [DOI] [PubMed] [Google Scholar]
- 270.Chin LS, Li L, Ferreira A, Kosik KS, Greengard P. Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9230–9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Ferreira A, Chin LS, Li L, Lanier LM, Kosik KS, Greengard P. Distinct roles of synapsin I and synapsin II during neuronal development. Mol Med. 1998;4:22–28. [PMC free article] [PubMed] [Google Scholar]
- 272.Gitler D, Takagishi Y, Feng J, et al. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004;24:11368–11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851–852. [DOI] [PubMed] [Google Scholar]
- 274.Ogiwara I, Miyamoto H, Tatsukawa T, et al. Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun Biol. 2018;1:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Sanders SJ, Murtha MT, Gupta AR, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Hosie AM, Dunne EL, Harvey RJ, Smart TG. Zinc-mediated inhibition of GABA(A) receptors: discrete binding sites underlie subtype specificity. Nat Neurosci. 2003;6:362–369. [DOI] [PubMed] [Google Scholar]
- 277.Geisler S, Schöpf CL, Stanika R, et al. Presynaptic α2δ−2 Calcium Channel subunits regulate postsynaptic GABAA receptor abundance and axonal wiring. J Neurosci. 2019;39:2581–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Chen Y-K, Chen C-Y, Hu H-T, Hsueh Y-P. CTTNBP2, but not CTTNBP2NL, regulates dendritic spinogenesis and synaptic distribution of the striatin–PP2A complex. Mol Biol Cell. 2012;23: 4383–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Doers ME, Musser MT, Nichol R, et al. iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014;23:1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Krey JF, Pasca SP, Shcheglovitov A, et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Costa V, Aigner S, Vukcevic M, et al. mTORC1 inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep. 2016;15:86–95. [DOI] [PubMed] [Google Scholar]
- 282.Kathuria A, Nowosiad P, Jagasia R, et al. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol Psychiatry. 2018;23:735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Huang G, Chen S, Chen X, et al. Uncovering the functional link between SHANK3 deletions and deficiency in neurodevelopment using iPSC-derived human neurons. Front Neuroanat. 2019; 13:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Molliver ME, Kostović I, van der Loos H. The development of synapses in cerebral cortex of the human fetus. Brain Res. 1973; 50:403–407. [DOI] [PubMed] [Google Scholar]
- 285.Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Menassa DA, Gomez-Nicola D. Microglial dynamics during human brain development. Front Immunol. 2018;9:1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Takouda J, Katada S, Nakashima K. Emerging mechanisms underlying astrogenesis in the developing mammalian brain. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:386–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Yamaguchi M, Seki T, Imayoshi I, et al. Neural stem cells and neuro/gliogenesis in the central nervous system: understanding the structural and functional plasticity of the developing, mature, and diseased brain. J Physiol Sci. 2016;66:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Levison SW, Goldman JE. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron. 1993;10:201–212. [DOI] [PubMed] [Google Scholar]
- 290.Marshall CAG, Suzuki SO, Goldman JE. Gliogenic and neurogenic progenitors of the subventricular zone: who are they, where did they come from, and where are they going? Glia. 2003;43: 52–61. [DOI] [PubMed] [Google Scholar]
- 291.Ge W-P, Miyawaki A, Gage FH, Jan YN, Jan LY. Local generation of glia is a major astrocyte source in postnatal cortex. Nature. 2012;484:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Bushong EA, Martone ME, Ellisman MH. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J Dev Neurosci. 2004;22:73–86. [DOI] [PubMed] [Google Scholar]
- 293.Robertson JM. Astrocyte domains and the three-dimensional and seamless expression of consciousness and explicit memories. Med Hypotheses. 2013;81:1017–1024. [DOI] [PubMed] [Google Scholar]
- 294.Petrelli F, Pucci L, Bezzi P. Astrocytes and microglia and their potential link with autism spectrum disorders. Front Cell Neurosci. 2016;10:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Richardson WD, Kessaris N, Pringle N. Oligodendrocyte wars. Nat Rev Neurosci. 2006;7:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Warf BC, Fok-Seang J, Miller RH. Evidence for the ventral origin of oligodendrocyte precursors in the rat spinal cord. J Neurosci. 1991;11:2477–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Rivers LE, Young KM, Rizzi M, et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Barres BA, Hart IK, Coles HS, et al. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70: 31–46. [DOI] [PubMed] [Google Scholar]
- 299.Trapp BD, Nishiyama A, Cheng D, Macklin W. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J Cell Biol. 1997;137:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Cohen RI, Rottkamp DM, Maric D, Barker JL, Hudson LD. A role for semaphorins and neuropilins in oligodendrocyte guidance. J Neurochem. 2003;85:1262–1278. [DOI] [PubMed] [Google Scholar]
- 301.Fox MA, Afshari FS, Alexander JK, Colello RJ, Fuss B. Growth conelike sensorimotor structures are characteristic features of post-migratory, premyelinating oligodendrocytes. Glia. 2006;53:563–566. [DOI] [PubMed] [Google Scholar]
- 302.Jakovcevski I, Filipovic R, Mo Z, Rakic S, Zecevic N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front Neuroanat. 2009;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Snaidero N, Möbius W, Czopka T, et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell. 2014;156:277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Sobottka B, Ziegler U, Kaech A, Becher B, Goebels N. CNS live imaging reveals a new mechanism of myelination: the liquid croissant model. Glia. 2011;59:1841–1849. [DOI] [PubMed] [Google Scholar]
- 305.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Gomez Perdiguero E, Klapproth K, Schulz C, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Monier A, Adle-Biassette H, Delezoide A-L, Evrard P, Gressens P, Verney C. Entry and distribution of microglial cells in human embryonic and fetal cerebral cortex. J Neuropathol Exp Neurol. 2007;66:372–382. [DOI] [PubMed] [Google Scholar]
- 308.Verney C, Monier A, Fallet-Bianco C, Gressens P. Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J Anat. 2010;217:436–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Casano AM, Peri F. Microglia: multitasking specialists of the brain. Dev Cell. 2015;32:469–477. [DOI] [PubMed] [Google Scholar]
- 310.Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. [DOI] [PubMed] [Google Scholar]
- 312.Tremblay M-È, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8:e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Reemst K, Noctor SC, Lucassen PJ, Hol EM. The indispensable roles of microglia and astrocytes during brain development. Front Hum Neurosci. 2016;10:566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Edmonson C, Ziats MN, Rennert OM. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol Autism. 2014;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Laurence JA, Fatemi SH. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum. 2005;4:206–210. [DOI] [PubMed] [Google Scholar]
- 316.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. [DOI] [PubMed] [Google Scholar]
- 317.Fatemi SH, Folsom TD, Reutiman TJ, Lee S. Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse. 2008;62:501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Madore C, Leyrolle Q, Lacabanne C, et al. Neuroinflammation in autism: plausible role of maternal inflammation, dietary omega 3, and microbiota. Neural Plast. 2016;2016:3597209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Theoharides TC, Tsilioni I, Patel AB, Doyle R. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl Psychiatry. 2016;6:e844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Wei H, Zou H, Sheikh AM, et al. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation. 2011;8:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. 2011;25:40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Masi A, Breen EJ, Alvares GA, et al. Cytokine levels and associations with symptom severity in male and female children with autism spectrum disorder. Mol Autism. 2017;8:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Molloy CA, Morrow AL, Meinzen-Derr J, et al. Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol. 2006;172:198–205. [DOI] [PubMed] [Google Scholar]
- 324.Morgan JT, Chana G, Pardo CA, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry. 2010;68:368–376. [DOI] [PubMed] [Google Scholar]
- 325.Suzuki K, Sugihara G, Ouchi Y, et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiat. 2013;70:49–58. [DOI] [PubMed] [Google Scholar]
- 326.Arshad M, Stanley JA, Raz N. Adult age differences in subcortical myelin content are consistent with protracted myelination and unrelated to diffusion tensor imaging indices. NeuroImage. 2016; 143:26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Deoni SCL, Zinkstok JR, Daly E, et al. White-matter relaxation time and myelin water fraction differences in young adults with autism. Psychol Med. 2015;45:795–805. [DOI] [PubMed] [Google Scholar]
- 328.Gonzalez-Gronow M, Cuchacovich M, Francos R, et al. Catalytic autoantibodies against myelin basic protein (MBP) isolated from serum of autistic children impair in vitro models of synaptic plasticity in rat hippocampus. J Neuroimmunol. 2015;287:1–8. [DOI] [PubMed] [Google Scholar]
- 329.Tierney E, Nwokoro NA, Porter FD, Freund LS, Ghuman JK, Kelley RI. Behavior phenotype in the RSH/Smith-Lemli-Opitz syndrome. Am J Med Genet. 2001;98:191–200. [DOI] [PubMed] [Google Scholar]
- 330.Stogsdill JA, Ramirez J, Liu D, et al. Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature. 2017; 551:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Marie C, Clavairoly A, Frah M, et al. Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc Natl Acad Sci U S A. 2018;115:E8246–E8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Maezawa I, Jin L-W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Derecki NC, Cronk JC, Lu Z, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012; 484:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Lioy DT, Garg SK, Monaghan CE, et al. A role for glia in the progression of Rett’s syndrome. Nature. 2011;475:497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Gupta S, Ellis SE, Ashar FN, et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun. 2014;5:5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Alonso MA, Weissman SM. cDNA cloning and sequence of MAL, a hydrophobic protein associated with human T-cell differentiation. Proc Natl Acad Sci U S A. 1987;84:1997–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Schaeren-Wiemers N, Valenzuela DM, Frank M, Schwab ME. Characterization of a rat gene, rMAL, encoding a protein with four hydrophobic domains in central and peripheral myelin. J Neurosci. 1995;15:5753–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Russo FB, Freitas BC, Pignatari GC, et al. Modeling the interplay between neurons and astrocytes in autism using human induced pluripotent stem cells. Biol Psychiatry. 2018;83:569–578. [DOI] [PubMed] [Google Scholar]
- 340.Williams EC, Zhong X, Mohamed A, et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum Mol Genet. 2014;23: 2968–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Domingues HS, Portugal CC, Socodato R, Relvas JB. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol. 2016;4:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Trujillo C, Gao R, Negraes PD, et al. Nested oscillatory dynamics in cortical organoids model early human brain network development. bioRxiv. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Lombardo MV, Lai M-C, Baron-Cohen S. Big data approaches to decomposing heterogeneity across the autism spectrum. Mol Psychiatry. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Toro R, Konyukh M, Delorme R, et al. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26:363–372. [DOI] [PubMed] [Google Scholar]
- 345.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012;4(3):a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Erickson CA, Davenport MH, Schaefer TL, et al. Fragile X targeted pharmacotherapy: lessons learned and future directions. J Neurodev Disord. 2017;9:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Hyman SE. The daunting polygenicity of mental illness: making a new map. Philos Trans R Soc Lond Ser B Biol Sci. 2018;373:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Hyman SE. How far can mice carry autism research? Cell. 2014; 158:13–14. [DOI] [PubMed] [Google Scholar]