

Abstract
HIV-1 entry into cells is mediated by the fusion protein gp41. Cholesterol plays an important role in this virus-cell fusion, but molecular structural information about cholesterol-gp41 interaction is so far absent. Here, we present experimental and computational data about cholesterol complexation with gp41 in lipid bilayers. We focus on the C-terminal region of the protein, which comprises a membrane-proximal external region (MPER) and the transmembrane domain (TMD). We measured peptide-cholesterol contacts in virus-mimetic lipid bilayers using solid-state NMR spectroscopy, and augmented these experimental data with all-atom molecular dynamics simulations. 2D 19F NMR spectra show correlation peaks between MPER residues and the cholesterol isooctyl tail, indicating that cholesterol is in molecular contact with the MPER-TMD trimer. 19F-13C distance measurements between the peptide and 13C-labeled cholesterol show that C17 on the D ring and C9 at the intersection of B and C rings are 7.0 Å from the F673 sidechain 4-19F. At high peptide concentrations in the membrane, the 19F-13C distance data indicate three cholesterol molecules bound near F673 in each trimer. Mutation of a cholesterol-recognition amino acid consensus (CRAC) motif did not change these distances, indicating that cholesterol binding does not require this sequence motif. Molecular dynamics simulations further identify two hotspots for cholesterol interactions. Taken together, these experimental data and simulations indicate that the helix-turn-helix conformation of the MPER-TMD is responsible for sequestering cholesterol. We propose that this gp41-cholesterol interaction mediates virus-cell fusion by recruiting gp41 to the boundary of the liquid-disordered and liquid-ordered phases to incur membrane curvature.
Keywords: REDOR, MPER, membrane proteins, 19F NMR, virus-cell fusion, binding stoichiometry
Introduction
Cholesterol is a central component of biological membranes. Cholesterol-rich regions of lipid bilayers provide sites of membrane protein interactions with each other and with intracellular proteins to carry out a myriad of biological functions [1, 2]. Many membrane proteins also specifically complex with cholesterol for function. For example, cholesterol binds G-protein coupled receptors (GPCRs) to affect their oligomerization [3], dynamics [4], and signaling [5]. Cholesterol binds PDZ domains of scaffold proteins to regulate protein networking and cell signaling [6, 7]. Cholesterol also interacts with the amyloid precursor protein and the Aβ peptide in lipid membranes [8–10], and these interactions have been suggested to be involved in the pathogenesis of Alzheimer’s disease [11].
The envelope glycoprotein (Env) of the human immunodeficiency virus 1 (HIV-1), the causative agent of the acquired immunodeficiency syndrome, mediates virus entry into cells. Env consists of a trimer of gp120 and gp41 heterodimers [12, 13]. Binding of gp120 to cell-surface receptors triggers large-scale conformational changes of gp41, which lead to fusion of the virus envelope with the plasma membrane or the endosomal membrane of the cell [14]. The gp41 conformational changes include the exposure and insertion of an N-terminal fusion peptide (FP) into the target cell membrane, while the protein remains anchored in the viral envelope by the C-terminal transmembrane domain (TMD). This extended conformation is metastable, and proceeds to bend into a hairpin, in so doing pulling the target cell membrane to the virus envelope. The trimer of hairpins, or six-helix bundle, marks the fully fused state [15–17]. The hydrophobic FP and TMD are structurally unknown in the post-fusion six-helix bundle; but they are believed to facilitate the formation of the six-helix bundle by disrupting the integrity of the target cell membrane and virus envelope [18, 19]. In addition to the FP and TMD, a membrane-proximal external region (MPER) N-terminal to the TMD also exerts a significant influence on membrane fusion and virus entry [20, 21]. The MPER harbors the epitopes of several broadly neutralizing antibodies [22–25] and binds to the lipid bilayer surface, approximately perpendicular to the TM helix [19].
Four lines of biochemical and biophysical evidence strongly suggest that cholesterol is important for gp41-mediated virus-cell fusion [26]. First, cholesterol depletion in the membrane severely impairs gp41-mediated virus entry [27, 28] as well as virus budding [29]. Second, peptide studies show that the FP, MPER, and TMD all have affinity for cholesterol-rich regions of the lipid membrane. Fluorescence microscopy and fusion assays show that the FP preferentially localizes to the edge of cholesterol-rich lipid domains, between liquid-ordered (Lo) and liquid-disordered (Ld) phases [30]. Similarly, MPER preferentially aggregates to cholesterol and sphingomyelin rich region of the membrane [31]. Third, a cholesterol-recognition amino acid consensus (CRAC) motif is present in the segment 679LWYIK683 between the MPER and the TMD [32]. The CRAC motif is characterized by a triad of a methyl-rich aliphatic Leu or Val residue, a central aromatic Tyr residue, and a basic Arg or Lys residue [33]. This motif runs from the N-terminus to the C-terminus of the protein sequence. Fluorescence spectroscopy, differential scanning calorimetry, and X-ray scattering data showed that this short peptide preferentially sorts to the cholesterol-rich region of the membrane [34–36]. Mutations of this CRAC motif retain fusion activity only if the mutant retains interactions with cholesterol [37]. Fourth, partition and leakage assays showed that a peptide containing the C-terminus of the MPER and the N-terminus of the TMD preferentially bound and destabilized cholesterol-rich membranes [38, 39].
Despite these biochemical data, direct molecular structural information about cholesterol interaction with gp41 has not been reported. In particular, the C-terminal segment of the protein, MPER-TMD, is anchored in the HIV virus envelope, which is well known to contain a high concentration of cholesterol [40]. Until recently, the MPER structure has largely come from solution NMR studies of the MPER peptide in cholesterol-free DPC micelles [24, 41]. Recently, the structure of the TMD and the C-terminal end of the MPER was determined in cholesterol-free lipid bicelles using solution NMR [42, 43]. However, these studies were inconclusive about whether the TMD is trimeric or monomeric in bicelles. We recently investigated the three-dimensional structural topology of the full-length MPER-TMD peptide (residues 665–704) in lipid bilayers using solid-state NMR [19]. By measuring both intermolecular and intramolecular distances, we found that MPER-TMD is trimeric in phospholipid bilayers, and the MPER lies on the membrane surface, bent from the TMD. This structural topology exists in a virus-mimetic membrane that contains POPC, POPE, POPS, sphingomyelin, and cholesterol.
Here, we investigate the intermolecular interactions between the MPER-TMD and cholesterol in virus-mimetic lipid membranes using solid-state NMR spectroscopy. Using fluorinated MPER-TMD and 13C-labeled cholesterol, we measured semi-quantitative distances between specific MPER residues and cholesterol atoms. We incorporated fluorine at two native MPER residues, F673 and W680. A mutant yeast strain was used to express 1-13C labeled cholesterol (1-13C CHOL) [44–46]. We find that cholesterol is in molecular contact with the MPER-TMD, but this complexation does not require the CRAC motif, because a mutant peptide without the motif exhibits the same peptide–CHOL distances. We also conducted molecular dynamics simulations to provide site-specific information about the hotspots in the peptide that interact with cholesterol, and to probe the stoichiometry of cholesterol binding over a wide range of protein concentrations.
Materials and Methods
Synthesis and purification of isotopically labeled gp41 MPER-TMD
The gp41(661–704) peptide used in this study corresponds to the MPER and TMD regions of HIV-1 clade D gp41. The amino acid sequence is LELDKWASL WNWFNITNWL WYIRLFIMIV GGLVGLRIVF AVLSI (Fig. 1a). This sequence differs slightly from the clade B gp41 sequence used in our recent study [19], by extending the N-terminus by four residues, and by having R683 instead of K683. The N-terminal extension is intended to allow future studies of antibody binding to MPER, while the use of R683 allows direct comparison of this study with previous solution NMR studies of clade D gp41 [42, 43]. Because of the common cationic nature of Arg and Lys, we do not expect any structural conclusion to be affected by this residue change. The 44-residue peptide was synthesized using a custom-designed flow synthesizer [47, 48] and Fmoc solid-phase peptide synthesis protocols. To investigate whether the CRAC motif is necessary for cholesterol binding, we also synthesized an AFI mutant peptide containing L679A, Y681F, R683I mutations. Both wild-type and mutant peptides incorporated 4-19F-F673 and 5-19F-W680 for distance measurements.
Figure 1.
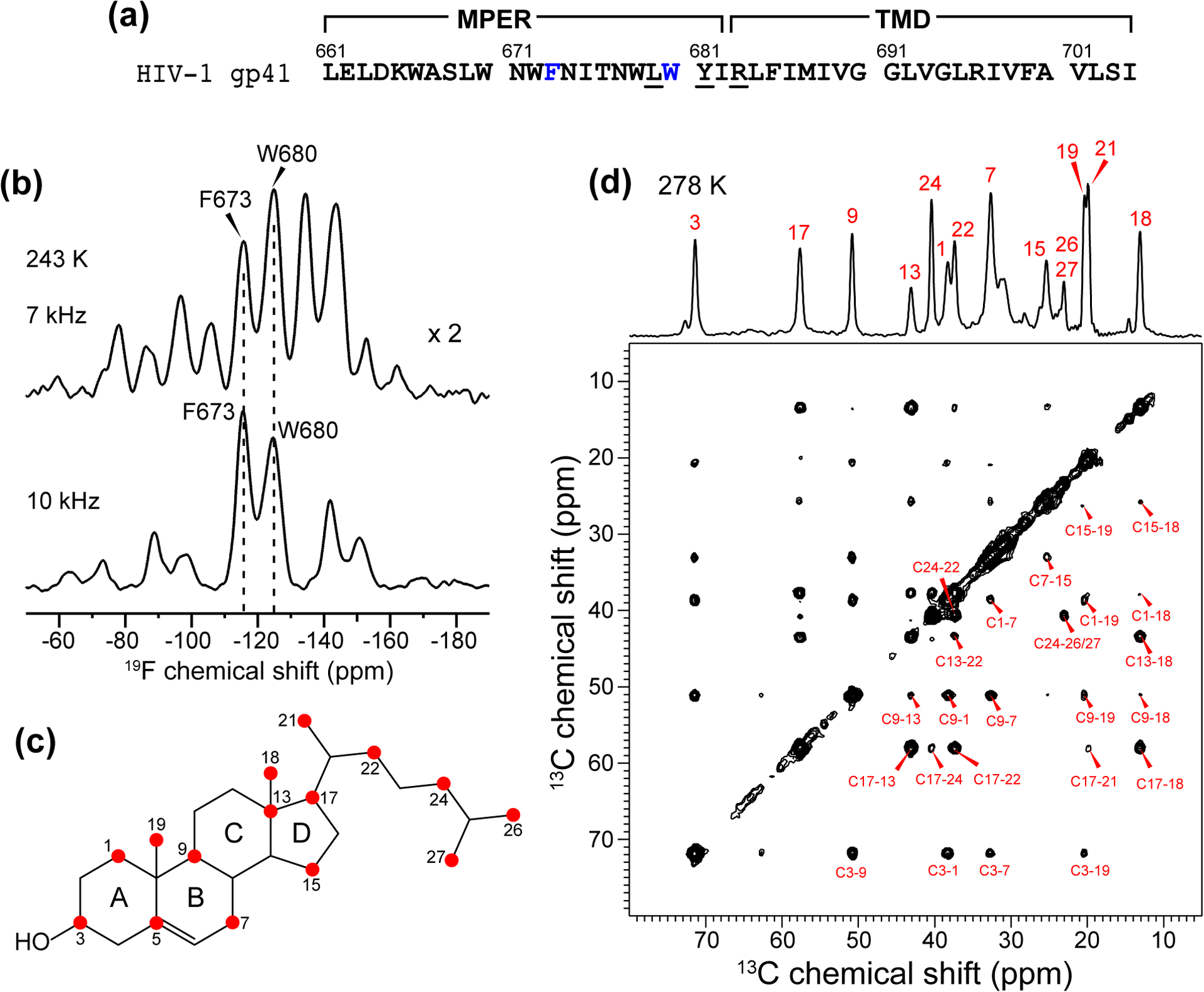
(a) Amino acid sequence of gp41 MPER-TMD. Fluorinated F673 and W680 are shown in blue, while the CRAC residues L679, Y681, and R683 are underlined. The AFI mutant contains L679A, Y681F, R683I mutations. (b) Low-temperature 1D 19F NMR spectra of wild-type MPER-TMD in the VM+ membrane. Comparison of the 7 kHz and 10 kHz MAS spectra distinguishes the centerbands and sidebands. (c) 13C-labeled sites in 1-13C CHOL.[44] (d) 2D 13C-13C correlation spectra of 1-13C CHOL in gp41-containing VM+ membranes. The spectrum was measured at 278 K using a 13C spin diffusion mixing time of 300 ms.
The gp41(661–704) peptides were synthesized on the 0.05 mmol scale using H-Rink amide ChemMatrix resin (0.1 g at 0.5 mmol/g loading size). The resin was swelled in the reaction vessel for 5 min in ~5 mL of N,N-dimethylformamide (DMF) at 70°C. A 10-fold excess of unlabeled amino acids and 4-fold excess of fluorinated amino acids were singly- and triply-coupled using coupling times of 50 and 70 s, respectively. After the final coupling step, the peptide was deprotected and cleaved from the resin by addition of TFA/phenol/H2O/TIPS solution (88 : 5 : 5 : 2 by volume) for 2.5 h. The resin was filtered off, and the crude peptide was precipitated and triturated three times using cold diethyl ether, then dried under vacuum overnight at room temperature. The dried crude peptide was dissolved in TFE (2,2,2-trifluoroethanol) and purified by preparative reverse-phase HPLC using a Vydac C18 column. A 10 min isocratic gradient (5% of channel A and 95% channel B) was followed by a linear gradient of 5–100% for channel A (channel A is 75 : 25 v/v acetonitrile : isopropyl alcohol and channel B is water) over 90 min at a flow rate of 10 mL/min. MALDI-MS analysis found the peptide mass to be 5317.9 Da for wild-type (WT) gp41(661–704) and 5215.3 Da for the AFI mutant, in good agreement with the calculated masses. The total yield of the synthesis and purification was ~7%.
Membrane sample preparation
1-13C CHOL was expressed from Saccharomyces cerevisiae strain RH6829 using 2-13C labeled acetate as the 13C source [44, 49, 50]. The sterol esters were saponified and the crude product was purified by HPLC, giving a final yield of ~30 mg per liter of culture. Heptafluorinated cholesterol (F7-CHOL) was purchased from Sigma. This cholesterol has the same interfacial area, molecular orientation, and phospholipid interaction as hydrogenated cholesterol based on pressure-area isotherm data [51].
The WT and mutant gp41 peptides were reconstituted into virus-mimetic phospholipid membranes (VM+), which consist of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), sphingomyelin (SM), and cholesterol (CHOL) [52, 53]. Three membrane samples were prepared. Sample 1 contains POPC : POPE : SM : F7-CHOL at a molar ratio of 16 : 16 : 16 : 21, while the peptide : total phospholipid and SM : CHOL molar ratio (P : L : C) was 1 : 16 : 7. Sample 2 has the same peptide and lipid concentrations, but contains 1-13C CHOL to permit measurements of 13C-19F distances. Sample 3 contains POPC : POPE : SM : 1-13C-CHOL at a molar ratio of 32 : 32 : 32 : 21, while the P : L : C ratio was 1 : 32 : 7. Comparison of sample 2 and sample 3 allows us to probe whether higher lipid concentrations reduce the fraction of cholesterol bound to the peptide. The molar concentration of cholesterol is 30 mol% in samples 1 and 2 and 18 mol% in sample 3.
All lipids and cholesterol were dissolved in 600 μl chloroform and 4 μl methanol. The peptide was dissolved in 300 μl TFE and mixed with the lipid solution. The solvents were removed by nitrogen gas, then the samples were dried under vacuum overnight. The dried powder was resuspended in pH 7.5 HEPES buffer (10 mM HEPES/NaOH, 1 mM EDTA, 0.1 mM NaN3) and dialyzed against pH 7.5 HEPES buffer for 3 days with six buffer changes to remove salt and residual TFA and TFE. The proteoliposomes were spun at 40,000 rpm using a Beckman SW60T rotor at 4°C for 6–10 h to obtain membrane pellets. The pellets were incubated in a desiccator until it reached a hydration level of ~40 wt% water and then spun into magic-angle spinning (MAS) rotors for solid-state NMR experiments.
Solid-state NMR spectroscopy
Solid-state NMR spectra were measured on a Bruker NMR spectrometer at 9.4 Tesla (400 MHz 1H Larmor frequency) using a 4 mm 1H/19F/13C MAS probe. 13C chemical shifts were externally referenced to the adamantane CH2 signal at 38.48 ppm on the TMS scale. 19F chemical shifts were referenced to the 19F signal of Teflon at −122 ppm on the CFCl3 scale. 1D 13C and 19F CP-MAS spectra were measured from 293 K to 238 K under 7 kHz to 10 kHz MAS. Typical radiofrequency (rf) field strengths were 62.5 kHz for 19F pulses, 50 kHz for 13C pulses, and 71.4 – 83.3 kHz for 1H heteronuclear decoupling.
2D 13C-13C correlation spectra were measured to confirm 13C-labeled sites in 1-13C CHOL. 2D 19F-19F correlation spectra of fluorinated gp41 and F7-CHOL were measured to investigate peptide-peptide and peptide-CHOL interactions. We used the CORD dipolar recoupling sequence [54] for 13C and 19F spin diffusion. The mixing times ranged from 100 ms to 750 ms. These 2D spectra were measured at 238 K under 10 kHz MAS.
19F-detected and 13C-detected 13C-19F REDOR experiments were conducted to measure gp41-CHOL spatial proximities. F673, W680-fluorinated peptide was combined with 1-13C CHOL in VM+ membranes. For each mixing time, a pair of spectra were measured, with and without inversion pulses on the unobserved channel [55]. The control experiment (S0) does not have inversion pulses on the unobserved channel and gives the full echo intensity, which controls for T2 relaxation of the observed nuclear spin. The dephased experiment (S) has inversion pulses on the unobserved channel to induce dipolar dephasing. The 19F-detected 13C-dephased REDOR experiments used 90°180°90° composite pulses on the unobserved 13C channel, whereas the 13C-detected REDOR experiments used 90°180°90° composite pulses on the unobserved 19F channel. These composite pulses compensate for rf field inhomogeneity [50, 56]. Both the 19F-detected and 13C-detected REDOR experiments were conducted at 238 K under 10 kHz MAS. This low temperature was necessary to immobilize the translational motion of the peptide and CHOL so that peptide-cholesterol distances can be measured. REDOR mixing times ranged from 0.6 ms to 8 ms for the 19F-detected REDOR experiments and 5 ms to 8 ms for the 13C-detected REDOR experiments.
The REDOR dephasing values, S/S0, were obtained from the ratio of the peak heights in the dephased and control spectra. This intensity ratio depends on dipolar coupling through the equation [55, 57]
| (1) |
where the phase ΔΦ of the recoupled dipolar interaction is proportional to the distance-dependent dipolar coupling constant DIS and the mixing time tmix, . The dipolar coupling constant is inversely proportional to the internuclear distance rIS to the third power and proportional to the gyromagnetic ratios γ of the two spins, . In Equation 1, β and α are the polar angle and azimuthal angle of the internuclear vector with respect to a rotor-fixed coordinate system whose z axis is along the rotor axis.
Uncertainties in the S/S0 values were propagated from the experimental uncertainties and εS of each REDOR spectrum according to the equation . The values of and εS are the inverse of the signal-to-noise ratios of the peaks.
The 13C-detected 13C-19F dephasing curves were fit using the SIMPSON program [58] assuming a two-spin system. An intensity scaling factor of 0.85 was applied to the simulated curves to compensate for 13C and 19F pulse imperfections. Best-fit distances were obtained by minimizing the root-mean-square deviations between the measured and simulated curves. The total measuring time of the 19F-detected 13C dephased REDOR spectra was 35 days to include multiple samples and multiple mixing times, while the total measuring time for the 13C-tectected REDOR experiments was 38 days.
Molecular dynamics simulations
We used the solid-state NMR structural model of gp41-TMD (PDB: 6DLN) [19] as the input structure for MD simulations. The peptide was embedded in a lipid bilayer using CHARMM-GUI [59, 60]. The bilayer consists of POPC, POPE, SM and CHOL at a molar ratio of 64 : 64 : 64 : 84, to which we inserted four gp41 trimers. Thus, the P : L : C molar ratio of the system is 1 : 16 : 7, which is identical to the composition of the high peptide-concentration samples in the NMR experiments.
The peptide-lipid system was equilibrated using the highly mobile membrane mimetic (HMMM) model [61] to allow efficient sampling of lipid distribution around gp41. The system size was 10.5 × 10.5 × 7.3 nm3. 36 Cl− ions were added to reach charge neutrality, then an additional 6 Na+ and Cl− ions were added to reach a salt concentration of 10 mM as used in the SSNMR samples. Protein, lipids, cholesterol and ions were described using the CHARMM36 force field [62], and water was described with TIP3P [63]. The protein structure model was energy minimized using the conjugate gradient method to remove any bad contacts between solvent and solute atoms. This was followed by a 200 ns constant-pressure constant-temperature simulation in which all heavy atoms of the protein were restrained. Three randomly equilibrated structures were then converted to full lipid models using CHARMM-GUI. These three full lipid systems were simulated for 500 ns in independent restraint-free simulations at constant pressure and constant temperature. The hydrogen atoms were constrained using the LINCS algorithm [64] to allow a 2 fs time step for integration. The temperature and pressure of the system in both the HMMM and full lipid models were controlled using the Nosè-Hoover thermostat [65] and Parrinello-Rahman barostat [66], respectively. All simulations were performed using the GROMACS-2018.3 package [67].
We also simulated a lower peptide concentration, where a single gp41 trimer was inserted into the lipid bilayer. This composition corresponds to a P : L : C molar ratio of 1 : 64 : 28. To ensure consistent sampling of cholesterol distributions in the two sets of simulations, the initial structure of the lower peptide concentration simulation was taken from a fully equilibrated snapshot of the higher-peptide concentration condition, but with three out of four gp41 trimers removed. Three independent simulations of at least 500 ns each were carried out. Results for a set of low peptide concentration simulations prepared independently of the higher-peptide concentration simulations are discussed in the Supporting Information.
Results
MPER-TMD contacts with cholesterol in lipid bilayers
To investigate cholesterol interaction with gp41, we reconstituted fluorinated MPER-TMD and 1-13C CHOL in the virus-mimetic (VM+) membrane. The cholesterol concentrations used in our samples were 18 mol% to 30 mol%, which are similar to the cholesterol concentrations in cell membranes [68] but lower than those in virus lipid envelopes [40]. Two peptide: lipid molar ratios, 1 : 16 and 1 : 32, were used in our study. These high peptide concentrations are required to provide sufficient sensitivity for the NMR spectra. Each HIV virion contains only about ten Env trimers; however, the spatial distribution of these trimers is not random, with local clustering observed in cryoelectron tomography data [69, 70]. Moreover, whether gp41 trimers cluster to mediate virus-cell fusion is not yet known. The equivalent fusion protein in the influenza virus, hemagglutinin, has been shown to cluster to at least three trimers to cause fusion [14, 71]. Similar clustering of multiple trimers has been suggested for the parainfluenza virus fusion protein F based on analytical ultracentrifugation data [72]. Therefore, the high peptide concentrations used in the current NMR experiments may be relevant for the local gp41 densities in the virus during fusion.
For the two MPER residues, the 19F isotropic chemical shift of 4F-F673 at −115 ppm is well resolved from the chemical shift of 5F-W680 at −125 ppm (Fig. 1b). The 19F NMR spectra measured under 7 kHz MAS gave multiple spinning sidebands, which fit to chemical shift anisotropies (CSAs) of δ = 57 ppm for 4F-F673 and δ = 46 ppm for 5F-W680. These CSA values are near the rigid limit for amino acid 4F-Phe (δ = 59 ppm) and 5F-Trp (δ = 51 ppm), indicating that the MPER is immobilized in the membrane at 243 K. The 13C spectrum of 1-13C CHOL (Fig. 1c) at 278 K shows narrow linewidths of 0.5 ppm, and all 13C chemical shifts are readily assigned using the 2D 13C-13C correlation spectrum (Fig. 1d) [44].
19F is an excellent probe of long-range distances up to ~2 nm due to its large gyromagnetic ratio [73, 74]. If the fluorinated F673 and W680 lie within ~2 nm of each other, then 19F–19F correlation peaks should be detectable in the 2D 19F-19F correlation spectra. Fig. 2a shows the 500 ms 2D 19F spin diffusion spectrum of MPER-TMD. A correlation peak between F673 and W680 is observed, indicating that the two fluorine atoms are within ~2 nm of each other. The cross-peak intensity is asymmetric, with the W680 5-19F peak in the ω1 dimension showing higher intensity than the F673 4-19F peak in the ω1 dimension (Fig. 2c). We tentatively attribute this asymmetry to longer T2 relaxation time of 5-19F Trp (~2 ms) than 4-19F Phe (~1.3 ms). The presence of a W680-F673 cross peak is consistent with the helical conformation of MPER. The solid-state NMR structural model of gp41(665–704) in lipid bilayers [19] gave an average intrahelical distance of 2.0 nm between the two fluorines (Fig. 2d), whereas the interhelical distances range from 1.9 to 2.6 nm. The DPC-micelle bound MPER has an intramolecular F673-W680 distance of 1.6 nm [24] (Fig. 2e). Since the intermolecular distances between the two residues are on average longer than the intramolecular distance, we attribute the cross peak mainly to intramolecular contact between the two sidechains.
Figure 2.
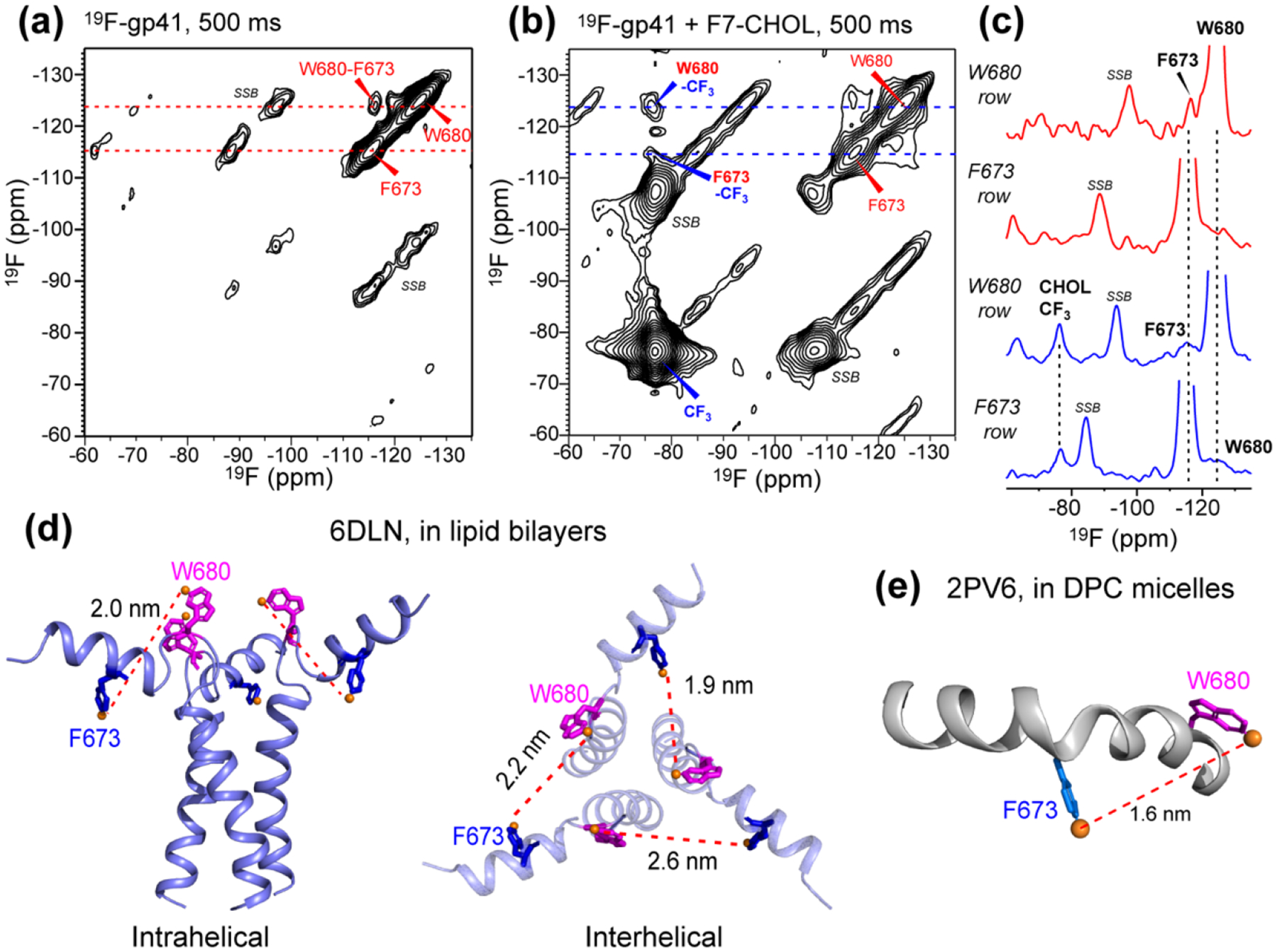
2D 19F-19F correlation spectra of membrane-bound gp41 with 500 ms mixing. (a) 2D spectrum of fluorinated gp41 in membranes containing 1-13C CHOL. A W680-F673 cross peak is observed. Spinning sidebands are denoted by SSB. (b) 2D 19F-19F correlation spectrum of gp41 in membranes containing F7-CHOL. Cross peaks between W680 and CHOL CF3 and between F673 and CHOL are observed. (c) 1D cross sections from the W680 and F673 rows in the 2D spectra of (a) and (b), showing protein-protein and protein-CHOL cross peaks. (d) Solid-state NMR structural model of trimeric MPER-TMD in lipid bilayers.[19] Intrahelical (left) and interhelical (right) distances between 4F-F673 and 5F-W680 are indicated. (e) Solution NMR structural model of MPER in DPC micelles, [24] showing a 1.6 nm distance between F673 Hζ and W680 Hζ3.
We next measured the 2D 19F-19F correlation spectrum of MPER-TMD in the VM+ membrane containing F7-CHOL, to investigate whether the fluorinated peptide are in molecular contact with cholesterol. With 500 ms mixing, the 2D spectrum showed clear correlation peaks between the cholesterol CF3 at −77 ppm and the 4F-F673 and 5F-W680 peaks (Fig. 2b, c). Therefore, the cholesterol tail is within nanometer contact with both MPER residues. Again, W680 has a higher cross peak with cholesterol than F673. These peptide-cholesterol correlation peaks set the stage for a more quantitative study of the distances and stoichiometry of cholesterol binding to gp41 using REDOR.
Using fluorinated MPER-TMD and 1-13C CHOL, we measured distance-dependent 19F-13C dipolar couplings. The REDOR experiments were conducted with both 19F detection of the peptide and 13C detection of cholesterol. Each 1-13C CHOL contains 15 13C-labeled sites whereas each peptide contains only two fluorine atoms (Fig. 3a). We first measured 19F-detected REDOR spectra, because the high sensitivity of 19F NMR and the simultaneous dephasing of each 19F by multiple 13C spins facilitate the observation of dipolar dephasing. We then switched to 13C-detected and 19F-dephased REDOR experiments to obtain more site-specific information about which CHOL carbons are close to the fluorinated residues. 13C-19F dipolar couplings are sensitive to distances up to ~1.0 nm [75, 76].
Figure 3.
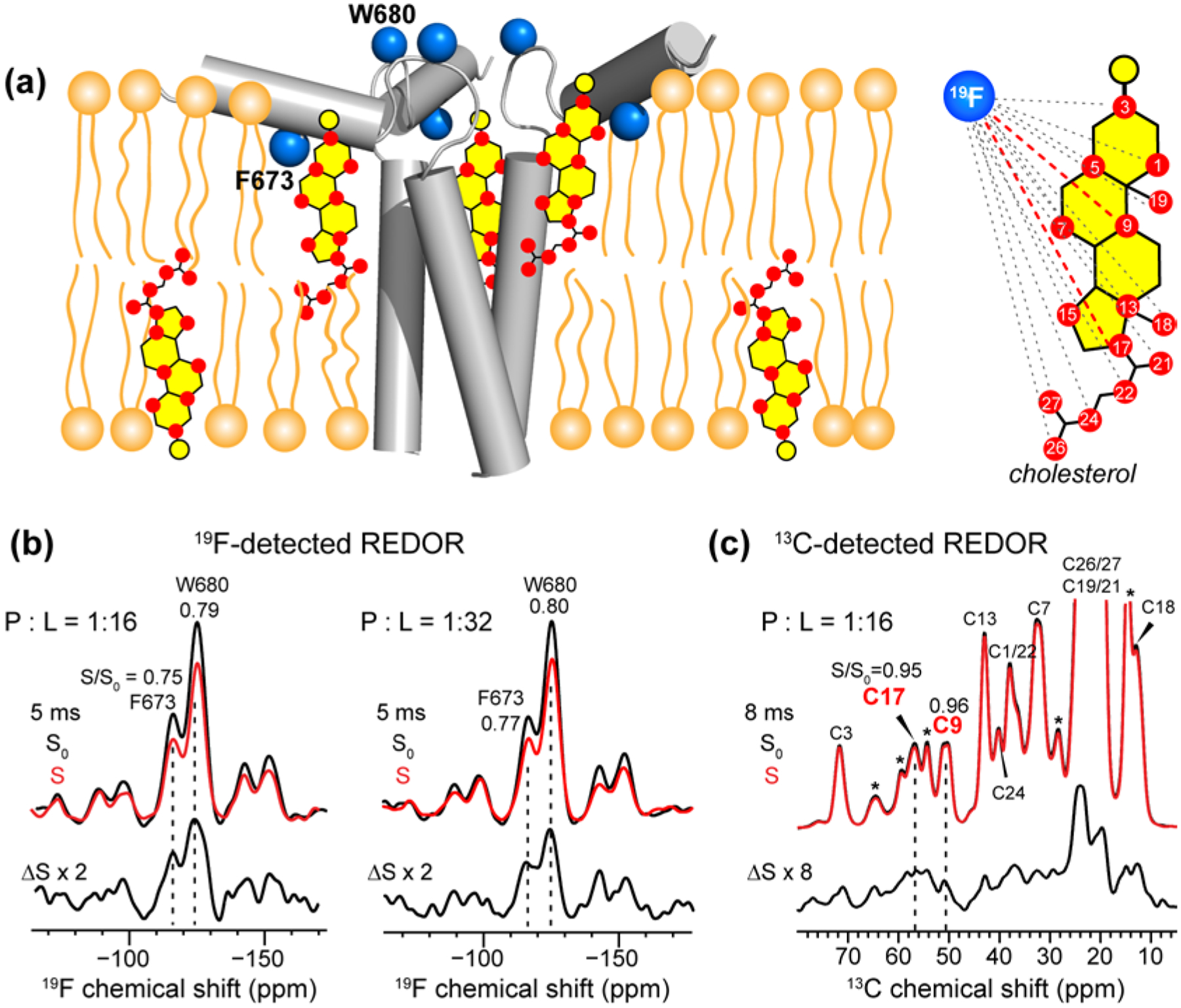
19F-detected and 13C-detected 13C-19F REDOR spectra to determine cholesterol-gp41 proximity in the VM+ membrane. (a) Schematic of fluorinated MPER-TMD trimer in VM+ membranes. 1-13C CHOL with 13C-labeled sites is illustrated. Each fluorine is coupled to multiple CHOL carbons. (b) Representative 19F-detected REDOR spectra, measured at 238 K under 10 kHz MAS. At 5 ms mixing, significant intensity differences are observed between the control (S0) and dephased (S) spectra, indicating that F673 and W680 are close to 1-13C CHOL. Two samples with protein/lipid molar ratios of 1 : 16 and 1 : 32 gave similar results. The S/S0 values are indicated for the two 19F peaks. (c) Representative 13C-detected REDOR spectra, measured at 238 K under 10 kHz MAS. At 8 ms mixing, clear intensity differences between the S0 and S spectra are observed, indicating that some of the cholesterol molecules are in molecular contact with the peptide. The dephasing values S/S0 for C17 and C9 are indicated. Asterisks indicate lipid natural abundance signals, some of which partially overlap with the cholesterol peaks.
Fig. 3b, c show representative 19F-detected and 13C-detected REDOR spectra. The spectra were measured at low temperature (238 K) in order to immobilize the peptide and cholesterol. The 19F-detected spectra show significant intensity differences between the control (S0) and dephased (S) spectra (Fig. 3b). With 5 ms mixing, the S/S0 intensity ratios are 0.75 for 4F-F673 and 0.79 for 5F-W680, indicating that both fluorinated sidechains are within a nanometer of CHOL carbons. Doubling the lipid concentration relative to the peptide did not change the REDOR dephasing, indicating that cholesterol has sufficient affinity to remain bound to the peptide even with two-fold dilution of the peptide. At P : L : C molar ratios of 1 : 16 : 7 and 1 : 32 : 7, the cholesterol concentrations are 30 mol% and 18 mol%, respectively. The former mimics the liquid-ordered (Lo) region of biological membranes whereas the latter mimics the boundary between the Lo and liquid-disordered (Ld) phases. Thus, the 19F-detected REDOR spectra indicate that gp41 interacts with cholesterol similarly in both micro-environments.
The 13C-detected REDOR spectra (Fig. 3c) gave more site-specific information about 13C-19F distances, because each cholesterol 13C signal is dephased by only two 19F spins. Compared to the 19F-detected spectra, the intensity difference (ΔS) between the control and dephased spectra is much smaller, corresponding to S/S0 values of 0.92 and higher. The high values are not surprising because cholesterol is in 7-fold excess to peptide monomers in the membrane, thus only a fraction of all cholesterol molecules is expected to be sufficiently close to the peptide to experience dipolar dephasing by the 19F spins. Despite this small fraction, because the 13C control spectrum has extremely high sensitivity, the difference intensity is precise. We focus on the S/S0 values of two cholesterol carbons, C17 and C9, because these two peaks are well resolved from lipid natural abundance 13C signals (Fig. S1) even under the line broadened conditions at low temperature. The S/S0 values are 0.95 for C17 and 0.96 for C9 at 8 ms REDOR mixing. Several other cholesterol 13C signals such as C3 and C18 show higher S/S0 values; however, their chemical shifts are very close to the chemical shifts of lipid peaks such as glycerol G2 and acyl chain ω, thus the S/S0 values are not exclusively due to cholesterol.
To investigate whether the CRAC motif is required for gp41-cholesterol interaction, we measured 19F-detected 13C-dephased REDOR spectra of the AFI mutant. The mutant peptide exhibits the same 19F-13C REDOR dephasing as the WT peptide (Fig. 4), thus indicating that the CRAC motif is not required for cholesterol complexation with gp41.
Figure 4.
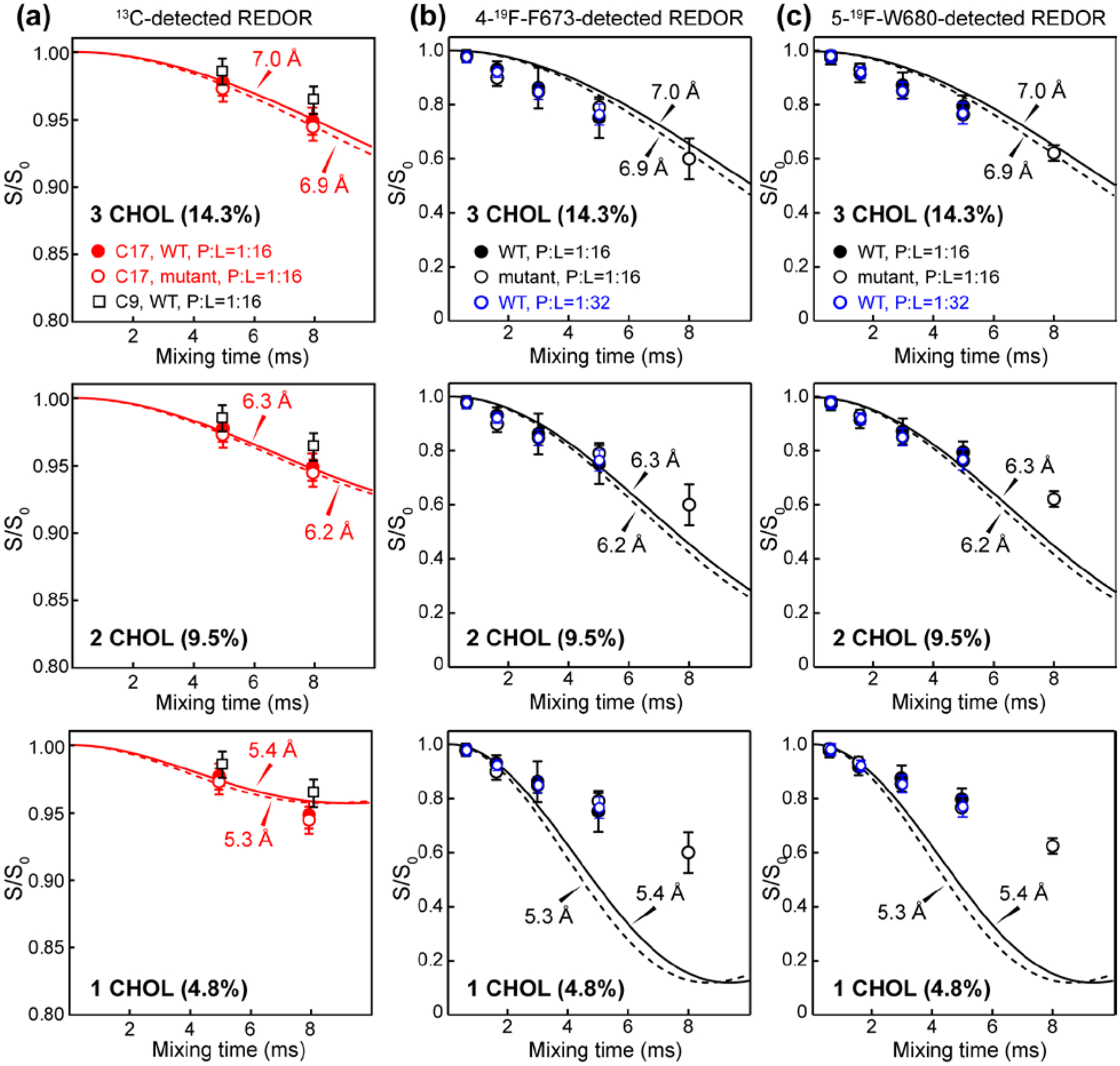
Cholesterol binding stoichiometry to trimeric MPER-TMD from 13C-19F REDOR analysis. (a) 13C-detected REDOR dephasing of C17 and C9. Results of the WT and mutant peptides are compared, and the C17 and C9 dephasing results are compared. (b) 19F-detected REDOR dephasing of 4F-F673. Results of the WT and mutant peptide at P:L = 1:16 and the WT peptide at P:L = 1:32 are compared. (c) 19F-detected REDOR dephasing of 5F-W680. Top row shows best-fit simulations using a model of three CHOL molecules bound per trimer. At a P : C ratio of 1 : 7, this number corresponds to 14.3% of all CHOL bound per trimer. Middle row shows best-fit simulations using a model of two CHOL molecules bound per trimer. This stoichiometry corresponds to an intensity scaling factor of 9.5% (= 2/21). Bottom row shows best-fit simulations using a model of one CHOL bound to each trimer. This stoichiometry corresponds to an intensity scaling of 4.8% (= 1/21).
Number of cholesterol molecules associated with the MPER-TMD trimer
We next sought to determine the number of CHOL molecules bound per trimer and the distances of the bound cholesterol to the peptide. We extract this information by analyzing the 13C-detected and 19F-detected 13C-19F REDOR spectra. Each 13C spin of 1-13C CHOL is dephased by two 19F spins, whose distances are not known simultaneously. However, we can approximate this three-spin system using a two-spin model, because dipolar couplings scale with internuclear distances r according to 1/r3, thus the 19F spin closer to the carbon will dominate the REDOR dephasing. Each cholesterol molecule is unlikely to be equidistant to both F673 and W680. This assumption is borne out by MD simulations (vide infra). Once the 13C-detected REDOR dephasing is fit to an effective two-spin distance, we then consider the 19F-detected REDOR dephasing. Here the dipolar coupling network is more complex, because each 19F is simultaneously dephased by multiple 13C spins. Each 1-13C CHOL contains 15 13C spins, many of which may be similarly close to a fluorine atom (Fig. 3a). Therefore, we consider the 19F-detected REDOR dephasing qualitatively, by requiring that the two-spin distance obtained from the 13C-detected REDOR data must be longer than the effective distance manifested in the 19F-detected REDOR dephasing.
Fig. 4a shows the 13C-19F dephasing curves of CHOL C17 and C9 atoms. Since the cholesterol concentration is 7-fold higher than peptide monomers, only a fraction of cholesterol molecules is expected to be within molecular contact with the MPER-TMD. Thus, fitting these REDOR curves requires two parameters, the number of cholesterol molecules in close proximity to the peptide and the 13C-19F distance. We first investigated the model of three cholesterol molecules tightly bound to each trimer. At the P : L : C molar ratio of 1 : 16 : 7, this stoichiometry corresponds to 3 out of 21, or 14.3%, of all cholesterol molecules bound. Thus, we fit the C17-19F REDOR dephasing using an intensity scaling factor of 14.3%. The best-fit distance for C17-19F dephasing is 7.0 Å. We then calculated the two-spin REDOR dephasing for this distance (Fig. 4b, c) without the intensity scaling factor, since all peptide 19F spins should be in close contact with some cholesterol molecules. The simulated dephasing curves decay more slowly than the measured 19F-detected REDOR dephasing. This is consistent with the fact that each 19F spin is dephased by multiple 13C spins. Therefore, the three-CHOL binding model is in good agreement with the experimental data. Mutating the CRAC residues caused minimal change to the dipolar dephasing. Similarly, diluting the peptide concentration two-fold did not significantly change the dephasing (Fig. 4b, c). Therefore, the gp41-cholesterol interaction is sufficiently specific for the peptide and cholesterol concentration ranges used here.
We next tested the two-CHOL binding model. Using an intensity scaling factor of 2/21 = 9.5%, we obtained a best-fit distance of 6.3 Å for C17-detected REDOR dephasing. Applying this distance to the 19F-detected F673 and W680 curves resulted in good apparent agreement with the measured dipolar dephasing. However, this apparent agreement is inconsistent with the fact that each 19F is coupled to multiple 13C spins. Thus, the two-CHOL stoichiometry is not consistent with the experimental data. Finally, we tested the single-CHOL binding model, using an intensity scaling factor of 1/21 = 4.8%. The best-fit distance from the C17-detected REDOR dephasing is 5.4 Å. This short distance resulted in a simulated 19F-13C REDOR curve that decays much faster than the experimental data, thus ruling out the single-CHOL binding stoichiometry.
Molecular dynamics simulations of cholesterol interactions with gp41
To obtain more site-specific information about the distance distribution, binding stoichiometry, and concentration dependence of gp41-cholesterol interactions, we performed all-atom molecular dynamics simulations. We took the solid-state NMR structural model of MPER-TMD [19] (PDB: 6DLN) as the initial structure, and constructed a molecular model of the peptide-containing virus-mimetic membrane using the experimental molar ratios of 3 : 16 : 16 : 16 : 21 for peptide : POPC : POPE : SM : cholesterol (Fig. S2). To efficiently sample the lipid and cholesterol distribution around the peptide, we used the HMMM model [61] where part of the POPC, POPE and SM lipid tails were removed to enhance their diffusion. The center of the lipid bilayer was replaced with an organic solvent 1,1-dicholorethane (DCLE). The peptide-containing HMMM system was equilibrated for 200 ns, then the truncated lipid tails were regrown to their full length. The full system was simulated for another 500 ns in three independent runs.
Fig. 5a shows a snapshot of the lipid membrane at the end of the 500 ns run for the high-concentration membrane (P : L : C = 1 : 16 : 7). Cholesterol is homogeneously distributed in the membrane, with multiple molecules in close contact with the MPER-TMD trimers. To quantify the peptide-cholesterol contacts, we calculated the number of cholesterol molecules that have any atom within 3 Å of any atom in the MPER-TMD. The value of 3 Å was chosen because it is the typical distance for both polar (e.g. hydrogen bonding) and hydrophobic interactions. With this criterion, we found approximately 8 cholesterol molecules associated with each gp41 trimer (Fig. S2b). Some of these cholesterol molecules are near the MPER while others pack close to the TMD (Fig. 5b). Thus, the simulations show a larger number of bound cholesterol than the experimental data. However, the REDOR data report cholesterol carbons near two specific MPER residues, F673 and W680, while simulations search for cholesterol molecules near both the MPER and the TMD. Thus, the REDOR data should represent a lower bound to the number of bound cholesterols to each trimer.
Figure 5:
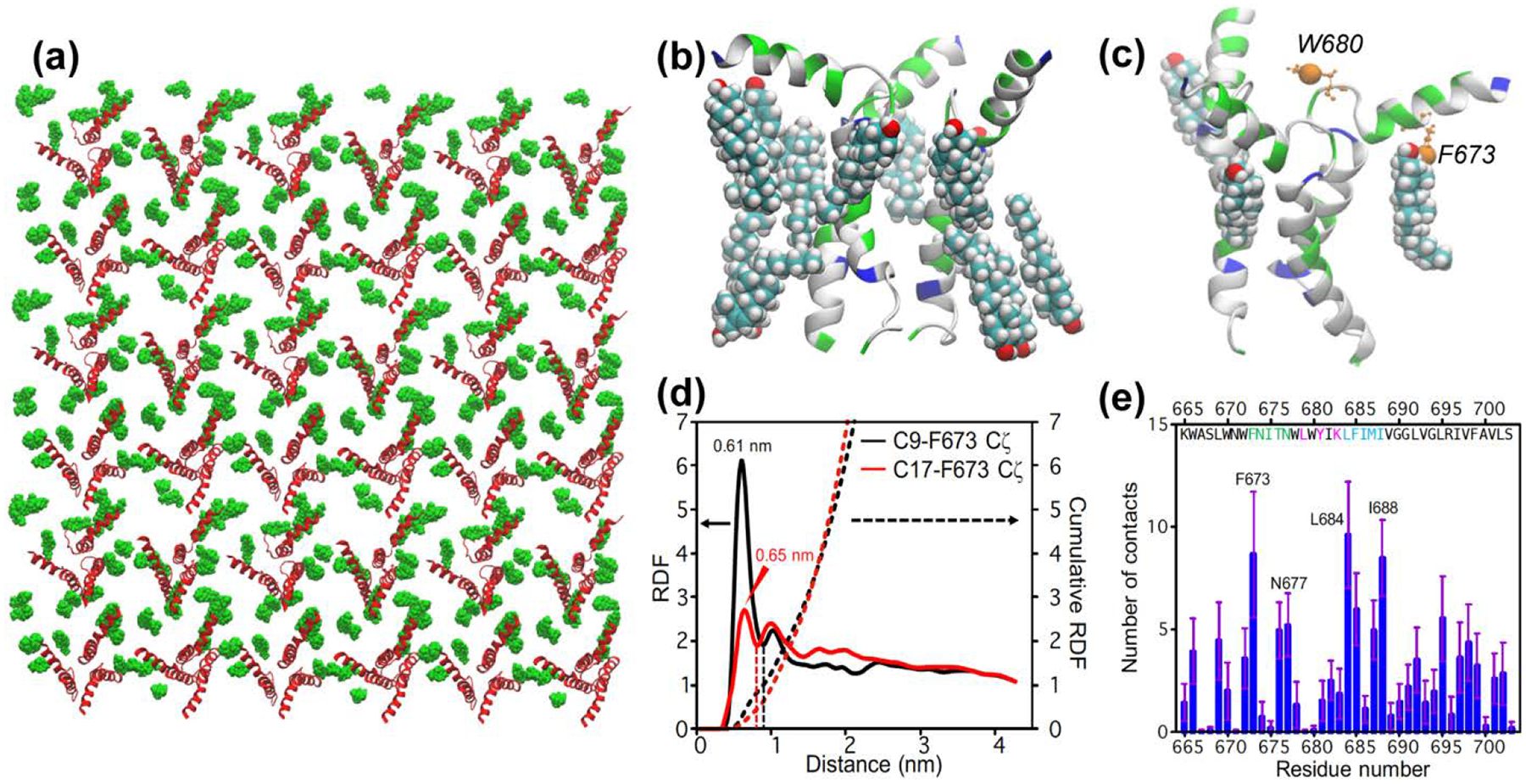
All-atom molecular dynamics simulation of CHOL-gp41 interactions at P : L : C = 1 : 16 : 7. (a) Top view of the equilibrated lipid membrane, showing protein (red) and cholesterol (green) molecules in the upper leaflet. (b) A representative structure showing cholesterol molecules within 3 Å of a trimer. (c) A representative snapshot showing the positions of F673 Cζ and W680 Cζ3 (orange spheres) in the MPER-TMD trimer. Three bound cholesterols whose C9 atoms are within 7.0 Å from F673 Cζ are shown. These sub-nanometer contacts can be detected by 13C-19F REDOR dipolar coupling measurements. (d) Radial distribution function (RDF) and cumulative sums (dotted lines) of cholesterol C9 and C17 from F673 Cζ. The value of the cumulative RDF up to the first minimum (dashed lines) for C9 and C17 is averaged to give the number of bound cholesterols per gp41 monomer, which is 0.75. Thus, 2.3 cholesterols are bound near the MPER per trimer. (e) Number of cholesterol atoms in close contact with each MPER-TMD residue.
To further compare the simulated and the experimental data, we calculated the radial distribution functions (RDFs) of C17 and C9 from F673 Cζ and W680 Cζ3 (Fig. 5c), which mimic the 4-19F and 5-19F atoms of the two residues. The results show that C9 and C17 are located at more than 1.0 nm away from W680 Cζ3 (Fig. S2c), suggesting that 5F-W680 contributes less to C9 and C17 detected REDOR dephasing than 4F-F673. In comparison, the C9 and C17 distances to F673 Cζ peak at 6.1 and 6.5 Å (Fig. 5d), respectively, in good agreement with the measured distances of 7.0 Å by REDOR. Integrating the RDFs up to the first minimum yielded a value of 1.0 for C9 and 0.5 for C17. Taking the average of the two values, and multiplying it by 3 for the three gp41 monomers, we obtained a stoichiometry of 2.3 cholesterols near MPER per trimer as measured from the F673 sidechain. This result provides support to the conclusion from the analysis of the REDOR data (Fig. 4).
To identify important peptide residues for cholesterol interaction, we computed the number of cholesterol atoms that are within 3 Å of each peptide residue. Fig. 5e shows a small number of contacts for the CRAC segment 679LWYIK683, while the highest numbers of atomic contacts are observed for the MPER segment 673FNITN677 and the TMD segment 684LFIMI688. This finding is consistent with the experimental data that the triple CRAC mutations (L679A, Y681F, and K683I) did not perturb cholesterol binding (Fig. 4). Interestingly, the TMD segment 683RLFIMIV689, which encompasses the second hotspot of cholesterol interaction, fulfills the so-called CARC motif [33] by having R683, F685, and V689. This CARC motif was predicted by molecular docking [77] to have affinity for cholesterol because the N-terminal Arg or Lys interact with the cholesterol hydroxyl group, the central aromatic residue interact with cholesterol via CH-π interactions, while the C-terminal Leu or Val interact with the isooctyl tail.
To further probe cholesterol-gp41 interactions, we repeated the simulations using a four-fold lower peptide to lipid ratio (P : L : C = 1 : 64 : 28). This reduction of protein concentration decreased the number of close contacts with cholesterol (Fig. 6a). Within 3 Å of any peptide atoms, there are 6 cholesterols (Fig. 6b, c). By integrating the RDF up to the first minimum (Fig. 6d), we obtained a value of 0.36 for C9 and 0.67 for C17. Taking the average of the two, and multiplying it by 3 to account for the three gp41 monomers, we obtained a stoichiometry of 1.5 cholesterol near MPER per gp41 trimer. Despite the smaller number of bound cholesterol, 673FNITN677 and 684LFIMI688 remain the two hotspots of cholesterol interaction with gp41 (Fig. 6e). We note that for the low peptide concentration, care needs to be exercised to properly sample the cholesterol distribution to avoid artificial clustering. Simulations prepared with a different protocol led to a slightly lower stoichiometry of 1.1 cholesterol near MPER per gp41 trimer (Fig. S3).
Figure 6:
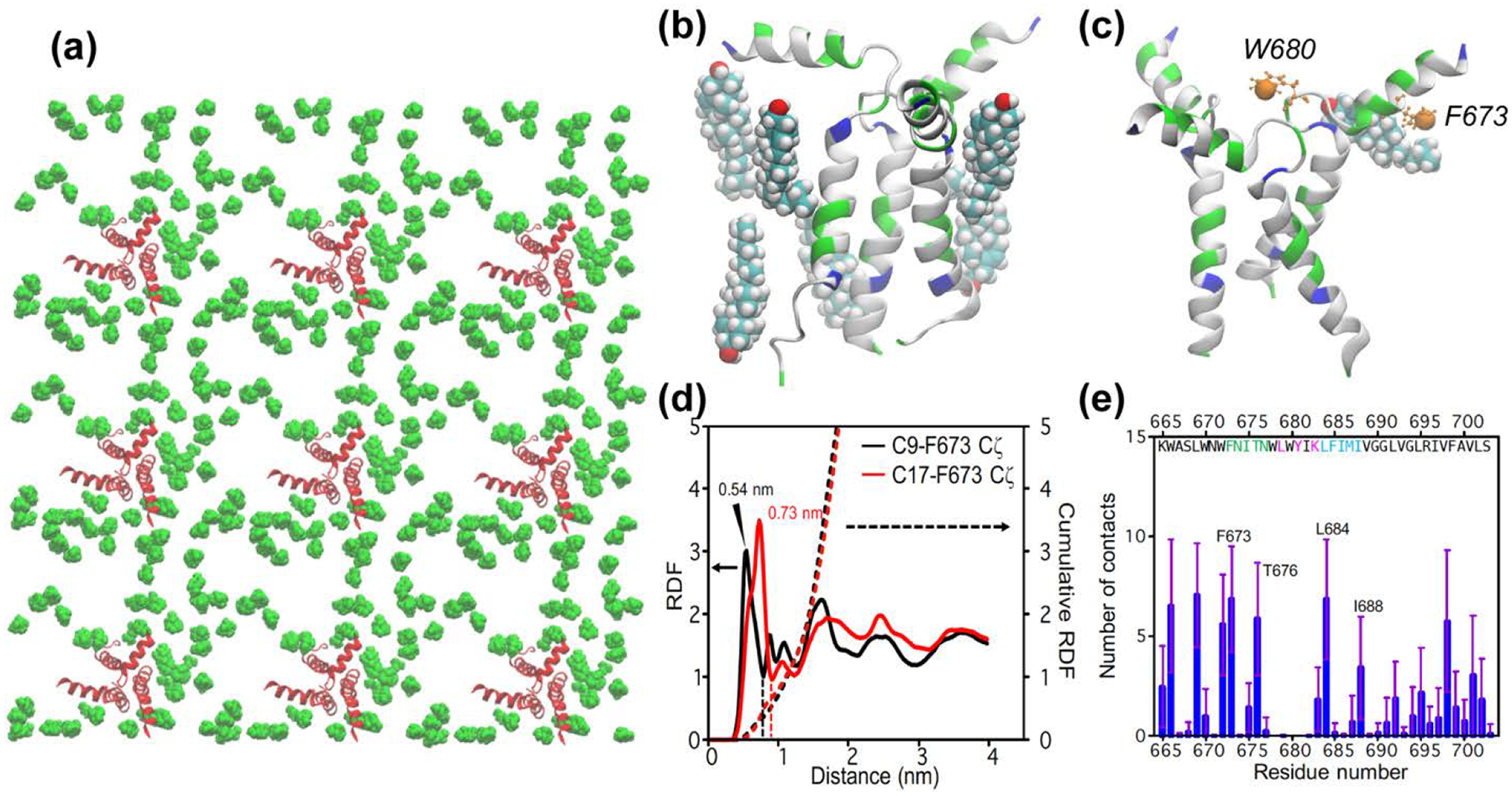
All-atom MD simulation of cholesterol-gp41 contacts at low protein concentrations (P : L : C = 1 : 64 : 28) (a) Top view of the equilibrated lipid membrane, showing only protein (red) and cholesterol (green) molecules in the upper leaflet. (b) A representative structure of cholesterol molecules within 3 Å of the MPER-TMD trimer. (c) A representative snapshot showing the location of W680 Cζ3 and F673 Cζ (orange spheres) in the MPER-TMD structure. Also shown is a bound cholesterol whose C9 atom is within 7.5 Å of F697 Cζ. (d) Radial distribution functions (solid lines) and cumulative sums (dotted lines) of cholesterol C9 and C17 from F673 Cζ. The value of the cumulative RDF up to the first minimum (dashed lines) for C9 and C17 is averaged to give the number of bound cholesterols per gp41 monomer, which is 0.52. Multiplied by a factor of 3, this gives a stoichiometry of 1.5 cholesterols near MPER for each gp41 trimer. (e) Number of cholesterol atoms in close contact with each MPER-TMD residue.
Fig. S4a shows a representative binding mode of cholesterol with only the MPER. The polar W666 and hydrophobic F673 of MPER bind to the head and tail of the cholesterol, respectively. Another representative binding mode (Fig. S4b) shows that the MPER and TMD can cooperatively bind a single cholesterol. In one case, the polar T676 of MPER and hydrophobic L684 of TMD sequester a cholesterol, and in another case, multiple hydrophobic residues of TMD bind the hydrophobic tail while the cholesterol headgroup is stabilized by the polar N677 of MPER. As expected, the cholesterols from the lower leaflet mostly bind to TMD through hydrophobic interactions. A representative snapshot (Fig. S4c) shows the tail of one cholesterol packed against V701 and I697 and the tail of another cholesterol packed with A700 and V693, while their headgroups remain in the lipid headgroup region of the membrane. For a discussion of cholesterol orientation upon interacting with different regions of the peptide, see Supporting Information.
Discussion
These solid-state NMR data and molecular dynamics simulations give molecular structural evidence about cholesterol complexation with the gp41 C-terminus in a native-like lipid bilayer. The two fluorine atoms provide nanometer distance rulers between MPER and cholesterol. Compared to radio, spin, photoaffinity, and fluorescently labeled cholesterol [78–80], 19F NMR of sparsely fluorinated peptides is much less perturbing. The 13C-19F distance data were measured at low temperature on frozen membranes. This low temperature is necessary to immobilize the rotational and translational diffusion of the peptide and cholesterol, which will average the dipolar couplings and prevent the measurement of intermolecular distances. The distance information obtained from these frozen membranes represents the equilibrium average of peptide-cholesterol proximity, without kinetic information about the on- and off-rates of cholesterol binding.
The peptide-cholesterol 19F-19F cross peaks (Fig. 2b) and 19F-13C REDOR data (Fig. 3) unambiguously show that cholesterol is in molecular contact with the MPER-TMD. 2D 19F-19F correlation spectra of model compounds [73, 74] indicate that 19F spin diffusion can detect internuclear distances of ~2 nm. Thus, the isooctyl tail of cholesterol is within ~2 nm of the MPER residues. Since the hydrophobic chain of cholesterol is embedded in the middle of the lipid bilayer, about 2 nm from the membrane surface, this result implies that cholesterol is sequestered directly under the “canopy” of the MPER helix, instead of being displaced horizontally from the MPER helix. This conclusion is supported by the 13C-19F REDOR data (Fig. 4a), which show that the C17-bearing D ring and the C9-bearing B/C ring junction of cholesterol are ~7.0 Å from F673 and W680 sidechains. Although the REDOR data do not directly indicate which of the two fluorines contributed more to the dipolar dephasing, MPER structural data so far show that F673 faces the membrane interior whereas W680 faces water [24]. Molecular dynamics simulations support this model, by finding a larger number of contacts between F673 and cholesterol than between W680 and cholesterol (Fig. 5e). Therefore, we tentatively attribute the 19F-13C REDOR dephasing predominantly to 4-19F F673. On the other hand, the 19F-detected 19F-13C REDOR data show only slightly higher S/S0 values for W680 than for F673 (Fig. 3b), suggesting that the average distance of cholesterol to W680 is not much longer than the distance to F673. Whether this similar average distance results from the F673 and W680 rotameric structures or due to the cholesterol distribution relative to the MPER will require future studies. Regardless of which residue accounts more for dipolar interaction with 13C-labeled cholesterol, the ~7.0 Å distance constrains the cholesterol C17 and C9 to be directly under the MPER helix, near a TMD helix.
The stoichiometry of CHOL binding to the MPER-TMD trimer depends on the distance range used to define the bound cholesterol. With 19F-13C REDOR experiments that detect the F673 sidechain, the data indicate three bound cholesterol molecules per trimer near F673. This stoichiometry was found in both the 30% CHOL membrane (P : L : C = 1 : 16 : 7) and the 18 mol% CHOL membrane (P : L : C = 1 : 32 : 7) (Fig. 4). This cholesterol concentration range spans the biologically relevant concentrations of cholesterol in Ld and Lo phases. The fact that cholesterol dilution did not change cholesterol complexation with the MPER-TMD suggests that the affinity of the peptide for cholesterol is stronger than the affinity of lipids for cholesterol.
Molecular dynamics simulations provided additional insights into the gp41-cholesterol interaction, by considering a much lower protein concentration than accessible by NMR experiments. At a P : L : C molar ratio of 1 : 64 : 28, the hotspots for cholesterol interactions remained the same, suggesting that the affinity of the 673FNITN677 and 684LFIMI688 segments for cholesterol is intrinsic to these amino acid sequences. However, the average number of cholesterols near MPER decreased to 1.5 per trimer (Fig. 6d), despite the fact that the cholesterol concentration is in 28-fold excess to the protein. To rationalize this changing cholesterol stoichiometries for the different protein concentrations, we note that at the high protein concentration used here, the gp41 trimers are spatially close to each other (Fig. 5a), thus they are expected to confine cholesterols and enhance their binding interactions through configurational entropy effects [81]. A previous coarse-grained simulations found that hemagglutinin clustering in multi-component lipid bilayers led to local enhancement of saturated lipids and cholesterols [82]. While a quantitative analysis will require a separate study, the different cholesterol binding stoichiometries observed at different protein concentrations suggest that elevated cholesterol binding in vivo may be correlated with MPER-TMD trimer clustering in the membrane. The number of Env trimers in each virion is 8–10, and the diameters of the HIV virus are 110–140 nm [40, 69]. These values translate to a biological P : L : C molar ratio of about 1 : 5500 : 4500. This extremely low protein concentration would suggest a bound cholesterol number of 1 per trimer; however, it is not known whether gp41 clusters locally in the virus envelope to carry out virus-cell fusion. If gp41 forms clusters, like hemagglutinin [71], then cholesterol may bind with the higher stoichiometry determined here.
Fig. 7 shows the structural model of trimeric MPER-TMD complexed with the three nearest cholesterol molecules obtained from MD simulations. The MPER segment 673FNITN677 and the TMD segment 684LFIMI688 before and after the CRAC segment together sequester a cholesterol molecule. These results indicate that the requirement for cholesterol binding to the C-terminus of gp41 is not the CRAC motif, but the helix-turn-helix topology of this C-terminal domain. Within this fold, a number of atomic interactions are potentially responsible for stabilizing the peptide-cholesterol complex. These include 1) hydrophobic interactions between the methyl-rich Leu and Ile residues in the TM helix and the methyl-rich rough face of cholesterol [50]; 2) aromatic interactions between the lipid-facing polar residues of MPER [24] and the cholesterol sterol rings; and 3) hydrogen bonding between Trp residues in the MPER and the cholesterol C3 hydroxyl group. Molecular dynamics simulations suggest that the TMD binds cholesterol mostly through hydrophobic interactions, whereas the MPER interacts with cholesterols through both polar and hydrophobic motifs. Although the TMD hotspot is part of a CARC motif when combined with K683 and V689, our simulations show that K683 and V689 themselves have only a small number of contacts with cholesterol (Fig. 5e). Future mutagenesis-based structural measurements will be necessary to fully understand whether this CARC motif plays a role in cholesterol binding to MPER-TMD. We also do not know the equilibrium dissociation constant of cholesterol to gp41. Measurement of this binding affinity by NMR requires low-concentration samples, which will have prohibitively low sensitivity. However, the affinity to the MPER region is likely weak, based on the simulated radial distribution functions. As shown in Figs. 5d and 6d, the peak value of the cholesterol-F673 radial distribution functions is in the range of 3–6, which corresponds to an attractive potential of mean force in the range of 1–2 kBT (recall that , in which w(r) is the potential of mean force). The modest strength of the interaction is likely a consequence of the competition between MPER-cholesterol and lipid-cholesterol interactions.
Figure 7.
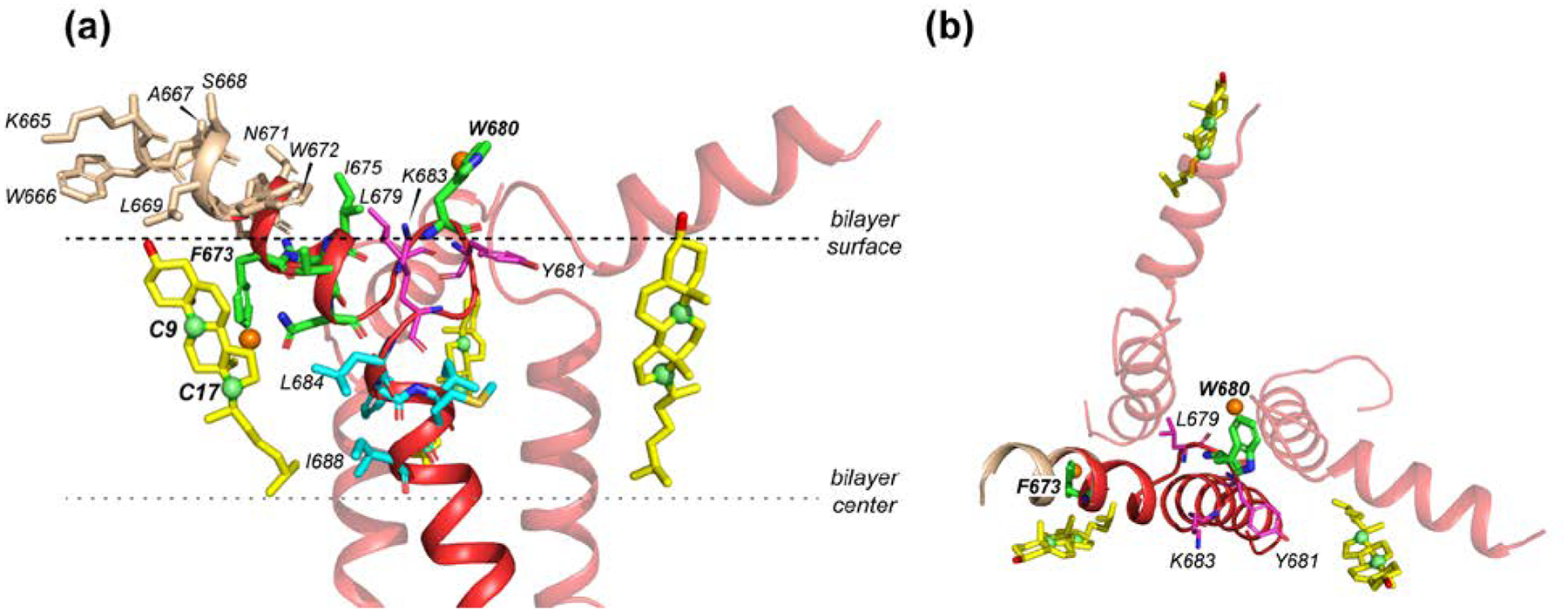
Solid-state NMR structural model (PDB: 6DLN) of trimeric MPER-TMD complexed with cholesterol molecules (yellow) in the upper leaflet of a lipid bilayer. (a) Side view. Residues with the largest numbers of contacts with cholesterol are shown in green for 673FNITN677 and cyan for 684LFIMI688. The CRAC residues L679, Y681 and K683 are shown in magenta. Fluorinated (orange spheres) F673 points to the membrane interior, close to a cholesterol, whereas 5F-W680 points to water, away from cholesterol. (b) Top view, showing the three nearest cholesterols bound to the MPER-TMD trimer. Two of the cholesterol molecules are associated with the middle of the MPER helix, in close proximity to F673, while the third cholesterol is associated with the N-terminal end of one MPER helix.
The MPER Trp residues are important for virus-cell fusion: their mutations severely reduce virus entry and membrane fusion [21, 23]. Our data suggest that this importance is not only due to Trp residues’ well known ability to tether membrane proteins to the membrane-water interface [83, 84], but also due to the involvement of Trp in recruiting cholesterol to the MPER helix. Indirect support for the requirement of both the MPER and TMD for complexing cholesterol is provided by differential scanning calorimetry data, which showed that full-length MPER (residues 665–683) did not cause cholesterol-rich domains, while the 679LWYIK683 peptide did [85]. Moreover, both the MPER and TMD are necessary for membrane fusion, while each helix alone has little ability to cause vesicle leakage at physiologically relevant cholesterol concentrations [38]. Even a truncated construct containing the C-terminal half of the MPER, the N-terminal half of the TMD, and the intervening LWYIR, is able to cause vesicle leakage at high cholesterol concentrations [39, 86]. These biochemical data thus indicate that both membrane fusion and cholesterol sequestration require joining part of the MPER and part of the TMD through the 679LWYIK683 turn.
The finding that gp41 complexation with cholesterol requires a helix-turn-helix fold is in excellent agreement with our recent results about the influenza M2 protein [44, 50]. Mutation of M2’s CRAC motif in a membrane-surface amphipathic helix did not change cholesterol binding. Instead, removal of the amphipathic helix abolished cholesterol contact with the TM helix. Therefore, the HIV-1 gp41 and influenza M2 studies suggest a general principle for cholesterol binding to membrane proteins, which is the combination of a membrane-spanning helix and a membrane-surface amphipathic helix. This combination is effective for sequestering cholesterol, because of the combined contributions from hydrophobic residues in the TM helix and polar residues in the membrane-surface amphipathic helix to stabilize the non-polar and polar groups of cholesterol, respectively.
How does cholesterol binding to gp41 mediate virus-cell fusion? We hypothesize that the extended membrane-surface MPER helices, together with the trimeric TMD stalk, create a strongly asymmetric wedge shape that should incur local membrane curvature [18, 87]. This local curvature and disorder prime the virus envelope to bend and merge with the opposing target cell membrane, which is bound by the N-terminal FP and fusion-peptide proximal region [88]. Second, cholesterol binding of gp41 may act as a mechanism for lateral partitioning of gp41 to the edge of the cholesterol-rich microdomain. Total internal reflection fluorescence microscopy and content mixing assays showed that the N-terminal FP of gp41 is preferentially located at the edge of Ld and cholesterol-rich Lo domains of lipid vesicles [30]. This boundary location of FP was hypothesized to be juxtaposed with the Ld-Lo boundary of the virus lipid envelope where the MPER-TMD resides. Such boundary matching between the two membranes may facilitate fusion, because the boundaries present fault lines for membrane deformation and line tension that can only be relieved by membrane fusion.
Supplementary Material
Acknowledgments
The authors thank Alexander Shcherbakov for help with the 13C-19F REDOR experiments. This work is supported by NIH grant GM066976 to M.H. The solid-state NMR instruments are supported by NIH grant P41 GM132079. For the computational work, T.M. is supported by the NSF grant DMS1661900, and Y.O. is supported by the NSF grant CHE 1829555 to Q.C. Computational resources from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF grant number ACI-1548562, are greatly appreciated. Part of the computational work was performed on the Shared Computing Cluster which is administered by Boston University’s Research Computing Services (URL: www.bu.edu/tech/support/research/).
References
- [1].Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. [DOI] [PubMed] [Google Scholar]
- [2].Jacobson K, Mouritsen OG, Anderson RG. Lipid rafts: at a crossroad between cell biology and physics. Nat. Cell Biol 2007;9:7–14. [DOI] [PubMed] [Google Scholar]
- [3].Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, et al. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GF. Electron crystallography reveals the structure of metarhodopsin I. EMBO J. 2004;23:3609–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sengupta D, Chattopadhyay A. Molecular dynamics simulations of GPCR-cholesterol interaction: An emerging paradigm. Biochim. Biophys. Acta 2015;1848:1775–1782. [DOI] [PubMed] [Google Scholar]
- [6].Sheng R, Chen Y, Yung Gee H, Stec E, Melowic HR, Blatner NR, et al. Cholesterol modulates cell signaling and protein networking by specifically interacting with PDZ domain-containing scaffold proteins. Nat. Commun 2012;3:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. Domains, motifs, and scaffolds: The role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem 2006;75:655–680. [DOI] [PubMed] [Google Scholar]
- [8].Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, et al. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science. 2012;336:1168–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yip CM, Elton EA, Darabie AA, Morrison MR, McLaurin J. Cholesterol, a modulator of membrane-associated Abeta-fibrillogenesis and neurotoxicity. J. Mol. Biol 2001;311:723–734. [DOI] [PubMed] [Google Scholar]
- [10].Di Scala C, Chahinian H, Yahi N, Garmy N, Fantini J. Interaction of Alzheimer’s beta-amyloid peptides with cholesterol: mechanistic insights into amyloid pore formation. Biochemistry. 2014;53:4489–4502. [DOI] [PubMed] [Google Scholar]
- [11].Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat. Neurosci 2003;6:345–351. [DOI] [PubMed] [Google Scholar]
- [12].Checkley MA, Luttge BG, Freed EO. HIV-1 Envelope Glycoprotein Biosynthesis, Trafficking, and Incorporation. J. Mol. Biol 2011;410:582–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wilen CB, Tilton JC, Doms RW. HIV: Cell Binding and Entry. Cold Spring Harb. Perspect. Med 2012;2:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gallo SA, Finnegan CM, Viard M, Raviv Y, Dimitrov A, Rawat SS, et al. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 2003;1614:36–50. [DOI] [PubMed] [Google Scholar]
- [15].Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. [DOI] [PubMed] [Google Scholar]
- [16].Harrison SC. Viral membrane fusion. Nat. Struct. Mol. Biol 2008;15:690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. [DOI] [PubMed] [Google Scholar]
- [18].Yao H, Lee MW, Waring AJ, Wong GC, Hong M. Viral fusion protein transmembrane domain adopts beta-strand structure to facilitate membrane topological changes for virus-cell fusion. Proc. Natl. Acad. Sci. U.S.A 2015;112:10926–10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kwon B, Lee M, Waring AJ, Hong M. Oligomeric Structure and Three-Dimensional Fold of the HIV gp41 Membrane-Proximal External Region and Transmembrane Domain in Phospholipid Bilayers. J. Am. Chem. Soc 2018;140:8246–8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Munoz-Barroso I, Salzwedel K, Hunter E, Blumenthal R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J. Virol 1999;73:6089–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bellamy-Mclntyre AK, Lay CS, Bar S, Maerz AL, Talbo GH, Drummer HE, et al. Functional links between the fusion peptide-proximal polar segment and membrane-proximal region of human immunodeficiency virus gp41 in distinct phases of membrane fusion. J. Biol. Chem 2007;282:23104–23116. [DOI] [PubMed] [Google Scholar]
- [22].Huarte N, Lorizate M, Kunert R, Nieva JL. Lipid modulation of membrane-bound epitope recognition and blocking by HIV-1 neutralizing antibodies. FEBS Lett. 2008;582:3798–3804. [DOI] [PubMed] [Google Scholar]
- [23].Salzwedel K, West JT, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol 1999;73:2469–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, et al. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28:52–63. [DOI] [PubMed] [Google Scholar]
- [25].Irimia A, Serra AM, Sarkar A, Jacak R, Kalyuzhniy O, Sok D, et al. Lipid interactions and angle of approach to the HIV-1 viral membrane of broadly neutralizing antibody 10E8: Insights for vaccine and therapeutic design. PLoS Pathog. 2017;13:e1006212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schroeder C Cholesterol-Binding Viral Proteins in Virus Entry and Morphogenesis. Subcell. Biochem 2010;51:77–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liao ZH, Graham DR, Hildreth JEK. Lipid rafts and HIV pathogenesis: Virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res. Hum. Retrovir 2003;19:675–687. [DOI] [PubMed] [Google Scholar]
- [28].Guyader M, Kiyokawa E, Abrami L, Turelli P, Trono D. Role for human immunodeficiency virus type 1 membrane cholesterol in viral internalization. J. Virol 2002;76:10356–10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nguyen DH, Hildreth JEK. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol 2000;74:3264–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yang ST, Kiessling V, Simmons JA, White JM, Tamm LK. HIV gp41-mediated membrane fusion occurs at edges of cholesterol-rich lipid domains. Nat. Chem. Biol 2015;11:424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Saez-Cirion A, Arrondo JLR, Gomara MJ, Lorizate M, Iloro I, Melikyan G, et al. Structural and functional roles of HIV-1 gp41 pre-transmembrane sequence segmentation. Biophys. J 2003;85:3769–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vincent N, Genin C, Malvoisin E. Identification of a conserved domain of the HIV-1 transmembrane protein gp41 which interacts with cholesteryl groups. Biochim. Biophys. Acta 2002;1567:157–164. [DOI] [PubMed] [Google Scholar]
- [33].Fantini J, Barrantes FJ. How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol 2013;4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Epand RF, Thomas A, Brasseur R, Vishwanathan SA, Hunter E, Epand RM. Juxtamembrane protein segments that contribute to recruitment of cholesterol into domains. Biochemistry. 2006;45:6105–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Greenwood AI, Pan JJ, Mills TT, Nagle JF, Epand RM, Tristram-Nagle S. CRAC motif peptide of the HIV-1 gp41 protein thins SOPC membranes and interacts with cholesterol. Biochim. Biophys. Acta 2008;1778:1120–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schwarzer R, Levental I, Gramatica A, Scolari S, Buschmann V, Veit M, et al. The cholesterol-binding motif of the HIV-1 glycoprotein gp41 regulates lateral sorting and oligomerization. Cell. Microbiol 2014;16:1565–1581. [DOI] [PubMed] [Google Scholar]
- [37].Vishwanathan SA, Thomas A, Brasseur R, Epand RF, Hunter E, Epand RM. Large changes in the CRAC segment of gp41 of HIV do not destroy fusion activity if the segment interacts with cholesterol. Biochemistry. 2008;47:11869–11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Apellaniz B, Garcia-Saez A, Nir S, Nieva JL. Destabilization exerted by peptides derived from the membrane-proximal external region of HIV-1 gp41 in lipid vesicles supporting fluid phase coexistence. Biochim. Biophys. Acta 2011;1808:1797–1805. [DOI] [PubMed] [Google Scholar]
- [39].Apellaniz B, Nieva JL. Fusion-competent state induced by a C-terminal HIV-1 fusion peptide in cholesterol-rich membranes. Biochim. Biophys. Acta 2015;1848:1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. U.S.A 2006;103:2641–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Song L, Sun ZY, Coleman KE, Zwick MB, Gach JS, Wang JH, et al. Broadly neutralizing anti-HIV-1 antibodies disrupt a hinge-related function of gp41 at the membrane interface. Proc. Natl. Acad. Sci. U.S.A 2009;106:9057–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dev J, Park D, Fu Q, Chen J, Ha HJ, Ghantous F, et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science. 2016;353:172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chiliveri SC, Louis JM, Ghirlando R, Baber JL, Bax A. Tilted, Uninterrupted, Monomeric HIV-1 gp41 Transmembrane Helix from Residual Dipolar Couplings. J. Am. Chem. Soc 2018;140:34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Elkins MR, Sergeyev IV, Hong M. Determining Cholesterol Binding to Membrane Proteins by Cholesterol (13)C Labeling in Yeast and Dynamic Nuclear Polarization NMR. J. Am. Chem. Soc 2018;140:15437–15449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shivapurkar R, Souza CM, Jeannerat D, Riezman H. An efficient method for the production of isotopically enriched cholesterol for NMR. J. Lipid Res 2011;52:1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Souza CM, Schwabe TM, Pichler H, Ploier B, Leitner E, Guan XL, et al. A stable yeast strain efficiently producing cholesterol instead of ergosterol is functional for tryptophan uptake, but not weak organic acid resistance. Metab. Eng 2011;13:555–569. [DOI] [PubMed] [Google Scholar]
- [47].Mijalis AJ, Thomas DA 3rd, Simon MD, Adamo A, Beaumont R, Jensen KF, et al. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol 2017;13:464–466. [DOI] [PubMed] [Google Scholar]
- [48].Simon MD, Heider PL, Adamo A, Vinogradov AA, Mong SK, Li X, et al. Rapid flow-based peptide synthesis. Chembiochem. 2014;15:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Elkins MR, Hong M. Elucidating ligand-bound structures of membrane proteins using solid-state NMR spectroscopy. Curr. Opin. Struct. Biol. 2019;57:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Elkins MR, Williams JK, Gelenter MD, Dai P, Kwon B, Sergeyev IV, et al. Cholesterol-binding site of the influenza M2 protein in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. U.S.A 2017;114:12946–12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kauffman JM, Westerman PW, Carey MC. Fluorocholesterols, in contrast to hydroxycholesterols, exhibit interfacial properties similar to cholesterol. J. Lipid Res 2000;41:991–1003. [PubMed] [Google Scholar]
- [52].Cady SD, Wang T, Hong M. Membrane-dependent effects of a cytoplasmic helix on the structure and drug binding of the influenza virus M2 protein. J. Am. Chem. Soc 2011;133:11572–11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Luo W, Cady SD, Hong M. Immobilization of the Influenza A M2 Transmembrane Peptide in Virus-Envelope Mimetic Lipid Membranes: A Solid-State NMR Investigation. Biochemistry. 2009;48:6361–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hou G, Yan S, Trebosc J, Amoureux JP, Polenova T. Broadband homonuclear correlation spectroscopy driven by combined R2(n)(v) sequences under fast magic angle spinning for NMR structural analysis of organic and biological solids. J. Magn. Reson 2013;232:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gullion T, Schaefer J. Rotational echo double resonance NMR. J. Magn. Reson 1989;81:196–200. [DOI] [PubMed] [Google Scholar]
- [56].Sinha N, Schmidt-Rohr K, Hong M. Compensation for pulse imperfections in rotational-echo double-resonance NMR by composite pulses and EXORCYCLE. J. Magn. Reson 2004;168:358–365. [DOI] [PubMed] [Google Scholar]
- [57].Pan Y, Gullion T, Schaefer J. Determination of C-N internuclear distances by rotational-echo double-resonance NMR of solids. J. Magn. Reson 1990;90:330. [Google Scholar]
- [58].Bak M, Rasmussen JT, Nielsen NC. SIMPSON: a general simulation program for solid-state NMR spectroscopy. J. Magn. Reson 2000;147:296–330. [DOI] [PubMed] [Google Scholar]
- [59].Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem 2008;29:1859–1865. [DOI] [PubMed] [Google Scholar]
- [60].Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 2016;12:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ohkubo YZ, Pogorelov TV, Arcario MJ, Christensen GA, Tajkhorshid E. Accelerating Membrane Insertion of Peripheral Proteins with a Novel Membrane Mimetic Model. Biophys. J 2012;102:2130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Huang J, MacKerell AD Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem 2013;34:2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983;79:926–935. [Google Scholar]
- [64].Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997;18:1463–1472. [Google Scholar]
- [65].Evans DJ, Holian BL. The Nose–Hoover thermostat. J. Chem. Phys 1985;83:4069–4074. [Google Scholar]
- [66].Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys 1981;52:7182–7190. [Google Scholar]
- [67].Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 1995;91:43–56. [Google Scholar]
- [68].Gerl MJ, Sampaio JL, Urban S, Kalvodova L, Verbavatz JM, Binnington B, et al. Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane. J. Cell Biol 2012;196:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhu P, Chertova E, JB Jr., Lifson JD, Arthur LO, Liu J, et al. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 2003;100:15812–15817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhu P, Liu J, Bess JJ, Chertova E, Lifson JD, Grisé H, et al. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. [DOI] [PubMed] [Google Scholar]
- [71].Danieli T, Pelletier SL, Henis YI, White JM. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol 1996;133:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Donald JE, Zhang Y, Fiorin G, Carnevale V, Slochower DR, Gai F, et al. Transmembrane orientation and possible role of the fusogenic peptide from parainfluenza virus 5 (PIV5) in promoting fusion. Proc. Natl. Acad. Sci. U. S. A 2011;108:3958–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Roos M, Wang T, Shcherbakov AA, Hong M. Fast Magic-Angle-Spinning (19)F Spin Exchange NMR for Determining Nanometer (19)F-(19)F Distances in Proteins and Pharmaceutical Compounds. J. Phys. Chem. B 2018;122:2900–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Roos M, Mandala VS, Hong M. Determination of Long-Range Distances by Fast Magic-Angle-Spinning Radiofrequency-Driven 19F–19F Dipolar Recoupling NMR. J. Phys. Chem. B 2018;122:9302–9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Shcherbakov AA, Hong M. Rapid Measurement of Long-Range Distances in Proteins by Multidimensional 13C-19F REDOR NMR under Fast Magic-Angle Spinning. J. Biomol. NMR 2018;71:31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Shcherbakov AA, Roos M, Kwon B, Hong M. Two-dimensional 19F-13C correlation NMR for 19F resonance assignment of fluorinated proteins. J. Biomol. NMR 2020;74:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Baier CJ, Fantini J, Barrantes FJ. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep 2011;1:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Scheidt HA, Muller P, Herrmann A, Huster D. The potential of fluorescent and spin-labeled steroid analogs to mimic natural cholesterol. J. Biol. Chem 2003;278:45563–45569. [DOI] [PubMed] [Google Scholar]
- [79].Gimpl G Cholesterol-protein interaction: methods and cholesterol reporter molecules. Subcell. Biochem 2010;51:1–45. [DOI] [PubMed] [Google Scholar]
- [80].Mukherjee S, Chattopadhyay A. Membrane organization at low cholesterol concentrations: a study using 7-nitrobenz-2-oxa-1,3-diazol-4-yl-labeled cholesterol. Biochemistry. 1996;35:1311–1322. [DOI] [PubMed] [Google Scholar]
- [81].Zhou HX, Rivas G, Minton AP. Macromolecular Crowding and Confinement: Biochemical, Biophysical, and Potential Physiological Consequences. Annu. Rev. Biophys 2008;37:375–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Parton DL, Tek A, Baaden M, Sansom MSP. Formation of Raft-Like Assemblies within Clusters of Influenza Hemagglutinin Observed by MD Simulations. PLoS Comp. Biol 2013;9:e1003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].de Jesus AJ, Allen TW. The role of tryptophan side chains in membrane protein anchoring and hydrophobic mismatch. Biochim. Biophys. Acta 2013;1828:864–876. [DOI] [PubMed] [Google Scholar]
- [84].Killian JA, von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem. Sci 2000;25:429–434. [DOI] [PubMed] [Google Scholar]
- [85].Epand RF, Sayer BG, Epand RM. The tryptophan-rich region of HIV gp41 and the promotion of cholesterol-rich domains. Biochemistry. 2005;44:5525–5531. [DOI] [PubMed] [Google Scholar]
- [86].Apellaniz B, Rujas E, Carravilla P, Requejo-Isidro J, Huarte N, Domene C, et al. Cholesterol-dependent membrane fusion induced by the gp41 membrane-proximal external region-transmembrane domain connection suggests a mechanism for broad HIV-1 neutralization. J. Virol 2014;88:13367–13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Yao H, Hong M. Conformation and Lipid Interaction of the Fusion Peptide of the Paramyxovirus PIV5 in Anionic and Negative-Curvature Membranes From Solid-State NMR. J. Am. Chem. Soc 2014;136:2611–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lee M, Morgan CA, Hong M. Fully hydrophobic HIV gp41 adopts a hemifusion-like conformation in phospholipid bilayers. J. Biol. Chem 2019;294:14732–14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.