

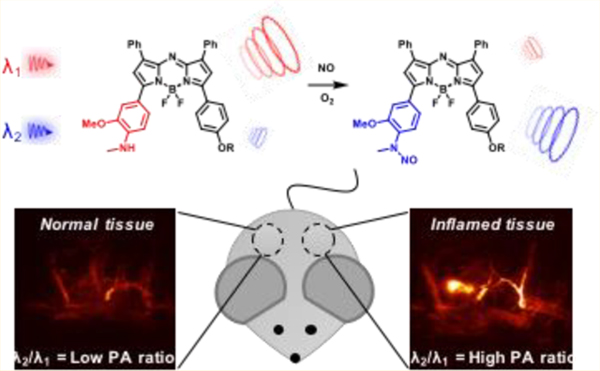
Abstract
Photoacoustic (PA) imaging is an emerging imaging modality that utilizes optical excitation and acoustic detection to enable high resolution at centimeter depths. The development of activatable PA probes can expand the utility of this technology to allow for detection of specific stimuli within live-animal models. Herein, we report the design, development, and evaluation of a series of Acoustogenic Probe(s) for Nitric Oxide (APNO) for the ratiometric, analyte-specific detection of nitric oxide (NO) in vivo. The best probe in the series, APNO-5, rapidly responds to NO to form an N-nitroso product with a concomitant 91 nm hypsochromic shift. This property enables ratiometric PA imaging upon selective irradiation of APNO-5 and the corresponding product, tAPNO-5. Moreover, APNO-5 displays the requisite photophysical characteristics for in vivo PA imaging (e.g., high absorptivity, low quantum yield) as well as high biocompatibility, stability, and selectivity for NO over a variety of biologically relevant analytes. APNO-5 was successfully applied to the detection of endogenous NO in a murine lipopolysaccharide-induced inflammation model. Our studies show a 1.9-fold increase in PA signal at 680 nm and a 1.3-fold ratiometric turn-on relative to a saline control.
Graphical Abstract
INTRODUCTION
Nitric oxide (NO) is a metastable free radical with key roles in the regulation of vascular tone,1 neuronal signaling,2 tumor progression,3 and immunology.4,5 Under normoxic conditions, NO is biosynthesized by nitric oxide synthases (NOS), which generate NO through the oxidation of L-arginine to L-citrulline. The NOS family includes an inducible, calcium-independent isoform (iNOS), which is primarily linked to the immune response, and constitutively expressed, calcium-dependent isoforms (ncNOS and ecNOS), which are linked to homeostatic maintenance.6 NO can also be enzymatically7 or nonenzymatically generated by the reduction of nitrite under hypoxic conditions, a process which has been observed in the ischemic heart and results in NO-mediated tissue damage.8 Due to its various roles in physiological and pathological processes, the identification, detection, and quantification of NO continues to be an active area of research. Since ex vivo samples (e.g., cultured cells, tissue slices, etc.) do not adequately mimic the complexities of in vivo samples (e.g., interaction between the cell and its surrounding environment), it is essential to develop new methods for the real-time detection of NO. However, noninvasive, in vivo detection of NO with high spatiotemporal resolution remains a profound challenge in the field. The ability to perform longitudinal studies with high-resolution in vivo imaging would provide exciting avenues for studying the role of NO as a neurotransmitter9,10 and elucidating its dichotomous role in cancer progression,3,11 where the location, source, and role of NO remain unclear. The current arsenal of NO donors12 and ex vivo analyses provides important preliminary information but has yet to address these outstanding questions.
Current methods for detecting NO are inadequate for in vivo applications. For example, the colorimetric Griess assay is useful for analyzing NO in cell lysates; however, this is performed indirectly via detection of nitrite and nitrate and requires acidic conditions that are not biocompatible.13 In contrast, techniques such as electron paramagnetic resonance (EPR) spectroscopy14 and magnetic resonance imaging (MRI)15 have been employed for NO detection in vivo at relevant imaging depths; however, these approaches are limited by low resolution and sensitivity, respectively.16 Amperometry displays high sensitivity (pM) but requires invasive procedures and can only detect NO in direct contact with the probe.17–19 Optical methods such as luminescence20 and fluorescence imaging21 are noninvasive and enable high resolution with high contrast at shallow imaging depths;16 as a result, a diverse palette of reaction-based fluorescent probes has been developed, primarily for cellular studies. In principle, many of the red-shifted and multiphoton congeners can be employed for in vivo imaging, but these studies require invasive surgical procedures,22 are confined to peripheral limbs,23 and/or suffer from limited resolution23,24 due to the scattering of emitted light within tissue. Several other fluorescent NO probes have been applied to living animals; however, in these examples, the specimen is sacrificed to enable harvesting and ex vivo imaging of the tissue of interest.25–29
Photoacoustic (PA) imaging is a noninvasive imaging modality with the potential to overcome these challenges. PA imaging couples the precision of optical imaging with the deep tissue capabilities of ultrasound imaging. Specifically, PA imaging relies on the ability of optically excited small molecules to release heat through nonradiative relaxation. By pulsing the excitation source, it is possible to generate fluctuating thermoelastic expansions that propagate through the tissue as ultrasound and can be measured by transducers. Since sound scatters 3 orders of magnitude less than light within tissue, PA imaging can generate high-resolution images at depths inaccessible to fluorescence imaging. Empirically, it has been shown that the spatial resolution of PA imaging is ~1/200 of the imaging depth. For example, at 8 cm depths, the resolution is estimated be ~400 μM, while at 1 cm the resolution would be predicted to be ~50 μm.30 Owing to the noninvasive nature of PA imaging, it is possible to conduct longitudinal experiments and acquire high-resolution deep tissue images within live animals. To date, analyte-specific acoustogenic probes have been developed for the detection of copper31 and calcium;32,33 however, the utility of these probes has only been demonstrated in tissue-mimicking systems and in translucent larval zebrafish. More recently, a stimulus-specific acoustogenic probe for hypoxia has been reported and applied for imaging hypoxia within murine tumor and ischemia models.34
Herein, we report the design, synthesis, characterization, and application of a series of five reaction-based PA probes for NO. While all members of this panel exhibited excellent in vitro selectivity for NO over other reactive nitrogen species, oxidants, and reactive carbonyl species (Figure S1), we employed our best probe, APNO-5, to noninvasively detect endogenous NO in a murine lipopolysaccharide (LPS)-mediated inflammation model using PA imaging. This work highlights the utility of small-molecule, reaction-based photoacoustic probes for in vivo analyte-specific detection.
RESULTS AND DISCUSSION
Acoustogenic Probe Design.
When developing acoustogenic probes for NO, it is essential to satisfy six design criteria to optimize in vivo performance. First, a probe must exhibit maximal absorption in the NIR-I (620–950 nm) or NIR-II (1000–1700 nm)35 window to ensure incident light penetrates deep within tissue without significant attenuation. Second, it should possess a large extinction coefficient (>104 M−1 cm−1) and low quantum yield, since absorptivity and nonemissive relaxation are proportional to PA signal intensity. Third, it must react with NO in a rapid and highly selective manner, since NO is present in low abundance (nM to low μM range) and has a short biological lifetime (seconds).36 Fourth, a probe should be optimized for ratiometric imaging, which is necessary to account for imaging artifacts (e.g., dye loading, heterogeneous distribution, and variability in imaging depth).37 Fifth, the probe must display favorable pharmacokinetic and pharmacodynamic properties (e.g., rapid uptake by the tissue of interest). Last, the probe should be biocompatible and produce minimal effects on the native biological system.38
To this end, we have rationally developed and synthesized a panel of Acoustogenic Probe(s) for Nitric Oxide (APNO). We began by incorporating the o-phenylenediamine (OPD) functionality39 onto the aza-BODIPY dye platform to afford APNO-1 (Figure 1). This scaffold was selected over the various other NIR absorbing dye platforms (e.g., cyanines) because of its well-documented photo- and chemostability. We rationalized that strategic positioning of the trigger would allow for NO-mediated disruption of the push–pull system, resulting in a concomitant hypsochromic (blue) shift in the wavelength of maximum absorbance (λmax). Empirically, a shift ≥50 nm is desirable for ratiometric imaging such that each form can be selectively irradiated to produce a corresponding PA signal.
Figure 1.

Structures, triggers, and sensing mechanisms for the panel of APNO (red) and tAPNO (blue).
Treatment of APNO-1 with NO resulted in a 55 nm λmax blueshift from 731 to 676 nm (Figure S2). When APNO-1 and turned-over APNO-1 (tAPNO-1) were excited at 740 and 680 nm, respectively, a ratiometric turn-on PA response of 7.4-fold was observed (Figure S3). Experimental excitation wavelengths differ from the reported λmax values due to a combination of instrumental range (680–900 nm) and experimentally larger PA ratio. Although the observed ratiometric response would be sufficient to detect most analyte targets, we deemed it essential to improve this property further due to the low biological abundance of NO. Moreover, the preparation of APNO-1 was hampered by decomposition of key synthetic intermediates which resulted in low overall yields.
In order to address these limitations, we turned our attention to developing probes that rely on NO-mediated deamination.40,41 Specifically, APNO-2 features an electron-rich o-aminophenol moiety that undergoes NO-mediated diazonium formation and subsequent homolytic bond cleavage to afford tAPNO-2 (Figure 1). It was rationalized that the removal of the strongly electron-donating aniline would have a larger effect on the λmax than triazole formation. Treatment of APNO-2 with NO resulted in a 59 nm blueshift from 735 to 676 nm (Figure S2) and 13.1-fold ratiometric turn-on response when irradiated at 750 and 680 nm (Figure S3). While this improvement in the dynamic range may have been sufficient, it was limited by insufficient reaction kinetics. Specifically, the reaction proceeds via an N-nitroso intermediate (which was detected via fluorescence and UV-vis) followed by slow diazonium formation, and decomposition to yield the product.40 These properties significantly complicate the interpretation of imaging results. Of note, we also prepared APNO-3, a constitutional isomer in which the aniline is not directly in conjugation with the π-system. While this probe also underwent a blueshift upon reaction with NO, the change in λmax was small and the probe suffered from pH dependence in the biological range (Figure S4). Taken together, the two deamination-based probes do not satisfy the necessary criteria for in vivo detection of NO.
In light of the rapid formation of N-nitroso intermediates, we reasoned that monoalkylation of a reactive aniline could both enhance the reactivity and block the necessary rearrangements for diazonium formation and deamination. This led to the development of APNO-4 (Figure 1) which features an N-methylaniline nucleophile trigger. Additionally, the hydroxyl group was replaced with a methoxy substituent for synthetic ease and to prevent possible pH dependence. While there are examples of photoinduced electron transfer (PET)-based fluorogenic probes relying on NO-responsive secondary anilines in the literature,28,29,42 to the best of our knowledge, APNO-4 represents the first probe (for any imaging modality) that employs the N-nitrosation for modulation of a push−pull system for ratiometric imaging. We observed rapid formation of the anticipated N-nitroso product (tAPNO-4) and a significant 86 nm blueshift of the λmax from 764 to 678 nm upon treatment with NO (Figure S2); however, tAPNO-4 was poorly soluble in aqueous solution. This resulted in inconsistent results due to aggregation and precipitation. To overcome this limitation, we developed APNO-5, a positively charged analogue featuring a PEGylated tetraalkylammonium group to improve solubility, prevent dye aggregation, and facilitate uptake by cells in vivo.
Synthesis.
Each APNO was synthesized via Paal−Knorr cyclization and coupling of two 4-nitro-1,3- diphenylbutan-1-one analogues, followed by boron chelation to afford the aza-BODIPY dye.43
The synthesis of APNO-5 began with o-aminophenol. Carbamate protection with carbonyldiimidazole followed by Friedel–Crafts acylation afforded acetophenone 2. Claisen–Schmidt condensation between 2 and benzaldehyde afforded chalcone 3 in good yields. Sequential Boc-protection and carbamate deprotection were performed in one pot under basic conditions, followed by methylation of the chalcone intermediate 4 with sodium hydride and methyl iodide to yield 5. Michael addition with the nitromethane anion under basic conditions provided the 4-nitro-1,3- diphenylbutan-1-one 6 in 86% yield. The asymmetric dipyrromethane core was formed in reasonable yields followed by boron chelation with boron trifluoride dietherate in the presence of Hünig’s base to afford 8 in 33% yield. Copper(I)-catalyzed alkyne–azide cycloaddition with trimethylammonium-PEG-N3 gave APNO-5 in quantitative yields (Scheme 1).
Scheme 1.

Synthesis of APNO-5
In Vitro Characterization of APNO-5.
As anticipated, APNO-5 reacted rapidly with NO to afford the N-nitroso product (tAPNO-5) along with a significant 91 nm blueshift of the λmax from 764 to 673 nm, the largest in the APNO series (Figure 2a).
Figure 2.

(a) Normalized absorbance spectra of 2 μM APNO-5 (red) and tAPNO-5 (blue). (b) APNO-5 (2 μM) fluorescence enhancement following 1 h treatment with excess (100 equiv) reactive metal (red), carbonyl (green), oxygen (blue), or nitrogen (purple) species. (c) PA spectra of 10 μM APNO-5 (red) and tAPNO-5 (blue). (d) PA images of 10 μM APNO-5 and tAPNO-5 at 680 and 770 nm. All spectra and assays were performed in ethanolic 20 mM potassium phosphate buffer (pH 7.4, 50% v/v). Data presented as mean ± standard deviation (n = 3) for parts b and c.
The extinction coefficient of APNO-5 was sufficiently large (3.2 × 104 M−1 cm−1) to ensure generation of strong PA signals in vivo (Table 1). Selective irradiation of APNO-5 and tAPNO-5 at 770 and 680 nm, respectively, resulted in a 18.6-fold ratiometric PA turn-on response in vitro (Figure 2c). Additionally, the improved aqueous solubility due to the appendd tetraalkylammonium PEG group enhanced its properties for ratiometric PA imaging. Insoluble aza-BODIPY dyes are known to form J-aggregates and nanoparticles, where the λmax is red-shifted relative to the nonaggregated form.44 Since our strategy relies heavily on selective excitation at specific wavelengths, it is essential that formation of wavelength-shifted aggregates is minimized. To verify this, we monitored the degree of aggregation as a function of surfactant concentration at a fixed dye concentration. At 2 μM, aggregation was attenuated at concentrations as low as 0.1% surfactant for APNO-5, whereas APNO-4 required ≥10% (Figure S5). Moreover, the tetraalkylammonium PEG appendage increased the water solubility of APNO-5 and tAPNO-5, as indicated by clogP values of 3.8 and 3.8, relative to 7.6 and 7.3 for APNO-4 and tAPNO-4 (Table S2). Since the calculation methods are unable to account for charge, APNO-5 and tAPNO-5’s predicted values should serve as overapproximations of the true values.45 This key structural feature was also required for APNO-5 to permeate deep into tissue after subcutaneous administration into live mice. Other congeners pooled at the injection site and showed minimal response owing to poor tissue uptake (data not shown).
Table 1.
Photophysical Characterization of APNO and tAPNO in Ethanolic 20 mM Potassium Phosphate Buffer (pH 7.4, 50% v/v)
| APNO | ƛmax (nm) | ε (M−1 cm−1) | ϕa | tAPNO | ƛmax (nm) | ε (M−1 cm−1) | ϕa |
|---|---|---|---|---|---|---|---|
| 1 | 731 | 3.3 × 104 | 0.0065 | 1 | 676 | 3.7 × 104 | 0.14 |
| 2 | 735 | 7.4 × 104 | 0.0006 | 2 | 676 | 6.9 × 104 | 0.12 |
| 3 | 679 | 2.2 × 104 | 0.0708 | 3 | 678 | n.d.b | n.d.b |
| 4 | 764 | 3.7 × 104 | 0.0003 | 4 | 678 | n.d.c | n.d.c |
| 5 | 764 | 3.2 × 104 | 0.0003 | 5 | 673 | 3.3 × 104 | 0.23 |
Measured relative to dimethoxy aza-BODIPY(ϕ = 0.32, CHCl3).
Not determined: tAPNO-3 displays pH dependence under these conditions.
Not determined: tAPNO-4 precipitated out of solution.
Next, we determined that the fluorescence quantum yields of APNO-5 and tAPNO-5 were sufficiently low (Table 1). This is ideal for PA imaging, since the quantum yield is inversely proportional to the intensity of the PA signal. Nevertheless, we conducted selectivity assays using fluorimetry due to its sensitivity and dynamic range. Treatment of APNO-5 with excess NO (100 equiv) resulted in a significant 174.4-fold fluorescence enhancement, whereas other reactive nitrogen species (RNS) (e.g., nitroxyl and peroxynitrite) did not elicit an appreciable response (Figure 2d). High selectivity is important because NO can rapidly convert into other RNS under biological conditions (besides N2O3, the hypothesized nitrosating species).28 Importantly, most reported NO triggers react with N2O3 rather than directly with NO. While this decreases sensitivity, it is suggested to prevent perturbation of NO signal transduction.39 When APNO-5 was treated with a panel of oxidants, no oxidation of the aza-BODIPY core nor the N-methylaniline trigger was observed. Finally, the reactivity of APNO-5 was assessed against a series of key biological carbonyl species including formaldehyde (FA), ascorbic acid (AA), dehydroascorbic acid (DHA), and glyoxal to test for side-reactivity and false turn-on via Schiff base formation (Figure 2b). Even with large stoichiometric excesses of each carbonyl, no cross-reactivity was observed.
We then determined the chemo- and photostability of APNO-5 in the interest of supporting in vivo imaging over extended periods (hours to days). First, the absorption spectra of a solution of APNO-5 were monitored over the course of 5 days. During this time, no significant decomposition of the probe was observed. Next, we subjected a solution of APNO-5 to continuous irradiation with an OPO laser at 770 nm for 60 min. Under these conditions, minimal photobleaching was observed (Figure S6). Interestingly, it was discovered that tAPNO-5 undergoes photoinduced denitrosylation when continuously irradiated; however, only moderate signal loss is observed (<30%) under standard imaging conditions (confirmed by UV–vis; data not shown). Moreover, since APNO-5 is released as the product during prolonged irradiation, it is reasonable to predict minimal decrease in sensitivity.
After confirming the requisite selectivity, solubility, and stability, the probe’s biocompatibility and subcellular localization were assessed. First, RAW 264.7 macrophages were incubated with APNO-5 (1 and 5 μM) for 3, 6, and 24 h. Biocompatibility was assessed for both membrane integrity (trypan blue assay) and cell metabolism (MTT assay). Both assays indicated that APNO-5 was minimally cytotoxic at a concentration of 5 μM, with greater than 79% viability observed after 24 h (Figure S7). Next the localization was determined via colocalization studies with organelle-specific dyes (LysoTracker Green, MitoTracker Green, and ER-Tracker Green) within RAW 264.7 macrophages. On the basis of the overlap between APNO-5 and each of the trackers, it was clear that APNO-5 broadly localizes throughout the cell (except the nucleus) with slight preference for the lysosome and mitochondria (Figure S8).
Noninvasive Photoacoustic Imaging of NO in a Murine Inflammation Model.
As mentioned previously, the noninvasive detection of NO in vivo remains a significant challenge despite the general availability of NO-specific fluorescent probes. To evaluate the utility of APNO-5 for the detection of NO in vivo, we turned to a LPS-induced murine inflammation model. This system was selected because mice show similar inflammatory responses to LPS as humans, making it a useful model for inflammation.46
Mice were treated with either LPS or a saline vehicle control via subcutaneous (s.c.) injection into their flank. Following background scans, the dye was administered in the same location as the treatment. APNO-5 and tAPNO-5 were irradiated at 770 and 680 nm, respectively, and the change in ratiometric signal (PAI680/PAI770) was monitored over time. Following administration, we observed significant pooling of the probe at the injection site. The initial broad absorbance band of APNO-5 in the injection solution resulted in a ratio of ~1.07 at 1 h. Diffusion of the PA signal shortly thereafter (2 h) indicated uptake into the surrounding tissue (Figure 3). At this time, we observed robust conversion of APNO-5 to afford tAPNO-5 in LPS-treated animals, as determined by an increase in the ratiometric PA signal to ~1.2, which remained constant for the remainder of the experiment. In contrast, the PA ratio for the saline-treated mice was ~0.9 and did not change throughout the experiment. The initial decrease in PA ratio for the control animal can be attributed to sharpening of APNO-5’s absorbance profile following uptake and solubilization within the tissue. This was confirmed by monitoring the absorbance profile in the absence and presence of rat liver microsomes, a source of membrane lipids and proteins for association, and the PA spectrum of APNO-5 in a mouse as a function of time (Figure S9). Taken together, this represents a 1.31-fold ratiometric turn on (PAI680/PAI770) and 1.89-fold signal enhancement at 680 nm (Figure 4) after 5 h.
Figure 3.

Representative PA images of APNO-5 responding to endogenous NO within a murine LPS-induced inflammation model. Images of APNO-5 (68 μg/kg) in a mouse flank following subcutaneous LPS or saline treatment. APNO-5 and tAPNO-5 were selectively imaged at 770 and 680 nm, respectively.
Figure 4.

Ratiometric imaging (PAI680/PAI770) of endogenously produced NO within an LPS-induced murine inflammation model over 5 h. Data presented as mean ± standard deviation (n = 8). Statistical analysis between LPS and saline treated mice was performed using 2-way ANOVA (α = 0.05). LPS and saline ratiometric signals were compared at each time point using Sidak’s multiple comparison test (α = 0.05). *, p < 0.05; **, p < 0.01.
To confirm that the signal was a result of APNO-5 nitrosation and not LPS-induced changes in endogenous chromophores (e.g., oxy-hemoglobin, deoxy-hemoglobin), PA spectra were acquired following LPS or saline treatment both in the presence and in the absence of dye. As expected, LPS induces a small increase in ratiometric signal in the absence of dye; however, it does not account for the total signal enhancement (Figure S10). This data, together with our in vitro results, provides compelling evidence that APNO-5 is capable of detecting endogenously generated NO in vivo. We propose that APNO-5 may be effective at even greater depths with the use of general administration (e.g., intravenous) and/or alternative instrumentation, since our specific PA tomographer’s imaging window is limited to 2.5 cm.
CONCLUSION
PA imaging is an emerging imaging technology that has the potential to perform real-time, noninvasive molecular imaging in deep tissue. In this work, we rationally developed a series of NO-responsive probes for PA imaging. Since we installed the most common aerobic NO triggers on a single dye platform, it was possible to systematically compare OPD-, deamination-, and N-nitrosation-based sensing mechanisms. It is clear that the OPD-based trigger is a reasonable approach if triazole formation can be coupled to a robust readout. This is in contrast to some reports implicating cross-reactivity with ascorbic acid and dehydroascorbic acid.47 This suggests that the side reaction is probe dependent, as we did not observe any response to these two carbonyl species, nor with glyoxal. While our work demonstrates that NO-mediated deamination can be expanded, it is limited by the potential formation of metastable intermediates that can complicate interpretation. We found the N-nitrosation trigger to be the optimal of the three triggers examined. When the N-alkyl amine is sufficiently nucleophilic, the reaction with NO is rapid and gives rise to a single product with predictable chemostability.12
The best probe in our series, APNO-5, exceeds all six of the essential criteria for optimal in vivo performance. This performance was evaluated in a well-established murine LPS-induced inflammation model: rapid conversion of APNO-5 to tAPNO-5 was observed exclusively following LPS treatment and not in the saline-treated control. Importantly, since PA imaging is noninvasive, it is possible to monitor a single animal throughout the entire time course of the experiment. This is in contrast to most other imaging modalities, which provide information at only a single time point because the animal must be sacrificed to harvest the tissue for ex vivo imaging.
In conclusion, this work represents one of very few studies that have successfully accomplished in vivo imaging of NO and the first using PA imaging. Compared to previous reports, our strategy does not require the surgical generation of an intravital window and extends the detection of NO to significantly deeper regions of the animal. Owing to the noninvasive nature of PA imaging and excellent performance of APNO-5, we anticipate our work will provide a means to study other NO-mediated biological processes. Current efforts are focused on applying these photoacoustic probes for NO to study the role of NO in tumor progression.
EXPERIMENTAL DETAILS
Materials and Synthetic Procedures.
All materials were purchased from Acros Organic, Airgas, AK Scientific, Alfa Aesar, Amersco, Cambridge Isotope Laboratories, Cayman Chemicals, Click Chemistry Tools, Combi-Blocks, Fischer Scientific, Gold Biotechnology, Life Technologies, Macron Fine Chemicals, McMaster-Carr, Oakwood Chemical, Sigma-Aldrich, or Thermo Scientific and used without further purification. Specific purchasing information can be found in the Supporting Information. Thin-layer chromatography (TLC) was performed on glass-backed TLC plates precoated with silica gel containing an UV254 fluorescent indicator (Macherey-Nagel). Compounds were visualized with a 254/365 nm hand-held UV lamp (UVP). Flash chromatography was performed using 230–400 mesh silica gel P60 (SiliCycle Inc.). Preparative TLC purification was performed on 1.00 mm thick TLC plates precoated with silica gel containing a UV254 fluorescent indicator (Macherey-Nagel). Non-commercially available anhydrous solvents were dried over 3 Å molecular sieves activated via heating under a vacuum at 300 °C. All glassware used in anhydrous reactions was flame-dried under a vacuum and cooled immediately prior to use. Additional detailed synthetic procedures can be found in the Supporting Information.
Benzo[d]oxazol-2(3H)-one (1).
A solution of 2-aminophenol (10.0 g, 92.0 mmol, 1 equiv) and di(1H-imidazol-1-yl)methanone (CDI) (37.0 g, 228.0 mmol, 2.5 equiv) in anhydrous MeCN (250.0 mL) was stirred at 70 °C for 4 h. When judged to be complete by TLC, the reaction was concentrated and suspended in EtOAc. Excess CDI was quenched with 2 M aq HCl, and the organic layer was washed sequentially with a 2 M solution of aq HCl (3×), sat. NaHCO3, and H2O (2×). The organic fraction was dried over Na2SO4 and concentrated to afford the product as an orange solid (10.5 g, 78.0 mmol, 85%), which was used without further purification. Rf = 0.39 (3:7 v/v EtOAc/hexanes). 1H NMR (500 MHz, DMSO-d6) δ 11.56 (bs, 1H), 7.29–7.24 (m, 1H), 7.17–7.11 (m, 1H), 7.10–7.04 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 154.43, 143.35, 130.40, 123.75, 121.80, 109.76, 109.48.
6-Acetylbenzo[d]oxazol-2(3H)-one (2).
The product was synthesized according to previously reported protocols.48 A flame-dried two-neck round-bottom flask was charged with AlCl3 (156 g, 1.16 mol, 10 equiv). The solid was heated to 45 °C under a nitrogen atmosphere and treated with dropwise addition of anhydrous DMF (26.0 mL). Compound 1 (15.7 g, 116.2 mmol, 1 equiv) was added portion-wise to the slurry. Acetyl choride (50.0 mL, 697.0 mmol, 6 equiv) was added dropwise via addition funnel then heated to 75 °C for 2 h. The reaction was slowly poured into ice, and the resultant precipitate was filtered and rinsed with H2O to afford the product as an orange solid (15.6 g, 88.0 mmol, 76%), which was used without further purification. Rf = 023 (1:1 v/v EtOAc/hexanes) 1H NMR (500 MHz, DMSO-d6) δ 12.04 (s, 1H), 7.83, (m, 2H), 7.20–7.17 (m, 1H), 2.56 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 196.30, 154.41, 143.28, 134.81, 131.21, 125.33, 109.33, 109.03, 26.60.
6-Cinnamoylbenzo[d]oxazol-2(3H)-one (3).
A solution of 2 (8.0 g, 45.3 mmol, 1 equiv) and benzaldehyde (4.8 g, 45.3 mmol, 1 equiv) in EtOH (100 mL) was treated with dropwise addition of a 10 M solution of aq KOH (13.6 mL, 136.0 mmol, 3 equiv). The reaction was allowed to stir at room temperature for 4 h. The slurry was concentrated to an orange solid and recrystallized in EtOH to yield the product as a yellow solid (8.3 g, 48.4 mmol, 68%). Rf = 0.48 (2:1:7 v/v acetone/MeOH/hexanes). 1H NMR (400 MHz, DMSO-d6) δ 12.11 (s, 1H), 8.10 (d, J = 1.4 Hz, 1H), 8.06 (dd, J = 8.2, 1.6 Hz, 1H), 7.98 (d, J = 15.6 Hz, 1H), 7.89 (dd, J = 6.7, 2.9 Hz, 2H), 7.73 (d, J = 15.5 Hz, 1H), 7.45 (m, 3H), 7.23 (d, J = 8.1 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 187.30, 154.49, 143.78, 143.50, 134.98, 134.76, 131.85, 130.58, 128.93, 128.91, 125.78, 125.74, 121.77, 109.60, 109.56, 109.54.
tert-Butyl (4-Cinnamoyl-2-hydroxyphenyl) Carbamate (4).
A suspension of 3 (0.9 g, 3.4 mmol, 1 equiv), K2CO3 (1.4 g, 10 mmol, 3 equiv), and di-tert-butyl dicarbonate (1.5 g, 7 mmol, 2 equiv) in MeOH (16 mL) was stirred at 45 °C for 3 h. The reaction was diluted with sat. NaHCO3 and extracted with EtOAc (3×). The combined organic layers were dried over Na2SO4, concentrated, and purified via silica gel column chromatography (3:7 v/v EtOAc/hexanes) to afford the product as a yellow solid (0.8 g, 2.3 mmol, 69%). Rf = 0.44 (3:7 v/v EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 8.51 (bs, 1H), 7.93–7.88 (m, 2H), 7.81 (d, J = 15.6 Hz, 1H), 7.67−7.59 (m, 3H), 7.56 (d, J = 15.7 Hz, 1H), 7.43–7.39 (m, 3H), 7.30 (s, 1H), 1.55 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 189.94, 153.28, 145.71, 145.04, 134.97, 132.88, 132.26, 130.76, 129.09, 128.70, 122.66, 121.71, 118.18, 116.10, 81.77, 28.42.
tert-Butyl (4-Cinnamoyl-2-methoxyphenyl)(methyl) Carbamate (5).
A 38 mL pressure flask under nitrogen was charged with 4 (0.6 g, 1.8 mmol, 1 equiv), anhydrous DMF (8 mL), MeI (1.2 g, 8.8 mmol, 5 equiv), and NaH (60 wt %, 2 g, 5.5 mmol, 3 equiv) at 0 °C. The reaction was allowed to warm to room temperature and was stirred for 1 h. The reaction was then heated to 70 °C for 3 h. The reaction was quenched with water and extracted with EtOAc (3×). The organic layers were combined, concentrated, and purified via silica gel column chromatography (3:17 v/v EtOAc/hexanes) to afford the product as a yellow solid (5.9 g, 1.6 mmol, 91%). Rf = 0.24 (1:9 v/v EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.85 (d, J = 15.7 Hz, 1H), 7.70–7.63 (m, 4H), 7.58 (d, J = 15.6 Hz, 1H), 7.43 (qd, J = 3.8, 1.7 Hz, 3H), 7.32 (d, J = 8.4 Hz, 1H), 3.94 (s, 3H), 3.21 (s, 3H), 1.45 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 189.15, 155.05, 154.77, 144.64, 137.50, 136.91, 134.71, 130.45, 128.84, 128.36, 128.32, 121.65, 121.20, 111.14, 79.86, 55.50, 36.65, 28.13.
1-(3-Methoxy-4-(methylamino)phenyl)-4-nitro-3-phenylbutan-1-one (6).
A solution of 5 (0.53 g, 1.4 mmol, 1 equiv) and nitromethane (0.87 g, 14 mmol, 10 equiv) in EtOH (3 mL) was treated with a 1 M solution of aq KOH (0.29 mL, 0.2 equiv). The reaction was stirred at 60 °C for 7 h. The reaction was diluted with brine, extracted with EtOAc (3×), and concentrated. The crude intermediate was dissolved in CH2Cl2 (8 mL) and cooled to 0 °C in an ice bath. Trifluoroacetic acid (2 mL) was added dropwise, and the reaction was allowed to stir at room temperature for 3 h. The reaction was quenched with sat. NaHCO3 and extracted with CH2Cl2 (3×). The combined organic fractions were dried over Na2SO4, concentrated, and purified via silica gel column chromatography (3:7 v/v EtOAc/hexanes) to afford the product as a brown solid (0.40 g, 1.2 mmol, 86%). Rf = 0.47 (3:7 v/v EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.45 (dd, J = 8.3, 1.9 Hz, 1H), 7.28 (d, J = 1.9 Hz, 1H), 7.27−7.16 (m, 6H), 6.40 (d, J = 8.3 Hz, 1H), 4.82 (s, 1H), 4.78 (dd, J = 12.5, 6.3 Hz, 1H), 4.60 (dd, J = 12.5, 8.5 Hz, 1H), 4.13 (tt, J = 8.3, 6.1 Hz, 1H), 3.79 (s, 3H), 3.30 (dd, J = 16.9, 6.2 Hz, 1H), 3.23 (dd, J = 16.9, 8.0 Hz, 1H), 2.85 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 194.87, 146.29, 144.41, 139.75, 129.11, 127.81, 127.60, 124.86, 124.62, 107.80, 106.78, 79.88, 55.69, 40.82, 40.00, 29.81.
(Z)-2-Methoxy-N-methyl-4-(4-phenyl-5-((3-phenyl-5-(4-(prop-2-yn-1-yloxy)phenyl)-2H-pyrrol-2-ylidene)amino)-1H-pyrrol-2-yl)aniline (7).
A suspension of 6 (0.40 g, 1.2 mmol, 1 equiv) and 35 (0.80 g, 2.5 mmol, 2 equiv) in n-butanol (25 mL) was heated to 110 °C to dissolve all solids. NH4OAc (1.4 g, 19 mmol, 15 equiv) was added in one portion and the reaction stirred at the same temperature for 5 h. The reaction was azeotroped with PhMe and dried to a blue-green solid. The product was extracted from brine with CH2Cl2 (3×). The combined organic fractions were dried over Na2SO4 and purified twice via silica gel column chromatography (3:7 v/v EtOAc/hexanes and then 4:1 CH2Cl2/hexanes) to afford the product as a dark green solid (340 mg, 0.60 mmol, 48%). Rf = 0.50 (3:7 v/v EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 8.07–8.03 (m, 4H), 7.67–7.63 (m, 2H), 7.52–7.48 (m, 2H), 7.43–7.33 (m, 5H), 7.32–7.27 (m, 1H), 7.24 (s, 1H), 6.91 (d, J = 2.0 Hz, 1H), 6.89 (s, 1H), 6.54 (d, J = 8.1 Hz, 1H), 4.66 (d, J = 2.5 Hz, 2H), 3.93 (s, 3H), 2.90 (s, 3H), 2.56 (t, J = 2.4 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 165.10, 157.81, 157.16, 146.83, 146.36, 142.93, 142.52, 141.97, 135.43, 134.75, 133.79, 129.30, 128.78, 128.26, 128.22, 128.12, 128.12, 127.09, 126.44, 125.66, 124.00, 120.73, 119.45, 115.39, 108.94, 108.27, 106.90, 78.46, 75.98, 55.98, 55.30, 29.91.
4-(5,5-Difluoro-1,9-diphenyl-7-(4-(prop-2-yn-1-yloxy)-phenyl)-5H-5l4,6l4-dipyrrolo[1,2-c:2′,1′-f][1,3,5,2]-triazaborinin-3-yl)-2-methoxy-N-methylaniline (8).
A solution of 7 (0.077 g, 0.14 mmol, 1 equiv) and N,N-diisopropylethylamine (0.36 mL, 2.1 mmol, 15 equiv) in anhydrous CH2Cl2 (5.5 mL) was cooled to 0 °C in an ice bath and treated portion-wise with boron trifluoride diethyl etherate (0.18 mL × 3, 4.3 mmol, 45 equiv) over 24 h. After each addition, the reaction was allowed to warm to room temperature. When complete, the reaction was diluted with sat. NaHCO3 and extracted with CH2Cl2 (3×). The combined organic fractions were dried over Na2SO4, concentrated, and purified via silica gel column chromatography (3:7 v/v EtOAc/hexanes) to afford the product as a dark purple solid (0.027 g, 0.045 mmol, 33%). Rf = 0.29 (3:7 v/v EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 8.11–8.03 (m, 7H), 7.96 (s, 1H), 7.78 (dd, J = 8.6, 2.0 Hz, 1H), 7.49–7.39 (m, 6H), 7.36 (t, J = 7.3 Hz, 1H), 7.07–7.02 (m, 2H), 6.93 (s, 1H), 6.64–6.57 (m, 1H), 4.76 (d, J = 2.4 Hz, 2H), 3.94 (s, 3H), 2.97 (s, 3H), 2.57 (q, J = 2.6 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 160.63, 158.78, 152.53, 147.06, 146.35, 143.84, 143.69, 143.52, 139.05, 133.54, 132.43, 131.07, 129.58, 129.44, 129.39, 129.08, 128.81, 128.77, 128.61, 128.55, 128.39, 127.42, 126.40, 120.09, 118.38, 116.69, 115.27, 114.76, 110.96, 108.45, 78.45, 75.96, 55.97, 55.78, 29.85. 11B NMR (161 MHz, CDCl3) δ 1.34 (t, J = 32.8 Hz). 19F NMR (471 MHz, CDCl3) δ –131.51 (dd, J = 66.0, 32.9 Hz).
APNO-5.
Compound 8 (0.046 g, 0.076 mmol, 1 equiv), tris(3-hydroxypropyltriazolylmethyl)amine (0.007 g, 0.015, 0.2 equiv), 39 (0.099 g, 0.25 mmol, 3.5 equiv), and copper sulfate pentahydrate (0.095g, 0.38 mmol, 5 equiv) were dissolved in degassed THF (3.8 mL), and then, sodium ascorbate (0.023 g, 0.11 mmol, 1.5 equiv) in degassed water (0.8 mL) was added under nitrogen. The reaction was allowed to stir at room temperature for 3 h. When the reaction was complete, the product was extracted with 3:7 v/v MeOH/CH2Cl2 (3×) from sat. NaI. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude residue was purified via preparatory TLC (1:9 v/v H2O/MeCN). The product was recovered in 1:9 v/v MeOH/CH2Cl2 with sonication, filtered, and dried to afford the product as a red solid (0.077 g, 0.076 mmol, 100%). Rf = 0.55 (1:9 v/v H2O/MeCN). 1H NMR (500 MHz, DMSO-d6) δ 8.26 (s, 1H), 8.25–8.22 (m, 2H), 8.16 (dd, J = 8.8, 2.0 Hz, 1H), 8.14–8.11 (m, 2H), 8.09 (d, J = 9.0 Hz, 2H), 8.06 (s, 1H), 7.99–7.96 (m, 1H), 7.56 (dd, J = 8.2, 6.6 Hz, 2H), 7.49 (td, J = 7.5, 5.7 Hz, 3H), 7.41–7.35 (m, 1H), 7.27 (s, 1H), 7.23–7.16 (m, 3H), 6.75 (d, J = 8.9 Hz, 1H), 5.27 (s, 2H), 4.57 (t, J = 5.2 Hz, 2H), 3.95 (s, 3H), 3.84 (t, J = 5.2 Hz, 2H), 3.82−3.77 (m, 2H), 3.58–3.51 (m, 5H), 3.50–3.48 (m, 5H), 3.06 (s, 9H), 2.94 (d, J = 5.2 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 159.42, 149.00, 147.30, 146.52, 145.96, 142.83, 142.61, 141.90, 135.72, 133.62, 132.04, 130.88, 130.19, 130.08, 129.55, 129.06, 128.99, 128.87, 128.53, 125.67, 125.49, 116.42, 116.35, 115.10, 109.53, 70.24, 70.04, 70.00, 69.88, 69.80, 69.14, 64.89, 64.49, 61.71, 56.10, 53.60, 53.57, 53.54, 49.93, 29.81. 11B NMR (161 MHz, DMSO-d6) δ 1.33 (t, J = 34.5 Hz). 19F NMR (471 MHz, DMSO-d6) δ–129.98 (dd, J = 67.0, 29.9 Hz). HR-MS calcd [M]+ 902.4258, found 902.4345.
Instruments.
1H, 13C, 11B, and 19F NMR spectra were acquired on Varian 400, Varian 500, or Carver B500 spectrometers. The following abbreviations were used to describe coupling constants: singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), multiplet (m), and broad singlet (bs). Spectra were visualized and analyzed using MestReNova (version 10.0). High-resolution mass spectra were acquired with a Waters Q-TOF Ultima ESI mass spectrometer and a Waters Synapt G2-Si ESI/LC-MS mass spectrometer. Ultraviolet–visible (UV–vis) measurements or spectra were recorded on a Cary 60 spectrometer or SpectraMax M2 plate reader (Molecular Devices). Fluorescence spectra were acquired on a QuantaMaster-400 scanning spectrofluorometer with micro fluorescence quartz cuvettes (Science Outlet). Refractive indices were measured using a RHB-32ATC Brix Refractometer. Cellular imaging was performed on either a EVOS FL epifluorescence microscope or a Zeiss LSM 700 confocal microscope. Photoacoustic imaging was performed using the Endra Nexus 128 photoacoustic tomography system (Ann Arbor, MI, USA), and the data were analyzed using either OsiriX (version 8.0) or Horos (version 3.0) imaging software. All other data analysis was performed using GraphPad Prism (version 6.0). A Mettler Toledo SevenCompact pH meter was used for pH measurements.
Photophysical Characterization.
Extinction coefficients and fluorescence quantum yields were acquired in experimental triplicates. tAPNO was generated by reacting APNO with NO (500 equiv) for greater than 1 h at room temperature (complete conversion was confirmed by UV–vis). Extinction coefficients were acquired by titrating or diluting compound into chloroform or ethanolic 20 mM potassium phosphate buffer (pH 7.4, 50% v/v) within the linear range (typically absorbance values 0.05–1.50). Fluorescence quantum yields were obtained using a modified method for relative fluorescence quantum yield.49 Briefly, samples were prepared and the absorbance and fluorescence were monitored (n = 3). Dye was sequentially titrated or diluted, and the absorbance was kept low (<0.1) to prevent secondary absorbance events. Quantum yields were calculated relative to dimethoxy aza-BODIPY (ϕ = 0.36, chloroform)50 or indocyananine green (ϕ = 0.106, DMSO).51 The refractive index of ethanolic 20 mM potassium phosphate buffer (pH 7.4, 50% v/v) was measured to be 1.3573.
Reaction of APNO with NO.
APNO was dissolved in ethanolic 20 mM potassium phosphate buffer (pH 7.4, 50% v/v). UV–vis and fluorescence measurements were acquired from 400 to 950 nm. The reactions were initiated by the addition of MAHMA-NONOate (prepared in degassed 10 mM aq KOH) at room temperature. The reaction with NO(g) was confirmed by treating APNO (20 μM) in ethanolic water (50% v/v) with NO(g) until a color change was observed. The reaction products for APNO-1, APNO-2, APNO-4, and APNO-5 were confirmed by ESI/LC-MS (Figures S11–S14).
Selectivity Studies.
Unless otherwise noted, APNO was preincubated at 37 °C in an ethanolic 20 mM potassium phosphate buffer (pH 7.4, 50% v/v) for at least 15 min. The initial fluorescence was measured followed by the addition of 100 equiv of various reactive metals, oxygen, nitrogen, and carbonyl species. The reaction was sealed and incubated at 37 °C for 1 h. Final measurements were recorded, and relative turn-on was determined by the sum of intensity over the total range of emission. Due to the rapid oxidation of Fe(II) in phosphate buffer, Fe(II) selectivity studies were performed in ethanolic 20 mM HEPES buffer (pH 7.4, 50% v/v). All metal solutions were prepared from their chloride salt in water with the exception of Fe(II) which was prepared from FeSO4·(H2O)7. Formaldehyde solutions were heated to 100 °C before use to ensure depolymerizing of any paraformaldehyde. Dehydroascorbic acid solutions were prepared by heating a solution at 65 °C in water. Perchlorate, nitrite, and nitrate solutions were prepared for their corresponding sodium salts in water. Superoxide anion was added as a solution of potassium superoxide in DMSO. Nitroxyl was generated in situ from a solution of Angeli’s salt in degassed 10 mM potassium hydroxide solution. NO was generated in situ from a solution of MAHMA-NONOate in degassed 10 mM potassium hydroxide. Hydroxyl radical was prepared using Mohr’s salt in degassed 1 M aqueous hydrochloric acid and hydrogen peroxide. Fe(II), nitroxyl, and hydroxyl radical selectivity assays were performed under nitrogen to prevent oxidation. Peroxynitrite was prepared according to previously reported literature.52 All other analytes were prepared by dilution from commercially available sources.
PA Spectra.
Tissue phantoms were prepared by mixing agarose (4 g) in a solution of 2% reduced fat milk (2 mL) and deionized H2O (78 mL). The mixture was heated in a microwave oven until a viscous gel was produced (30 s); the gel was then mixed and reheated for an additional 15 s to ensure homogeneity. The gel was transferred to a custom Teflon mold made to fit within the Endra bowl system with hypodermic steel tubing inserted to prepare channels for placing FEP tubes (0.06 in. diameter) containing the samples. After cooling the gel for a minimum of 1 h at 4 °C, the phantom was removed from the mold. When imaging, sample solutions (200 μL) were pipetted into FEP tubing (0.08 in. diameter, cut to 10 cm long). The tubing was then inserted into the phantom and sealed by folding over the ends and securing with additional tubing (0.12 in. diameter, cut to 5 mm long). Images were acquired using the Step and Shoot mode with 120 angles and 10 pulses per angle. APNO (10 μM) was dissolved in an ethanolic 20 mM potassium phosphate buffer (pH 7.4, 50% v/v). tAPNO was generated by reacting APNO with NO (2.5 mM MAHMA NONOate, 500 equiv NO) for greater than 1 h at room temperature (complete conversion was confirmed by UV–vis). PA spectra were measured in the same solvent at 10 nm intervals, and signal was integrated over the total range of signal. The mean signal for each FEP tube was averaged for each sample. Fresh samples were prepared for each tAPNO-5 wavelength to limit photoinduced cleavage of the N-nitroso.
Photostability Studies.
APNO-5 or tAPNO-5 solutions (20 μM) were prepared in potassium phosphate saline with CrEL (0.1% v/v). tAPNO-5 was generated according to the same procedure as previously mentioned. PA images were acquired at the wavelength corresponding to the maximal ratiometric PA response using 120 angles and 10 pulses per angle for a total of 60 min. The mean signal was acquired, the average PAI was obtained for the two tubes, and relative signals were calculated according to the initial PA signal.
PA Imaging of the LPS-Induced Murine Inflammation Model.
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of the University of Illinois at Urbana–Champaign, following the principles outlined by the American Physiological Society on research animal use. Six to eight week old BALB/c mice were obtained from Jackson Laboratory. Lipopolysaccharide was dissolved in sterile saline at a concentration of 1 mg/mL. Hair was removed from the flanks using depilatory cream, and then, mice were challenged subcutaneously with either 4 mg/kg of LPS or an equivolumetric amount of saline in the flank. After 3.5 h, mice were anaesthetized using isoflurane, and background PA signals were acquired at 680 and 770 nm. After an additional 0.5 h, a 17 μM solution of APNO-5 in sterile saline containing 0.5% DMF was subcutaneously injected into the same flank at 68 μg/kg (80 μL for 20 g mouse). PA signals were monitored every hour for 5 h in triplicate at 680 and 770 nm. Average PA intensities were acquired over the total volume of interest (1 cm3), and the triplicates were averaged. PA spectra were acquired after 5 h with excitations every 10 nm under the same conditions as previously described, either with or without the administration of APNO-5.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Chemistry-Biology Interface Training Grant (T32 GM070421 to C.J.R. and E.Y.Z.), the National Science Foundation Graduate Research Fellowship Program (NGE-1144245 to E.Y.Z.), the Springborn Fellowship (to E.Y.Z.), the Alfred P. Sloan fellowship (FG-2017-8964 to J.C.), and the Neurotechnology for Memory and Cognition Award (to M.D.J.). Major funding for the 500 MHz Bruker CryoProbe was provided by the Roy J. Carver Charitable Trust (Muscatine, Iowa; Grant No. 15–4521) to the School of Chemical Sciences NMR Lab. The Q-Tof Ultima mass spectrometer was purchased in part with a grant from the National Science Foundation, Division of Biological Infrastructure (DBI-0100085). We acknowledge Professor Elvira de Mejia (Food Science and Human Nutrition, UIUC) for providing RAW 264.7 macrophage cells. We also acknowledge the Core Facilities at the Carl R. Woese Institute for Genomic Biology for access to the Zeiss LSM 700 Confocal Microscope and corresponding software.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b10783.
Experimental details, including detailed synthetic procedures, spectral data, and supplemental in vitro and in vivo characterization (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Zhao Y; Vanhoutte PM; Leung SW S. J. Pharmacol. Sci. 2015, 129, 83. [DOI] [PubMed] [Google Scholar]
- (2).Yun HY; Dawson VL; Dawson TM Mol. Psychiatry 1997, 2, 300. [DOI] [PubMed] [Google Scholar]
- (3).Fukumura D; Kashiwagi S; Jain RK Nat. Rev. Cancer 2006, 6, 521. [DOI] [PubMed] [Google Scholar]
- (4).MacMicking J; Xie QW; Nathan C. Annu. Rev. Immunol. 1997, 15, 323. [DOI] [PubMed] [Google Scholar]
- (5).Bogdan C. Nat. Immunol. 2001, 2, 907. [DOI] [PubMed] [Google Scholar]
- (6).Nathan C; Xie Q. Cell 1994, 78, 915. [DOI] [PubMed] [Google Scholar]
- (7).Dungel P; Penzenstadler C; Ashmwe M; Dumitrescu S; Stoegerer T; Redl H; Bahrami S; Kozlov AV Sci. Rep. 2017, 7, 12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zweier JL; Wang P; Samouilov A; Kuppusamy P. Nat. Med. 1995, 1, 804. [DOI] [PubMed] [Google Scholar]
- (9).Zhou L; Zhu D-Y Nitric Oxide 2009, 20, 223. [DOI] [PubMed] [Google Scholar]
- (10).Vincent SR Prog. Neurobiol. 2010, 90, 246. [DOI] [PubMed] [Google Scholar]
- (11).Floyd RA; Kotake Y; Towner RA; Guo W-X; Nakae D; Konishi YJ Toxicol. Pathol. 2007, 20, 77. [Google Scholar]
- (12).Wang PG; Xian M; Tang X; Wu X; Wen Z; Cai T; Janczuk AJ Chem. Rev. 2002, 102, 1091. [DOI] [PubMed] [Google Scholar]
- (13).Tsikas DJ Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2007, 851, 51. [DOI] [PubMed] [Google Scholar]
- (14).Komarov A; Mattson D; Jones MM; Singh PK; Lai CS Biochem. Biophys. Res. Commun. 1993, 195, 1191. [DOI] [PubMed] [Google Scholar]
- (15).Sharma R; Seo J-W; Kwon SJ Nanomater. 2014, 2014, 523646. [Google Scholar]
- (16).Hong H; Sun J; Cai W. Free Radical Biol. Med. 2009, 47, 684. [DOI] [PubMed] [Google Scholar]
- (17).Shim JH; Lee Y. Anal. Chem. 2009, 81, 8571. [DOI] [PubMed] [Google Scholar]
- (18).Jo A; Do H; Jhon GJ; Suh M; Lee Y. Anal. Chem. 2011, 83, 8314. [DOI] [PubMed] [Google Scholar]
- (19).Li Y; Hu K; Yu Y; Rotenberg SA; Amatore C; Mirkin MV J. Am. Chem. Soc. 2017, 139, 13055. [DOI] [PubMed] [Google Scholar]
- (20).Li H; Wan A. Analyst (Cambridge, U. K.) 2015, 140, 7129. [DOI] [PubMed] [Google Scholar]
- (21).Nagano T; Yoshimura T. Chem. Rev. 2002, 102, 1235. [DOI] [PubMed] [Google Scholar]
- (22).Kashiwagi S; Tsukada K; Xu L; Miyazaki J; Kozin SV; Tyrrell JA; Sessa WC; Gerweck LE; Jain RK; Fukumura D. Nat. Med. 2008, 14, 255. [DOI] [PubMed] [Google Scholar]
- (23).Li H; Zhang D; Gao M; Huang L; Tang L; Li Z; Chen X; Zhang X. Chem. Sci. 2017, 8, 2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Huo Y; Miao J; Han L; Li Y; Li Z; Shi Y; Guo W. Chem. Sci. 2017, 8, 6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sasaki E; Kojima H; Nishimatsu H; Urano Y; Kikuchi K; Hirata Y; Nagano TJ Am. Chem. Soc. 2005, 127, 3684. [DOI] [PubMed] [Google Scholar]
- (26).Dong X; Heo CH; Chen S; Kim HM; Liu Z. Anal. Chem. 2014, 86, 308. [DOI] [PubMed] [Google Scholar]
- (27).Mao Z; Feng W; Li Z; Zeng L; Lv W; Liu Z. Chem. Sci. 2016, 7, 5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mao Z; Jiang H; Li Z; Zhong C; Zhang W; Liu Z. Chem. Sci. 2017, 8, 4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mao Z; Jiang H; Song X; Hu W; Liu Z. Anal. Chem. 2017, 89, 9620. [DOI] [PubMed] [Google Scholar]
- (30).Wang LV; Hu S. Science 2012, 335, 1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li H; Zhang P; Smaga LP; Hoffman RA; Chan JJ Am. Chem. Soc. 2015, 137, 15628. [DOI] [PubMed] [Google Scholar]
- (32).Mishra A; Jiang Y; Roberts S; Ntziachristos V; Westmeyer GG Anal. Chem. 2016, 88, 10785. [DOI] [PubMed] [Google Scholar]
- (33).Roberts S; Seeger M; Jiang Y; Mishra A; Sigmund F; Stelzl A; Lauri A; Symvoulidis P; Rolbieski H; Preller M; Deán Ben XL; Razansky D; Orschmann T; Desbordes S; Vetschera P; Bach T; Ntziachristos V; Westmeyer GG J. Am. Chem. Soc. 2017, DOI: 10.1021/jacs.7b03064. [DOI] [PubMed] [Google Scholar]
- (34).Knox HJ; Hedhli J; Kim TW; Khalili K; Dobrucki LW; Chan J. Nat. Commun. 2017, 8, 1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jiang Y; Upputuri PK; Xie C; Lyu Y; Zhang L; Xiong Q; Pramanik M; Pu K. Nano Lett. 2017, 17, 4964. [DOI] [PubMed] [Google Scholar]
- (36).Kelm M. Biochim. Biophys. Acta, Bioenerg. 1999, 1411, 273. [DOI] [PubMed] [Google Scholar]
- (37).Zhuang Y; Xu Q; Huang F; Gao P; Zhao Z; Lou X; Xia F. ACS Sens. 2016, 1, 572. [Google Scholar]
- (38).Reinhardt CJ; Chan J. Biochemistry 2017, DOI: 10.1021/acs.biochem.7b00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kojima H; Nakatsubo N; Kikuchi K; Kawahara S; Kirino Y; Nagoshi H; Hirata Y; Nagano T. Anal. Chem. 1998, 70, 2446. [DOI] [PubMed] [Google Scholar]
- (40).Shiue TW; Chen YH; Wu CM; Singh G; Chen HY; Hung CH; Liaw WF; Wang YM Inorg. Chem. 2012, 51, 5400. [DOI] [PubMed] [Google Scholar]
- (41).Beltrán A; Burguete MI; Abánades DR; Pérez-Sala, D.; Luis, S. V.; Galindo, F. Chem. Commun. 2014, 50, 3579. [DOI] [PubMed] [Google Scholar]
- (42).Miao J; Huo Y; Lv X; Li Z; Cao H; Shi H; Shi Y; Guo W. Biomaterials 2016, 78, 11. [DOI] [PubMed] [Google Scholar]
- (43).Grossi M; Palma A; McDonnell SO; Hall MJ; Rai DK; Muldoon J; Oshea DF J. Org. Chem. 2012, 77, 9304. [DOI] [PubMed] [Google Scholar]
- (44).Chen Z; Liu Y; Wagner W; Stepanenko V; Ren X; Ogi S; rthner F. Angew. Chem., Int. Ed. 2017, 56, 5729. [DOI] [PubMed] [Google Scholar]
- (45).Tetko IV; Gasteiger J; Todeschini R; Mauri A; Livingstone D; Ertl P; Palyulin VA; Radchenko EV; Zefirov NS; Makarenko AS; Tanchuk VY; Prokopenko VVJ Comput.Aided Mol. Des. 2005, 19, 453. [DOI] [PubMed] [Google Scholar]
- (46).Copeland S; Warren HS; Lowry SF; Calvano SE; Remick D. Clin. Diagn. Lab. Immunol. 2005, 12, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zhang X; Kim WS; Hatcher N; Potgieter K; Moroz LL; Gillette R; Sweedler JV J. Biol. Chem. 2002, 277, 48472. [DOI] [PubMed] [Google Scholar]
- (48).Liacha M; Yous S; Poupaert JH; Depreux P; Aichaoui H. Monatsh. Chem. 1999, 130, 1393. [Google Scholar]
- (49).Wü C; Grabolle M; Pauli J; Spieles M; Resch-Genger U. Nat. Protoc. 2013, 8, 1535. [DOI] [PubMed] [Google Scholar]
- (50).Zhao W; Carreira EM Angew. Chem., Int. Ed. 2005, 44, 1677. [DOI] [PubMed] [Google Scholar]
- (51).Brouwer AM Pure Appl. Chem. 2011, 83, 2213. [Google Scholar]
- (52).Uppu RM Anal. Biochem. 2006, 354, 165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.