

ABSTRACT
Son of Sevenless (SOS), one of guanine nucleotide exchange factors (GEFs), activates Ras. We discovered that the allosteric domain of SOS yields SOS to proceed a previously unrecognized autoactivation kinetics. Its essential feature is a time-dependent acceleration of SOS feedback activation with a reaction initiator or with the priming of active Ras. Thus, this mechanistic autoactivation feature explains the notion, previously only conjectured, of accelerative SOS activation followed by the priming of active Ras, an action produced by another GEF Ras guanyl nucleotide-releasing protein (RasGRP). Intriguingly, the kinetic transition from gradual RasGRP activation to accelerative SOS activation has been interpreted as an analog to digital conversion; however, from the perspective of autoactivation kinetics, it is a process of straightforward RasGRP-mediated SOS autoactivation. From the viewpoint of allosteric protein cooperativity, SOS autoactivation is a unique time-dependent cooperative SOS activation because it enables an active SOS to accelerate activation of other SOS as a function of time. This time-dependent SOS cooperativity does not belong to the classic steady-state protein cooperativity, which depends on ligand concentration. Although its hysteretic or sigmoid-like saturation curvature is a classic hallmark of steady-state protein cooperativity, its hyperbolic saturation figure typically represents protein noncooperativity. We also discovered that SOS autoactivation perturbs the previously predicted hysteresis of SOS activation in a steady state to produce a hyperbolic saturation curve. We interpret this as showing that SOS allostery elicits, through SOS autoactivation, cooperativity uniquely time-dependent but not ligand concentration dependent.
KEYWORDS: Allosteric regulation, cooperativity, Ras, SOS, RasGRP, logistic
Introduction
Ras GTPases modulate various cellular signaling cascades critical to cell survival, differentiation, and growth[1]. Son of Sevenless (SOS) is one of the guanine nucleotide exchange factors (GEFs) of Ras GTPases [2–5]. GEFs activate Ras by enhancing the guanine nucleotide exchange of Ras to produce the active GTP-bound Ras (Ras•GTP); GTPase-activating proteins counteract GEFs by stimulating the intrinsically slow rate of GTP hydrolysis of Ras•GTP to populate the inactive form of Ras (Ras•GDP)[5]. Misregulation of SOS deregulates Ras activity that is often linked to diseases, including Noonan syndrome and hereditary gingival fibromatosis Type 1 [6–12].
Several other Ras GEFs also are known. These include Ras guanyl nucleotide-releasing protein (RasGRP) [2,3,13–16]. However, unlike these other GEFs, SOS is unique because its activity has been shown to be regulated allosterically through its membrane binding interactions [17–19]. SOS also is shown to be autoinhibited, which limits its allosteric activation [17–19].
SOS consists of several domains that function for its catalysis, allosteric regulation, membrane binding interactions, and autoinhibition (Figure 1). Cell division cycle 25 (Cdc25) and Ras exchanger motif (REM) are, respectively, the catalytic and allosteric domains of SOS[19]. The membrane-binding Histone (H) domain in combination with Dbl-homology (DH) and Pleckstrin-homology (PH) domains have been suggested as responsible for the autoinhibition of SOS [17–19]. Similarly, it has also been suggested that the PH domain interacts with phosphatidylinositol 4,5-bisphosphate (PIP2) in a membrane to unlock SOS autoinhibition[19].
Figure 1.
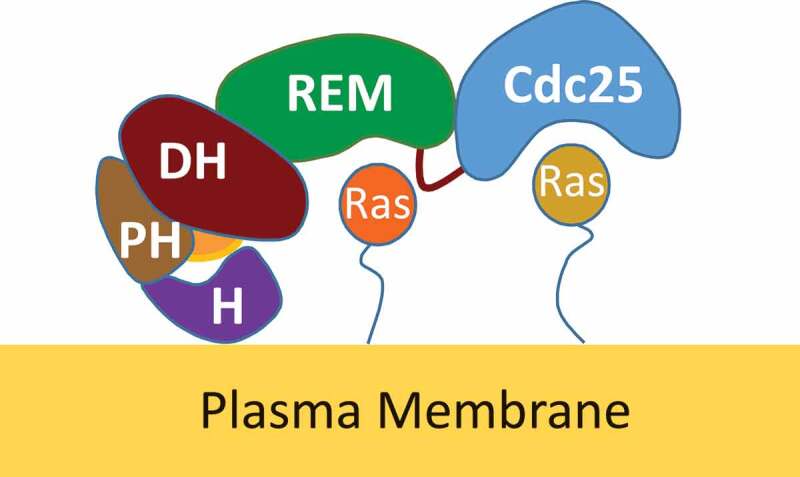
SOS binding interactions with allosteric and substrate Ras proteins. An illustrative cartoon depicts SOS domains from the N- to C-terminus (left to right): H (purple), PH (orange), DH (magenta), REM (green), and Cdc25 (blue) domains. The H domain interacts directly with a membrane that helps SOS to anchor on a membrane. The DH domain functions with the H and PH domains to control access of the allosteric Ras in the REM site. The REM domain that binds the allosteric Ras (red) controls the catalytic function of the catalytic Cdc25 domain that binds the substrate Ras (orange)
Previous structural-based kinetic studies suggested that the allosteric activation of SOS occurs through the action of its unique positive feedback loop. This loop couples with the membrane-binding mediated release of autoinhibition in cells (Figure 2) [20–22]. The action of the previously proposed SOS positive feedback loop [21,22] to activate SOS then seems to operate in such a way that only a fraction of active SOS is necessary to produce active Ras•GTP. This active Ras•GTP then circles back as a ligand to target the allosteric site of other inactive SOS proteins. It also is assumed that the feedback targets of SOS proteins are bound to the PIP2-containing membrane; thus, although they are inactive, they are not autoinhibited. In turn, this targeting of the allosteric site induces SOS conformational changes to produce more of the active SOS proteins (Figure 2). Activation of this positive feedback loop requires priming by active Ras•GTP. The source of this priming Ras is RasGRP [21,22]. Accordingly, of these two GEFs, RasGRP is first activated to produce the priming Ras. The action of the priming Ras with the positive feedback loop then activates the SOS. The unique sequence of this activation of SOS preceded by activation of RasGRP is termed a bimodality of SOS with RasGRP [22,23]. Because of the distinctive pattern of accelerative SOS activation following gradual RasGRP activation[19], this bimodaility feature has been compared with a conversion from an analog RasGRP to a digital SOS response [22,23].
Figure 2.
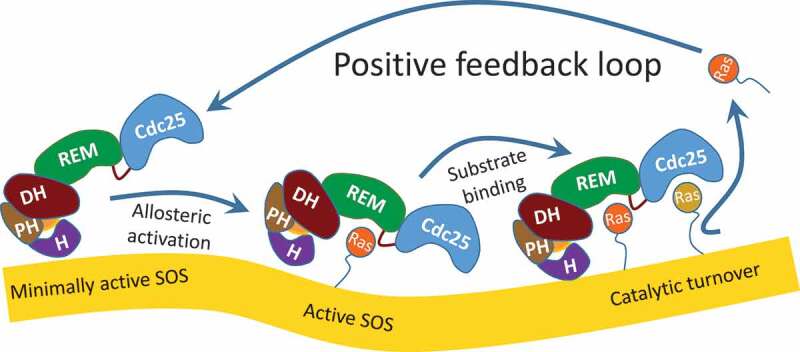
SOS activation by positive feedback loop action. This depicts the sequence of the generic positive feedback loop-mediated SOS activation. The binding of an active Ras on the allosteric REM site of the minimally active allosteric Ras-deficient SOS (left) produces an allosterically activated SOS (middle). The activated SOS then produces active Ras (right), which returns to the allosteric REM site of the minimally active allosteric Ras-deficient SOS (left), thereby activating SOS
A recent fluctuation dynamic study proposed an alternative SOS allosteric activation mechanism without reference to any role of the proposed priming Ras[24]. The study showed that only a few of the SOS molecules on the confined local-membrane region – and none in the bulk assay solution – were allosterically activated in the absence of priming Ras. This study also proposed a unique facet of the distinctive stochastic fluctuations of SOS activity such as a short- and long-lived active SOS molecule. Altogether, the researchers proposed the following single molecular mechanism for allosteric SOS activation without the presence of the priming active Ras[24]: the fluctuation of stochastic activity, but not the average kinetic rate, of the long-lived active SOS is responsible for the allosteric SOS activation within the local confined membrane region[24]. However, this study also was unsuccessful in clarifying the activation mechanism for propagation of the features of allosteric SOS in a small, confined region.
A computational study also predicted a hysteretic or sigmoid-like (S shape) saturation curvature for SOS activation in a steady state[23]. The study also proposed that the predicted hysteresis of steady-state SOS activation was because of the SOS allostery associated with the action of the positive feedback loop[23]. The hysteretic saturation curve typically represents classic protein cooperativity as a function of allosteric ligand concentration [25–28]. The protein cooperativity herein refers to the fact that the protein allosteric ligand binding affects the ligand binding of another protein or to more proteins. Therefore, the predicted positive feedback loop action-induced hysteresis of SOS activation in a steady state suggests a potential classic allosteric SOS cooperativity.
Nevertheless, all of these previous studies of allosteric SOS features have left unanswered several fundamental questions: What is the exact kinetic mechanism of the SOS activation associated with the positive feedback loop action? In the activation of the positive SOS feedback loop, what are the exact mechanistic features associated with priming Ras in the control of SOS activity? How is the mechanical function of the priming Ras in SOS activation associated with the proposed bimodality? Why is SOS activated quicker than RasGRP? Why is the priming Ras required for the allosteric SOS activation only in bulk kinetic conditions[19]. and not in stochastic fluctuation dynamic conditions within a local confined membrane region?[24] Finally, it is unclear whether the occurrence of the predicted steady-state SOS hysteresis [23] accurately accounts for the positive feedback loop action of active Ras•GTP on activation of allosteric SOS. This concern arises because the positive feedback loop action over time produces the ligand active Ras•GTP; thus, the concentration of the ligand active Ras•GTP is changed during activation of the allosteric SOS. This change in the ligand concentration as a function of time is incompatible with the steady-state condition applied to the activation of the allosteric SOS in the computational study. Therefore, the previous model used to predict hysteretic allosteric SOS activation in a steady state through its being coupled with the positive feedback loop fails to accurately describe the features of this activation and how it is associated with the feedback loop.
We discovered a unique kinetic mechanism of SOS activation that explains how the positive feedback loop action activates SOS. Our finding also explains the necessity for the priming active Ras [21,22] for SOS activation and for the previously proposed bimodality of the accelerative digital SOS activation with gradual activation of the analog RasGRP [23,29]. As for the role of the priming Ras in SOS allostery, further analysis of our results also explains the discrepancy between the study result under bulk kinetic conditions [19] and the study result under stochastic fluctuation dynamic conditions within a local confined membrane region[24]. Our discovery also introduces the notion of an unprecedented and uniquely time-dependent SOS cooperativity. An extension of our findings with this new notion also holds clues about the effect of this time-dependent SOS cooperativity on the previously predicted steady-state hysteretic SOS activation. Therefore, our finding calls for a new mechanistic view of the distinctive Ras activity regulated by SOS and of the effects of this activity on biological cell signaling through Ras proteins.
Materials and methods
Protein constructs
Human-origin SOS constructs, SOS possessing the catalytic Cdc25 and allosteric REM domains (SOScat) and the catalytic Cdc25 and allosteric REM domains as well as the membrane-binding H, DH, and PH domains (SOSmemb-cat) were expressed in, and purified from, insect sf9 by using the pIEx vector (Novagen). We have also previously used this sf9 system to purify p120GAP[30]. Human-origin wt and various mutant Ras proteins (Kirsten Ras, 1–181) were expressed in and purified from E. coli. The mutant proteins included combinations of the allosteric site-specific (Y64A [17,18]) and/or uniform lipid vesicle-forming Ras (C118S [19]). One example of these is Y64A/C118S Ras, which binds only to the SOS allosteric site and produces a uniform lipid vesicle-tethered Ras.
Preparation of allosterically activated SOS constructs
In this study, these SOS constructs were often preloaded with the allosteric ligand Y64A Ras (either Y64A Ras•GTP or Y64A Ras•GDP). It binds specifically to the SOS allosteric REM site but not to the catalytic Cdc25 site [17,18]. When necessary, the REM site of these SOS constructs was preloaded with Y64A Ras•GTP to produce partly or fully active SOS. The proportions of the SOS REM site pre-loaded with Y64A Ras•GTP are conveniently expressed as “mole percent (%) activated SOS”: 0% with Y64A Ras•GTP (or the allosteric Ras-deficient) has been shown to be 0% activated SOS; 5% with Y64A Ras•GTP has been shown to be 5% activated SOS; 25% with Y64A Ras•GTP has been shown to be 25% activated SOS; 50% with Y64A Ras•GTP has been shown to be 50% activated SOS; and 100% with Y64A Ras•GTP has been shown to be 100% activated SOS. Note that as discussed in the Threshold of SOS autoactivation in the Results section, the fraction of the partly activated SOS proteins also functions as the reaction initiator for SOS autoactivation (i.e. wt Ras•GTP).
Preparation of the solution and lipid vesicle-tethered ras substrates
Two different substrate Ras figures – Ras in solution and Ras on a lipid vesicle – were used for the SOS catalysis assays. Although the substrate Ras figures differed, the fundamental assay schemes of SOS catalysis were identical. The routine solution substrate Ras preloaded with the fluorescence-tagged GTP or GDP was prepared according to the established method [19,31,32]. The lipid vesicle-tethered Ras preloaded with the fluorescence-tagged GTP or GDP also was prepared by strict adherence, with one exception, to a previously published method (Protocol Exchange (2010) doi:10.1038/nprot.2010.155; Direct coupling of Ras to preformed maleimide-functionalized lipid membranes by Jodi Gureasko, William J. Galush, Holger Sondermann, Jay T. Groves & John Kuriyan; http://www.nature.com/protocolexchange/protocols/439#/procedure). The exception was that we used an Avanti Mini-Extruder with a 0.1 μM membrane to produce lipid vesicles and a column packed with Superose 6 (1.5 cm × 15 cm, GE Healthcare), instead of Sepharose CL-4B, to remove unmodified Ras from Ras-tethered lipid vesicles. Within these analyses, we used one batch of the lipid vesicle-tethered with Ras that lacks PIP2.
Kinetic analyses
Ras, as a substrate and a ligand, respectively, binds to the catalytic Cdc25 and to the allosteric REM site of SOS (Figure 1). Ras exists in two forms: active GTP- and inactive GDP-bound Ras (Ras•GTP and Ras•GDP, respectively). Although slow, the GTP on Ras•GTP is intrinsically hydrolyzed to produce Ras•GDP. Thus, we used the nonhydrolysable GTP analog 5‘-guanylyl imidodiphosphate (GppNHp) instead of GTP. However, to avoid confusion, we labeled GppNHp as GTP throughout this article. Accordingly, two key Ras figures, active Ras•GTP and inactive Ras•GDP, respectively, have been used as the SOS substrates and ligands.
Kinetic assay schemes: SOS catalyzes nucleotide exchange of the Ras-bound GDP or GTP with a fresh solution GTP or GDP. Fluorescence mant-tagged GDP- and GTP-bound Ras (Ras•GDP* and Ras•GTP*, respectively) were used for the SOS kinetic studies. The GDP* or GTP* complex was, respectively, preloaded on Ras to produce Ras•GDP* and Ras•GTP*. Also, the Ras•GDP* and Ras•GTP* complex was either in solution or anchored on the lipid vesicle. For those of the Ras activation and inactivation assays as well as the Ras activity reservation assay (see below) – unless otherwise necessary – the pseudo first-order reaction condition was employed by using an excess of solution GTP or GDP.
Ras activation assay
The assay scheme for the SOS-mediated Ras activation assay is as following: inactive Ras•GDP* + GTP → active Ras•GTP + GDP*. For the SOS-mediated Ras activation assay, SOS was introduced into the assay solution containing the inactive Ras•GDP* complex in solution or with it tethered to the lipid vesicle in the presence of the excess fresh solution GTP. Through the time-dependent SOS-mediated facilitation of the nucleotide exchange of the inactive Ras-bound GDP* with a fresh solution GTP, the inactive Ras-bound GDP* was replaced with the solution GTP to produce an active Ras•GTP and free GDP*. The mant fluorescence intensity of the inactive Ras-bound form (i.e. Ras•GDP*) was higher than that of the released free form (i.e. GDP*). Thus, during the SOS-mediated Ras activation process, the mant fluorescence intensity declined proportionally to that of the consumption of inactive Ras•GDP* to produce active Ras•GTP and GDP*. Accordingly, the time-dependent declination of the mant fluorescence is directly coupled with Ras activation that reflects the catalytic action of SOS.
Ras inactivation assay: The assay scheme for the SOS-mediated Ras inactivation assay is to be: active Ras•GTP* + GDP → inactive Ras•GDP + GTP*. The method of the SOS-mediated Ras inactivation assay was identical to that of the SOS-mediated Ras activation assay, except that the active Ras•GTP* complex in solution or tethered to the lipid vesicle and the fresh solution GDP were used instead of the inactive Ras•GDP* complex and the excess fresh solution GTP. The mant fluorescence intensity of the active Ras-bound form (i.e. Ras•GTP*) exceeded that of the released free form (i.e. GTP*). Thus, as in the case of the SOS-mediated Ras inactivation assay, the mant fluorescence intensity declined during the catalytic action of SOS for the inactivation of Ras. Therefore, as in the case of the Ras activation assay, the time-dependent mant fluorescence declination is proportional to Ras inactivation that reflects the catalytic action of SOS.
Data analyses
Note that the time-dependent declination of the mant fluorescence intensities reflects the displacement of the Ras bound GTP* or GDP* with GDP or GTP by the catalytic action of SOS over time. Therefore, the graphic configuration of Figure 3(c), but not 3B, was immediately applicable to analyze the time-dependent kinetic action of SOS catalysis. Accordingly, the formulas established for graphic Figure 3(c) were used to determine the features of the time-dependent kinetics of the SOS-mediated nucleotide exchange of Ras in both solution and on a lipid vesicle. However, in the case of the titration analysis of the SOS action at Ras concentrations, the graphic configuration of Figure 3(a) and its relevant formulas were applicable to analysis of the ligand concentration-dependent kinetic action of SOS catalysis.
Figure 3.
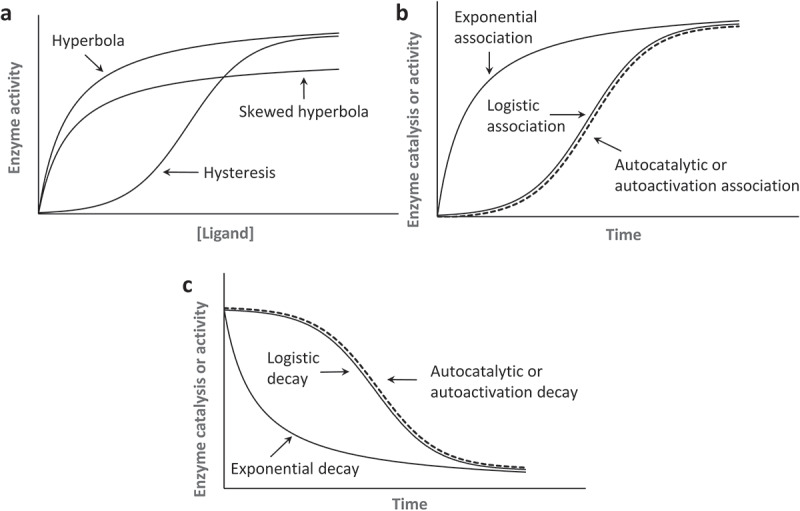
Kinetic curves relevant to enzyme kinetics. (a). Hyperbolic and hysteretic saturation curvatures that are function of a ligand concentration are shown. To show the plot of the hyperbolic saturation curve, we used a one-site equilibrium binding equation of θ = [L]/(Kd + [L]) in which θ can be a fraction of the receptor protein ligand complex or enzyme activity; [L] denotes the ligand concentration; and Kd is a dissociation constant. For the hysteretic saturation curve (positive cooperativity) plot, we used the Hill equation of θ = [L]n/(Kd + [L]n) with a Hill coefficient n of 2. To show the skewed hyperbolic saturation curve (negative cooperativity) plot, we used the Hill equation of θ = [L]n/(Kd + [L]n) with a Hill coefficient n of – 2 . It is notable that when n = 1, the Hill equation essentially becomes a one-site equilibrium binding equation. When enzyme activity is directly linked to the receptor ligand binding interaction, [L] and Kd can be replaced with [S] and Km, respectively. This replacement makes θ an enzyme activity in which [S] and Km denote, respectively, the substrate concentration and a Michaelis-Menten constant. (b). Exponential, logistic, and autocatalytic or autoactivation association curvatures are shown. To illustrate the plot of the exponential and logistic association, respectively, we used the integrated rate law of γ = 1 – e−kt and γ = 1/(1 + e−kt) in which γ can be a fraction of an active enzyme, k equals a first-order rate constant, and t denotes time. For the autocatalytic or autoactivation plot, the integrated rate law of γ = 1/[1 + ([A]i/[A]a)•e−kt] was used in which [A]i and [A]a denote, respectively, the inactive and active enzyme fractions. (c). Exponential, logistic, and autocatalytic or autoactivation decay curvatures that are functions of time also are shown. For the plot of the exponential decay, we used the integrated rate law of γ = e−kt. We used the integrated rate law of γ = 1–1/(1 + e−kt) to plot logistic decay and the integrated rate law of γ = 1–1/[1 + ([A]i/[A]a)•e−kt] to plot autocatalytic or autoactivation decay. Note that the logistic and the autocatalytic or autoactivation associations are essentially the same when the ratio of the parameters [A]i and [A]a (i.e. [A]i/[A]a) is 1. The same is true for the logistic and autocatalytic decay
Each kinetic study was repeated at least three times. Student t-tests for various null hypotheses (H0s) and alternative hypotheses (HAs) with a p < 0.05 were performed with Prism software to examine whether each of the rate constant, ligand concentration required for 50% maximal binding (K0.5), or Hill coefficient values estimated from the triplicate data set differed from the corresponding value from another data set.
Results
Allostery and cooperativity
A broad definition of protein allostery is that an effector ligand controls enzyme activity by binding to the allosteric site of an enzyme [27,28,33–38]. A state in which the allosteric ligand and the enzyme substrate are the same is termed homotropic allostery; when they differ, the allostery feature is described as heterotropic[36].
Ligand concentration-dependent allosteric kinetic features
When enzyme activity is a function of ligand concentration at a fixed time, the ligand concentration is the only function variable for the enzyme activity [27,28,33–37,39]. When the steady state is unperturbed, such as through a feedback loop action (see below), a hyperbolic curve can describe the dependence of the enzyme activity on an allosteric ligand concentration (Figure 3(a)). When this allosteric binding of the ligand with the enzyme shifts the steady state of other enzymes in favor of binding with the allosteric effector ligand to produce more of the active enzymes, the resulting change in the enzyme activity takes on a hysteretic or sigmoid-like (S shape) saturation curvature as a function of the ligand concentration [40] (Figure 3(a)). This is known to be positive cooperativity, and the hysteretic saturation curve is a hallmark of its presence [27,33–37]. Negative cooperativity produces a skewed hyperbolic saturation curve; it reflects that the enzyme has been diverted from favoring binding with the allosteric ligand (Figure 3(a)). Notably, because we consider only homotropic allostery within this study, the ligand concentration is in all cases essentially equivalent to the substrate concentration for enzyme kinetics.
Time-dependent allosteric kinetic features
When the allosteric ligand concentration is not in a steady state with the receptor protein, the classic ligand concentration-dependent protein allosteric kinetic features [27,28,33–37,39] (above) do not apply. Regulation of a feedback loop-mediated enzyme activity is one such case in which ligand production is linked with the enzyme catalysis (Figure 2). Thus, the ligand concentration is not in a steady state but increased or decreased during the enzymatic action. The protein allostery coupled with the enzyme catalyzed-feedback loop is time dependent because the enzyme catalysis that modulates the ligand concentration is a function of time. However, unlike with the classic ligand concentration-dependent allosteric kinetic features (above), the mechanistic features of this time-dependent feedback loop protein allostery and their roles in cell functions have received little acknowledgement or study.
Without the allosteric ligand-binding interaction, enzyme activity is constant. When such constancy is the case – although it could be dependent on the reaction order – the enzyme catalysis proceeds through a time-dependent exponential function (Figure 3(b))[40]. However, when something such as enzyme ligand-binding interactions change the enzyme activity during the enzymatic reaction process, the time-dependent enzyme kinetics become complicated. This complication is because “the enzyme activity“ per se becomes time dependent so that “the change in the enzyme activity” also is a function of time along with “the enzyme catalysis.” The enzyme activity is defined as the quantity of the active enzyme[40]. Accordingly, although the notions of the change in the enzyme activity and the enzyme catalysis are both time dependent, they have fundamental differences: “the time-dependent change in enzyme activity” refers to a change in the “quantity” of active enzyme present over time, but “the enzyme catalysis” denotes the increase in the “rate” of a chemical reaction by an active enzyme as a function of time.
Among the several applicable time-dependent curvatures, a logistic function of time is of special interest because its mechanistic feature is relevant to, and thus reflects, time-dependent allosteric SOS activation. Like the hysteretic saturation curvature, the logistic curvature of time also is sigmoid-like (S shaped). When the logistic function of time is applied to an allosteric enzymatic action, the enzymatic action starts out slow, undergoes an exponential spurt, and then resumes a slower enzymatic action (Figure 3(b))[41]. An exemplary depiction with the trajectory of the time-dependent logistic enzymatic action is as follows (Figure 4): At Time Unit 1, only 1 enzyme is active (a reaction initiator), and it initiates a cumulative (or positive derivative) enzyme activation process by producing 10 turnover products that in turn allosterically activate 10 enzymes; subsequently, at Time Unit 2, the newly activated 10 enzymes produce 100 (10 × 10) turnover products that further allosterically activate 100 enzymes; at Time Unit 3, the newly activated 100 enzymes then produce 1000 (100 × 10) turnover products that allosterically activate 1000 enzymes. This cumulative chain of events in allosteric enzyme activity continues incrementally through the positive feedback mechanism until it encounters boundaries (e.g. encounters the quantity limitations of substrates or enzymes) that limit the accelerative propagation of enzyme activation.
Figure 4.
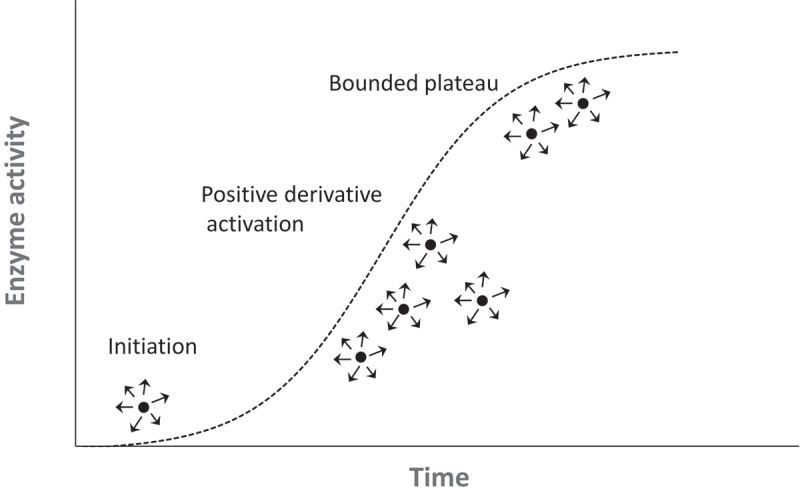
Time-dependent logistic enzyme activation process. This illustrates a time-dependent cumulative enzyme activation. Each small solid circle depicts an enzyme, and arrows with circles indicate the production of enzymatic reaction products. (a) reaction initiator (or reaction trigger) initiates logistic enzyme activation, which then progressively activates other enzymes until the process is bounded when enzyme substrates limit the activation process
Logistic function variations and their applications
As a default in time-dependent logistic enzyme action (see above), one enzyme number in the logistic process is, as denoted as a reaction initiator (see above), always active to spontaneously initiate the cumulative enzyme activation process. However, the time-dependent logistic process of many natural processes does not always start spontaneously to initiate a cumulative chain of events. This is because these natural processes often lack a reaction initiator (or reaction trigger) to initiate the logistic process. To accommodate the logistic natural processes that require a reaction initiator, an autocatalytic function has been established[41], in which the autocatalytic function is an extension of the logistic function of time that includes a reaction initiator unit.
However, enzyme autocatalysis that follows the autocatalytic function describes the unusual “enzyme catalysis”[41] but not “the change in the enzyme activity” as a function of time[40]. As noted elsewhere, the notion of “the enzyme catalysis” differs from that of “the change in the enzyme activity.” Therefore, to correctly describe the change in the enzyme activity that nevertheless follows the autocatalytic function, we have introduced a new term autoactivation. Autoactivation essentially describes a process that follows a logistic function of time with a reaction initiator that is applied to the time-dependent change in enzyme activity but not to enzyme catalysis. All the logistic functions of time as well as autocatalytic and autoactivation functions share the same equation components, yet the autocatalytic and autoactivation functions possess an additional parameter term – the “reaction initiator” – that is expressed as the fraction ratio of the inactive and active enzymes (see Figure 3(b) legend).
The model reactions described above for the exponential as well as the logistic function and its extension functions make a case for the increment of products throughout time (Figure 3(b)). However, when the model reactions are for the consumption of the enzyme substrates, the slopes of these functions are decreased throughout the duration time (Figure 3(c)). However, regardless of the increased product or decreased substrate concentration quantities, they all depict the same time-dependent kinetic or mechanistic phenomena. Nonetheless, the function of time that decreases the enzyme substrate over time (Figure 3(c)) is relevant to the SOS kinetic experiments (see the section on SOS autoactivation).
Time-dependent kinetic analyses of SOS catalysis
To examine the mechanistic features of the positive feedback loop action of the SOS and its link to SOS activation, we performed time-dependent kinetic analyses of SOS activation.
Essentially, two human-origin SOS1-based constructs – SOScat and SOSmemb-cat – have been used in this study. SOScat possesses the SOS catalytic domain Cdc25 and the allosteric domain REM. SOScat is useful for exploring the fundamental allosteric kinetic features of SOS, because SOScat is capable of functioning on the solution Ras as well as on membrane-anchored Ras regardless of the presence of PIP2 in the membrane. SOSmemb-cat contains the Cdc25 and REM domains as well as the H, DH, and PH domains. However, SOSmemb-cat is capable only of functioning on Ras on the PIP2-containing membrane[19]. Therefore, a kinetic study with SOSmemb-cat and its comparison to SOScat is useful to identify the specific role of a membrane with and without PIP2 in the SOS allostery.
SOS autoactivation
To determine the mode of allosteric SOS activation, we performed a Ras activation assay by using 0% activated SOScat with Ras in solution and also with it tethered to the lipid vesicle as described in Materials and Methods.
By accounting for the pseudo first-order kinetic assay conditions, the solution of Ras activation kinetics by SOScat is expected to follow the first-order exponential-decay curvature. However, Figure 5(a) shows that the effect of the time-dependent kinetic features of SOScat on the activation of Ras in solution is not exponential but unexpectedly logistic. Figure 3(c) illustrates the key difference in the kinetic features of the time-dependent logistic and exponential functions. Although the reaction rate of the logistic function is slow at first and then accelerates until bounded or limited, the reaction rate of the exponential decay function is fast at first and then decelerates. The only way to follow the logistic function in enzyme catalysis is to change the enzyme activity per se throughout the assay time period. This subsequent analysis suggests that SOScat is activated as a function of time during the activation of Ras in solution.
Figure 5.
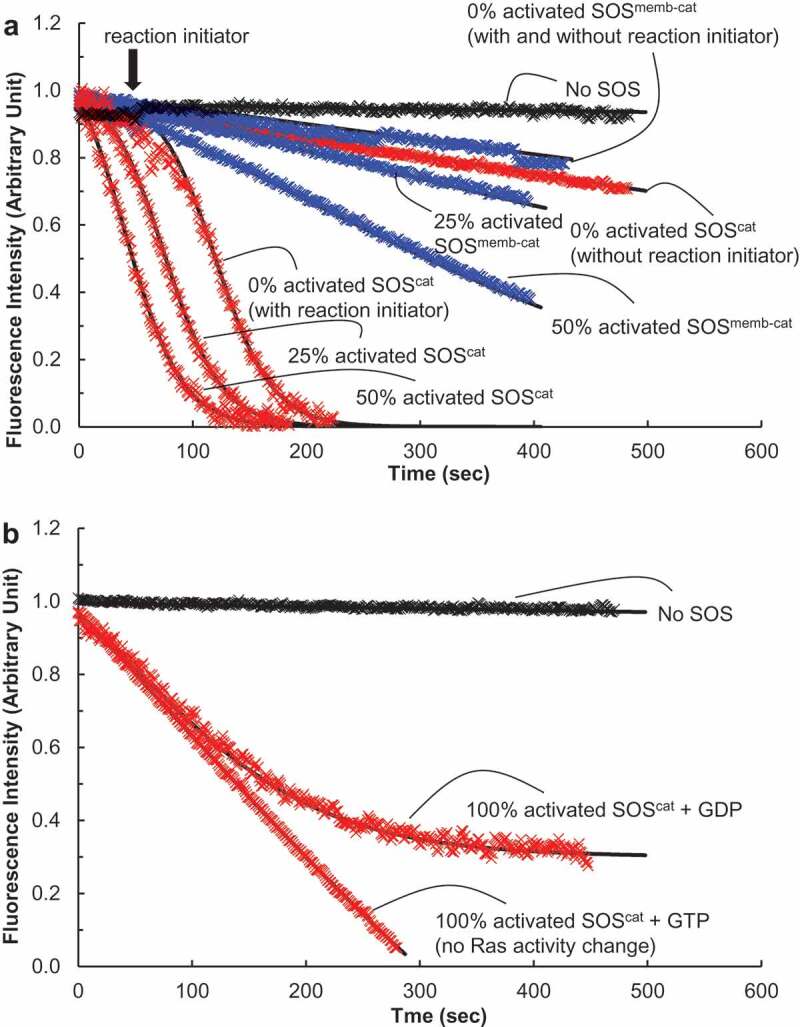
Autoactivation of SOS with solution Ras. We performed fluorescence mant-based Ras activation and inactivation assays on the catalytic activity of SOScat (in red) or SOSmemb-cat (in blue) for the Ras solution. The assay buffer for this analysis consisted of 5 mM MgCl, 10 mM MgCl2, 1 mM EDTA, and 10 mM TrisHCl (pH 7.4). (a). The Ras activation assay was initiated by addition of the various forms of SOScat or SOSmemb-cat (1 μM) to an assay cuvette containing the solution wt Ras that is preloaded with mant fluorescence-tagged GDP* (wt Ras•GDP*, 1 μM) in the presence of excess GTP (1 mM) in an assay buffer. This assay produces wt Ras•GTP (Ras activation). We used a LS 55 Fluorescence Spectrometer (excitation wavelength of 360 nm and emission wavelength of 440 nm) to monitor the change in the mant fluorescence by the SOS-mediated displacement of the wt Ras•GDP* with the abundant free GTP to produce wt Ras•GTP. When necessary, as indicated by an arrow, an autoactivation reaction initiator wt Ras•GTP (0.1 μM, final concentration) was added over the course of the assay with 0% activated SOS. The fraction of the partly activated SOS (e.g. 25% activated SOS, see the Materials and Methods section for its preparation) is equivalent to the reaction initiator wt Ras•GTP for initiation of SOS autoactivation. Therefore, for the assay with the partly activated SOS, we omitted use of the reaction initiator wt Ras•GTP. All the data were fit to Equation (1), and the k values of 0% activated SOScat (without the reaction initiator), 0% activated active SOScat (with the reaction initiator), 25% activated SOScat, and 50% activated SOScat were estimated, respectively, as >1700, 117, 78, and 42 s. The k values of 0% activated SOSmemb-cat (without the reaction initiator), 0% activated SOSmemb-cat (with the reaction initiator), 25% activated SOSmemb-cat, and 50% activated SOSmemb-cat also were estimated, respectively, as >20,000, >20,000, 629, and 346 s. (b). The Ras inactivation assay was initiated by the addition of 100% activated SOScat (1 μM) to an assay buffer. The LS 55 Fluorescence Spectrometer was also used to monitor the change in the mant fluorescence by the SOS-mediated displacement of the solution wt Ras preloaded with mant fluorescence-tagged GTP* (wt Ras•GTP*, 1 μM) with the abundant free GDP (1 mM) in the assay buffer to produce wt Ras•GDP (Ras inactivation). As a control, an abundant free GTP (1 mM) instead of the abundant free GDP (1 mM) was used. The catalytic action of SOS on wt Ras•GTP* with the abundant free GTP that produces wt Ras•GTP does not change Ras activity (both of the substrate wt Ras•GTP* and the product wt Ras•GTP are the same active Ras). Therefore SOS action on this control experimental condition results in Ras activity preservation (no Ras activity change)
Figure 5(a) also shows that the time-dependent logistic kinetics of SOScat on the activation of Ras in solution are triggered when a fraction of wt Ras•GTP is present at the initial reaction mixture. However, the presence of wt Ras•GTP is necessary at the outset; otherwise the time-dependent logistic kinetics of SOScat on activation of the Ras solution does not happen. As introduced in the section on Logistic function variations and their applications, the natural kinetic process that succeeds the time-dependent logistic function often requires a reaction initiator known to be autocatalytic[41]; and an application of the autocatalytic function for the allostery of enzyme action equipped with a reaction initiator is termed autoactivation. Accordingly, the added wt Ras•GTP that initiates SOS activation (Figure 5(a)) is counted as a reaction initiator for SOS autoactivation. SOS that lacks allosteric wt Ras•GTP has minimal SOS activity, but allosteric wt Ras•GTP-bound SOS has full SOS activity [17–19]. Thus, in consideration of the experimental conditions in conjunction with the autoactivation features of the time-dependent logistic curvature (Figure 3(c)) and the necessity of the reaction initiator, we conjectured that SOS is autoactivated as follows: (i) the initial supply of the reaction initiator wt Ras•GTP produces the allosteric wt Ras•GTP-bound active SOS; (ii) this allosterically activated SOS in turn accelerates the production of wt Ras•GTP; (iii) the wt Ras•GTP produced increasingly binds to the remaining unoccupied part of the allosteric site of SOS to generate more and more of the active allosteric wt Ras•GTP-bound SOS; and (iv) the wt Ras•GTP-mediated cycle of activation of SOS continues until all the SOS is fully activated. Taken together, the observed “time-dependent and reaction initiator-mediated acceleration of the catalytic action of SOS” fits into Equation (1), which represents the process of autoactivation (Figure 3(c)).
| 1 |
denotes the fraction of SOS active at given time t, and k is an autoactivation constant. and are, respectively, the fraction concentrations of inactive and active SOS at time 0. The ratio of with determines the lag time of the process.
Figure 6 also shows that, with the reaction initiator wt Ras•GTP, the time-dependent logistic kinetic feature of SOScat also is exhibited during the activation of Ras tethered to the lipid vesicle. However, without the reaction initiator wt Ras•GTP, the time-dependent logistic kinetic feature of SOScat with Ras tethered to the lipid vesicle was not observed. Ras in solution and tethered to the lipid vesicle represent, respectively, Ras in cytoplasm and on a membrane. Therefore, this result in conjunction with SOScat autoactivation with Ras in solution (above) leads to a suggestion that the unique SOS kinetic features of autoactivation are preserved without regard to whether Ras is in cytosol or anchored on the membranes in cells.
Figure 6.
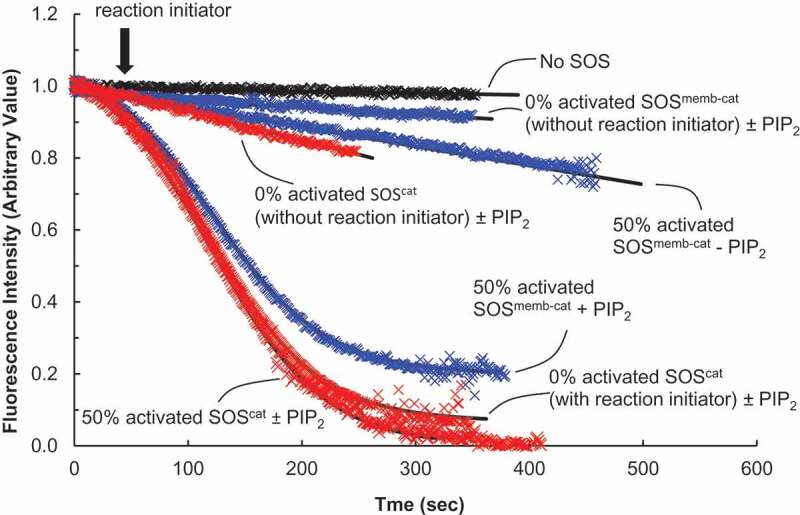
Autoactivation and autoinhibition of SOS with lipid vesicle-tethered Ras. We also performed fluorescence mant-based Ras activation assays of the catalytic activity of SOScat (in red) or SOSmemb-cat (in blue) for the lipid vesicle-tethered Ras. The assay buffer for this analysis was the same as the one used in Figure 5. We initiated the assay by addition of the various forms of SOScat or SOSmemb-cat (1 μM) to an assay cuvette containing the lipid vesicle-tethered wt Ras loaded with mant fluorescence-tagged GDP* (wt Ras•GDP*, ~0.1 × 103 Ras•GDP molecules/μm2) in the presence of excess GTP (1 mM) in an assay buffer. We used a LS 55 Fluorescence Spectrometer to monitor the changes in mant fluorescence intensity caused by the SOS-mediated displacement of the vesicle-tethered wt Ras•GDP* with the abundant free GTP that produces the vesicle-tethered wt Ras•GTP. The data obtained were fit to Equation (1). As with Figure 5, when necessary, and as indicated by an arrow, an autoactivation reaction initiator, wt Ras•GTP (0.1 μM, final concentration), was added during the assay with 0% activated SOS. The autoactivation reaction initiator was not added to the partly activated SOS (e.g. 50% activated SOS, see the Materials and Methods section for its preparation). This is because the partly activated SOS initiates SOS autoactivation without wt Ras•GTP, the reaction initiator. The k values of 0% activated SOScat for the lipid vesicle-tethered wt Ras•GDP* – whether or not PIP2 was present – were determined to be > 1400 s. The k values of 0% activated SOScat with a reaction initiator for the lipid vesicle-tethered wt Ras•GDP* – whether or not PIP2 was present – were determined to be 82 s. The k value of 50% activated SOScat for the lipid vesicle-tethered wt Ras•GDP* – whether or not PIP2 was present – was determined to be 58 s. The k values of 0% activated SOSmemb-cat for the lipid vesicle-tethered wt Ras•GDP* – whether or not PIP2 was present – were determined to be > 1600 s. Finally, the k values of 50% activated SOSmemb-cat for the PIP2-lacking lipid vesicle-tethered and the PIP2-containing lipid vesicle-tethered wt Ras•GDP* were determined, respectively, to be > 1500 and 147 s
The SOS autoactivation we observed as depicted above is unprecedented, yet it explains the mechanistic features of the positive feedback loop action of SOS. And because the cumulative SOS catalytic product facilitates SOS activation throughout the time, this SOS autoactivation in fact delivers the time-dependent SOS cooperativity. However, as introduced earlier, this time-dependent cooperativity is not the same as the classic steady-state protein cooperativity.
Threshold of SOS autoactivation
The necessity of a reaction initiator is one of the key signatures of autoactivation. Discovering the dependency of SOS activation on a reaction initiator also aided us in recognizing this activation occurs through an autoactivation process (see above). In SOS autoactivation, the reaction initiator as denoted as in Equation (1) can be either wt Ras•GTP or the fraction necessary to activate SOS. This is because wt Ras•GTP in turn binds to SOS to produce the partly activated SOS that also triggers SOS autoactivation. SOS autoactivation with Ras in solution and tethered to the PIP2-containing lipid vesicle was consistently triggered by not only the direct addition of the reaction initiator wt Ras•GTP but also by the presence of a certain fraction of activated SOS, such as 50% activated SOScat. Intriguingly, some researchers have speculated that this essential reaction initiator was a priming Ras [21,22]. Notwithstanding their speculation, it is also notable that adding more of the reaction initiator or increasing the quantity of the activated SOS shortens the delay (lag) time necessary for SOS autoactivation to reach its maximum speed (Figure 5(a)). This is because the reaction initiator wt Ras•GTP (i.e. ) is an inverse element of the lag time determination factor in Equation (1). Nevertheless, it is notable that the autoactivation of SOScat with Ras in solution (e.g. 50% activated SOScat in Figure 5) has a shorter lag time than that of SOScat with Ras tethered to the lipid vesicle (e.g. 50% activated SOScat with and without PIP2 in Figure 6). This faster activation occurs because unlike with the concentration of solution Ras used (1 μM), the Ras density tethered to the lipid vesicle (0.1 × 103 Ras•GDP molecules/μm2) for this assay was far less than the optimal density of Ras tethered to the lipid vesicle that we needed for SOS catalysis[19]. Therefore, if one uses a denser lipid vesicle-tethered Ras (> 0.5 × 103 Ras•GDP molecules/μm2), the autoactivation lag time of SOScat with the lipid vesicle-tethered Ras is shorter than that of the result shown. However, this shorter lag time increases the difficulty of tracing the kinetic trail of SOS autoactivation with the denser Ras tethered to the lipid vesicle. Therefore, exclusively for presentation purposes, we used the earlier lower-density Ras tethered to the lipid vesicle for the assay of the SOS-mediated activation of Ras tethered to the lipid vesicle.
The inverse correlation of the quantity of the reaction initiator with the lag time of SOS activation suggests a trigger threshold of SOS autoactivation regardless of whether SOS autoactivation with Ras is in solution or tethered to the lipid vesicle. Determination of this threshold is essential to assessing precisely how SOS activity is regulated in cells. According to Equation (1), in reflects a trigger threshold amount of SOS autoactivation. As noted elsewhere, the reaction initiator can be either wt Ras•GTP or a fraction of the activated SOS. Therefore, the trigger threshold amount of SOS autoactivation should be expressed, depending on experimental conditions as either a mole percentage of wt Ras•GTP or as a fraction of the partly activated SOS (e.g. 25% activated SOS, see Materials and Methods section). We determined the exact threshold amount of reaction initiator necessary to initiate SOS autoactivation by performing several Ras activation assays in which we used SOScat with Ras in solution and tethered to the lipid vesicle in combinations with various concentrations of the reaction initiator wt Ras•GTP in the presence of excess GTP (Figure 7). Our results indicate that triggering SOS autoactivation (less than 60 s lag time) with either Ras in solution or tethered to the lipid vesicle requires >10% and 1% (mole percentage) of Ras, respectively, in the form of wt Ras•GTP in solution or tethered to the lipid vesicle. This difference is not unexpected because SOS activity has been shown to increase up to 500 fold through its binding on the lipid vesicle[19]. Accordingly, the SOS with Ras tethered to a lipid vesicle produces the reaction initiator wt Ras•GTP (i.e. ) faster than the SOS with Ras in solution. The results essentially suggest that the initiation threshold for SOS with the membrane-bound Ras is >10 fold lower than with cytosolic Ras. The results add an unprecedented notion that, in addition to the cell signaling molecules such as PIP2[19], the SOS membrane-binding interaction per se contributes to SOS activation because it lowers the SOS autoactivation initiation threshold and thus aids initiation of autoactivation. The results also imply that – because at least 1% of the reaction initiator wt Ras•GTP is required to initiate SOS autoactivation on a membrane – another Ras GEF, such as RasGRP [21,22], is truly necessary to initiate SOS autoactivation. This implication thus supports the role of RasGRP on Ras activation [21,22].
Figure 7.
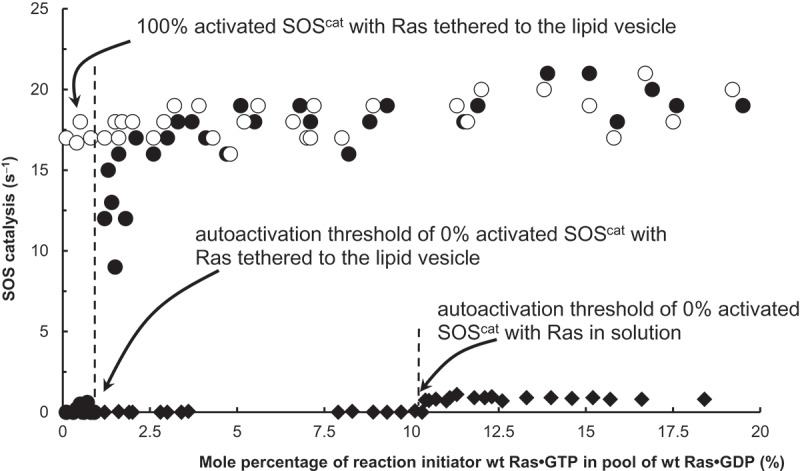
Threshold units of solution and lipid vesicle-tethered Ras for SOS autoactivation. We also performed fluorescence mant-based assays of the catalytic activity of SOScat for Ras in a solution and tethered to the lipid vesicle. The assay was initiated by addition of 0% activated SOScat (1 μM) to an assay cuvette containing the solution wt Ras loaded with mant fluorescence-tagged GDP* (wt Ras•GDP*, 1 μM, final concentration) mixed with various amounts of the reaction initiator (the solution wt Ras•GTP*, 0–20%) in the presence of excess GTP (1 mM) in an assay buffer. A mixture of the lipid vesicle-tethered wt Ras•GDP* (~3.0 × 103 Ras•GDP molecules/μm2, final density) with the reaction initiator (the lipid vesicle-tethered wt Ras•GTP*, 0–20%) also was used for this assay instead of the solution Ras mixture. We used a LS 55 Fluorescence Spectrometer to monitor the mant fluorescence changes by the SOS-mediated exchange of the solution or vesicle-tethered wt Ras•GDP* with the abundant free GTP to produce the solution or vesicle-tethered wt Ras•GTP, respectively. SOScat activities at a reaction time of 60 s were recorded and then plotted against the mole percentage of the reaction initiator wt Ras•GTP in a pool of wt Ras•GDP (%). A fully activated (100% activated) SOScat was used as a control for this assay
Autoinhibition of SOS autoactivation: It has been shown that the H/PH/DH domains autoinhibit SOS activation and that the SOS membrane-binding interaction in the presence of PIP2 thwarts this autoinhibition[19]. To examine the effect of this autoinhibition on SOS autoactivation, we used SOSmemb-cat with Ras tethered to a lipid vesicle to perform a Ras activation assay as described in Materials and Methods. SOSmemb-cat possesses the autoinhibiting H/PH/DH domains in addition to the catalytic Cdc25 and allosteric REM domains.
Figure 6 shows that SOSmemb-cat with a reaction initiator exhibits autoactivation only with Ras tethered to a lipid vesicle that contains PIP2. In comparison, no autoactivation of SOSmemb-cat was observed with Ras in solution (Figure 5(a)). The results suggest that the autoinhibition H/PH/DH domains of SOSmemb-cat prevent SOS autoactivation with both Ras in solution and with Ras tethered to a lipid vesicle. The results also suggest that when SOS docks on a membrane, SOS autoinhibition is only released if the membrane contains PIP2, and thus SOS autoactivation is initiated. Unlike SOSmemb-cat, SOScat lacks these H/PH/DH domains. Thus, SOScat is not autoinhibited. This lack of an autoinhibition feature in SOScat explains why PIP2 is unnecessary in a membrane to initiate autoactivation of SOScat (Figure 6).
Taking these factors into consideration, we postulate that autoactivation of cytosolic SOS is limited in cells. However, when SOS binds to a membrane that contains PIP2 and when a sufficient reaction initiator like wt Ras•GTP is present on this membrane, the combination triggers SOS autoactivation that yields full SOS activity in cells.
SOS autoinactivation: We used SOScat with Ras in solution as described in Materials and Methods to also examine the mode of allosteric SOS inactivation. We used a fully active (100% activated) SOScat for this assay. It was produced by preloading of the GTP*-bound allosteric site-specific Y64A Ras [17,18] on SOScat (see Materials and Methods).
The fully active SOScat kinetics on Ras inactivation follow an exponential function (Figure 5(b)). Because SOScat is initially fully activated in the Ras inactivation assay, further SOS autoactivation coupled with a logistic function of time was not expected. However, the exponential function observed was shown to be to some extent decelerated as a function of time compared with that of the control fully active SOScat kinetics on Ras activity preservation (no Ras activity change, Figure 5(b)). We inferred that this unusual exponential feature of the SOS-mediated Ras inactivation is because the reverse of autoactivation, which we term autoinactivation, occurred in the process of the SOS-mediated Ras inactivation that gradually declines SOS activity as a function of time. The potential mechanism of this gradual decline may be that: (i) the initial catalytic action of the 100% activated SOScat produces wt Ras•GDP; (ii) in turn, this Ras•GDP replaces the allosteric Y64A Ras•GTP of the 100% activated SOScat to produce allosteric wt Ras•GDP-bound SOScat; (iii) this newly produced less active SOS reduces production of wt Ras•GDP, which further delays SOS inactivation as a function of time; and (iv) the delay of SOS inactivation allows SOS to proceed Ras inactivation without significant deceleration. This notion suggests that, although SOS allostery enables SOS autoinactivation, it does not significantly enhance SOS inactivation. In fact, it tends to deter it.
Nonetheless, it is noteworthy that the cellular GTP fraction (GTP/(GTP + GDP)) is ~90%[42]. Therefore, the kinetic conditions necessary to induce SOS autoinactivation are unlikely to occur in vivo. Hence, although this autoinactivation is one of the intriguing kinetic features of SOS, we suspect it has little biological relevance.
Steady-state kinetic analyses of SOS catalysis: To assess any effect of the SOS autoactivation on potential SOS cooperativity under the steady-state conditions, 0% and 5% activated SOScat were titrated with various concentrations of the lipid vesicle-tethered Ras•GDP in the presence of excess GTP (Figure 8). Unlike 0% activated SOScat, 5% activated SOScat in the titration solution enables SOS to be autoactivated, whereas 0% activated active SOScat in the titration solution is incapable of SOS autoactivation. These titration analyses also sought to provide insight into the link between the time-dependent and steady-state cooperativity of SOS.
Figure 8.
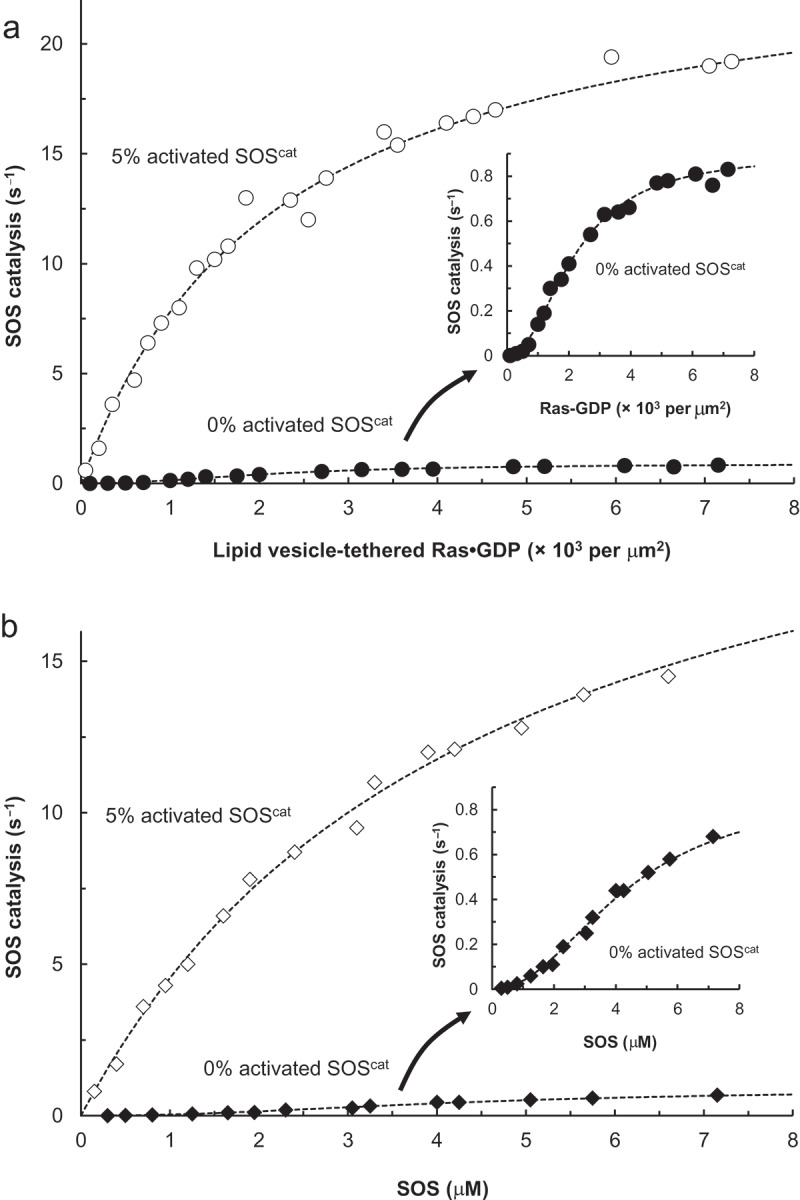
SOS autoactivation-mediated SOS cooperativity. SOScat titration with the lipid vesicle-tethered wt Ras•GDP* and reverse titration were performed in the presence of excess GTP. When necessary, we used 5% activated SOScat instead of 0% activated SOScat. Unlike with 0% activated SOS, the partly activated SOS (e.g. 5% activated SOS, see the Materials and Methods section for its preparation) initiates SOS autoactivation without wt Ras•GTP, the reaction initiator. (a). 0% or 5% activated SOScat (1 μM) was titrated with various densities of the lipid vesicle-tethered Ras•GDP* (0–8.0 × 103 Ras•GDP molecules/μm2) in the presence of excess free GTP (1 mM). The resultant SOS-mediated Ras nucleotide exchange of each titration was monitored as indicated in Figure 5. GraphPad Prism software was used to calculate and plot the apparent kcat values of each titration that reflected Ras•GTP production (s−1) against Ras•GDP (molecules/μm2) and fit them to a hyperbolic saturation or a Hill plot. A hyperbolic saturation curve was used to determine the Km of SOScat for the lipid vesicle-tethered as 2.4 × 103 molecules/μm.2 The Hill coefficient and half-maximal effective substrate concentration (K0.5) values of the hysteretic saturation curvature titration were, respectively, 2.1 and 0.9 × 103 molecules/μm.2 (b). The lipid vesicle-tethered Ras•GDP* (1.0 × 103 Ras•GDP molecules/μm2) was titrated with various amounts of 0% or 5% activated SOScat (0–10 μM) in the presence of excess free GTP (1 mM); the corresponding apparent kcat (s−1) values were calculated as indicated in A and plotted against SOScat (μM). GraphPad Prism software was also used to fit the titration plots to a hyperbolic saturation or a Hill plot. The SOS concentration needed to yield one half of the maximal velocity for the catalysis of the lipid vesicle-tethered Ras•GDP* (1.0 × 103 Ras•GDP molecules/μm2) was determined to be 1.9 μM. The hyperbolic saturation curvature was used for this determination. Intriguingly, the estimated Hill coefficient of the hysteretic saturation curvature titration of B also converges to 2.1. Insets: Expansions of the hysteretic saturation curvature in both A and B are shown
The titration curvature of 0% activated SOScat with the lipid vesicle-tethered Ras•GDP in the presence of excess GTP is hysteretic (Figure 8(a) inset) and almost identical to that of the titration of 0% activated SOScat with the lipid vesicle-tethered Ras•GDP in the presence of excess GDP[19]. The same titration, but with 5% activated SOScat showed an unexpected clear hyperbola (Figure 8(a)). The shapes of the reverse titrations of the lipid vesicle-tethered Ras•GDP and an excess GTP mixture with 0% and 5% activated SOScat also exhibit, respectively, hysteretic and hyperbolic saturation curvatures (Figure 8(b)). In the reverse titrations, the lipid vesicle-tethered Ras•GDP was the receptor and SOScat was a titrant; this reverse titration scheme is the same computational set that predicts steady-state SOScat activation hysteresis[23]. Note that again, although the hysteretic and the hyperbolic saturation curvatures resemble autoactivation and exponential curvatures, both are a function of the ligand (or substrate) concentration, but the autoactivation and exponential curvatures are a function of time. Therefore, the hysteretic and hyperbolic saturation curvatures (Figure 8) describe steady-state kinetics but not time-dependent kinetics.
Note that, although the allosteric Ras•GTP-bound SOS exhibits a full SOS activity, the activity of the allosteric Ras•GDP-bound SOS also is slightly higher than that of the allosteric Ras-deficient SOS (SOS activity: allosteric ligand Ras-deficient SOS (i.e. 0% activated SOS) < allosteric ligand Ras•GDP-bound SOS ≪ allosteric ligand Ras•GTP-bound SOS (i.e. 100% activated SOS) [3,17,19]. Accordingly, we interpreted the result of the hysteretic saturation titration with 0% activated SOScat as demonstrating: (i) because of a lack of a reaction initiator, SOS autoactivation was not initiated, and thus only Ras•GDP, but not Ras•GTP, exists; (ii) the subsequent dominant binding of Ras•GDP on the allosteric site of 0% activated SOScat only slightly increases the SOS activity that exhibits a hysteretic saturation curvature.
The hyperbolic curvature of the titration that appeared during the conditions of producing Ras•GTP with 5% activated SOScat was unexpected, yet intriguing. The best explanation for the hyperbolic feature of titration under conditions of SOS autoactivation is that: (i) 5% activated SOScat initiates SOS autoactivation and then produces more and more Ras•GTP that generates the fully active allosteric Ras•GTP-bound SOScat; and the fully allosterically activated SOS in turn maximizes SOS catalysis at the given substrate concentrations (i.e. the lipid vesicle-tethered Ras•GDP) that exhibit a hyperbola. However, its hyperbolic appearance contradicts the previous prediction of hysteresis in SOScat activation[23].
These results of titration suggest that SOS autoactivation essentially perturbs the steady state between the receptor SOS and its allosteric ligand Ras, resulting in the production of a fully active SOS at each titration point. Thus, the signature of protein cooperativity, its hysteretic saturation curvature, goes unobserved under the titration conditions of SOS. Taken together, although the SOS allosteric feature is committed to the cooperativity of SOS, it is nevertheless unnecessarily dictating the steady-state cooperativity of SOS. Such a notion has not been substantiated in the field of the protein cooperativity.
Discussion
SOS is a multidomain allosteric enzyme that catalyzes a Ras nucleotide exchange, resulting in activation of Ras[19]. We have shown that SOS activation occurs through autoactivation. Both SOS autoactivation and the previously proposed SOS positive feedback loop model [21–23] describe a complex SOS allostery in which SOS activation occurs through the SOS allosteric binding of its own catalytic product (active wt Ras•GTP). However, the mechanistic features of the positive feedback loop of SOS were previously unclear. Notably, the notion of a positive or negative feedback loop that mediates the regulation of protein activity is a convenient and generic way of referring to the circuits of the general cell signaling network [43–51]. It is not a reference to an effect of the specific-time dependent mechanistic ligand-binding interaction on enzyme action. Nor is it a reference to a specific kinetic feature of enzyme action.
In essence, autoactivation, an extended form of the logistic function, explains the previously unsolved mechanistic features of SOS catalysis that included (i) the Ras priming process, (ii) SOS bimodality with RasGRP, and (iii) accelerative SOS activation (changing the analog RasGRP activity to digital SOS activity). These are further explained as follows: (i) The initiation feature of SOS autoactivation requires a reaction initiator such as active Ras (e.g. , Equation (1)), which explains the previously observed priming process of SOS activation [21,22]. (ii) The bimodality of SOS with RasGRP, in which the activation of SOS is succeeded by activation of RasGRP[23], also is explained by the role of RasGRP as the producer of the reaction initiator of active Ras for the initiation of this autoactivation. Therefore, RasGRP activation proceeds earlier than that of SOS activation. (iii) It also is noteworthy that the previously proposed comparison of signal conversion from analog RasGRP activity to digital SOS activity [23,29] reflects the conversion of the typical kinetic activation of RasGRP to the logistic accelerative kinetic activation of SOS autoactivation. In plain autoactivation kinetic terms, this conversion is simply a process of the RasGRP-mediated logistic burst of activation of SOS autoactivation. Nonetheless, this logistic accelerative autoactivation process is obviously an effective and speedy way of activating an enzyme compared with one that relies on a typical first- or second-order kinetic process.
In SOS autoactivation, the reaction initiator as denoted as in Equation (1) can be either wt Ras•GTP or the activated SOS. This is because wt Ras•GTP in turn binds to SOS to produce the activated SOS that also triggers SOS autoactivation. Our result for the threshold number of reaction initiator units necessary for SOS autoactivation provides mechanistic details of the previously proposed role of the priming Ras in the SOS allostery [21,22]. Moreover, it also apparently contradicts the report of the recent fluctuation dynamic study[24]. That study showed that, although no reaction initiator was added, SOS allosteric activation was observed in a small confined-membrane region but not in the bulk assay solution. With the introduction of the stochastic fluctuation dynamics of SOS activity – a short- and a long-lived active SOS – the researchers then proposed a single molecular mechanism for allosteric SOS activation.
In that proposal, a few stochastic long-lived active SOS catalyze allosteric SOS activation on a local small-membrane region without the presence of the reaction initiator. We would interpret this proposed mechanism in view of a conventional mole percentage in conjunction with the results of our threshold number determination: A change of 2 moles in 1,000 moles results in only a 0.2% change. However, a change of 2 moles in 100 moles results in a 2% mole percentage change. Thus, achieving a 2% threshold within a small local area, such as a confined-membrane region, is possible; consequently, strictly speaking, our results do not diverge from those of these earlier researchers. We reason that, in accounting for the reaction of initiator-dependent SOS autoactivation, the proposed few stochastic long-lived active SOS would be equivalent to the reaction initiator ( in Equation (1) or a priming Ras [21,22]). Therefore, we postulate that the activation of the long-lived, active SOS dependent-allosteric SOS [24] also follows the SOS autoactivation process. This postulation opens the possibility that, on a small scale such as in a local, confined small-membrane region[24], SOS autoactivation can be initiated even without RasGRP. Further studies are necessary to determine whether small-scale stochastic SOS autoactivation occurs in cells. If it does, how is it regulated, and how does it differ from the RasGRP-mediated SOS autoactivation process in cell signaling events?
The time-dependent acceleration configuration of SOS autoactivation also suggests an unprecedented perspective on time-dependent SOS cooperativity. This time-dependent cooperativity that autoactivation mediates differs from the previously established classic steady-state-based protein cooperativity [27,33–37]. Yet again, this classic cooperativity is a function of ligand (substrate) concentration, but time-dependent cooperativity is a function of time. As an extension of the autoactivation-mediated time-dependent SOS cooperativity, we have also discovered that the SOS autoactivation delivers the steady-state cooperativity of SOS that shapes the hyperbola. This observation departs from the previous computational view of the steady-state SOS activity hysteresis[23]. Unlike the hysteretic saturation curvature, the hyperbolic saturation figure reflects steady-state binding or kinetics without the presence of protein cooperativity. Our best explanation for this result is that the autoactivation-mediated time-dependent SOS cooperativity perturbs the hysteretic steady-state cooperativity of SOS. This perturbation results in the production of a hyperbolic saturation configuration that reflects full SOS activity at any ligand concentration.
The fundamental kinetic features of this SOS autoactivation are identical to those of the autocatalysis observed in a metaloenzyme carbon monoxide dehydrogenase[52]. Therefore, this SOS autoactivation may not be rare. We anticipate that many positive homotropic regulations [35] follow the kinetics of logistic accelerative autoactivation. Consequently, we expect the application of this work to be another meaningful way in which the use of this unique biochemical and biophysical kinetic approach contributes to our understanding of the cell and molecular biology associated with many uncharacterized positive allosteric homotropic regulations.
Acknowledgments
This work was supported by NIH under grant 1R15AI096146-01A1 to J.H.
Funding Statement
This work was supported by the National Institute of Allergy and Infectious Diseases [1R15AI096146-01A1].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Wennerberg K, Rossman KL, Der CJ.. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. [DOI] [PubMed] [Google Scholar]
- [2].Bonfini L, Karlovich CA, Dasgupta C, et al. The Son of sevenless gene product: a putative activator of Ras. Science. 1992;255:603–606. [DOI] [PubMed] [Google Scholar]
- [3].Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, et al. The structural basis of the activation of Ras by Sos. Nature. 1998;394:337–343. [DOI] [PubMed] [Google Scholar]
- [4].Nimnual A, Bar-Sagi D.. The two hats of SOS. Science‘s STKE. 2002;2002:pe36. [DOI] [PubMed] [Google Scholar]
- [5].Bos JL, Rehmann H, Wittinghofer A.. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. [DOI] [PubMed] [Google Scholar]
- [6].Gelb BD, Tartaglia M. Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Hum Mol Genet. 2006;15(Spec No 2):R220–R226. [DOI] [PubMed] [Google Scholar]
- [7].Roberts AE, Araki T, Swanson KD, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70–74. [DOI] [PubMed] [Google Scholar]
- [8].Hart TC, Zhang Y, Gorry MC, et al. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am J Hum Genet. 2002;70:943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Roberts AE, Allanson JE, Tartaglia M, et al. Noonan syndrome. Lancet. 2013;381:333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Prendiville TW, Gauvreau K, Tworog-Dube E, et al. Cardiovascular disease in Noonan syndrome. Arch Dis Child. 2014;99:629–634. [DOI] [PubMed] [Google Scholar]
- [11].Cordeddu V, Yin JC, Gunnarsson C, et al. Activating mutations affecting the Dbl homology domain of SOS2 yndrome. Hum Mutat. 2015;36:1080–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gelb BD, Roberts AE, Tartaglia M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog pediatr cardiol. 2015;39:13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Arnaout AH, Dawson PM, Soomro S, et al. HER2 (c-erbB-2) oncoprotein expression in colorectal adenocarcinoma: an immunohistological study using three different antibodies. J Clin Pathol. 1992;45:726–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ahmed AE, Abdel-Aziz AH, Abdel-Rahman SZ, et al. Pulmonary toxicity of acrylonitrile: covalent interaction and effect on replicative and unscheduled DNA synthesis in the lung. Toxicology. 1992;76:1–14. [DOI] [PubMed] [Google Scholar]
- [15].Ebinu JO, Bottorff DA, Chan EY, et al. RasGRP, a Ras guanyl nucleotide- releasing protein with calcium- and diacylglycerol-binding motifs. Science. 1998;280:1082–1086. [DOI] [PubMed] [Google Scholar]
- [16].Bottorff D, Ebinu J, Stone JC. RasGRP, a Ras activator: mouse and human cDNA sequences and chromosomal positions. Mamm Genome. 1999;10:358–361. [DOI] [PubMed] [Google Scholar]
- [17].Margarit SM, Sondermann H, Hall BE, et al. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. [DOI] [PubMed] [Google Scholar]
- [18].Sondermann H, Soisson SM, Boykevisch S, et al. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119:393–405. [DOI] [PubMed] [Google Scholar]
- [19].Gureasko J, Galush WJ, Boykevisch S, et al. Membrane-dependent signal integration by the Ras activator Son of sevenless. Nat Struct Mol Biol. 2008;15:452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Boykevisch S, Zhao C, Sondermann H, et al. Regulation of ras signaling dynamics by Sos-mediated positive feedback. Curr Biol. 2006;16:2173–2179. [DOI] [PubMed] [Google Scholar]
- [21].Roose JP, Mollenauer M, Ho M, et al. Unusual interplay of two types of Ras activators, RasGRP and SOS, establishes sensitive and robust Ras activation in lymphocytes. Mol Cell Biol. 2007;27:2732–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jun JE, Rubio I, Roose JP. Regulation of ras exchange factors and cellular localization of ras activation by lipid messengers in T cells. Front Immunol. 2013;4:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Das J, Ho M, Zikherman J, et al. Digital signaling and hysteresis characterize ras activation in lymphoid cells. Cell. 2009;136:337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Iversen L, Tu HL, Lin WC, et al. Molecular kinetics. Ras activation by SOS: allosteric regulation by altered fluctuation dynamics. Science. 2014;345:50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kurganov BI, Dorozhko AK, Kagan ZS, et al. The theoretical analysis of kinetic behaviour of “hysteretic” allosteric enzymes. I. The kinetic manifestations of slow conformational change of an oligomeric enzyme in the Monod, Wyman and Changeux model. J Theor Biol. 1976;60:247–269. [DOI] [PubMed] [Google Scholar]
- [26].Frieden C. Slow transitions and hysteretic behavior in enzymes. Annu Rev Biochem. 1979;48:471–489. [DOI] [PubMed] [Google Scholar]
- [27].Neet KE. Cooperativity in enzyme function: equilibrium and kinetic aspects. Methods Enzymol. 1980;64:139–192. [DOI] [PubMed] [Google Scholar]
- [28].Qian H. Cooperativity and specificity in enzyme kinetics: a single-molecule time-based perspective. Biophys J. 2008;95:10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chakraborty AK, Das J, Zikherman J, et al. Molecular origin and functional consequences of digital signaling and hysteresis during Ras activation in lymphocytes. Sci Signal. 2009;2:pt2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wey M, Lee J, Kim HS, et al. Kinetic mechanism of formation of hyperactive embryonic Ras in cells. Biochemistry. 2016;55:543–559. [DOI] [PubMed] [Google Scholar]
- [31].Lenzen C, Cool RH, Prinz H, et al. Kinetic analysis by fluorescence of the interaction between Ras and the catalytic domain of the guanine nucleotide exchange factor Cdc25Mm. Biochemistry. 1998;37:7420–7430. [DOI] [PubMed] [Google Scholar]
- [32].Heo J, Prutzman KC, Mocanu V, et al. Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J Mol Biol. 2005;346:1423–1440. [DOI] [PubMed] [Google Scholar]
- [33].Pauling L. The oxygen equilibrium of hemoglobin and its structural interpretation. Proc Natl Acad Sci U S A. 1935;21:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. [DOI] [PubMed] [Google Scholar]
- [35].Koshland DE Jr., Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. [DOI] [PubMed] [Google Scholar]
- [36].Kuo LC. Allosteric cofactor-mediated enzyme cooperativity: a theoretical treatment. Proc Natl Acad Sci U S A. 1983;80:5243–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ge H, Qian M. Theoretical analysis of the relationship between positive/negative cooperativity and enzyme activation/inhibition. Interdiscip Sci. 2009;1:204–213. [DOI] [PubMed] [Google Scholar]
- [38].Qian H, Shi PZ. Fluctuating enzyme and its biological functions: positive cooperativity without multiple states. J Phys Chem A. 2009;113:2225–2230. [DOI] [PubMed] [Google Scholar]
- [39].Wyman J. The turning wheel: a study in steady states. Proc Natl Acad Sci U S A. 1975;72:3983–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Segel IH. Enzyme kinetics. New York: A Wiley-Interscience publication; 1993. [Google Scholar]
- [41].Aa F, Rg P. Kinetics and mechanism. New York: Wiley; 1953. [Google Scholar]
- [42].Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. [DOI] [PubMed] [Google Scholar]
- [43].McAdams HH, Arkin A. Stochastic mechanisms in gene expression. Proc Natl Acad Sci U S A. 1997;94:814–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–387. [DOI] [PubMed] [Google Scholar]
- [45].Barkai N, Leibler S. Circadian clocks limited by noise. Nature. 2000;403:267–268. [DOI] [PubMed] [Google Scholar]
- [46].Kholodenko BN, Kiyatkin A, Bruggeman FJ, et al. Untangling the wires: a strategy to trace functional interactions in signaling and gene networks. Proc Natl Acad Sci U S A. 2002;99:12841–12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ferrell JE Jr. Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr Opin Cell Biol. 2002;14:140–148. [DOI] [PubMed] [Google Scholar]
- [48].Kussell E, Kishony R, Balaban NQ, et al. Bacterial persistence: a model of survival in changing environments. Genetics. 2005;169:1807–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bashor CJ, Helman NC, Yan S, et al. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science. 2008;319:1539–1543. [DOI] [PubMed] [Google Scholar]
- [50].Harding AS, Hancock JF. Using plasma membrane nanoclusters to build better signaling circuits. Trends Cell Biol. 2008;18:364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kholodenko BN, Hancock JF, Kolch W. Signalling ballet in space and time. Nat Rev Mol Cell Biol. 2010;11:414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Heo J, Halbleib CM, Ludden PW. Redox-dependent activation of CO dehydrogenase from Rhodospirillum rubrum. Proc Natl Acad Sci U S A. 2001;98:7690–7693. [DOI] [PMC free article] [PubMed] [Google Scholar]