

ABSTRACT
During development of the brain, neuronal circuits are formed through the projection of axons and dendrites in response to guidance signals. Rho GTPases (Rac1/RhoA/Cdc42) are major regulators of axo-dendritic outgrowth and guidance due to their role in controlling actin cytoskeletal dynamics, cell adhesion and motility. Functional redundancy of Rho GTPase-regulated pathways in neuronal development can mask the roles of specific GTPases. To examine potential Rho GTPase redundancy, we utilized a recently isolated hypomorphic mutation in a Caenorhabditis elegans Rac1 protein – CED-10(G30E) – which reduces the GTP binding and inhibits axon outgrowth of the PVQ interneurons. Here, we show that the CDC-42-specific guanine nucleotide exchange factor UIG-1 acts in parallel to CED-10/Rac1 to control PVQ axon outgrowth. UIG-1 performs this function in a cell-autonomous manner. Further, we found that transgenic expression of CDC-42 can compensate for aberrant CED-10(G30E)-regulated signalling during PVQ axon outgrowth. Together, our study reveals a previously unappreciated function for CDC-42 in PVQ axon outgrowth in C. elegans.
KEYWORDS: Axon outgrowth, Rho GTPases, Rac, CDC-42, guanine nucleotide exchange factor
Introduction
Rho GTPase proteins (Rho, Rac and Cdc42) act as molecular switches to control diverse neurodevelopmental events [1,2]. During brain development, appropriate projection of axons and dendrites is essential for the formation of neuronal circuits. Inappropriate extension of these projections during brain development is linked with several neurological disorders including autism spectrum disorders [3]. The correct coordination of actin filament networks at the growth cones of extending axons provides mechanical support to enable movement [4]. Therefore, as key regulators of actin cytoskeletal dynamics, Rho GTPases plays a prominent role in axon outgrowth and guidance [1,2]. Rho GTPases regulate the protrusive behaviour of axons through multiple interweaved signalling networks to dynamically remodel the actin cytoskeleton within growth cones. This enables the forward progression of axons through the extension and retraction of filopodial and lamellipodial structures[4]. Rho GTPases act as molecular switches during actin cytoskeletal remodelling to precisely control these regulatory networks. The activity of Rho GTPases is dependent on their guanine-binding status [5]. Extracellular and intracellular signalling control Rho GTPase switching between active GTP-bound and inactive GDP-bound states. These signals regulate specific Rho GTPase-regulatory molecules: guanine nucleotide exchange factors (GEFs), which exchange bound GDP for GTP, thereby activating Rho GTPases; and GTPase-activating proteins (GAPs) that inactivate Rho GTPases by enhancing intrinsic GTPase activity [5,6].
A previous study from our lab showed that in the Caenorhabditis elegans Rac1 homologue CED-10, an amino acid substitution in the highly conserved Switch 1 region confers a non-redundant function in axon outgrowth but not guidance [7]. The ced-10(rp100) hypomorphic mutation, which causes a glycine to glutamic acid (G30E) substitution, reduces CED-10 GTP binding and inhibition of axon outgrowth autonomously in the PVQ neurons [7]. Unlike other ced-10 hypomorphic alleles, rp100 does not cause defects in fecundity and apoptotic corpse engulfment pointing to a potential tissue- and/or pathway-specific effect of this mutation [7]. We also used the ced-10(rp100) mutation to confirm that CED-10 and MIG-2/RhoG act in parallel to control axon outgrowth [7]. Therefore, the ced-10(rp100) mutation may possibly be used as a tool to identify other key players in Rho GTPase signalling in the control of axon outgrowth.
In this work, we investigated whether UIG-1, a CDC-42-specific guanine nucleotide exchange factor [8], acts to control axon outgrowth of the PVQ interneurons in C. elegans. CDC-42 is a well-known small GTPase that involved in axon guidance, cell polarity and cell migration [9–11]. UIG-1 has previously been shown to regulate apoptotic corpse engulfment and muscle formation; however, a role in neuronal development has yet to be reported [8,12,13]. We find that in a wild type background UIG-1 is not required for PVQ development. However, using ced-10(rp100) mutant animals as a sensitized background, we found that loss of UIG-1 causes an almost 100% increase in penetrance of axon outgrowth defects. This suggests that UIG-1, and its regulation of CDC-42, functions in parallel to CED-10/Rac1 to control PVQ axon outgrowth. We further show that UIG-1 is expressed in multiple tissues and neurons, including the PVQs, and functions cell-autonomously in the PVQ neurons to control axon outgrowth. Finally, we found that CDC-42 can promote PVQ axon outgrowth as CDC-42 transgenic expression is able to suppress the PVQ outgrowth defects caused by the ced-10(rp100) mutation. Together our data reveal a previously unappreciated role for UIG-1 and CDC-42 in controlling PVQ axonal development.
Results and discussion
The UIG-1/CDC-42-specific GEF controls PVQ axon outgrowth, but not guidance, in parallel to CED-10/Rac1
We study the bilaterally symmetric PVQ interneurons (PVQL and PVQR) in C. elegans to identify molecules that control axon outgrowth and guidance. In wild-type animals, the embryonically born PVQ neurons are located in the posterior and extend axons in an anterior direction into the ipsilateral side of the ventral nerve cord (VNC) (Figure 1(a)). We visualize the PVQ neurons by driving fluorescent proteins under the control of the sra-6 or npr-11 promoters (Figures 1 and 2). Previously, we showed that the Rac GTPases CED-10 and MIG-2 act in parallel to control PVQ axon outgrowth and guidance [7]. This analysis revealed that the GEF UNC-73/Trio is a major upstream activator of these Rac GTPases during PVQ axon outgrowth [7]. However, the defects observed in unc-73 mutant animals are not fully penetrant, which suggested to us that additional Rho GTPases may also function to control PVQ outgrowth. We therefore examined whether CDC-42 signalling contributes to PVQ outgrowth.
Figure 1.
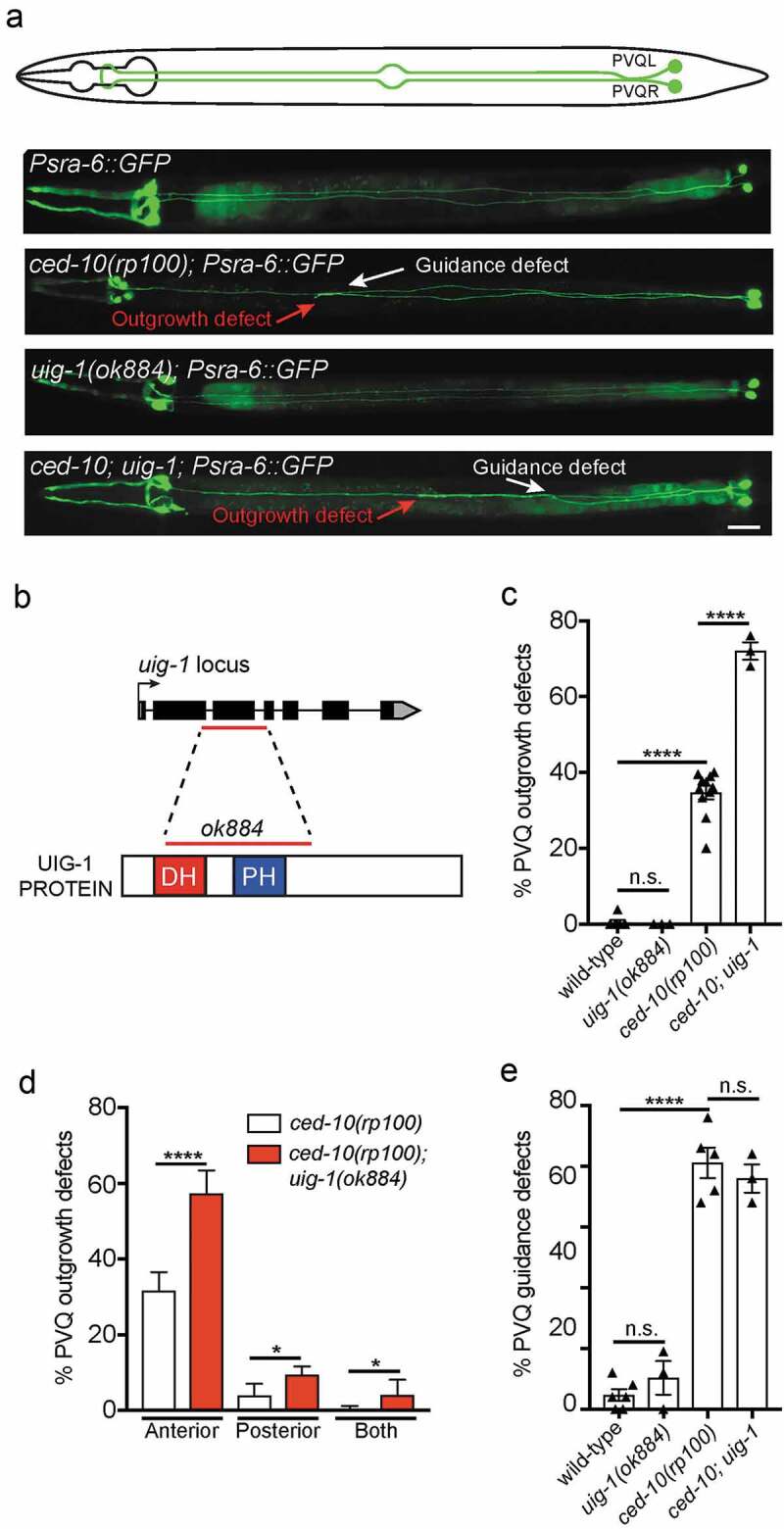
UIG-1/CDC42 GEF acts in parallel to CED-10/Rac1 to control PVQ axon outgrowth
(a) Ventral views of animals expressing the transgene oyIs14(Psra-6::gfp) to visualize the anatomy of the PVQ neurons (PVQL and PVQR). In wild-type adult animals (schematic and upper fluorescent image), the PVQ axons extend axons from the posterior into the ipsilateral side of the VNC. Representative examples of ced-10(rp100), uig-1(ok884) and ced-10(rp100); uig-1(ok884) mutant animals are shown. In ced-10(rp100) and ced-10(rp100); uig-1(ok884) mutant animals, PVQ axons exhibit premature termination of outgrowth (red arrow) and inappropriate axonal guidance (white arrow). Posterior to the right. Scale bar 20 μm. (b) Genetic and protein domain structures of UIG-1 showing that the ok884 lesion (red line) deletes both the DBL homology (DH) and pleckstrin homology (PH) domains. (c) ced-10(rp100) mutant animals exhibit PVQ outgrowth defects (~35% penetrance). Loss of UIG-1 enhances these defects to ~70%. Data are expressed as mean ±S.E.M. and statistical significance was assessed using t-test. ****<0.0001, comparing wild type and ced-10(rp100) animals, n.s. not statistically significant. Each triangle represents independent scoring replicates, n > 25 in each scoring replicate. (d) Phenotypic classification of PVQ outgrowth defects in ced-10(rp100) and ced-10(rp100); uig-1(ok884) mutant animals. Loss of UIG-1 enhances the penetrance and expressivity of PVQ outgrowth defects (red bars), with more animals having stopped axons in the posterior and with both axons affected. ****<0.0001, *<0.05. n > 25 in each scoring replicate. (e) ced-10(rp100) mutant animals exhibit PVQ guidance defects (~60% penetrance). Loss of UIG-1 does not enhance the penetrance of guidance defects. ****<0.0001, n.s. not statistically significant. Each triangle represents independent scoring replicates, n > 25 in each scoring replicate.
Figure 2.
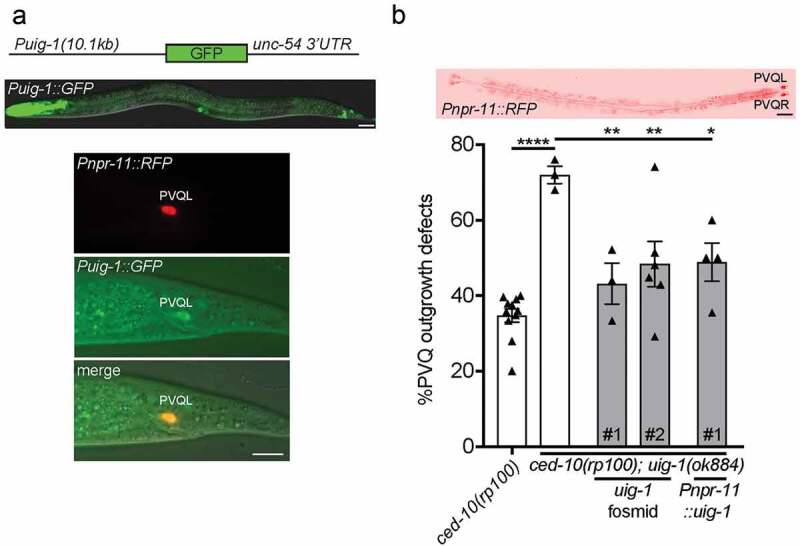
UIG-1/CDC42 GEF acts cell-autonomously to control PVQ axon outgrowth
(a) A 10.1kb uig-1 promoter drives GFP (Puig-1::GFP) expression in the pharynx, vulva, hindgut and neurons in the head and tail (upper image). Expression of Puig-1::GFP in the PVQ neurons is confirmed by co-localization with the PVQ-specific Pnpr-11::RFP reporter. Posterior to the right. Scale bars 20 μm. (b) Transgenic expression of a fosmid housing the uig-1 locus (WRM0623bF03) or PVQ-specific driven expression of uig-1 cDNA rescues the PVQ outgrowth defects of ced-10(rp100); uig-1(ok884) animals. ****<0.0001, comparing ced-10(rp100) and ced-10(rp100); uig-1(ok884) animals; **<0.01, *<0.05 comparing the transgenic rescue lines to ced-10(rp100); uig-1(ok884) animals. Each triangle represents independent scoring replicates, n > 25 in each scoring replicate. # refers to independent transgenic lines. Top image confirms PVQ-specific expression driven by the npr-11 promoter. Posterior to the right. Scale bar 20 μm.
Previous in vitro experiments showed that the C. elegans GEF UIG-1 specifically activates CDC-42, and has no effect on the GTPase activity of C. elegans RHO-1, CED-10, MIG-2 or mammalian RhoA and Rac1 [8]. Therefore, to examine the potential function for CDC-42 signalling in PVQ outgrowth, we analysed UIG-1, a GEF that exhibits specific activity for CDC-42 in both C. elegans and mammals [8]. We used the uig-1(ok884) deletion mutant which deletes a DNA region that encodes the Dbl-homology (DH) and Pleckstrin homology(PH) domains, which is required for GEF activity (Figure 1) [8]. Therefore, uig-1(ok884) is a likely null allele, however is viable and therefore enables an analysis of PVQ development in C. elegans larvae. In a wild type background, we found that UIG-1 has no detectable role in PVQ development (Figure 1(a,c–e)). As previously reported, we found that ced-10(rp100) mutants exhibit ~35% penetrance in PVQ outgrowth defects (Figure 1) [7]. When we generated a ced-10(rp100); uig-1(ok884) compound mutant, we discovered that the penetrance of PVQ outgrowth defects increased from ~35% to ~70% (Figure 1(c)). This enhancement was also observed when comparing the penetrance of PVQ outgrowth defects in ced-10(n3246) (5%) to ced-10(n3246); uig-1(ok884) (17%) animals (n > 75, *<0.05). We next asked whether the severity of PVQ axon outgrowth defects of ced-10(rp100) mutant animals is enhanced by the uig-1(ok884) mutation by examining the location of the prematurely terminated PVQ axons and whether one of both PVQ axons were terminated (Figure 1(d)). We found that loss of UIG-1 caused an increase in the number of axons terminating prior to the vulva at the mid-body (Figure 1(d)). In addition, we observed an enhancement of the number of animals in which both axons terminated prematurely (Figure 1(d)). We next assessed whether UIG-1 also plays a role in PVQ axon guidance in parallel to CED-10 (Figure 1(e)). We found that uig-1(ok884) mutant animals have similar background PVQ guidance defects to wild type and loss of UIG-1 does not enhance the PVQ guidance defects of ced-10(rp100) mutant animals (Figure 1(e)). We attempted to examine potential redundant functions between UIG-1 and UNC-73/GEF or MIG-2/RhoG; however, these compound mutants resulted in lethality, precluding analysis (data not shown). Together, our data reveal that UIG-1, a CDC-42-specific GEF, function in parallel to CED-10 to control PVQ axon outgrowth.
UIG-1 acts cell-autonomously to control PVQ axon outgrowth
Rho GTPases are important controllers of intracellular growth cone dynamics. Therefore, direct regulators of these molecular switches, such as UIG-1, likely control axon outgrowth cell-autonomously. To investigate this, we examined the expression pattern of uig-1 by driving green fluorescent protein (GFP) under the control of a 10.1kb uig-1 promoter (Figure 2(a)). We found that the uig-1 promoter drives expression in the pharynx, vulva, hindgut and a subset of neurons in the head and tail of C. elegans (Figure 2(a)). To examine whether uig-1 is expressed in the PVQ neurons, we crossed the Puig-1::GFP reporter strain into a PVQ-specific red fluorescent protein (RFP) reporter (Pnpr-11::RFP) strain. We found that Puig-1::GFP is indeed expressed, albeit weakly, in the PVQ neurons (Figure 2(a)).
Next, we performed rescue experiments to confirm the function of uig-1 in regulating PVQ axon outgrowth. First, we expressed a uig-1-containing fosmid in the ced-10(rp100); uig-1(ok884) double mutant and found that PVQ axon outgrowth defects were restored to levels not significantly different from the ced-10(rp100) single mutant (Figure 2(b)). This confirms that loss of uig-1 is responsible for the phenotypic enhancement we observe in PVQ outgrowth defects. To examine the potential cell-autonomous function for uig-1, we used the PVQ-specific npr-11 promoter to drive uig-1 cDNA in ced-10(rp100); uig-1(ok884) mutant animals (Figure 2(b)). We found that the cell-specific expression of uig-1 also suppressed PVQ outgrowth defects to that of the ced-10(rp100) single mutant (Figure 2(b)). These data show that UIG-1 acts cell-autonomously to control PVQ axon outgrowth.
CDC-42 transgenic expression can compensate for defective CED-10 signalling during PVQ axon outgrowth
As UIG-1 is a GEF that specifically functions to activate CDC-42 in C. elegans and mammals [8], we examined the role of CDC-42 in PVQ axon outgrowth (Figure 3). We analysed the cdc-42(gk388) deletion allele, which removes upstream regulatory sequences, the first exon and part of the first intron [12]. cdc-42(gk388) homozygous mutants are sterile, therefore we used a balancer chromosome to maintain gk388 in a heterozygous state [12]. We scored homozygous cdc-42(gk388) animals derived from heterozygous mothers and found that PVQ axon outgrowth was normal (Figure 3(a)). Next, we generated a ced-10(rp100); cdc-42(gk388) balanced double mutant to ask whether the reduction of CDC-42 enhances PVQ axon outgrowth defects of the ced-10(rp100) mutant. We found that ced-10(rp100); cdc-42(gk388) homozygous mutant animals derived from mothers that were heterozygous for cdc-42(gk388) did not enhance the PVQ axon outgrowth defects of ced-10(rp100) animals (Figure 3(a)). The lack of PVQ axon outgrowth enhancement in the ced-10(rp100); cdc-42(gk388) double mutant could be because of maternal contribution of wild-type cdc-42 mRNA to the embryo from heterozygous mothers. To circumvent this issue, we generated mosaic animals in which a genetically unstable cdc-42-rescuing fosmid, or a PCR product of the cdc-42 genomic region, was supplied to cdc-42(gk388) homozygous mothers (Figure 3(b)). Here, we analysed PVQ axon outgrowth of ced-10(rp100); cdc-42(gk388) and ced-10(rp100) animals in which cdc-42 expression was either absent or transgenically expressed specifically in the PVQ neurons (Figure 3(b)). We found that transgenic expression of cdc-42 in the PVQs of either ced-10(rp100); cdc-42(gk388) or ced-10(rp100) animals suppresses PVQ axon outgrowth defects (Figure 3(b)). The fact that we did not observe the enhancement of PVQ axon outgrowth defects in mosaic animals that lack the cdc-42 rescuing transgene in the PVQ neurons could be due to maternal contribution from transgenic mothers. Our data therefore suggest that CDC-42 can compensate for defective CED-10/Rac1 signalling during PVQ development.
Figure 3.
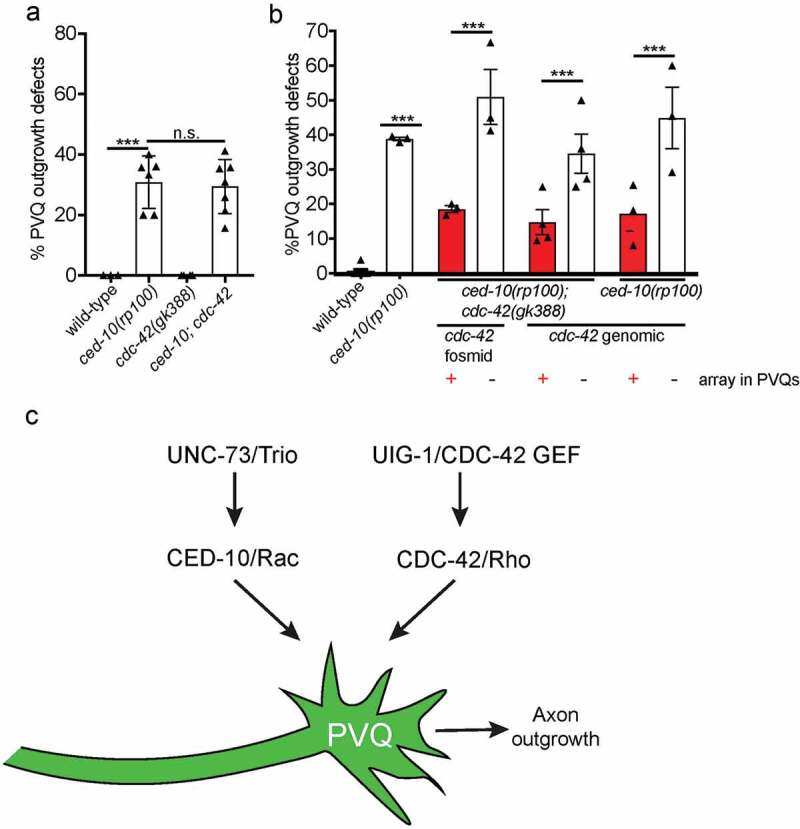
Transgenic expression of CDC-42 can compensate for defective CED-10/Rac1 signalling
(a) cdc-42(gk388) null mutant animals (from balanced heterozygous mothers) have no detectable PVQ outgrowth defects and do not enhance outgrowth defects of ced-10(rp100) mutant animals. Data are expressed as mean ±S.E.M. and statistical significance was assessed using t-test. ****<0.0001, comparing wild-type and ced-10(rp100) animals, n.s. not statistically significant. Each triangle represents independent scoring replicates, n > 25 in each scoring replicate. (b) Unstable transgenic arrays containing either cdc-42-containing fosmid (WRM0620aE11) or a cdc-42 genomic PCR product, Psra-6::mCherry and Pelt-2::gfp were established in ced-10(rp100); cdc-42(gk388); oyIs14 or ced-10(rp100); oyIs14 animals. PVQ outgrowth was scored in animals in which the array was present in the PVQs (Psra-6::mCherry, Pelt-2::gfp positive) or missing from the PVQs (Psra-6::mCherry negative, Pelt-2::gfp positive). Presence of cdc-42-containing arrays suppress the PVQ outgrowth defects of ced-10(rp100); cdc-42(gk388) and ced-10(rp100) mutant animals. Each triangle represents independent scoring replicates, n > 20 in each scoring replicate. (c) CED-10/Rac functions in parallel CDC-42/Rho to control PVQ axon outgrowth. UNC-73/Trio and UIG-1/CDC-42 GEF act upstream of these Rho GTPases in a positive manner.
This study reveals that UIG-1, a CDC-42-specific GEF, acts in parallel to CED-10/Rac1 to cell-autonomously control axon outgrowth of the PVQ interneurons in C. elegans (Figure 3(c)). We further showed that that CED-10/Rac1 and CDC-42 functionally compensate for each other during PVQ axon outgrowth. This posits that the expression of these prominent regulators of cytoskeletal remodelling may be modified during development and after axon injury to enable faithful axon outgrowth. As Rac1 and CDC-42 widespread regulators of cell behaviour, such functional compensation may also occur in other cellular and organismal contexts.
Materials and methods
C. elegans strains and genetics
All C. elegans strains were maintained at 20°C on NGM plates seeded with Escherichia coli OP50 bacteria, unless otherwise stated [14]. Strains were generated using standard genetic procedures and are available on request. All strains were backcrossed to N2 at least three times before scoring or generating compound mutants. Genotypes were confirmed using PCR genotyping to detect genetic lesions.
The following mutant strains and transgenic reporters were used in this study (in order of appearance in Figures): Figure 1: rpEx1624[Psra-6::GFP], ced-10(rp100); oyIs14[Psra-6::GFP], uig-1(ok884); rpEx1624[Psra-6::GFP], ced-10(rp100); uig-1(ok884); rpEx1624[Psra-6::GFP]. Figure 2: kyIs321[Pnpr-11::RFP]; rpEx1778[Puig-1::GFP], ced-10(rp100); uig-1(ok884); rpEx1624[Psra-6::GFP]; rpEx1784[uig-1 fosmid line 1], ced-10(rp100); uig-1(ok884); rpEx1624[Psra-6::GFP]; rpEx1788[uig-1 fosmid line 2], ced-10(rp100); uig-1(ok884); rpEx1624[Psra-6::GFP]; rpEx1785[Pnpr-11::uig-1 line 1]. Figure 3: cdc-42(gk388)/mIn1[mIs14 dpy-10(e128)]; oyIs14[Psra-6::GFP], ced-10(rp100); cdc-42(gk388)/mIn1[mIs14 dpy-10(e128)]; oyIs14[Psra-6::GFP], cdc-42(gk388); ced-10(rp100); oyIs14; rpEx1786[cdc-42 fosmid line 1], cdc-42 (gk388); ced-10(rp100); oyIs14; rpEx1787[cdc-42 genomic line 1], ced-10(rp100); oyIs14; rpEx1789[cdc-42 genomic line 1].
Neuroanatomical observation and scoring
Neurons were scored, blinded to genotype, in L4/young adult stages, unless otherwise stated. The Psra-6::GFP transgene was used to enable visualization of the PVQ neurons. All neuronal scoring was repeated at least in triplicate on independent days, n = 75, unless otherwise stated.
C. elegans plasmid generation
Puig-1::gfp
A 10,112bp uig-1 promoter was amplified from fosmid number WRM0623bF03 and inserted using restriction-free cloning into pPD95.75 GFP vector digested with HindIII and BamHI using the oligos:
5ʹ-GAAATGAAATAAGCTTTCTGAACCTTAATGACCTTTCTTG-3ʹ and
5ʹ-CCGGGGATCCTCTAGAGGCTTCGAGTGGATACGATTG-3ʹ.
Pnpr-11::uig-1 cDNA
uig-1 cDNA was amplified from an oligodT amplified cDNA library using the oligos:
5ʹ-aaaagctagcATGTCTTCGGTAACAACCG-3ʹ and
5ʹ- ttttccatggCTAAGCATTCTCGTATTGTCG-3ʹ. The uig-1 cDNA was ligated into a Pnpr-11-containing plasmid with a pPD49.26 backbone using NheI and NcoI. The npr-11 promoter was a kind gift from Cori Bargmann.
Fluorescence microscopy
Animals were mounted on 5% agarose pads and immobilized using 50mM NaN3. Examination and imaging of neurons were performed using an automated fluorescence microscope (Zeiss, AXIO Imager M2) and ZEN software (version 3.1).
Microinjections and transgenic animals
Transgenic animals were generated as previously described [15]. Rescue plasmids were injected directly into mutant strains. The WRM0623bF03 fosmid, spanning the entire uig-1 locus, was injected at 1ng/μl. Puig-1::GFP was injected at 50ng/μl. For cell-autonomous rescue, Pnpr-11::uig-1 cDNA was injected at 0.2ng/μl. Pmyo-2::mCherry was injected at 5ng/μl as a co-injection marker in all transgenic rescue animals.
Mosaic analysis
For mosaic analysis, transgenic animals were generated by injecting the cdc-42 fosmid (WRM0620aE11) at 5ng/μl (or for cdc-42 genomic rescue, 0.1ng/μl of a cdc-42 genomic PCR), Psra-6::mCherry at 30ng/μl and Pelt-2::gfp at 10ng/μl into the ced-10(rp100); cdc-42(gk388)/mIn1; oyIs14 strain. A transgenic line was selected that exhibited rescue of the PVQ axon outgrowth defects. Transgenic animals from this line were then scored for the phenotypic rescue of the PVQ defects in the presence and absence of the rescuing extrachromosomal array in the PVQ neurons by detection of Pmyo-2::mCherry fluorescence.
Statistical analysis
Statistical analysis was carried out using Graphpad Prism 7 using one-way ANOVA with Sidak’s Multiple Comparison Test. Values are expressed as mean ±SEM Values <0.05 were considered statistically significant.
Acknowledgments
We thank members of Pocock laboratory and Steffen Nørgaard for comments on the manuscript. Some strains used in this study were provided by the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). We thank Cori Bargmann for the kind gift of the npr-11 promoter. This work was supported by funding from a Monash Biomedicine Discovery Fellowship, NHMRC Project Grant (GNT1105374), NHMRC Senior Research Fellowship (GNT1137645), and a Victorian Endowment for Science, Knowledge and Innovation Fellowship (VIF23) to R.P.
Funding Statement
This work was supported by the National Health and Medical Research Council [GNT1105374]; National Health and Medical Research Council [GNT1137645]; Veski [VIF23].
Author contributions
W.C. and R.P. designed the research; W.C. and S.D. performed the research; W.C. and S.D. and R.P. analysed data, and R.P. wrote the paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. [DOI] [PubMed] [Google Scholar]
- [2].Govek EE, Newey SE, Van Aelst L.. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. [DOI] [PubMed] [Google Scholar]
- [3].Wegiel J, Kuchna I, Nowicki K, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010;119:755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. [DOI] [PubMed] [Google Scholar]
- [5].Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. [DOI] [PubMed] [Google Scholar]
- [6].Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. [DOI] [PubMed] [Google Scholar]
- [7].Norgaard S, Deng S, Cao W, et al. Distinct CED-10/Rac1 domains confer context-specific functions in development. PLoS Genet. 2018;14:e1007670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hikita T, Qadota H, Tsuboi D, et al. Identification of a novel Cdc42 GEF that is localized to the PAT-3-mediated adhesive structure. Biochem Biophys Res Commun. 2005;335:139–145. [DOI] [PubMed] [Google Scholar]
- [9].Lucanic M, Cheng HJ. A RAC/CDC-42-independent GIT/PIX/PAK signaling pathway mediates cell migration in C. elegans. PLoS Genet. 2008;4:e1000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Demarco RS, Struckhoff EC, Lundquist EA. The Rac GTP exchange factor TIAM-1 acts with CDC-42 and the guidance receptor UNC-40/DCC in neuronal protrusion and axon guidance. PLoS Genet. 2012;8:e1002665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gotta M, Abraham MC, Ahringer J. CDC-42 controls early cell polarity and spindle orientation in C. elegans. Curr Biol. 2001;11:482–488. [DOI] [PubMed] [Google Scholar]
- [12].Hsieh HH, Hsu TY, Jiang HS, et al. Integrin alpha PAT-2/CDC-42 signaling is required for muscle-mediated clearance of apoptotic cells in Caenorhabditis elegans. PLoS Genet. 2012;8:e1002663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Neukomm LJ, Zeng S, Frei AP, et al. Small GTPase CDC-42 promotes apoptotic cell corpse clearance in response to PAT-2 and CED-1 in C. elegans. Cell Death Differ. 2014;21:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mello CC, Kramer JM, Stinchcomb D, et al. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. Embo J. 1991;10:3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]