

ABSTRACT
Endoplasmic reticulum stress (ERS)-mediated autophagy is indispensable for modulation of replication and pathogenesis of numerous mammalian viruses. We have previously shown that classical swine fever virus (CSFV) infection induces ERS-mediated autophagy for maintaining viral replication both in vivo and in vitro, however, the underlying mechanism remains unclarified. Here we found that CSFV infection activates the PERK pathway-dependent complete autophagy to promote viral replication in cultured PK-15 and 3D4/2 cells. Likewise, our results also suggested the essential roles of the IRE1/GRP78-mediated complete autophagy in CSFV replication in vitro. Furthermore, we suggested that CSFV infection induces activation of the PERK and IRE1 pathway for potential immunoregulation via promoting transcription of proinflammatory cytokine (IFN-γ and TNF-α) genes in the CSFV-infected cells. Finally, pharmacological treatment of PERK- or IRE1-pathway regulators, and the corresponding SiRNAs interventions did not affect the viabilities of the cells, excluding the potential interference elicited by altered cell viabilities. Taken together, our results suggest that CSFV infection induces complete autophagy through activation of the PERK and IRE1 pathway to facilitate viral replication in cultured cells, and modulation of proinflammatory cytokines may be a potential mechanism involved in this event. Our findings will open new horizons for molecular mechanisms of sustainable replication and pathogenesis of CSFV, and lay a theoretical foundation for the development of ERS-autophagy-targeting therapeutic strategies for clinical control of CSF.
KEYWORDS: Autophagy, classical swine fever virus, endoplasmic reticulum stress, IRE1, PERK, proinflammatory cytokines, virus replication
Introduction
The endoplasmic reticulum (ER), an important membranous organelle in eukaryotic cells, is responsible for biosynthesis, folding, and maturation of a large number of secretory and membrane proteins [1,2]. Disturbance of ER homeostasis by several factors, such as microbial infections, imbalance of calcium, accumulation of unfolded/misfolded proteins, can eventually lead to the endoplasmic reticulum stress (ERS), and consequently triggers an adaptive mechanism called the unfolded protein response (UPR) to reinstate normal ER function [3,4]. The UPR is relayed through three ER transmembrane sensors including protein kinase R-like ER kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor-6 (ATF-6). These sensors were generally bound to and sequestered by a crucial ER chaperone glucose-regulated protein 78 (GRP78/HSPA5/BiP hereafter referred to as GRP78). Upon ERS, GRP78 dissociates from and activates the sensors, and then the UPR cascades are initiated to re-establish the ER homeostasis through attenuating global protein translation, enhancing ER functions, and activating the ER-associated protein degradation (ERAD) or autophagy to eliminate the misfolded/unfolded proteins [3,5]. Nevertheless, during prolonged or excess ERS, the UPR pathways are insufficient to restore ER homeostasis, and eventually result in apoptotic cell death [6–8].
Macroautophagy (hereafter called autophagy) typically functions as a pro-survival mechanism of cells and is responsible for lysosomal degradation and recycling of cytoplasmic components [9,10]. During the autophagic process, cytosolic cargoes including the damaged organelles and the unfolded/misfolded proteins are targeted and sequestered into double-membrane vesicles (called autophagosomes), which further fuse with the lysosomes to form autophagolysosomes for degradation of the wrapped contents [11,12]. Although autophagy generally acts significant roles in restoring normal cellular functions from adverse conditions [13], in other cases, the excessive or uncontrolled autophagic processes can cause cell death [14,15]. Evidences have revealed that the dysregulated autophagy is involved in many human cancers, aging, and neurodegenerative diseases [16–18], showing great potential and significance of the research.
Although ERS and autophagy are different biological processes in cells, both of them have been shown to modulate cellular function and fate (survival or death), and the crosstalk between ERS and autophagy has been extensively studied [19,20]. Accumulating evidence has suggested the induction of autophagy through activation of the Ca2+-dependent AMPK pathway and the UPR pathways [21,22]. All of the PERK, ATF-6, and IRE1 pathways have been reported to be involved in autophagy induction in the context of different diseases; nevertheless, which pathway is employed for induction of autophagy, and the UPR-dependent cell fate seemingly depends on the nature and duration of the stress, and even the cell type [20,23]. The essential roles of the ERS-mediated autophagy have been implicated in many human and animal diseases, especially in viral diseases. The effects of ERS-mediated autophagy on the replication and infection of viruses, as well as their underlying mechanisms, may vary in response to infections with different viruses. Dengue virus (DENV), hepatitis C virus (HCV), and Japanese encephalitis virus (JEV) all belong to the Flaviviridae family and are of great significance in human and animal health. HCV induces activation of the three UPR pathways (PERK, IRE1, and ATF-6 pathway) to trigger autophagy for sustainable replication in the infected hepatocytes [24]. In vitro and in vivo infection of DENV enhances the autophagic activity and viral replication via activation of the PERK and IRE1 pathways [25]. Although JEV infection also activates the three UPR pathways in neuronal cells, it seems to activate autophagy through the ATF-6 sensor and X-box protein 1 (XBP1), a primary target of IRE1, thus inhibiting viral replication [26]. Therefore, it can be seen that molecular mechanisms associated with ERS-mediated autophagy are complicated and still need further clarification.
Classical swine fever virus (CSFV), also a member of the Flaviviridae family, is the causative agent of classical swine fever (CSF) and causes an acutely fatal disease of pigs, characterized clinically by high fever, hemorrhagic necrotizing inflammation in multiple organs of pigs with high mortality, which threatens the pig industry worldwide [27,28]. Although CSFV has been extensively studied for decades by many researchers, the mechanisms associated with the pathogenesis of CSFV remain unclarified. We have previously shown that CSFV infection induces ERS-mediated autophagy for sustaining virus replication both in vivo and in vitro [29]; however, the underlying mechanisms remain largely unknown. Here we revealed that CSFV infection triggers complete autophagy through activation of the PERK and IRE1 pathway to facilitate viral replication in cultured cells, which may be a potential strategy hijacked by CSFV for immune escape. We also found that CSFV infection induces activation of PERK and IRE1 pathway for potential immunoregulation via promoting transcription of proinflammatory cytokines, suggesting a potential mechanism associated with the regulation of CSFV replication by ERS-mediated autophagy. Our findings will provide new insights into the molecular mechanisms of CSFV replication and the development of novel antiviral strategies against CSF.
Materials and methods
Cell, virus, and plasmid
Experiments were performed on the pig kidney cell line PK-15 (ATCC, CCL-33) and macrophage cell line 3D4/2 (ATCC, CRL-2845), which were, respectively, grown in the Dulbecco’s modified Eagle’s medium (DMEM, Gbico, C11995500BT) and 1640 medium (Gbico, C11875500BT), containing 10% (v/v) fetal bovine serum (FBS, Gbico, 10091148) and 1% (v/v) penicillin-streptomycin (Gibco, 15140122). Cells were maintained in a Heracell 150i incubator (Thermo Fisher Scientific) at 37°C with 5% CO2. The CSFV-Shimen strain was stocked in our laboratory and proliferated in the cultured PK-15 cells. Virus titers were determined with 50% tissue culture infective dose (TCID50) assays. The multiplicity of infections (MOIs) was calculated on the basis of virus titers and cell density when seeded. An autophagy dual-fluorescence reporter plasmid (mRFP-GFP-LC3) for the detection of autophagy flux was our laboratory stock.
Reagents and antibodies
The 4μ8 c (Sigma, SML0949), BiP inducer X (BIX, Sigma, SML1073), salubrinal (SAL, Sigma, SML0951), PERK Inhibitor I (GSK2606414/GSK, Calbiochem, 516535), and PERK activator CCT020312 (CCT, Calbiochem, 324879) were dissolved in dimethyl sulfoxide (DMSO, Sigma, D2650) to make stock solutions (10 mM), and further diluted properly in culture medium before treatment of the cells. Opti-MEM I Reduced Serum Medium (31985088) was purchased from Gibco. Lipofectamine 3000 (L3000015) was obtained from Thermo Fisher Scientific.
The primary antibodies used in our study were specific for phosphor (p)-PERK (Thr980) (Cell Signaling, 3179), total PERK (Cell Signaling, 3192), p-eIF2α (S51) (Bioworld, BS4787), total eIF2α (Bioworld, BS3651), GRP78 (Santa Cruz, sc-13968), ATG5 (Novus Biologicals, NB110-53818), SQSTM1/p62 (Cell Signaling, 39749), LC3B (Cell Signaling, 2775), CSFV-E2 (MEDIAN/JBT, 9011), Tubulin (Beyotime, AT819), and CSFV-Npro (kindly donated by professor Xinglong Yu, Hunan Agricultural University, China). The secondary antibodies including Alexa Fluor 488-labeled goat anti-mouse IgG(H + L) (A0428), 7-Amino-4-methylcoumarin-3-acetic acid-NHS ester (AMCA)-labeled goat anti-mouse IgG (H + L) (A0413), horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG(H + L) (A0208) and goat anti-mouse IgG(H + L) (A0216) were products obtained from Beyotime Biotechnology.
Virus infection and biochemical intervention
PK-15 or 3D4/2 cells were plated one day before virus infection. At approximately 80% confluence, cells were infected with CSFV (1 MOI) or an equal amount of medium and then incubated at 37°C for 1.5 hours (h). Then, the cells were washed twice with PBS, and further cultured in DMEM or 1640 containing 2% (v/v) FBS at 37°C and 5% CO2 for the indicated time until sample collection began. As for biochemical intervention, cells were, respectively, pretreated with PERK activator CCT (1 μM), PERK inhibitor GSK (1 μM), eIF2α dephosphorylation inhibitor SAL (1 μM), IRE1 inhibitor 4μ8 c (10 μM), GRP78 inducer BIX (10 μM), or 0.1% (v/v) DMSO for 1 h, and then infected with CSFV as above described. The cells were further grown in a maintenance medium containing the indicated chemicals. At 24 hpi (Hours post-infection), samples from each group were collected for the following assays.
Transfection and RNA interference
Cells at 60%~70% confluence were subject to plasmid transfection with lipofectamine 3000. For each well of 12-well plates, 2 μL lipofectamine 3000 and 1.5 μg plasmid mRFP-GFP-LC3 (with 3 μL P3000 reagent) were, respectively, diluted in 100 μL Opti-MEM medium; after incubation for 5 min at room temperature, the two mixtures were mixed in a 1:1 (v/v) ratio. 10 min later, cells were washed 3 times with PBS, and then inoculated with the DNA-lipid complex, and incubated in the CO2 incubator for 4 ~ 6 h, followed by CSFV infection if necessary.
The small interfering RNA (SiRNA) sequences targeted for the porcine PERK, IRE1, and GRP78 genes, as well as a negative control SiRNA (SiNC), was obtained from Sangon Biotech (Shanghai, China). A 20 μM stock solution of SiRNA was prepared. SiRNA sequences used in our study were as follows: 5ʹ-CCAGAGAAGUGGCAAGAAATT-3ʹ (sense strand) and 5ʹ-UUUCUUGCCACUUCUCUGGTT-3ʹ (antisense strand) for SiPERK; 5ʹ-CCAGAAGGAACUAGAGAAATT-3ʹ (sense strand) and 5ʹ-UUUCUCUAGUUCCUUCUGGTT-3ʹ (antisense strand) for SiIRE1; 5ʹ-UGGCAAAGAUGUUCGGAAATT-3ʹ (sense strand) and 5ʹ-UUUCCGAACAUCUUUGCCATT-3ʹ (antisense strand) for SiGRP78; 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ (sense strand) and 5ʹ-ACGUGACACGUUCGGAGAATT-3ʹ (antisense strand) for SiNC. The protocol of SiRNA transfection was consistent with that of plasmid transfection, except that P3000 reagent was excluded in the diluted SiRNA mixture. After transfection, the cells were further grown in maintenance medium for 12 h, followed by CSFV infection if necessary. The efficiency of gene knockdown was assessed by Western blot.
qRT-PCR assays
The qRT-PCR assays used in our study were performed as previously described [29]. Briefly, total RNA was extracted from the cells of each group using TRIzol reagent (Invitrogen, 15596026). One μg of each RNA was reversely transcribed into cDNA for detecting relative mRNA expression of the indicated genes. The qRT-PCR assays were performed on a Bio-Rad real-time PCR machine (1855200). Each sample was set up in triplicate and repeated twice independently. Relative mRNA expressions of the specific genes were determined using the 2−ΔΔCT method and normalized to the Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. The specific primers for qRT-PCR used in our study are listed in Table 1.
Table 1.
Primer information
| Genes | Sequence (5ʹ ~ 3ʹ) | GenBank Nos. | Size (bp) |
|---|---|---|---|
| CSFV-NS5B | Forward: CCTGAGGACCAAACACATGT Reverse: TGGTGGAAGTTGGTTGTGTCTG |
AY775178.2 | 174 |
| GRP78 | Forward: CCTACTCGTGCGTTGGGGT Reverse: GACGGCGTGATGCGGTT |
XM_001927795.7 | 79 |
| ATF4 | Forward: CAAGACAGCAGCCACTCGGTA Reverse: TCAGAGCTTCGTTCTTCTTTTCC |
NM_001123078.1 | 98 |
| CHOP | Forward: AGGTGCTGTCCTCAGATGAAAATG Reverse: AGAGGCAGGGTCAAGAGTGGTG |
NM_001144845.1 | 95 |
| ATG5 | Forward: GGTTTGAATATGAAGGCACACCA Reverse: TGTGATGTTCCAAGGAAGAGCTG |
AM087014.1 | 98 |
| ATG12 | Forward: CTTCTTCCGCTTCAGTTTCC Reverse: TGTGTCTCCCACAGCCTTTA |
NM_001190282.1 | 92 |
| Beclin1 | Forward: AGGAGCTGCCGTTGTACTGTTCT Reverse: TGCTGCACACAGTCCAGGAA |
NM_001044530.1 | 94 |
| IFN-γ | Forward: CCAGGCCATTCAAAGGAGCATGGA Reverse: GGCTTTGCGCTGGATCTGCAGA |
NM_213948.1 | 139 |
| TNF-α | Forward: TGGCCCAAGGACTCAGATCAT Reverse: TCGGCTTTGACATTGGCTACA |
EU682384.1 | 76 |
| GAPDH | Forward: TGGAGTCCACTGGTGTCTTCAC Reverse: TTCACGCCCATCACAAACA |
NM_001206359.1 | 121 |
Western blot analysis
Cells collected from each group were lysed on ice with cell lysis buffer (Beyotime. P0013) containing 2% (v/v) protease and phosphatase inhibitor cocktails (50×, Beyotime, P1050). After centrifugation at 13 000 r/min for 15 min at 4°C, the supernatant was obtained and then subject to protein quantification with a BCA protein assay kit (Beyotime, P0012). Aliquots of the supernatant were mixed with 5× SDS-PAGE loading buffer and boiled for 10 min, followed by Western blot analysis as previously described [29]. Primary antibodies used in our study were specific for p-PERK, total PERK, p-eIF2α, total eIF2α, GRP78, ATG5, p62, LC3B, Npro, and tubulin, respectively. Tubulin was used as a loading control. The grayscale value of the specific bands was analyzed with ImageJ software for semi-quantitation of protein expression.
Confocal fluorescence microscopy
To analyze the effect of biochemical pretreatment or SiRNA intervention on autophagic flux in the CSFV-infected cells, PK-15 cells cultured in glass-bottom cell culture dishes (Φ15 mm, 801002, NEST) were pretreated as described above with the indicated chemicals, and then transfected with mRFP-GFP-LC3, or were co-transfected with mRFP-GFP-LC3 and the indicated SiRNAs, followed by CSFV infection. The cells were further cultured in maintenance media for 24 h, and then subject to indirect immunofluorescence staining and confocal microscopy. Briefly, cells were washed twice with PBS and fixed with pre-cooled absolute ethanol for 30 min at −20°C, and then permeabilized with 0.3% Triton X-100 solution for 10 min at room temperature. After blocking with 5% bovine serum albumin (BSA) in PBS for 1 h at 37°C, the cells were incubated overnight at 4°C with mouse anti-CSFV E2 monoclonal antibodies at a dilution of 1:200 (v/v), followed by a 1 h of incubation in PBS containing AMCA-labeled goat anti-mouse IgG(H + L) antibodies (1:200, v/v). The fluorescent signals were observed under a TCS SP8 confocal fluorescence microscope (Leica). GFP degrades in the acidic environment of the lysosome while RFP does not. Therefore, yellow spots merged by green and red fluorescence indicate the formation of autophagosomes (incomplete autophagy), while red spots indicate the formation of autophagolysosomes and increased autophagy flux (complete autophagy). The average numbers of fluorescent spots in 10 cells from each group were analyzed with ImageJ software. Blue spots indicate a positive expression of CSFV-E2 proteins.
TCID50 assays
The cells and supernatants of each treatment were collected for virus titration measured with TCID50 assays, which were performed as described in our previous studies [29,30]. Briefly, cells seeded in 96-well plates were inoculated with serial 10-fold dilutions of CSFV for 1.5 h at 37°C. After further incubation for 48 h at 37°C, the cells were subject to an indirect immunofluorescence assay exploiting mouse anti-CSFV E2 antibody and Alexa Fluor 488-labeled goat anti-mouse IgG(H + L) antibody, to assess the expression of viral structural proteins. Viral titers were determined in accordance with the Reed–Muench method [31] and shown as Log10(TCID50/mL). Each treatment was set up in four replicates and repeated twice independently.
Cell viability assays
Viabilities of the PK-15 and 3D4/2 cells were measured by the CCK-8 assays as previously described [29]. Briefly, cells seeded in 96-well plates (1 × 104 cells/well) one day before the experiment was pre-treated with the indicated concentrations of chemicals or SiRNAs. After further incubation for 24 h, cells were incubated with 10% (v/v) CCK-8 in fresh medium for 1 h at 37°C. The absorbance at 450 nm was detected with a Ledetect 96 microplate reader (LABEXIM). Each sample was assayed in triplicate and repeated twice independently.
Statistical analysis
The data are shown as the mean ± standard deviation (SD) of two independent experiments and were analyzed with one-way ANOVA or two-way ANOVA tests. P values of less than 0.05 were considered to be statistically significant.
Results
CSFV infection induces the PERK pathway-dependent complete autophagy to promote viral replication in cultured cells
We have previously shown that CSFV infection induces ERS-mediated autophagy both in vivo and in vitro [29], while the underlying mechanisms remain unclear. We have also previously revealed activation of the PERK and IRE1 pathways during CSFV infection [29,30]. Therefore, the PERK and IRE1 pathways become our preference for exploring potential mechanisms involved in ERS-mediated autophagy during CSFV infection.
Firstly, the effect of pretreatment with PERK activator CCT020312 (CCT) and inhibitor GSK2606414 (GSK) on autophagic activities in the CSFV-infected PK-15 and 3D4/2 cells were analyzed by qRT-PCR and Western blot assays, respectively. We found that CCT pretreatment increased while GSK pretreatment decreased the relative mRNA expression of the activating transcription factor 4 (ATF4) and DNA-damage inducible transcript 3 (DDIT3/CHOP: hereafter called CHOP) genes, and autophagy-related 5 (ATG5) and BECN1/Beclin1 (hereafter called Beclin1) genes in the CSFV-infected PK-15 cells (Figure 1(a)), implying the necessity of the PERK signaling for induction of autophagy. In the meantime, CCT treatment not only significantly upregulated the phosphorylation level of PERK and its downstream eukaryotic initiation factor 2α (eIF2α) but also enhanced the expression of autophagy protein ATG5 and the conversion of LC3-I to LC3-II, with a concomitant decrease of SQSTM1/p62 (Sequestosome 1, hereafter called p62) expression in the CSFV-infected PK-15 cells; by contrast, GSK treatment inhibited the phosphorylation of PERK and eIF2α, the expression of ATG5, and the conversion of LC3-I to LC3-II, along with the increased expression of p62 in the CSFV-infected cells (Figure 1(b,c)). These results further confirmed the essential role of the PERK pathway in CSFV-induced autophagy. Additionally, we found that pretreatment with eIF2α dephosphorylation inhibitor salubrinal (SAL) elevated the phosphorylation level of eIF2α in the CSFV-infected cells, which not only increased the transcription level of ATF4 and CHOP genes but also promoted the transcription of ATG5 and Beclin1 genes, the expression of ATG5 protein, as well as the conversion of LC3-I to LC3-II, with a concomitant decrease of p62 expression (Figure 1(a–c)). We obtained similar results in the cultured 3D4/2 cell lines (Figure 2(a–c)). Collectively, the above results suggest that activation of the PERK pathway increases while inhibition of the PERK pathway reduces autophagic activities in the CSFV-infected cells.
Figure 1.
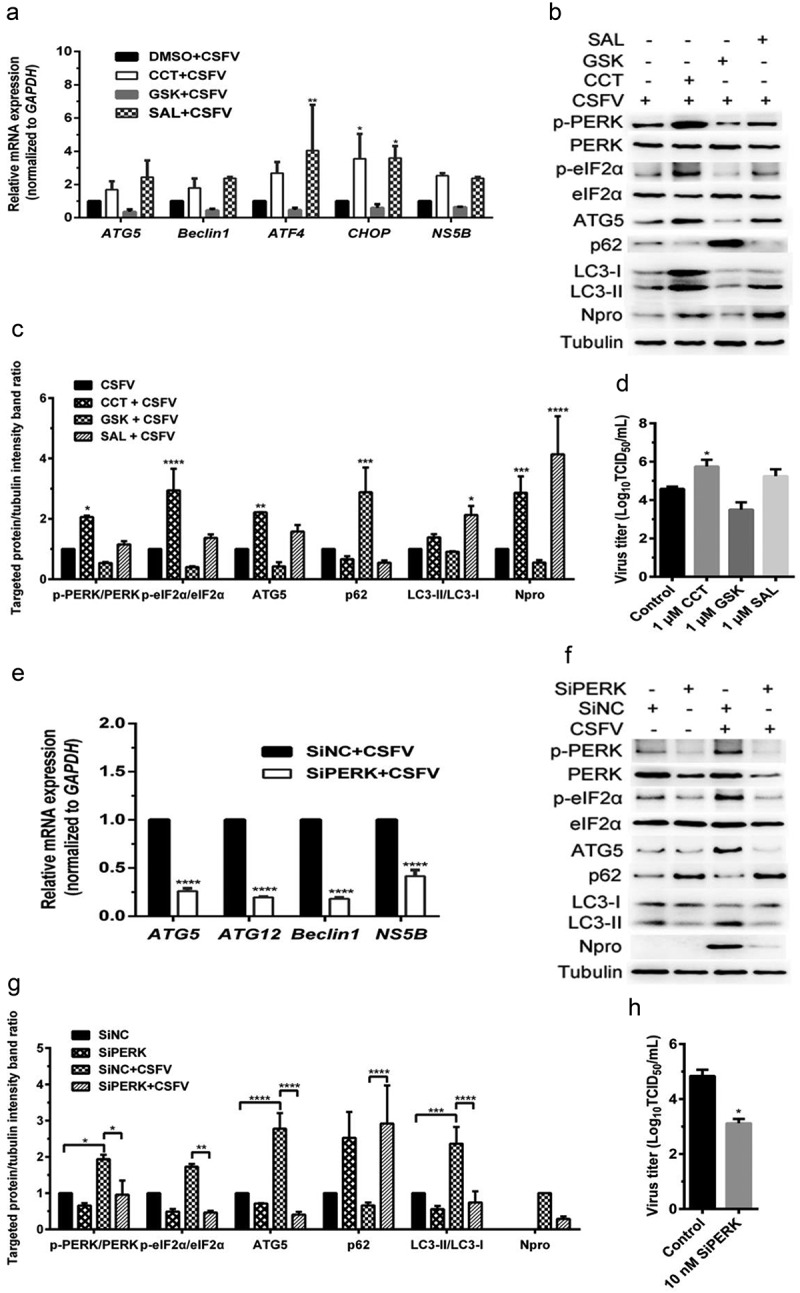
CSFV infection causes the PERK pathway-mediated autophagy to promote viral replication in cultured PK-15 cells. (a–d) PK-15 cells cultured in 12-well plates were respectively pretreated with 1 μM CCT, 1 μM GSK, 1 μM SAL or equal amount of DMSO for 1 h, and then subject to CSFV infection (1 MOI) for 1.5 h. The cells were further cultured in the presence of the chemicals for 24 h, and then collected for qRT-PCR (a), Western blot (b and c) and TCID50 assays (d), respectively. For qRT-PCR (a), relative quantification of ATG5, Beclin1, ATF4, CHOP and NS5B genes were assessed using the 2−ΔΔCT method and normalized to GAPDH. Two-way ANOVA tests: *, P< 0.05; **, P< 0.01. For Western blot (b), samples were prepared as described in the “materials and methods” for Western blot analyses using specific antibody against p-PERK, total PERK, p-eIF2α, total eIF2α, ATG5, p62, LC3B, Npro and tubulin. Grayscale value of the bands were analyzed with ImageJ software, and generations of images for protein quantification were performed with GraphPad Prism 6 software. Two-way ANOVA tests: *, P< 0.05; **, P< 0.01; ***, P< 0.001;****, P< 0.0001 (c). For virus titration (d), the cells and supernatants of each treatment were collected for TCID50 assays through an indirect immunofluorescence assay using mouse anti-CSFV E2 antibody. Virus titers were expressed as Log10(TCID50/mL). Two-way ANOVA tests: *, P< 0.05. (e–h) PK-15 cells cultured in 12-well plates were respectively transfected with 10 nM SiPERK or 10 nM SiNC for 4 ~ 6 h, and further incubated at 37°C for 12 h prior to a 1.5 h of incubation with 1 MOI of CSFV. The cells were further cultured for 24 h, and then collected for detection of relative mRNA expression of ATG5, ATG12, Beclin1 and NS5B genes by qRT-PCR (e), and determination of protein expression of p-PERK, total PERK, p-eIF2α, total eIF2α, ATG5, p62, LC3B, Npro , and tubulin through Western blot analysis (f and g), as well as measurement of virus titers with TCID50 assays (h). All the data shown are expressed as mean ± SD values of two independent experiments, and are analyzed by two-way ANOVA tests: *, P< 0.05; **, P< 0.01; ***, P< 0.001;****, P< 0.0001
Figure 2.
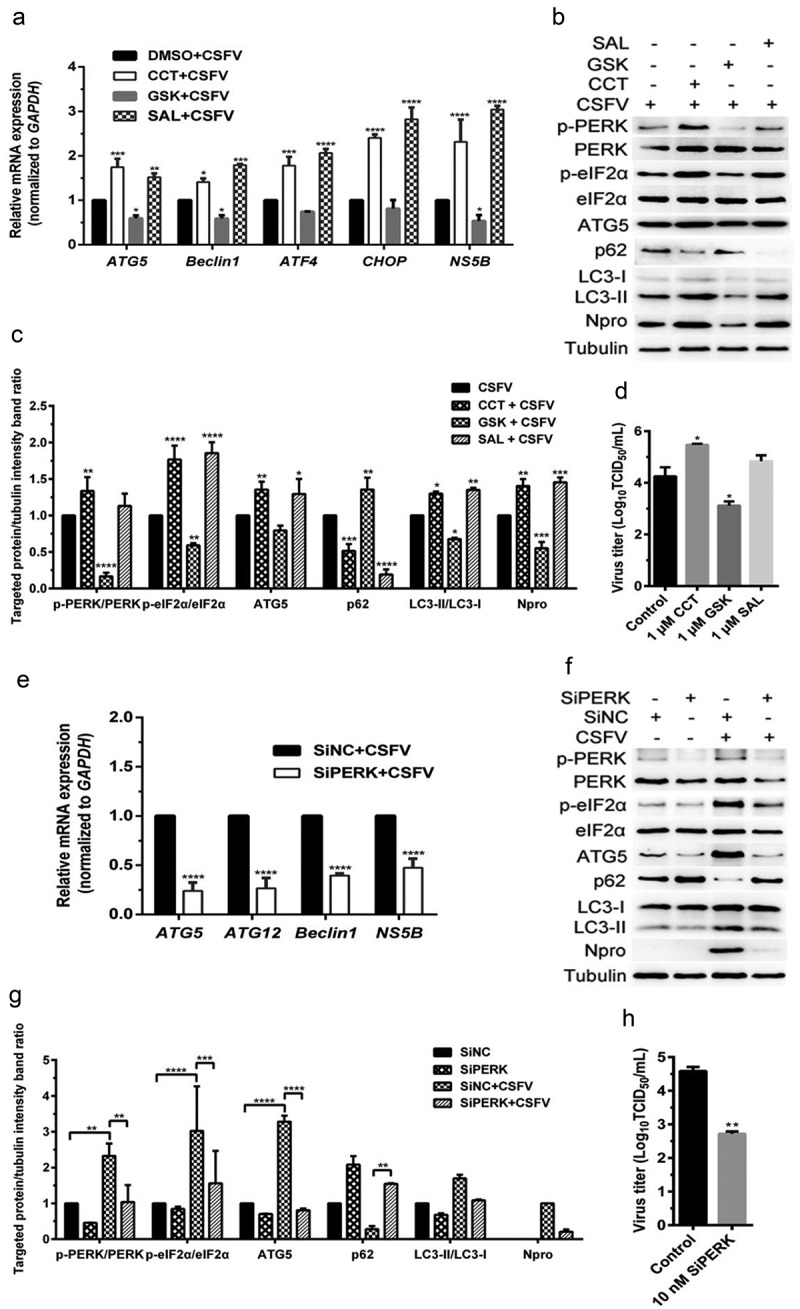
CSFV infection causes the PERK pathway-mediated autophagy to promote viral replication in cultured 3D4/2 cells. (a–d) 3D4/2 cells were treated with PERK pathway regulators and CSFV infection as described in the legend of Figure 1, and then collected for detection of relative mRNA expression of ATG5, Beclin1, ATF4, CHOP and NS5B genes by qRT-PCR (a), and determination of protein expression of p-PERK, total PERK, p-eIF2α, total eIF2α, ATG5, p62, LC3B, Npro, and tubulin through Western blot analysis (b and c), as well as measurement of virus titers with TCID50 assays (d). (e–h) 3D4/2 cells were treated with SiPERK and CSFV infection as described in the legend of Figure 1, and then collected for detection of relative mRNA expression of ATG5, ATG12, Beclin1 and NS5B genes by qRT-PCR (e), and determination of protein expression of p-PERK, total PERK, p-eIF2α, total eIF2α, ATG5, p62, LC3B, Npro and tubulin through Western blot analysis (f and g), as well as measurement of virus titers with TCID50 assays (h). All the data shown are expressed as mean ± SD values of two independent experiments. Two-way ANOVA tests: *, P< 0.05; **, P< 0.01; ***, P< 0.001;****, P< 0.0001
We further found that the PERK gene silencing by transfection of PERK-targeting SiRNA not only reduced the transcription level of ATG5, ATG12, and Beclin1 genes but also inhibited the expression of p-PERK and p-eIF2α, as well as ATG5 expression and LC3-I to LC3-II conversion, along with increased p62 expression in the CSFV-infected PK-15 and 3D4/2 cells (Figures 1(e,g) and 2(e,g)), suggesting that blockage of the PERK pathway reduced autophagic activities in the CSFV-infected cells. Moreover, we detected the autophagy flux by visualizing the expression of autophagy dual-fluorescence reporter plasmid (mRFP-GFP-LC3) in PK-15 cells undergoing pretreatment with PERK regulators or SiPERK intervention prior to CSFV infection through confocal immunofluorescence microscopy and found that both CCT and SAL pretreatment improved autophagy flux, while both GSK pretreatment and the PERK gene silencing reduced autophagy flux in CSFV-infected PK-15 cells as, respectively, demonstrated by the increased or decreased numbers of yellow (autophagosomes) and red fluorescent dots (autophagolysosomes) in cells (Figure 3), further confirming that the PERK pathway plays a crucial role in the complete autophagy induced by CSFV infection.
Figure 3.
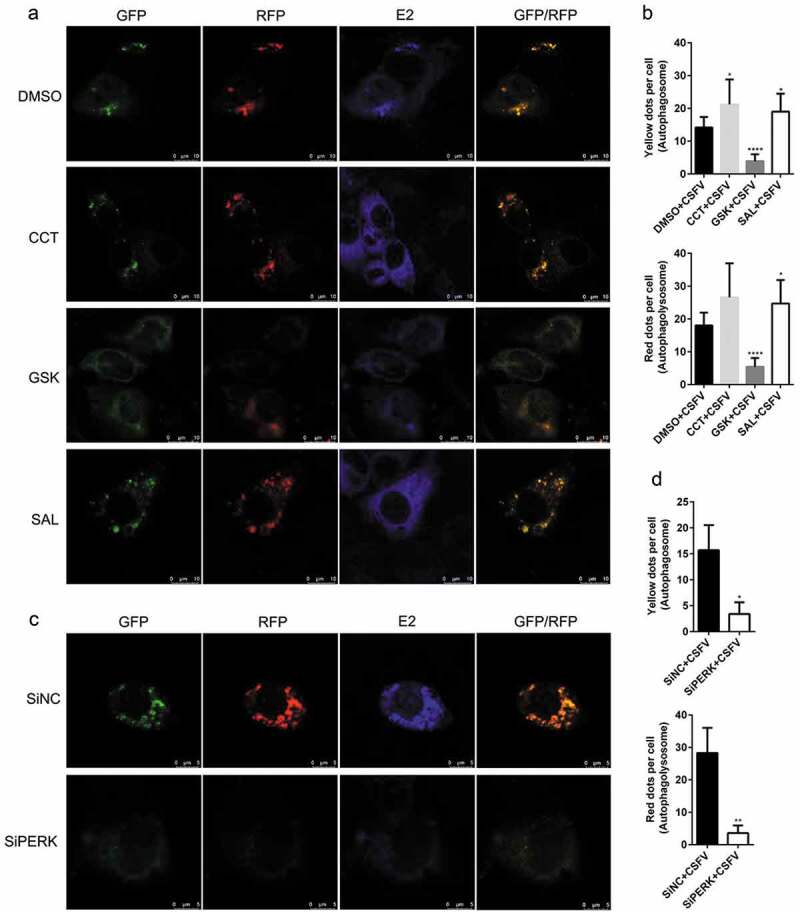
Modulation of the PERK pathway alters autophagy flux in the CSFV-infected PK-15 cells. (a) PK-15 cells cultured in glass-bottom dishes for confocal microscopy were pretreated with 1 μM CCT or 1 μM GSK or 1 μM SAL or equal amount of DMSO for 1 h, and then subject to mRFP-GFP-LC3 transfection for 4 ~ 6 h, followed by a 1.5 h of incubation with 1 MOI of CSFV. The cells were further cultured in the presence of the chemicals for 24 h, and then collected for indirect immunofluorescence staining and confocal fluorescence microscopy as described in the “Materials and Methods”. Several visual fields were selected for image acquisition, and the representative images were shown. GFP degrades in the acidic environment of lysosome while RFP does not. Therefore, yellow spots (overlapped by green and red fluorescence) indicate formation of autophagosomes (incomplete autophagy); however, red spots indicate formation of autophagolysosomes and increased autophagy flux (complete autophagy). Blue spots indicate positive expression of CSFV-E2 proteins. Scale bar: 10 μm. (b) The data show the quantification of autophagosomes (yellow) and autophagolysosomes (red) by counting the average number of fluorescent dots in ten cells with ImageJ software. One-way ANOVA: *, P< 0.05; ****, P< 0.0001. (c) PK-15 cells cultured in confocal dishes were respectively co-transfected with 1.5 μg mRFP-GFP-LC3 plasmid and 10 nM SiPERK or 10 nM SiNC for 4 ~ 6 h, and further incubated at 37°C for 12 h prior to a 1.5 h of incubation with 1 MOI of CSFV. The cells were further cultured for 24 h and then collected for indirect immunofluorescence staining and confocal fluorescence microscopy. Scale bar: 5 μm. (d) Statistical analysis of the effect of PERK gene silencing on autophagy flux in the CSFV-infected PK-15 cells. Student’s t-test: *, P< 0.05; **, P< 0.01
In addition, our results also showed that both CCT and SAL pretreatment increased, while both GSK treatment and the PERK gene silencing reduced copy numbers of CSFV NS5B genes, expression of Npro and E2 protein, and virus titers in the CSFV-infected cells (Figures 1–3(a,c)); suggesting a significant role of the PERK pathway in CSFV replication in vitro. Collectively, our data obtained above suggest that CSFV infection induces the PERK pathway-mediated complete autophagy to promote viral replication in cultured cells.
CSFV infection induces IRE1/GRP78-dependent complete autophagy to facilitate viral replication in cultured cells
The IRE1 pathway is another classical UPR pathway, and GRP78, one of the downstream molecules of IRE1, is a widely recognized marker protein for monitoring ERS [30,32]. Since we have shown that CSFV infection-induced activation of the PERK and IRE1 pathway and that the PERK pathway was essential for CSFV-induced autophagy, thus promoting viral replication in cultured cells, we further investigated the role of the IRE1 pathway in CSFV-induced autophagy and viral replication. The effect of an IRE1-specific inhibitor 4μ8 c and a potent BiP/GRP78 inducer X (BIX) on autophagic activities in the CSFV-infected PK-15 and 3D4/2 cells were measured by qRT-PCR and Western blot assays, respectively. Results showed that pretreatment with 4μ8 c significantly decreased the relative mRNA expression levels of GRP78, ATG5, ATG12, and Beclin1 genes in the CSFV-infected PK-15 cells (Figure 4(a)). Simultaneously, 4μ8 c treatment not only significantly downregulated the expression levels of GRP78 protein but also inhibited the expression of ATG5 and the conversion of LC3-I to LC3-II, with a concomitant increase of p62 expression in the CSFV-infected PK-15 cells (Figure 4(b,c)), suggesting the reduction of autophagic activities by inhibition of the IRE1 sensor. Contrastively, BIX treatment remarkably increased the transcriptional and translational levels of GRP78 in CSFV-infected PK-15 cells, which not only significantly upregulated the transcriptional levels of ATG5, ATG12, and Beclin1 genes, but also enhanced the expression of ATG5 protein and the conversion of LC3-I to LC3-II, accompanied with inhibition of p62 expression (Figure 4(a–c)). Similar results were also obtained in the cultured 3D4/2 cells (Figure 5(a–c)). The above results suggest that inhibition of IRE1 reduces, while activation of GRP78 increases autophagic activities in the CSFV-infected cells.
Figure 4.
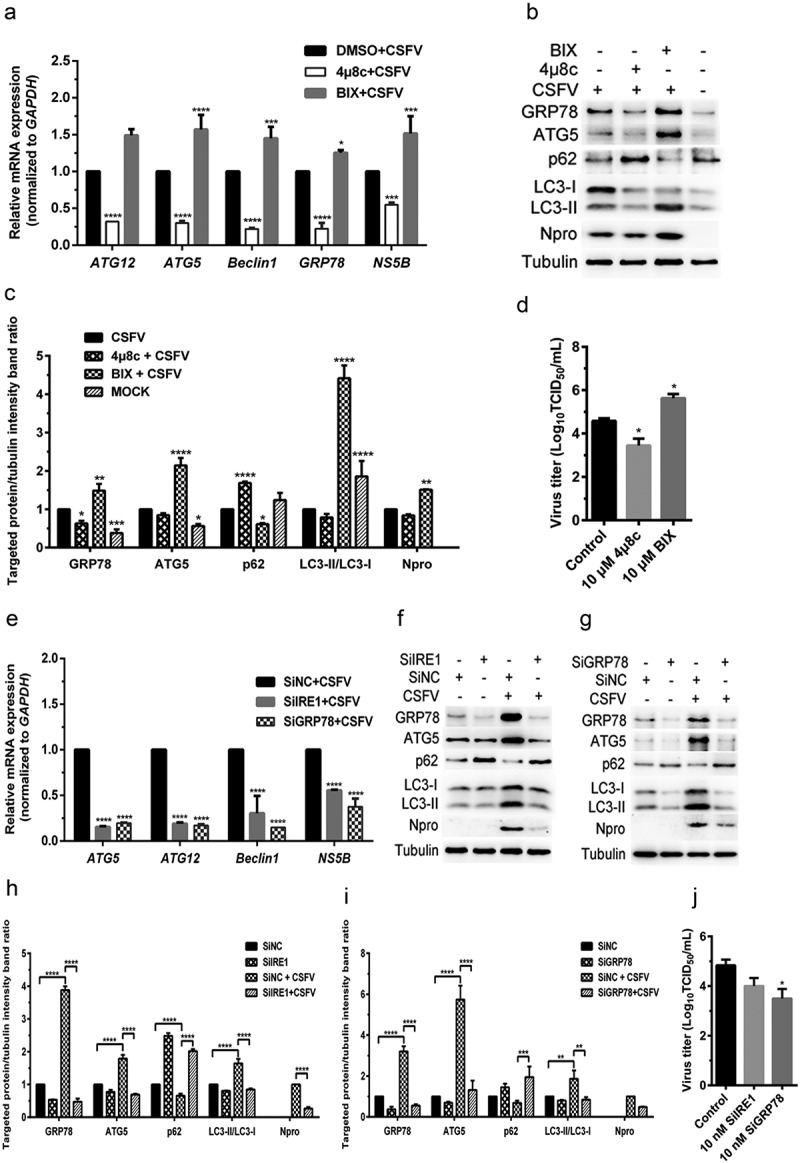
CSFV infection causes IRE1/GRP78-mediated autophagy to promote viral replication in cultured PK-15 cells. (a–d) PK-15 cells cultured in 12-well plates were respectively pretreated with 10 μM 4μ8 c, 10 μM BIX or equal amount of DMSO for 1 h, and then subject to CSFV infection (1 MOI) for 1.5 h. The cells were further cultured with the chemicals for 24 h, and then collected for detection of relative mRNA expression of GRP78, ATG5, ATG12, Beclin1 and NS5B genes by qRT-PCR (a), and determination of protein expression of GRP78, ATG5, p62, LC3B, Npro and tubulin through Western blot analysis (b and c), as well as measurement of virus titers with TCID50 assays (d). (e–j) PK-15 cells cultured in 12-well plates were respectively transfected with 10 nM SiIRE1, 10 nM SiGRP78 or 10 nM SiNC for 4 ~ 6 h, and further incubated at 37°C for 12 h prior to a 1.5 h of incubation with 1 MOI of CSFV. The cells were further cultured for 24 h, and then collected for detection of relative mRNA expression of ATG5, ATG12, Beclin1 and NS5B genes by qRT-PCR (e), and determination of protein expression of GRP78, ATG5, p62, LC3B, Npro and tubulin through Western blot analysis (f–i), as well as measurement of virus titers with TCID50 assays (j). All the data shown are expressed as mean ± SD values of two independent experiments. Two-way ANOVA tests: *, P< 0.05; **, P< 0.01; ***, P< 0.001; ****, P< 0.0001
Figure 5.
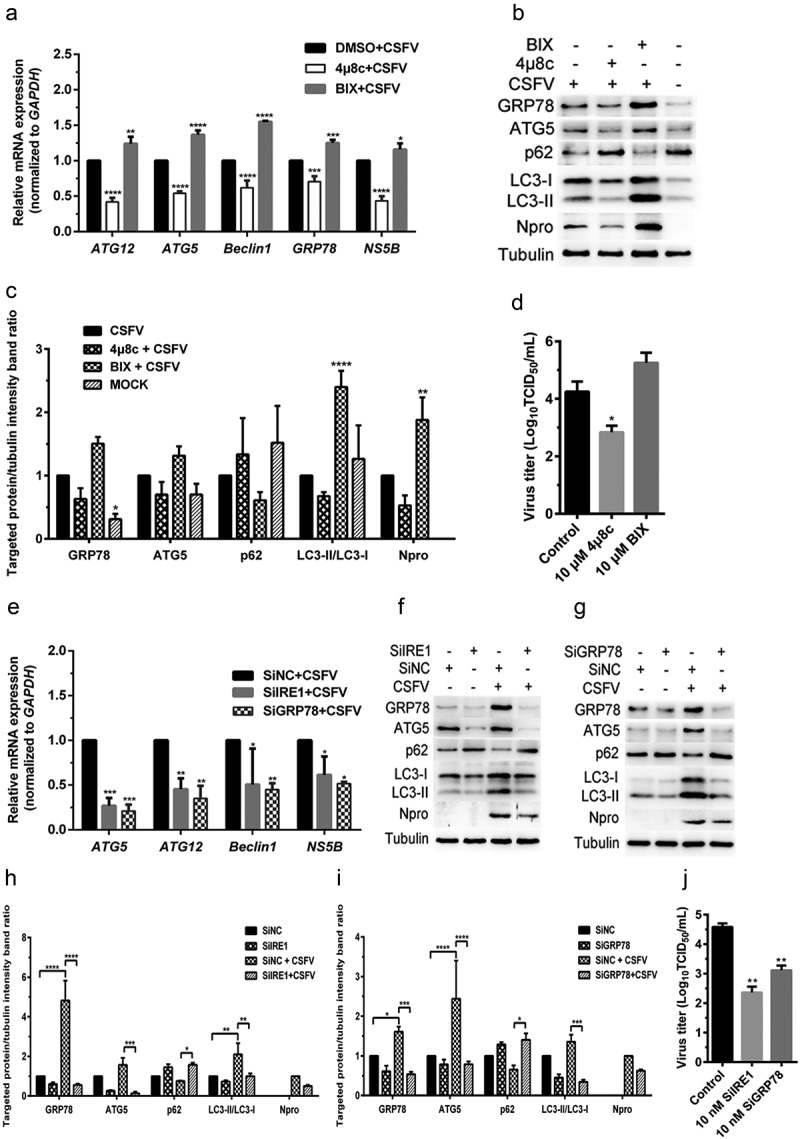
CSFV infection causes IRE1/GRP78-mediated autophagy to promote viral replication in cultured 3D4/2 cells. (a–d) 3D4/2 cells were treated with IRE1/GRP78 regulators and CSFV infection as described in the legend of Figure 4, and then collected for detection of relative mRNA expression of GRP78, ATG5, ATG12, Beclin1 and NS5B gene by qRT-PCR (a), and determination of protein expression of GRP78, ATG5, p62, LC3B, Npro and tubulin through Western blot analysis (b and c), as well as measurement of virus titers with TCID50 assays (d). (e–j). 3D4/2 cells treated with SiIRE1/SiGRP78 and CSFV infection as described in the legend of Figure 4, and then collected for detection of relative mRNA expression of ATG5, ATG12, Beclin1 and NS5B gene by qRT-PCR (e), and determination of protein expression of GRP78, ATG5, p62, LC3B, Npro and tubulin through Western blot analysis (f–i), as well as measurement of virus titers with TCID50 assays (j). All the data shown are expressed as mean ± SD values of two independent experiments. Two-way ANOVA tests: *, P< 0.05; **, P< 0.01; ***, P< 0.001; ****, P< 0.0001
Furthermore, we found that either the IRE1 or the GRP78 gene silencing by transfection of IRE1- or GRP78-targeting SiRNA significantly inhibited the transcription of ATG5, ATG12 and Beclin1 genes, the expression GRP78 and ATG5, and the conversion of LC3-I to LC3-II, with a concomitant increase of p62 expression in the CSFV-infected PK-15 and 3D4/2 cells (Figures 4(e–i) and 5(e–i)), suggesting that blockage of the IRE1/GRP78 pathway reduces autophagic activities in CSFV-infected cells. Additionally, we also found that BIX significantly increased, while both 4μ8 c pretreatment and the IRE1/GRP78 gene silencing reduced autophagy flux in the CSFV-infected PK-15 cells as, respectively, demonstrated by the increased or decreased numbers of yellow (autophagosomes), and red fluorescent dots (autophagolysosomes) in cells (Figure 6), further confirming the essential role of the IRE1/GRP78 pathway in CSFV-induced complete autophagy.
Figure 6.
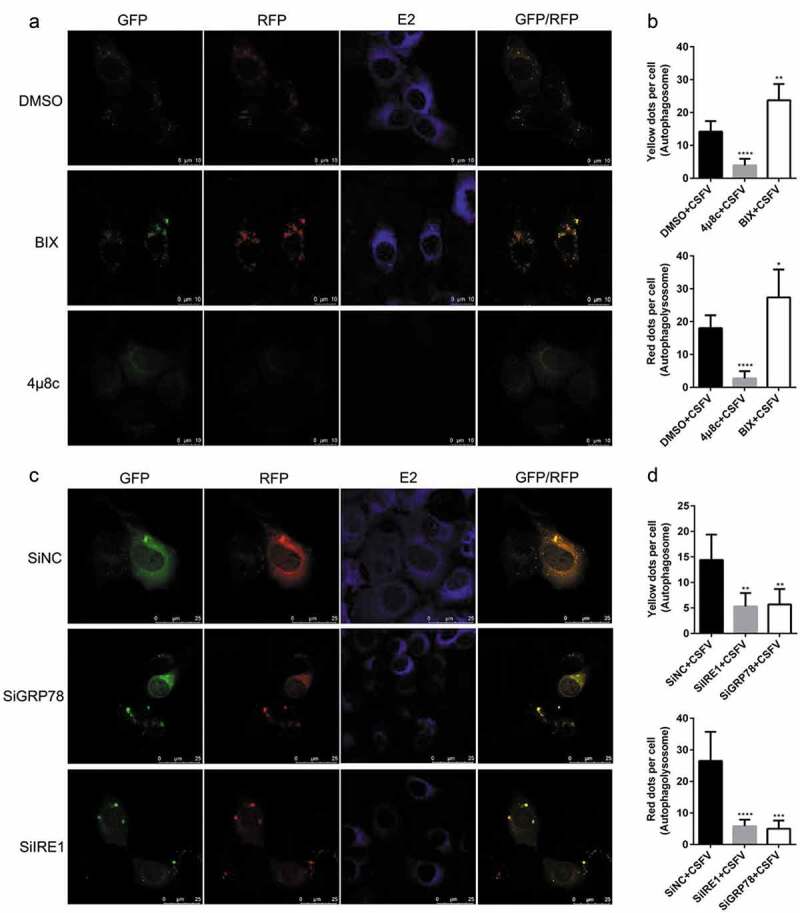
Modulation of the IRE1/GRP78 pathway alters autophagy flux in the CSFV-infected PK-15 cells. (a) PK-15 cells cultured in confocal dishes were pretreated with 10 μM 4μ8 c, 10 μM BIX or equal amount of DMSO (0.1%, v/v) for 1 h, and then subject to mRFP-GFP-LC3 transfection for 4 ~ 6 h, followed by a 1.5 h of incubation with 1 MOI of CSFV. The cells were further cultured in the presence of the chemicals for 24 h, and then collected for indirect immunofluorescence staining and confocal fluorescence microscopy. Yellow spots indicate formation of autophagosomes (incomplete autophagy), and red spots indicate formation of autophagolysosomes and increased autophagy flux (complete autophagy). Blue spots indicate positive expression of CSFV-E2 proteins. Scale bar: 10 μm. (b) The data show the quantification of autophagosomes (yellow) and autophagolysosomes (red) by counting the average number of fluorescent dots in ten cells with ImageJ software. One-way ANOVA: *, P< 0.05; **, P< 0.01; ****, P< 0.0001. (c) PK-15 cells cultured in confocal dishes were respectively co-transfected with 1.5 μg mRFP-GFP-LC3 plasmid and 10 nM SiIRE1, 10 nM SiIRE1 or 10 nM SiNC for 4 ~ 6 h, and further incubated at 37°C for 12 h prior to a 1.5 h of incubation with 1 MOI of CSFV. The cells were further cultured for 24 h and then collected for indirect immunofluorescence staining and confocal fluorescence microscopy. Scale bar: 25 μm. (d) Statistical analysis of the effect of IRE1/GRP78 gene silencing on autophagy flux in the CSFV-infected PK-15 cells. One-way ANOVA: **, P< 0.01; ***, P< 0.001; ****, P< 0.0001
In addition, we also found that both 4μ8 c pretreatment and the IRE1/GRP78 gene silencing reduced, while BIX treatment increased copy numbers of CSFV NS5B genes, expression of Npro and E2 protein, and virus titers in the CSFV-infected cells (Figures 4–6(a,c)), indicating a significant role of the IRE1/GRP78 pathway in CSFV replication in vitro. Taken together, our results suggest that CSFV elicits the IRE1/GRP78 pathway-mediated complete autophagy to facilitate virus replication.
The PERK and IRE1 pathways are associated with transcriptional regulation of proinflammatory cytokine genes during CSFV infection
Increasing evidence have revealed that many stressors-induced cellular ERS can promote the expression of inflammatory cytokines and then participate in the regulation of innate immunity [33,34]. Many pro-inflammatory cytokines, such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), can promote induction of autophagy [35,36]. Therefore, we further investigated the relationship between CSFV-induced ERS and proinflammatory cytokines. The effect of the PERK and IRE1-pathway regulators, and PERK/IRE1/GRP78 gene silencing on the transcription of proinflammatory cytokine genes were investigated by qRT-PCR assays. On the one hand, we found that CCT or SAL treatment increased the relative mRNA expression levels of IFN-γ and TNF-α mRNA in the CSFV-infected PK-15 cells (Figure 7(a)). Although the transcription of the TNF-α gene seemed not to be affected by CCT or SAL treatment in CSFV-infected 3D4/2 cells, the relative mRNA expression levels of IFN-γ were significantly increased (Figure 7(b)). By contrast, GSK treatment inhibited the transcriptional levels of IFN-γ and TNF-α mRNA in both cells infected with CSFV (Figure 7(a,b)). Further study found that the PERK gene silencing significantly suppressed the transcriptional levels of IFN-γ and TNF-α mRNA in the CSFV-infected PK-15 and 3D4/2 cells (Figure 7(e,f)). These results suggest that the PERK pathway could be involved in the transcriptional regulation of pro-inflammatory cytokine genes for potential immunoregulation in the CSFV-infected cells.
Figure 7.
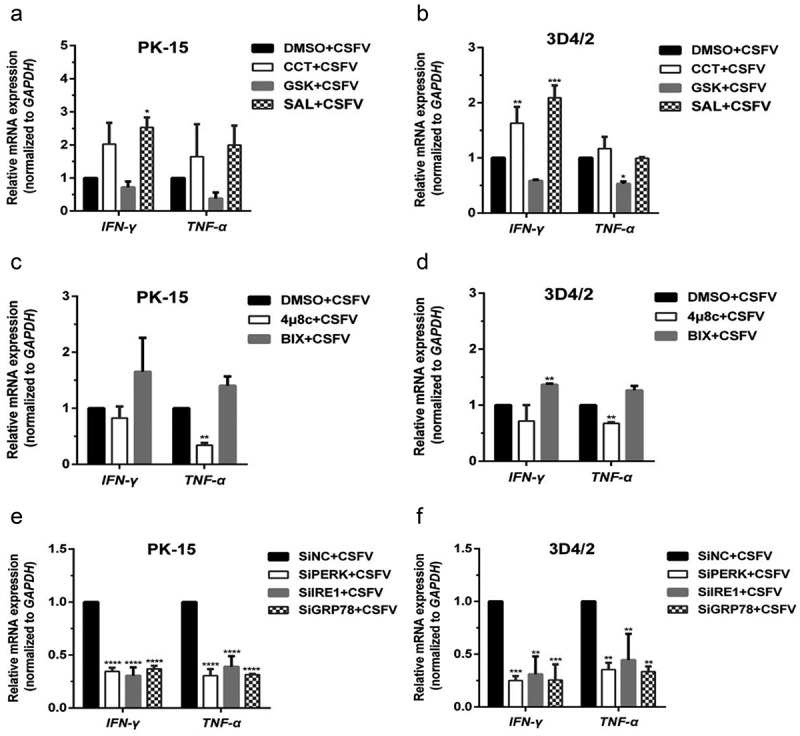
Pharmacological regulation and gene silencing of the PERK/IRE1 pathway molecules change transcription of proinflammatory cytokine genes in the CSFV-infected PK-15 and 3D4/2 cells. (a and b) PK-15 (a) and 3D4/2 cells (b) cultured in 12-well plates were respectively pretreated with 1 μM CCT, 1 μM GSK, 1 μM SAL or equal amount of DMSO (0.1%, v/v) for 1 h, and then subject to a 1.5 h of incubation with 1 MOI of CSFV. After further incubation in the presence of the chemicals for 24 h, cells were collected for detection of relative mRNA expression of pro-inflammatory cytokine genes (IFN-γ and TNF-α) through qRT-PCR assays. (c and d) PK-15 (c) and 3D4/2 cells (d) cultured in 12-well plates were respectively pretreated with 10 μM 4μ8 c, 10 μM BIX or equal amount of DMSO (0.1%, v/v) for 1 h, and then subject to a 1.5 h of incubation with 1 MOI of CSFV. After further incubation in the presence of the chemicals for 24 h, cells were collected for determination of transcriptional levels of IFN-γ and TNF-α genes by qRT-PCR. (e and f) PK-15 (e) and 3D4/2 cells (f) cultured in 12-well plates were respectively transfected with 10 nM SiPERK, 10 nM SiIRE1, 10 nM SiGRP78 or 10 nM SiNC for 4 ~ 6 h, and further incubated at 37°C for 12 h prior to a 1.5 h of incubation with 1 MOI of CSFV. The cells were further cultured for 24 h, and then collected for detection of relative mRNA expression of IFN-γ and TNF-α genes by qRT-PCR. The relative mRNA expression of the target genes were assessed by using the 2−ΔΔCT method and normalized to the GAPDH gene. Each template was ran in triplicate. All the data shown are expressed as mean ± SD values of two independent experiments. Two-way ANOVA tests: *, P< 0.05; **, P< 0.01; ***, P< 0.001; ****, P< 0.0001
On the other hand, our results also showed that BIX treatment increased the relative mRNA expression levels of IFN-γ and TNF-α mRNA in CSFV-infected PK-15 and 3D4/2 cells (Figure 7(c,d)). However, either 4μ8 c treatment or the IRE1/GRP78 gene silencing inhibited the transcription of IFN-γ and TNF-α mRNA in both cells infected with CSFV (Figure 7(c–f)). These results indicate important roles of the IRE1/GRP78 pathway in the transcriptional regulation of pro-inflammatory cytokine genes in the CSFV-infected cells. Comprehensively, our results suggest that CSFV infection induces ERS-driven activation of the PERK and IRE1 pathway for potential immunoregulation via promoting the expression of proinflammatory cytokines. This process might be associated with CSFV-induced autophagy and its influence on viral replication, which needs further investigations.
Pretreatment with the PERK and IRE1-pathway regulators, and the indicated SiRNA interventions do not affect cell viabilities
To determine whether the pharmacological treatment of ERS regulators or the indicated SiRNA interventions affected autophagic activities of cells, and the capability of CSFV replication by altering cell viabilities, the CCK8 assays were performed to analyze the effects of the PERK and IRE1-pathway regulators, and the PERK/IRE1/GRP78-targeting SiRNAs on the viability of PK-15 and 3D4/2 cells. Results showed that pretreatment with the indicated chemicals or transfection of SiRNAs had no significant effects on the viability of PK-15 (Figure 8(a)) and 3D4/2 cells (Figure 8(b)).
Figure 8.
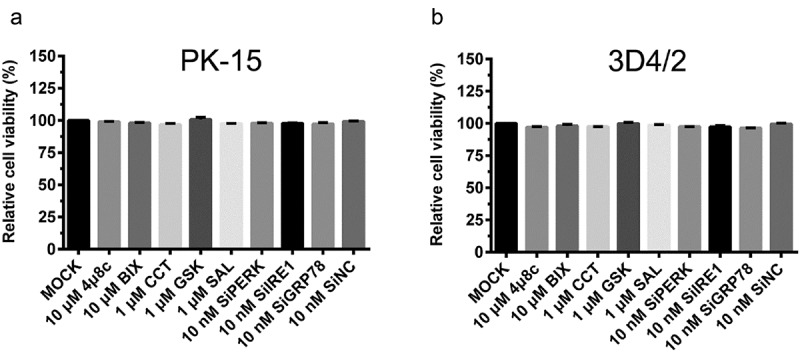
The effect of PERK and IRE1-pathway regulators, as well as PERK/IRE1/GRP78 gene silencing on cell viability of PK-15 and 3D4/2 cells. The cell viability of PK-15 (a) and 3D4/2 (b) cells were detected by the CCK-8 assays after treatments with the indicated concentrations of chemicals or SiRNAs. After further incubation for 24 h, cells were incubated with 10% (v/v) CCK-8 diluted in fresh medium for 1 h at 37°C. The absorbance at 450 nm was measured with a Ledetect 96 microplate reader. Each sample was assayed in triplicate. The data represent the mean ± SD of two independent experiments and are analyzed with one-way ANOVA tests
In summary, our results suggest that CSFV infection induces ERS and then triggers autophagy through activation of the PERK-eIF2α-ATF4-CHOP and the IRE1/GRP78 pathway, thus promoting replication of CSFV in cultured PK-15 and 3D4/2 cells (Figure 9), which may be a potential strategy hijacked by CSFV for immune escape. Furthermore, we also show that CSFV infection induces ERS-driven activation of the PERK and IRE1 pathway for potential immunoregulation via promoting transcription of proinflammatory cytokines, suggesting a possible mechanism involved in the regulation of CSFV replication by ERS-mediated autophagy. Our findings will open new horizons for molecular mechanisms of sustainable replication and pathogenesis of CSFV, and lay a theoretical foundation for the development and implementation of ERS- and autophagy-targeting therapeutic strategies for clinical control of CSF.
Figure 9.
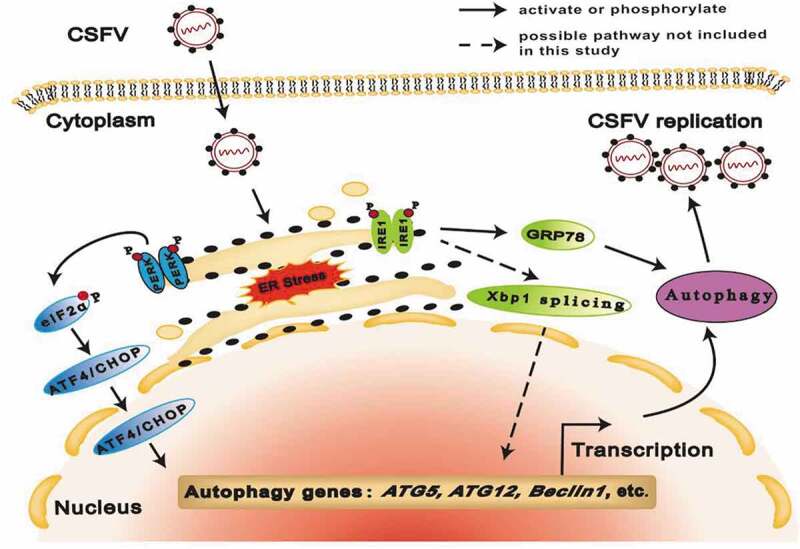
Proposed model for CSFV-induced autophagy via cellular PERK and IRE1 pathway. CSFV infection induces ERS and then triggers autophagy through the PERK-eIF2α-ATF4-CHOP and the IRE1/GRP78 pathway, thus facilitating viral replication in cultured cells
Discussion
The ERS-mediated autophagy can be a specific strategy hijacked by viruses for efficient infection [21,25]; nevertheless, its effects on viral replication and pathogenicity, as well as its underlying mechanisms differ in response to different virus infections. Flavivirus infections generally induce ERS and consequently activates the UPR pathways, due to their preferred replication at the ER-derived membranous structures [37–39]. Although HCV, DENV, JEV, and CSFV are all members of the Flaviviridae family, their potential mechanisms involved in the regulation of ERS-mediated autophagy and viral replication vary. We have previously shown that CSFV infection in vivo and in vitro causes ERS-mediated autophagy to promote viral replication [29]. Here we further revealed that CSFV infection induces ERS-driven activation of the PERK and IRE1 pathways, and then triggers autophagy to enhance viral replication. Our and other researchers’ findings together support the fact that, although the UPRs and autophagy generally function as a pro-survival mechanism of cells, many viruses have evolved specific strategies to regulate UPR-mediated autophagy for sustaining effective replication in host cells.
Generally, the PERK signaling is the preferentially activated pathway elicited by ERS [32]. Once activated, the phosphorylated PERK directly phosphorylates eIF2α, which leads to global attenuation of protein translation, thus potentially restraining viral replication [3,40]. However, the PERK pathway can be hijacked for autophagy induction and replication of viruses, including HCV, DENV, porcine circovirus type 2, porcine epidemic diarrhea virus, and Seneca valley virus [24,25,41–43]. We have previously reported that CSFV infection induces activation of the PERK-eIF2α-ATF4-CHOP pathway and complete autophagy both in vivo and in vitro [29,30]. Here we found that CSFV infection induces the PERK-eIF2α pathway-mediated autophagy to promote virus replication. It was also supposed that the PERK-eIF2α pathway selectively enhanced the expression of viral proteins for an efficient virus life cycle, although this remains further investigations. ATF4 and CHOP, two key transcription factors downstream of the PERK-eIF2α pathway, are selectively translated and typically regulate genes that encode ER chaperones, redox-, and autophagy-related proteins to restore ER homeostasis [44]. In our study, the PERK-eIF2α pathway-driven expressions of ATF4, CHOP, and autophagic proteins were observed in CSFV-infected cells, suggesting the possible involvement of ATF4 and CHOP in the ERS-mediated autophagy during CSFV infection, while the underlying mechanisms need to be identified further.
IRE1 is typically recognized as the most evolutionarily conserved ERS sensor [32]. The phosphorylated IRE1 splices the XBP1 mRNA to generate an active transcription factor XBP1s, which is involved in several pro-survival events in favor of protein homeostasis by inducing the expression of UPR-related genes [40,45]. The IRE1-XBP1s axis has also been shown to induce autophagy in endothelial cells [46]. However, IRE1 rather than XBP1 is essential for ATG5-dependent autophagy induced by IBV [47]. Our previous studies have shown that CSFV infection activates the IRE1-XBP1s pathway [29,30]. Here we further suggested the essential role of the IRE1-dependent autophagy in CSFV replication. It can be inferred from our and others’ studies that IRE1 plays vital roles in autophagy induction under virus infections or stress conditions.
GRP78, a major ER chaperone with anti-apoptotic abilities, is required for control of the ERS status and cellular homeostasis [48]. Diverse roles of GRP78 in the replication of different viruses have been reported [30,42,49]. Nevertheless, its underlying mechanisms remain largely unknown. Increasing evidence have shown that GRP78 is involved in autophagy induction in several cancer cells [50,51]; however, GRP78-mediated autophagy in the context of virus infections were rarely reported. Here we found that GRP78-mediated autophagy is required for CSFV replication. Since no present evidence has suggested the potential transcription factor activity of GRP78, the mechanism through which GRP78 influences the transcription and translation of autophagy-related genes remains unclear and needs further clarification.
A number of studies have revealed that complete autophagy was required for efficient replication of viruses including HCV, foot-and-mouth disease virus, and CSFV [52–54]. Here we found that activation of the PERK and IRE1 pathways are crucial for autophagic flux either by detecting p62 degradation or by monitoring fusion of autophagosomes with lysosomes in CSFV-infected cells. Our finding highlights the importance of the PERK and IRE1 pathways in CSFV-induced complete autophagy, which is similar to previous studies suggesting critical roles of the PERK and IRE1 pathways in the induction of complete autophagy [24,53]. Nevertheless, its underlying mechanisms need to be explored further.
Autophagy and apoptosis are two different but very important cross-talking processes in cells undergoing CSFV infection. Our previous studies have shown that CSFV infection in vitro and in vivo can induce autophagy [29,54,55]. Further studies suggest that autophagy induces apoptosis and death of T lymphocytes in the spleen of CSFV-infected pigs [55], which is potentially associated with CSFV-caused immunosuppression and pathogenesis in the infected pigs. However, our in vitro study reveals that CSFV induces autophagy to inhibit apoptosis by limiting reactive oxygen species (ROS)-dependent RIG-I-like receptor (RLR) signaling, thus facilitating viral persistent infection [56]. In a recently published paper, we have also suggested that lactate dehydrogenase B (LDHB) inhibition induces mitophagy to promote cell survival and virus replication by inhibiting apoptosis in cultured cells [57]. These findings may explain the reason why CSFV induces apoptosis in several types of tissue cells in infected pigs, while it is nonpathogenic to the cultured cells in vitro. It is likely that pathogenesis of CSFV is related to the relationship between autophagy and apoptosis during CSFV infection. Whether CSFV employs the ERS-mediated autophagy to enhance viral replication is associated with suppression of apoptosis is an intriguing assumption and remains to be studied further.
Accumulating studies have shown that cellular ERS promotes the expression of pro-inflammatory cytokines (such as IFN-γ and TNF-α) and then participates in the regulation of innate and adaptive immunity [33,34]. Both IFN-γ and TNF-α belong to the Th1-type cytokines and play significant roles in the regulation of cellular immune responses to intracellular microbial infections [58,59]. Cytokines have also been implicated in the regulation of autophagy. Th1-type cytokines such as IFN-γ and TNF-α generally induce autophagy in macrophages, while Th2-type cytokines (such as IL-4 and IL-13) often exert an inhibitory effect on autophagy [35,36,60]. Here we found that the PERK and IRE1 pathways play a crucial role in the transcriptional regulation of IFN-γ and TNF-α genes, suggesting a potential link between the PERK/IRE1-mediated autophagy and cellular immune regulation during CSFV infection, while the underlying mechanism needs further investigations. Whether CSFV employs the PERK/IRE1-mediated autophagy to sustain virus replication through regulation of expression of pro-inflammatory cytokines, also remains to be explored. Addressing these questions will shed new light on the development of new antiviral strategies for controlling CSF.
Acknowledgments
We thank professor Xinglong Yu (Hunan Agricultural University, China) very much for kindly providing the rabbit anti-CSFV Npro antibodies.
Funding Statement
This research was supported by grants from the National Key Research and Development Program of China [Nos. 2017YFD0500600 and 2016YFD0500700], the National Natural Science Foundation of China [Nos. 31472200, 31672590 and U1405216], the Science and Technology Program of Guangzhou, China [201803020005], the Key Research Projects of Universities in Guangdong Province [2019KZDXM026], the Science and Technology Program of Guangdong, China [2019B020211003], and the 111 Project [D20008].
Abbreviations
- ATF4/6
activating transcription factor 4/6
- ATG5/12
autophagy related 5/12
- BIX
BiP inducer X
- BSA
bovine serum albumin
- CCT
CCT020312
- CSF (V)
classical swine fever (virus)
- DDIT3/CHOP
DNA-damage inducible transcript 3
- DENV
dengue virus
- DMSO
dimethyl sulfoxide
- eIF2α
eukaryotic initiation factor 2α
- ER(S)
endoplasmic reticulum (stress)
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- GSK
GSK2606414
- HCV
hepatitis C virus
- hpi
hours post-infection
- HRP
horseradish peroxidase
- HSPA5/GRP78/BiP
heat shock protein family A member 5
- IFN-γ
interferon-γ
- IL-4/13
interleukin-4/13
- IRE1
inositol-requiring enzvme 1
- JEV
Japanese encephalitis virus
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MOI
multiplicity of infection
- mRFP
monomeric red fluorescent protein
- PERK
PKR-like ER kinase
- SQSTM1/p62
sequestosome 1
- SAL
salubrinal
- SiRNA
small interfering RNA
- TCID50
fifty pecent tissue culture infective dose
- TNF-α
tumor necrosis factor-α
- UPR
unfolded protein response
- XBP1(s)
(spliced) X-box binding protein 1
Disclosure statement
The authors declare that they have no conflict of interest.
References
- [1].Pires Da Silva J, Monceaux K, Guilbert A, et al. SIRT1 protects the heart from er stress-induced injury by promoting eEF2K/eEF2-dependent autophagy. Cells. 2020;9(2):426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mehrbod P, Ande SR, Alizadeh J, et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence. 2019;10(1):376–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. 2017;13(11):681–696. [DOI] [PubMed] [Google Scholar]
- [4].Kapoor A, Sanyal AJ. Endoplasmic reticulum stress and the unfolded protein response. Clin Liver Dis. 2009;13(4):581–590. [DOI] [PubMed] [Google Scholar]
- [5].Kong FJ, Ma LL, Guo JJ, et al. Endoplasmic reticulum stress/autophagy pathway is involved in diabetes-induced neuronal apoptosis and cognitive decline in mice. Clin Sci (Lond). 2018;132(1):111–125. [DOI] [PubMed] [Google Scholar]
- [6].Iurlaro R, Muñoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. Febs J. 2016;283(14):2640–2652. [DOI] [PubMed] [Google Scholar]
- [7].Wang S, Binder P, Fang Q, et al. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. Br J Pharmacol. 2018;175(8):1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Forman MS, Lee VM, Trojanowski JQ. ‘Unfolding’ pathways in neurodegenerative disease. Trends Neurosci. 2003;26(8):407–410. [DOI] [PubMed] [Google Scholar]
- [9].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. [DOI] [PubMed] [Google Scholar]
- [10].Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20(3):460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Deretic V, Klionsky DJ. Autophagy and inflammation: a special review issue. Autophagy. 2018;14(2):179–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gao L, Jauregui CE, Teng Y. Targeting autophagy as a strategy for drug discovery and therapeutic modulation. Future Med Chem. 2017;9(3):335–345. [DOI] [PubMed] [Google Scholar]
- [14].Liu Y, Shoji-Kawata S, Sumpter RM Jr., et al. Autosis is a Na+, K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110(51):20364–20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22(3):367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Levine B. Cell biology: autophagy and cancer. Nature. 2007;446(7137):745–747. [DOI] [PubMed] [Google Scholar]
- [17].Sayak KM, Chunjuan S, Xiaoping Q, et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014;10(11):1989–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mokarram P, Ahmadi M. Autophagy and cancer research in Iran. Autophagy. 2019;15(11):2039–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. [DOI] [PubMed] [Google Scholar]
- [20].Bhardwaj M, Leli NM, Koumenis C, et al. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin Cancer Biol. 2019. (Online ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rashid HO, Yadav RK, Kim HR, et al. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zaouali MA, Boncompagni E, Reiter RJ, et al. AMPK involvement in endoplasmic reticulum stress and autophagy modulation after fatty liver graft preservation: a role for melatonin and trimetazidine cocktail. J Pineal Res. 2013;55(1):65–78. [DOI] [PubMed] [Google Scholar]
- [23].Ding W, Ni H, Gao W, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282(7):4702–4710. [DOI] [PubMed] [Google Scholar]
- [24].Dash S, Chava S, Aydin Y, et al. Hepatitis C virus infection induces autophagy as a prosurvival mechanism to alleviate hepatic ER-stress response. Viruses. 2016;8(5):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee YR, Kuo SH, Lin CY, et al. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci Rep. 2018;8(1):489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sharma M, Bhattacharyya S, Sharma KB, et al. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J Gen Virol. 2017;98(5):1027–1039. [DOI] [PubMed] [Google Scholar]
- [27].Moennig V. The hog cholera virus. Comp Immunol Microbiol Infect Dis. 1992;15(3):189–201. [DOI] [PubMed] [Google Scholar]
- [28].Lohse L, Nielsen J, Uttenthal A. Early pathogenesis of classical swine fever virus (CSFV) strains in Danish pigs. Vet Microbiol. 2012;159(3–4):327–336. [DOI] [PubMed] [Google Scholar]
- [29].Zhu E, Chen W, Qin Y, et al. Classical swine fever virus infection induces endoplasmic reticulum stress-mediated autophagy to sustain viral replication in vivo and in vitro. Front Microbiol. 2019;10:2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].He W, Xu H, Gou H, et al. CSFV infection up-regulates the unfolded protein response to promote its replication. Front Microbiol. 2017;8:2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27(3):493–497. [Google Scholar]
- [32].Fung TS, Torres J, Liu DX. The emerging roles of viroporins in ER stress response and autophagy induction during virus infection. Viruses. 2015;7(6):2834–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Reverendo M, Mendes A, Arguello RJ, et al. At the crossway of ER-stress and proinflammatory responses. Febs J. 2019;286(2):297–310. [DOI] [PubMed] [Google Scholar]
- [34].Di Conza G, Ho PC. ER stress responses: an emerging modulator for innate immunity. Cells. 2020;9(3):695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ge Y, Huang M, Yao YM. Autophagy and proinflammatory cytokines: interactions and clinical implications. Cytokine Growth Factor Rev. 2018;43:38–46. [DOI] [PubMed] [Google Scholar]
- [36].Yuan Y, Ding D, Zhang N, et al. TNF-alpha induces autophagy through ERK1/2 pathway to regulate apoptosis in neonatal necrotizing enterocolitis model cells IEC-6. Cell Cycle. 2018;17(11):1390–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Su H, Liao C, Lin Y. Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J Virol. 2002;76(9):4162–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ambrose RL, Mackenzie JM. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J Virol. 2011;85(6):2723–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pena J, Harris E. Dengue virus modulates the unfolded protein response in a time-dependent manner. J Biol Chem. 2011;286(16):14226–14236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Renata S, Reed JC. ER stress-induced cell death mechanisms. BBA-Mol Cell Res. 2013;1833(12):3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hou L, Dong J, Zhu S, et al. Seneca valley virus activates autophagy through the PERK and ATF6 UPR pathways. Virology. 2019;537:254–263. [DOI] [PubMed] [Google Scholar]
- [42].Zhou Y, Qi B, Gu Y, et al. Porcine circovirus 2 deploys PERK pathway and GRP78 for its enhanced replication in PK-15 Cells. Viruses. 2016;8(2):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zou D, Xu J, Duan X, et al. Porcine epidemic diarrhea virus ORF3 protein causes endoplasmic reticulum stress to facilitate autophagy. Vet Microbiol. 2019;235:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13(8):477–491. [DOI] [PubMed] [Google Scholar]
- [45].Li S, Kong L, Yu X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit Rev Microbiol. 2015;41(2):150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Margariti A, Li H, Chen T, et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem. 2013;288(2):859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fung TS, Liu DX. The ER stress sensor IRE1 and MAP kinase ERK modulate autophagy induction in cells infected with coronavirus infectious bronchitis virus. Virology. 2019;533:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shima K, Klinger M, Schütze S, et al. The role of endoplasmic reticulum-related BiP/GRP78 in interferon gamma-induced persistent Chlamydia pneumoniae infection. Cell Microbiol. 2015;17(7):923–934. [DOI] [PubMed] [Google Scholar]
- [49].Win NN, Kanda T, Nakamoto S, et al. Inhibitory effect of Japanese rice-koji miso extracts on hepatitis A virus replication in association with the elevation of glucose-regulated protein 78 expression. Int J Med Sci. 2018;15(11):1153–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen S, Wu J, Jiao K, et al. MicroRNA-495-3p inhibits multidrug resistance by modulating autophagy through GRP78/mTOR axis in gastric cancer. Cell Death Dis. 2018;9(11):1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang Y, Wu H, Li Z, et al. A positive feedback loop between GRP78 and VPS34 is critical for GRP78-mediated autophagy in cancer cells. Exp Cell Res. 2017;351(1):24–35. [DOI] [PubMed] [Google Scholar]
- [52].Wang J, Kang R, Huang H, et al. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy. 2014;10(5):766–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sun P, Zhang S, Qin X, et al. Foot-and-mouth disease virus capsid protein VP2 activates the cellular EIF2S1-ATF4 pathway and induces autophagy via HSPB1. Autophagy. 2018;14(2):336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pei J, Zhao M, Ye Z, et al. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy. 2014;10(1):93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gou H, Zhao M, Fan S, et al. Autophagy induces apoptosis and death of T lymphocytes in the spleen of pigs infected with CSFV. Sci Rep. 2017;7(1):13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Pei J, Deng J, Ye Z, et al. Absence of autophagy promotes apoptosis by modulating the ROS-dependent RLR signaling pathway in classical swine fever virus-infected cells. Autophagy. 2016;12(10):1738–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fan S, Wu K, Zhao M, et al. LDHB inhibition induces mitophagy and facilitates the progression of CSFV infection. Autophagy. 2020:1–20. DOI: 10.1080/15548627.2020.1823123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–342. [DOI] [PubMed] [Google Scholar]
- [59].Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol. 2017;17(1):49–59. [DOI] [PubMed] [Google Scholar]
- [60].Subbian S, O’Brien P, Kushner NL, et al. Molecular immunologic correlates of spontaneous latency in a rabbit model of pulmonary tuberculosis. Cell Commun Signal. 2013;11(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]