

ABSTRACT
Aspergillus flavus (A. flavus) is one of the most important model environmental fungi which can produce a potent toxin and carcinogen known as aflatoxin. Aflatoxin contamination causes massive agricultural economic loss and a critical human health issue each year. Although a functional vacuole has been highlighted for its fundamental importance in fungal virulence, the molecular mechanisms of the vacuole in regulating the virulence of A. flavus remain largely unknown. Here, we identified a novel vacuole-related protein in A. flavus, the ortholog of phosphatidylinositol-3-phosphate-5-kinase (Fab1) in Saccharomyces cerevisiae. This kinase was located at the vacuolar membrane, and loss of fab1 function was found to affect the growth, conidia and sclerotial development, cellular acidification and metal ion homeostasis, aflatoxin production and pathogenicity of A. flavus. Further functional analysis revealed that Fab1 was required to maintain the vacuole size and cell morphology. Additional quantitative proteomic analysis suggested that Fab1 was likely to play an important role in maintaining vacuolar/cellular homeostasis, with vacuolar dysregulation upon fab1 deletion leading to impaired aflatoxin synthesis in this fungus. Together, these results provide insight into the molecular mechanisms by which this pathogen produces aflatoxin and mediates its pathogenicity, and may facilitate dissection of the vacuole-mediated regulatory network in A. flavus.
KEYWORDS: Aspergillus flavus, Fab1, vacuole, aflatoxin production, pathogenicity
Introduction
Aspergillus flavus (A. flavus) is one of the most important species in the Aspergillus genus because it can cause both noninvasive and invasive systematic aspergillosis in immunocompromised individuals, animals and economically important crops [1,2]. A. flavus produces a range of potent carcinogens and toxins collectively known as aflatoxins, creating concerns regarding their potential for environmental contamination [3]. It has been estimated that approximately 10% of food samples collected between 2007 and 2012 were estimated to contain aflatoxins [4]. When these foods are ingested or when A. flavus is able to grow without immunological constraint, acute toxicity in addition to a long-term increase in cancer risk can occur in affected individuals [5]. Owing to the economic and food safety issues in agriculture and medicine, much attention has been directed at the study of aflatoxin synthesis and pathway regulation through the genetics, molecular biology and biochemistry methods. Understanding the molecular mechanisms of aflatoxin biosynthesis and pathway regulation will enable the development of innovative approaches for reducing aflatoxin production in important agricultural crops and human exposure to aflatoxins in populations at high risk for aflatoxicosis. Although a wide variety of factors can induce or inhibit aflatoxin synthesis in Aspergillus [6–10], the detailed regulatory mechanisms controlling aflatoxin production in A. flavus are still fragmentary.
Recent studies have shown that fungal vacuoles/vesicles are versatile organelles, involved in regulating various cellular processes [11]. Mutations affecting vacuole function and vacuole membrane homeostasis can lead to defects in fungal virulence [12–14]. In Candida albicans, mutations of a subset of vacuolar proteins affected hyphal morphogenesis, growth, and virulence [15]. Additionally, a lack of the vacuolar transport chaperone Vtc4 in Ustilago maydis significantly decreased its virulence on maize [16], and deletion of the vesicle-vacuole fusion protein Vb1 influences aflatoxin synthesis in A. parasiticus [17]. Notably, previous efforts have focused on the role of vacuoles and vesicles in the secondary metabolism of A. flavus [17], suggesting that a functional vacuole plays an important role in fungal growth and virulence. However, very little is known about the regulatory mechanism of vacuoles in regulating Aspergillus metabolism, particularly the functions of vacuole-related genes.
Accumulating evidence has revealed that phosphatidylinositol lipids play important roles in various cellular processes [18]. Among them, phosphatidylinositol 3,5-bisphosphate (PtdIns-3,5-P2), which predominantly locates in the endosome and vacuole, has been implicated in membrane homeostasis in various types of eukaryotic cells [19,20]. PtdIns-3,5-P2 is generated from phosphatidylinositol 3-phosphate (PI-3-P) by the phosphoinositide phosphate kinase Fab1. Previous effects have stated that Fab1 kinase is highly conserved in many fungi and may play an important regulatory role in maintaining the normal vacuole function and morphology by regulating the PtdIns-3,5-P2 levels in phosphatidylinositol metabolism [21,22]. The ability of Fab1 to regulate vacuole size, protein sorting, osmotic balance and the trafficking of some substrates in yeast has been well established [19,20]. Notably, defects in the dynamic regulation of PtdIns-3,5-P2 are linked to human diseases [22]. In the human pathogenic fungus C. glabrata, PtdIns-3,5-P2 levels regulated by Fab1 are closely related to membrane trafficking and vacuole homeostasis, and C. glabrata Fab1 plays important roles in the intracellular survival and virulence of this fungus [23]. Despite the studies of Fab1 having had some success, the molecular details of how this enzyme regulates vacuolar functions and aflatoxin synthesis in Aspergillus are still lacking.
To fill this gap, we have made an attempt to unravel the functional role of the A. flavus putative orthologue of S. cerevisiae PtdIns-3,5-P2 kinase Fab1 in a variety of cellular processes. Our functional studies revealed that deletion of Fab1 influences the growth, development, aflatoxin production, pathogenicity, and vacuole/vesicle function of A. flavus. Further quantitative proteomic analysis suggested that Fab1 may maintain vacuolar/cellular homeostasis to facilitate aflatoxin synthesis in A. flavus. Together, these results provide insight into the mechanisms of A. flavus pathogenicity and aflatoxin biosynthesis.
Results
Discovery and validation of a novel vacuole-related protein
Through NCBI BLAST (Basic Local Alignment Search Tool) searching, we identified an A. flavus protein with significant similarity to yeast (Saccharomyces cerevisiae) PtdIns-3,5-P2 kinase (Fab1 protein) (Fig. S1). The coordinates of the putative fab1 were 4,215,543–4,220,895 from NW_002477237.1 fragment in the A. flavus genome, with no annotation information (Fig. S2). To further confirm the existence of the putative fab1, we performed transcriptional expression analysis by using previously published RNA-Seq data. As shown in Figure S2, the additional transcriptomic data also supported the presence of this novel gene in the A. flavus genome. Notably, a highly similar homolog of this protein was discovered in A. oryzae and designated as a putative phosphatidylinositol-3-phosphate 5-kinase (Fig. S3A). Moreover, analysis of the protein structural domain revealed that this family of proteins contains a number of highly conserved domains, including a zinc finger structure (FYVE), as well as Cpn60_TCP1 and phosphatidylinositol phosphate kinase domains (PIPKc) (Figure 1(a)), which correspond to phosphatidylinositol phosphate kinases in most eukaryotic cells [19]. These analyses thus suggested that this novel protein may function as a phosphatidylinositol phosphate kinase (named as Fab1) involved in phosphatidylinositol metabolism. We hypothesized that Fab1 in A. flavus may catalyze the synthesis of phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,5-bisphosphate, which are essential lipid molecules in a range of cellular processes [21]. Phylogenetic analysis further revealed that this protein was highly conserved among different species, including Aspergillus species, Elaphomyces granulatus, Penicillium arizonense, Penicillium coprophilum, Penicillium polonicum, Penicillium vulpinum and Talaromyces atroroseus (Fig. S3B). To further validate the novel protein, we assessed fab1 expression at the transcript level and our result revealed significant up-regulation under three distinct growth conditions (Figure 1(b)).
Figure 1.
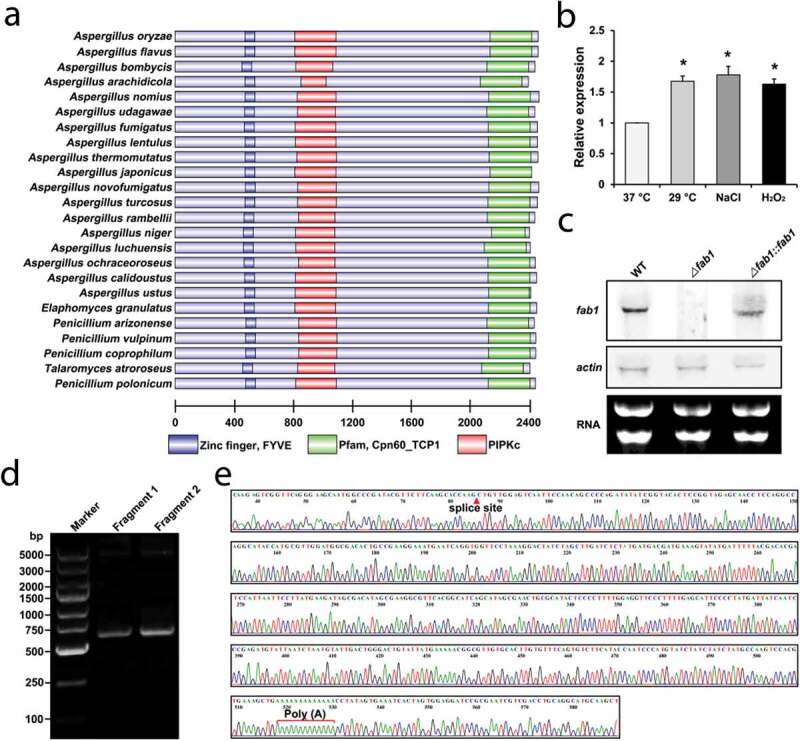
Fab1 identification and validation. (a) Domain analysis of Fab1 in different species. (b) Relative fab1 mRNA levels under different growth conditions. Data are presented as means ±S.D. and represent the results of three independent experiments. Statistically significant differences are indicated: *p < 0.05. (c) Validation of fab1 transcript quantification via northern blotting. (d) PCR validation of fab1 fragment clones by 3’RACE. The second round of PCR was performed with the gene-specific forward primers and the RACE adaptor primers. The PCR products were analyzed by electrophoresis. Fragment1 and Fragment2 are two independent biological replicates. (e) Sequencing validation of amplified fragment from fab1 by 3’RACE. The amplified fragment by 3’RACE included the splice site and ploy (A) of fab1.
Fab1 regulates growth, conidia, and sclerotial development
To elucidate the functional role of Fab1, we disrupted fab1 by gene replacement mutagenesis using a fusion PCR approach (Fig. S4A). Mutant (∆fab1) and complementation (∆fab1::fab1) strains of A. flavus were generated and validated by PCR and sequencing (Fig. S4B). We also confirmed whether fab1 was expressed in these strains via northern blotting (Figure 1(c)). In order to verify the fab1 3’ untranslated region (UTR), total A. flavus RNA was subjected to a 3’-RACE (rapid amplification of cDNA ends) experiment and the resultant 3’-RACE PCR product was sequenced to verify the splicing sites and poly(A) tail of fab1 (Figure 1(d,e)).
We next measured the growth and morphology of WT, ∆fab1 and ∆fab1::fab1 strains of A. flavus grown on yeast extract-sucrose agar (YES) and potato dextrose agar (PDA) agar media. After growth on YES and PDA media for 5 days, the ∆fab1 strain exhibited smaller diameter colonies relative to the WT and ∆fab1::fab1 strains (Figure 2(a,b) and Fig. S5A, B), suggesting that fab1 deletion reduced the growth rate of this fungus. Consistently, the ∆fab1 strain failed to form conidiophores according to microscopic examination, whereas this effect was restored in the ∆fab1::fab1 strain (Figure 2(c) and Fig. S5C), suggesting that fab1 is important for conidial formation in A. flavus. We additionally confirmed this result via measuring the expression of two transcription factor-encoding genes important for conidiation (brlA and abaA) [24,25] via qPCR during vegetative growth. We found the expression of both of these genes was significantly reduced by >90% in the ∆fab1 strain relative to in the WT and ∆fab1::fab1 strains (Fig. S5D). These results thus indicated that the conidiation pathway was affected by fab1 deletion, leading to downregulation of brlA and abaA expression.
Figure 2.
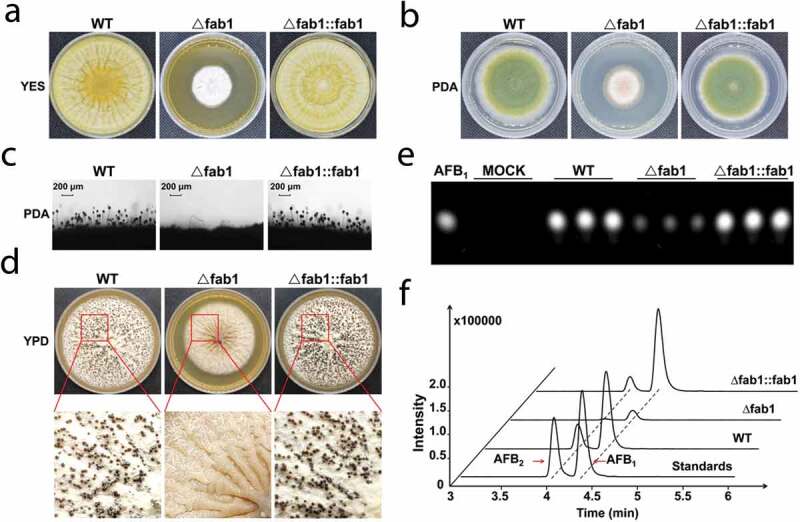
Morphological phenotypes of different A. flavus strains. Colony morphology of WT, ∆fab1 and ∆fab1::fab1 strains grown on YES (a) or PDA (b) media at 37°C for 5 days. (c) Conidiophores morphology of WT, ∆fab1 and ∆fab1::fab1 strains following a 12 h incubation. (d) Phenotypic characterization of WT, ∆fab1 and ∆fab1::fab1 strains grown on YPD medium at 37°C for 9 days. (e) TLC assay-mediated assessment of aflatoxin B1 (AFB1) production by the WT, ∆fab1 and ∆fab1::fab1 strains in YES liquid media cultured at 37°C for 5 days. (f) MS analysis of aflatoxin production by WT, ∆fab1 and ∆fab1::fab1 strains. All data represent the results of three independent experiments
As sclerotia are important means of resisting stress conditions, we assessed sclerotial formation when the strains were grown on sclerotia-inducing YPD medium at 37°C for 9 days. Unlike in the WT strain, sclerotia in the ∆fab1 strain were completely absent; this phenotype was reversed in the ∆fab1::fab1 strain (Figure 2(d) and Fig. S5E), suggesting that a loss of fab1 led to impairing sclerotial formation. We additionally assayed the expression of the sclerotia-associated nsdC, nsdD and nsdR and found that both nsdC and nsdR were twofold downregulated in the ∆fab1 strain, while nsdD was downregulated by 10-fold relative to in the WT and ∆fab1::fab1 strains (Fig. S5F). These results thus together clearly indicated that Fab1 is essential for A. flavus growth, conidial formation, and sclerotial production.
Fab1 is essential for aflatoxin biosynthesis and pathogenicity
The synthesis of phosphatidylinositol bisphosphate by Fab1 on the vacuole has the potential to mediate homeostatic regulation of the vacuolar membrane and to regulate proximal ion channels within these vacuoles [26,27]. As such, we hypothesized that vacuolar dysregulation arising as a result of the loss of fab1 would result in impaired aflatoxin export. To test this hypothesis, we assessed aflatoxin production via thin layer chromatography (TLC) and mass spectrometry (MS) analysis. TLC analyses indicated markedly reduced aflatoxin B1 production from the ∆fab1 strain relative to from the WT and ∆fab1::fab1 strains (Figure 2(e)), with the levels reduced by >10-fold relative to the WT strain (Fig. S5G). We then validated this result via high-performance liquid chromatography (HPLC) coupled to a Xevo TQ MS machine. This analysis confirmed that only trace levels of aflatoxin B1 were detectable and minimal aflatoxin B2 was evident in the ∆fab1 strain, whereas normal production of these aflatoxins was observed for the WT and ∆fab1::fab1 strains (Figure 2(f) and Fig. S5H). This thus further suggested that Fab1 was likely to influence the biosynthesis of aflatoxins and their consequent export by controlling vacuolar membrane homeostasis.
As aflatoxin contamination of food sources is a significant source of concern, we next explored the impact of fab1 deletion on the pathogenicity of A. flavus. We observed difference growth dynamics for these strains when they were used to inoculate peanut seeds; the growth, sporulation, and invasion of the ∆fab1 strain were markedly less effective than in the WT and ∆fab1::fab1 strains (Figure 3(a)). Consistent with our previous results, the ∆fab1 strain tended to produce fewer conidia than the WT and ∆fab1::fab1 strains (Figure 3(b)). Previous studies showed that aflatoxin is important for seed infection [28–30]. We subsequently assessed aflatoxin production in these samples, revealing that the WT and ∆fab1::fab1 strains, but not the ∆fab1 strain produced aflatoxin B1 (Figure 3(c)). Further MS analyses confirmed that the ∆fab1 strain failed to produce detectable aflatoxins, whereas normal aflatoxin production was evident for the WT and ∆fab1::fab1 strains (Figure 3(d)). We similarly analyzed the invasive growth of these A. flavus strains on maize seeds, revealing that aflatoxin production and conidial formation were significantly reduced for the ∆fab1 strain relative to the WT and ∆fab1::fab1 strains (Figure 3(e–h)). Together, these findings indicated that fab1 in A. flavus may play a regulatory role in seed infection.
Figure 3.
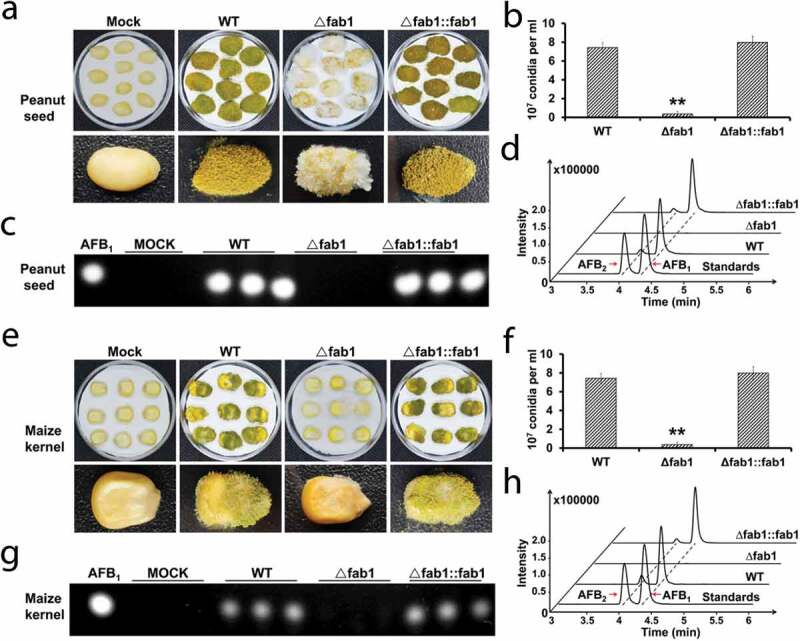
Infection analysis of seeds with different A. flavus strains. (a) Host colonization of WT, ∆fab1 and ∆fab1::fab1 strains. Peanut seeds were inoculated with WT, ∆fab1 and ∆fab1::fab1 strains and cultured at 29°C for 7 days. (b) Conidia amounts produced from the infected peanut seeds. (c) TLC assay results for aflatoxin B1 (AFB1) levels in extracts from infected peanut seeds. (d) MS analysis of aflatoxin production from infected peanut seeds. (e) Maize seeds were inoculated with WT, ∆fab1, or ∆fab1::fab1 strains and cultured at 29°C for 7 days. (f) Conidia amounts produced from the infected maize seeds. (g) TLC was used to detect the aflatoxin B1 (AFB1) production in the infected maize seeds. (h) MS analysis of aflatoxin production from the infected maize seeds. Data are presented as means ±S.D. and represent the results of three independent experiments. **p < 0.01
It has been reported that enzymes involved in aflatoxin biosynthesis including Nor-1, Ver-1, and OmtA are localized to the vacuoles and vesicles in A. parasiticus [31]. Thus, we hypothesized that fab1 deletion would similarly impact aflatoxin biosynthesis in A. flavus. By staining these cells with neutral red dye, while we observed normal vesicle formation in the WT strain, we saw no vesicles formation in ∆fab1 cells when grown on aflatoxin-inducing medium (YES) for 3 days (Figure 4(a) and B). To further confirm this finding, we isolated the vesicle-vacuole fraction from A. flavus cells as previously described [32]. We then performed TLC to measure aflatoxin levels in these subcellular fractions, further confirming that fab1 deletion led to a near total loss of aflatoxin levels in this vesicle-vacuole fraction. Our results suggested that aflatoxins biosynthesis and transport may be disrupted in the ∆fab1 strain (Figure 4(c) and Fig. S6). This same finding was also evident in the cytosolic and secreted fractions obtained from these cells. Based on these findings and previous reports, we, therefore, concluded that Fab1 is likely to play a central role in maintaining vacuolar/cellular homeostasis, with vacuolar dysregulation upon fab1 deletion leading to impaired aflatoxin synthesis, storage, and export, in agreement with previous reports [17].
Figure 4.
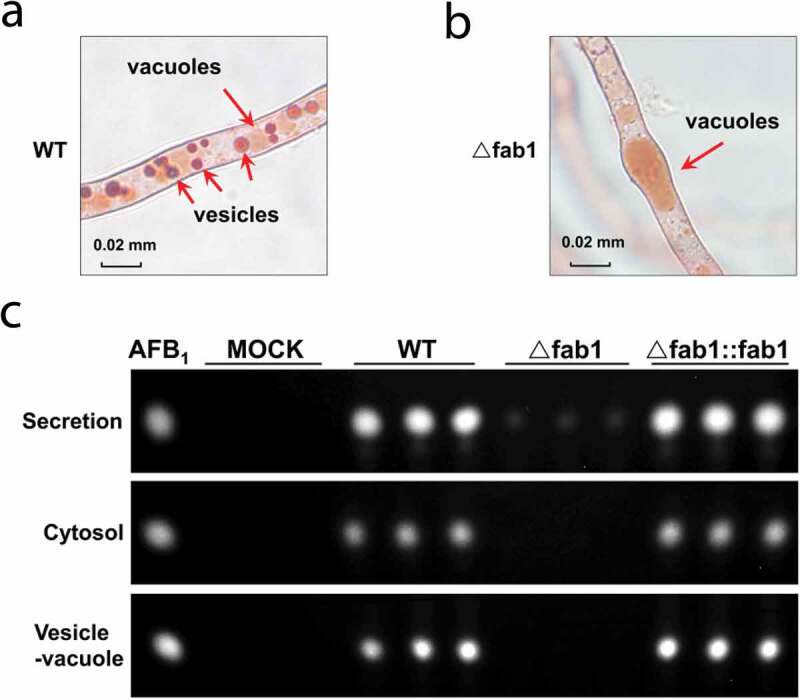
Aflatoxin production from different fraction of A. flavus strains. (a-b) Microscopic analysis of vacuole-vesicles stained with neutral red commonly used for staining of the vacuoles in vivo. (c) TLC assessment of aflatoxin B1 (AFB1) production from different fractions of WT, ∆fab1, and ∆fab1::fab1 strains
Deletion of fab1 impacts vacuolar phenotype
In yeast, Fab1 is associated with the vacuolar and pre-vacuolar compartments [33]. In order to assess Fab1 subcellular localization in A. flavus, we first produced a Fab1-GFP fusion protein that was validated by PCR (Fig. S7A) and western-blotting analysis with an anti-GFP antibody (Fig. S7B). Subsequently, the WT strain expressing the Fab1-GFP fusion protein was stained with the membrane-specific dye FM4-64. As illustrated in Figure 5(a), strong fluorescence was detected in the fungal cells by laser scanning confocal microscopy (LSCM) analysis during conidial germination and hyphal growth. Notably, the green fluorescence signals (indicated by GFP) were clearly observed on the vacuolar membrane of the wild-type strain, whereas hyphal cells also showed obvious red signals (fluorescence of FM4-64) on the vacuolar membrane when the strains were stained with the membrane-specific dye. As a result, these results indicated that Fab1 protein is localized to the vacuolar membrane (Figure 5(a)), in agreement with previous fungal studies [33].
Figure 5.
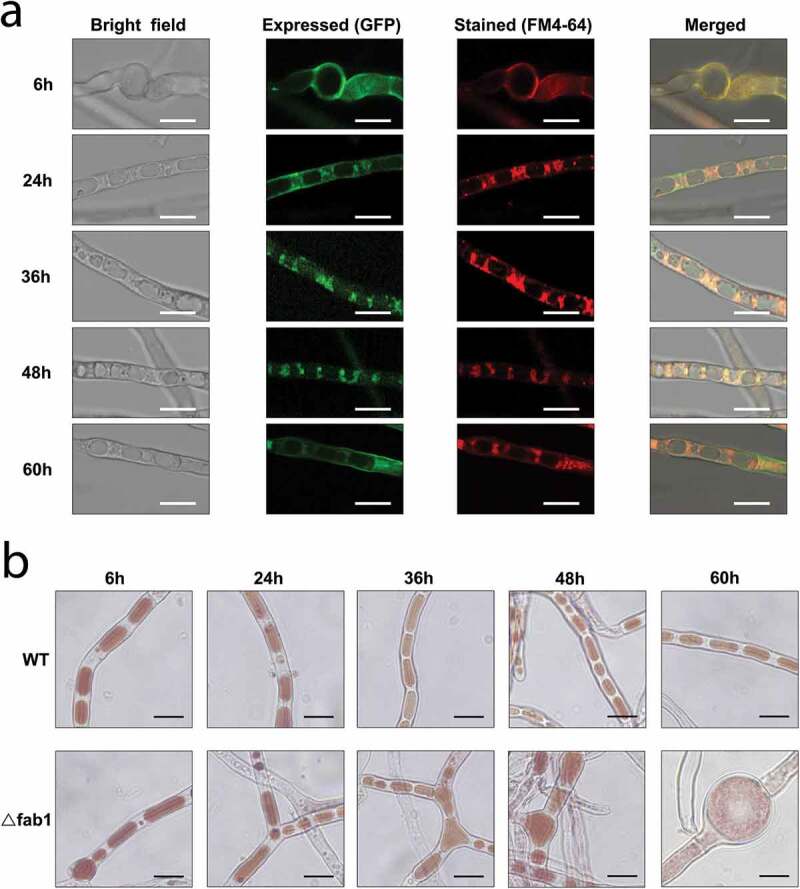
Localization of Fab1 in A. flavus strains. (a) Microscopic images of the WT strains expressing Fab1-GFP fusion protein. The strains were cultured in YES medium for 6, 24, 36, 48 and 60 h. The vacuolar membranes were stained with fluorochrome FM4-64. The bright, expressed, stained and merged images were shown. Scale bars represented 15 µm. (b) Bright-field microscopy of WT and ∆fab1 strains stained with neutral red commonly used for staining of the vacuole in vivo. The WT and ∆fab1 strains were cultured in YES medium for 6, 24, 36, 48 and 60 h. Scale bars represented 15 µm
Vacuoles were previously shown to be essential for fungal growth, differentiation, symbiosis, and pathogenesis [34]. We observed marked increases in the vacuole size in ∆fab1 fungi, with the vacuoles exhibiting swelling to fill most of the cell volume when the strains were grown under aflatoxin inducing conditions (YES) (Fig. S8). We next confirmed this vacuolar phenotype via staining the cells with neutral red dye when the cells were grown on aflatoxin-inducing medium (YES) during different growth stages. As shown in Figure 5(b), we observed the presence of significantly more large vacuoles in the ∆fab1 strain relative to in the WT control during conidial germination and hyphal growth. Surprisingly, the vacuole occupied most of the total volume of the fungal cell in ∆fab1 strain grown on YES medium for 60 h. The substantial vacuolar enlargement observed in the ∆fab1 strain had the potential to drive substantial cellular swelling. Our results suggested that the formation of large vacuoles was linked to fab1 deletion, and the swelling was presumably associated with the well-documented role of proteins in this family as regulators of vacuolar/cellular homeostasis and turnover of the vacuolar membrane [33,34]. We speculated that Fab1 is vital for normal vacuole function and morphology in A. flavus.
Deletion of fab1 impacts cellular acidification and metal ion homeostasis
Previous studies have proven that fungal vacuoles are acidic organelles, involved in many aspects of cellular processes, including cellular homeostasis, membrane trafficking, signaling, and nutrition, as well as stress responses [34]. To further determine whether deletion of fab1 would affect the cellular acidification, we measured the vacuolar and cytosolic pH in the WT, ∆fab1 and ∆fab1::fab1 strains. These strains were stained with a specific fluorescent dye (BCECF, 2,7-bis(2-carboxyethyl)-5,6-carboxyfluorescein-acetoxymethyl ester) to compare the changes of pH in different strains, and the vacuolar pH was quantified from the collected hyphal cells as previous reports [35]. As expected, the vacuolar pH in the stained ∆fab1 strain was quantified as 5.76, which was significantly more acidic than that of the WT (pH 6.13) and ∆fab1::fab1 (pH 6.13) strains (p < 0.01) (Figure 6(a–c)). Furthermore, we observed the similar changes of the cytosolic pH in the WT (pH 7.15), ∆fab1 (pH 5.79) and ∆fab1::fab1 (pH 6.95) strains (Figure 6(a–c)). These results suggested a significant acidification of vacuole and cytoplasm in the absence of fab1.
Figure 6.
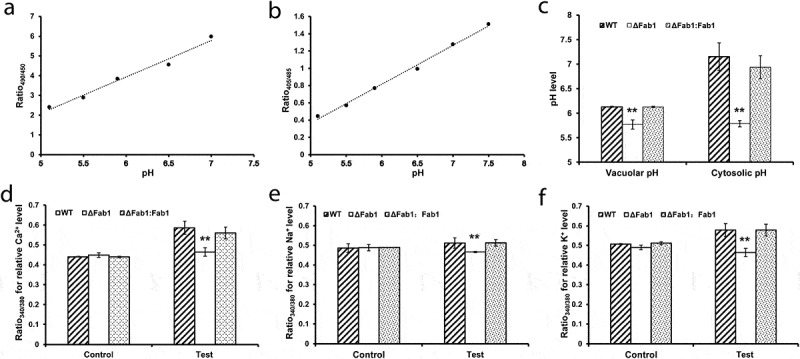
Impacts of fab1 deletion on cellular acidification and metal ion homeostasis. (a) Calibration curve showing the ratio of fluorescence intensity from excitation at 490 nm to intensity from excitation at 450 nm at different pH values. (b) Calibration curve showing the ratio of fluorescence intensity from excitation at 405 nm to intensity from excitation at 485 nm at different pH values. Wild-type cells were stained with BCECF-AM fluorescent dye and then equilibrated with buffers at varied pH for 60 min. A linear trendline is shown. (c) Vacuolar and cytosolic pH in WT, ∆fab1 and ∆fab1::fab1 strains. All strains were stained with BCECF-AM fluorescent dye and the fluorescence ratios were measured and converted to pH using the calibration curves. (d-f) Relative levels of intracellular Ca2+, Na+ and K+ in WT, ∆fab1 and ∆fab1::fab1 strains. All strains were stained with the specific fluorescent indicator dyes Fura-2-AM, SBFI-AM and PBFI-AM, respectively. The relative concentrations of Ca2+, Na+ and K+ in the cytoplasm were calculated based on the fluorescence ratio (Ratio 340/380). The unstained strains were used as controls. Data are presented as means ±S.D. and represent the results of three independent experiments. **p < 0.01
In addition, cellular metal ions (Ca2+, Na+ and K+) in the WT, ∆fab1 and ∆fab1::fab1 strains were assessed by using specific fluorescent indicator dyes, including Ca2+-specific fluorescent probe Fura-2-AM (Fura-2 acetoxymethyl ester), Na+-specific fluorescent probe SBFI-AM (sodium-binding benzofuran isophthalate acetoxymethyl ester) and K+-specific fluorescent probe PBFI-AM (potassium-binding benzofuran isophthalate acetoxymethyl ester), according to previously described methods [36–39]. Upon binding metal ions, these specific fluorescence dyes can exhibit a fluorescence absorption shift from 380 to 340 nm of excitation. As a result, the relative Ca2+, Na+ and K+ concentrations were evaluated and quantified as the ratio of fluorescence intensities assessed at the excitation wavelengths of 340/380 nm. As shown in Figure 6(d–f), we observed obvious differences of intracellular metal ions concentrations in the WT, ∆fab1 and ∆fab1::fab1 strains. The ∆fab1 strain showed significantly decreased relative levels of intracellular Ca2+, Na+ and K+ concentrations compared to the WT and ∆fab1::fab1 strains (p < 0.01), indicating significant changes in cellular homeostasis in the absence of fab1. Together, our observations suggested that fab1 deletion resulted in severe defect in vacuolar and cellular acidification, as well as cellular metal ion homeostasis.
Identification of proteins regulated by Fab1
A variety of methodologies now support relative quantification measurements in proteomics, and one of the most commonly used strategies through MS is stable isotope labeling of peptides using isobaric reagents, such as tandem mass tag (TMT). To this end, we next examined the molecular basis of Fab1 function by TMT-based quantitative proteomic analysis to identify proteins regulated by Fab1 (Figure 7(a)). Finally, a total of 4,928 proteins were identified by proteomic analysis. Among them, 4,292 proteins were identified based upon the presence of at least two unique peptides and 3,821 proteins (>89%) were quantifiable (Figure 7(b) and Table S1). The large number of identified proteins reproducibly quantified in all samples led to very strong correlations for these fungal strains (Figure 7(c)). We constructed a heatmap showing the relative levels of quantifiable proteins in the WT and ∆fab1 A. flavus strains, with 289 proteins exhibiting a > 1.2-fold change between samples (∆fab1/WT) (Figure 7(d) and Table S2). Of these differentially expressed proteins (DEPs), 163 were up-regulated and 126 were down-regulated.
Figure 7.
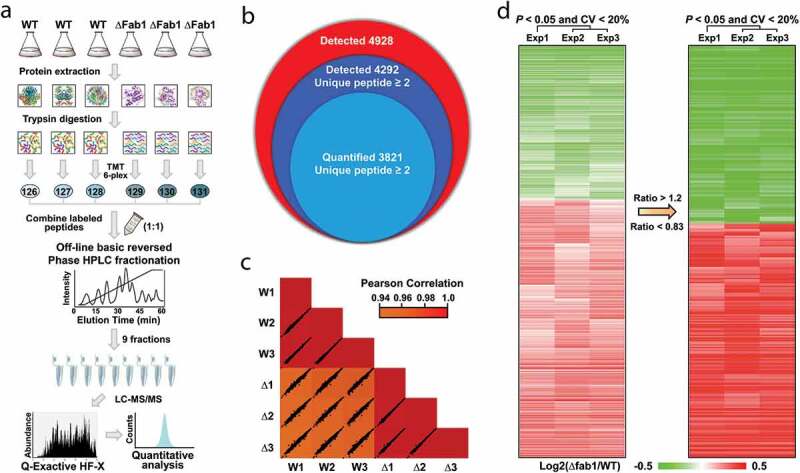
TMT-based quantitative proteomic analysis. (a) TMT strategy overview. (b) Venn diagram of identified and quantified proteins. (c) Comparison of TMT-based quantification intensities within three replicates, with correlations calculated via a Pearson correlation analysis. (d) Heatmap showing the ratio of quantifiable proteins between WT with Δfab1 strains
Functional annotation and cellular localization of Fab1-regulated proteins
To further investigate the function of Fab1-regulated proteins, we next explored the biological roles of the proteins based upon their gene ontology (GO) functional annotations. A large portion of these proteins were associated with “metabolic processes,” including phosphorus metabolism. As Fab1 appears to be associated with the vacuolar and pre-vacuolar compartments, and is a key regulator of vacuolar membrane homeostasis in A. flavus, it is perhaps unsurprising that these Fab1-regulated proteins were involved in transport and stress-response activities (Figure 8(a) and Table S3). Consistent with such roles, most of these proteins were found in the cytoplasm, cell membrane, and endoplasmic reticulum (Figure 8(a) and Table S3). Over 50 up-regulated DEPs were located on the integral component of the membrane, further indicating the importance of Fab1 in normal membrane homeostasis. Further enrichment analyses demonstrated that these DEPs were significantly enriched in metabolic processes, including metabolism of secondary metabolite glycerolipids and the metabolism of glycerophospholipid (Table S4). Interestingly, we also observed enrichment for the protein export, peroxisome, and MAPK signaling pathways, supporting a role for Fab1 in counteracting stress, due to the disruption of vacuolar/cellular homeostasis (Table S4).
Figure 8.
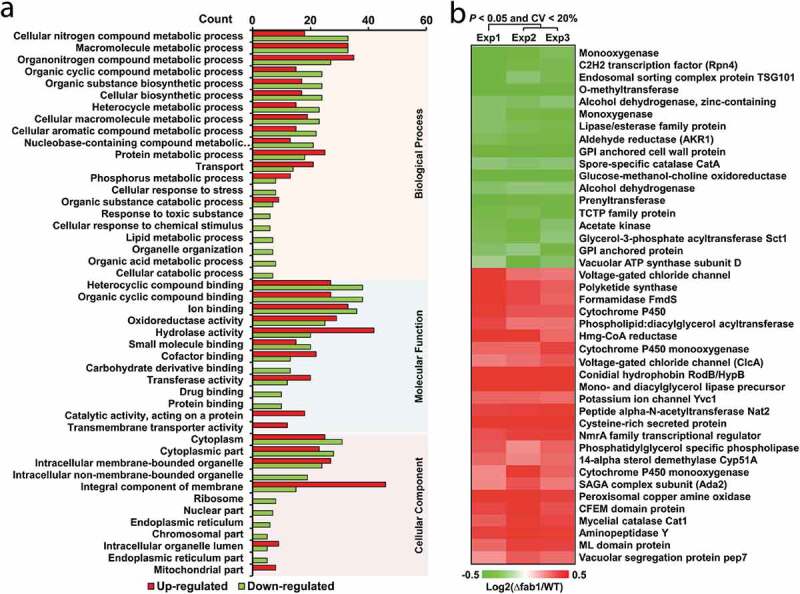
Functional analysis of Fab1-regulated proteins. (a) Functional annotation of differentially expressed proteins (DEPs). (b) Heatmap highlighting the distribution of DEPs associated with development and secondary metabolite production in WT and Δfab1 strains
Using the MultiLoc tool to assess protein subcellular localization, we found that most of the DEPs were located in the cytosol (Up: 64, Down: 63), followed by the extracellular (Up: 30, Down: 12) and mitochondria (Up: 17, Down: 8) compartments. Interestingly, many of these DEPs were located in the peroxisomes (Up: 14, Down: 12), vacuole (Up: 17, Down: 5), endoplasmic reticulum (Up: 6, Down: 4), and Golgi (Up: 3, Down: 0) (Fig. S9A and Table S5). A previous study showed that Fab1 is linked not only to vacuoles, but also to the pre-vacuolar compartment, which is important for protein trafficking from the Golgi and the plasma membrane to the vacuole in eukaryotic cells [40]. In the present work, we observed robust localization of Fab1 to the vacuolar membrane of A. flavus, in line with a previous fungal study [33]. Therefore, as unexpected, proteins involved in such trafficking activity were found to be significantly regulated by vacuolar dysregulation upon fab1 deletion.
Fab1-regulated proteins involved in secondary metabolite production
Consistent with the functional annotations, the DEPs were mapped to KEGG pathways and we found a subset of DEPs to be involved in development and secondary metabolite production in A. flavus (Figure 8(b)). For example, proteins associated with development, such as conidial hydrophobin RodB/HypB [41], glucose-methanol-choline oxidoreductase [42], spore-specific catalase CatA [43], a TCTP family protein [44], were also differentially expressed in the ∆fab1 strain. We further identified 14 proteins involved in aflatoxin biosynthesis, including monooxygenase, polyketide synthase, o-methyltransferase, alcohol dehydrogenase, aldehyde reductase, acetate kinase, lipase/esterase, Spt-Ada-Gcn5 acetyltransferase (Ada2) [45], and cysteine-rich secreted protein [46]. We also observed significant changes in the levels of two vacuole-associated proteins and four proteins linked with phosphatidylinositol metabolism, further supporting the roles of these proteins in vacuolar homeostasis.
Confirmation of differentially regulated proteins by parallel reaction monitoring and RT-PCR analysis
Owing to the high sensitivity and robustness, parallel reaction monitoring (PRM) has recently emerged as the “gold standard” MS technique for targeted proteomic approaches, which are routinely used for quantitative analysis of targeted proteins [47,48]. To independently validate our quantitative results, we next conducted a PRM assay for 15 of the identified DEPs. Most of the selected DEPs were represented by either one or two unique peptides, with 28 total peptides represented (Table S6). Relative to the WT strain, five of these DEPs were down-regulated and the remaining 10 were significantly upregulated in the ∆fab1 strain (Fig. S9B and Table S7). The PRM analysis revealed results nearly identical to our TMT-based analysis, confirming quality and quantitative accuracy of our MS data.
To further validate these findings, we subjected 41 of the DEPs to RT-PCR-mediated comparisons of transcript abundance between the WT and ∆fab1 A. flavus strains. We observed expression patterns consistent with our proteomic results for more than half of these genes, whereas discrepancies between the mRNA and protein abundance were observed for the remaining genes (Fig. S9C). This weak correlation between the mRNA and protein abundance suggests the potential for the proteins posttranscriptional regulation of these genes. Together, these results revealed that Fab1 may influence aflatoxin biosynthesis via maintaining vacuolar/cellular homeostasis by regulating diverse vacuole-related proteins.
Discussion
Fungal vacuoles/vesicles play a crucial role in the regulation of various cellular processes. In this study, we identified a novel phosphoinositide kinase (Fab1) in the vacuole of A. flavus. Experimental evidence revealed that Fab1 was essential for the growth, development, cellular acidification and metal ion homeostasis, aflatoxin production and pathogenicity of A. flavus. Further functional studies of Fab1 suggested that this kinase plays a crucial role in the maintenance of vacuole size and cell morphology in this fungus. Our results provide insight into the mechanisms of aflatoxin synthesis and A. flavus pathogenicity, highlighting potential novel strategies for controlling aflatoxin contamination.
The Fab1 protein identified in this work is highly conserved among different fungi (Figure 1(a) and Fig. S3B). Originally identified in yeast [49], this kinase can phosphorylate phosphatidylinositol-3-phosphate (PI-3-P) to phosphatidylinositol 3,5-bisphosphate (PtdIns-3,5-P2), which is closely linked to cellular stress responses [33]. PtdIns-3,5-P2 further regulates vacuole and cellular homeostasis [26,27], with reduced levels of PtdIns-3,5-P2 being associated with serious neurodegenerative diseases [50–52]. Notably, this lipid kinase appears to be an increasingly attractive target for drug development, particularly in the fields of cancer and other proliferative diseases, and in suppressing inflammatory and immune responses [53]. As such, Fab1 is necessary for regulating PtdIns-3,5-P2 levels in vivo, potentially playing essential roles in a wide range of processes. In agreement with previous results [33], we found that Fab1 localized to the vacuole membrane in A. flavus, where it appeared to play a key regulatory role in this fungus. Given the importance of PtdIns(3,5)P2, loss of fab1 in A. flavus led to significant defects in growth and development, as expected and observed in other organisms [20,54–58]. Interestingly, fab1 deletion results in altered cellular morphology without affecting the virulence of Candia albicans [59]. In this study, we generated evidence that Fab1 was able to impact aflatoxin production, suggesting this gene to be associated with the regulation of A. flavus virulence.
Based on our findings and past reports, we were able to construct a hypothetical model in which Fab1 regulates aflatoxin synthesis (Figure 9). Peroxisomes and mitochondria can serve as a source of the large amounts of acetyl-CoA necessary for aflatoxin synthesis, flowing from peroxisomes into vacuoles [5]. Early aflatoxin synthesis may also occur in peroxisomes (Figure 9). Consequently, acetyl-CoA synthesis and flux may be regulated by enzymes within these organelles, influencing early aflatoxin biosynthesis. The most important enzymes for the mid- and late stages of aflatoxin biosynthesis are also known to transit to the vacuoles from the nucleus, Golgi, and endoplasmic reticulum via autophagic and cytoplasm-to-vacuole-targeting (Cvt) pathways [5]. The DEPs in these organelles may also impact gene expression and transport mechanisms, thereby altering aflatoxin synthesis. Notably, enzymes linked with secondary metabolite production are often localized to vesicles and vacuoles, and the same is true for those associated with fungal aflatoxin production [31,60,61]. In Beauveria bassiana, the vacuole localized protein 4 (VLP4) are required for the autophagy, development, and virulence, suggesting the potential role of vacuolar protein in fungal virulence [62]. In this study, altered expression of the vacuole/vesicle-associated proteins and of other transport-associated proteins thus can impact aflatoxin biosynthesis, transport, and excretion. Together, we conclude that fab1 deletion may alter PtdIns-3,5-P2 levels, further disrupting vacuolar homeostasis and impacting the whole metabolic network in A. flavus.
Figure 9.
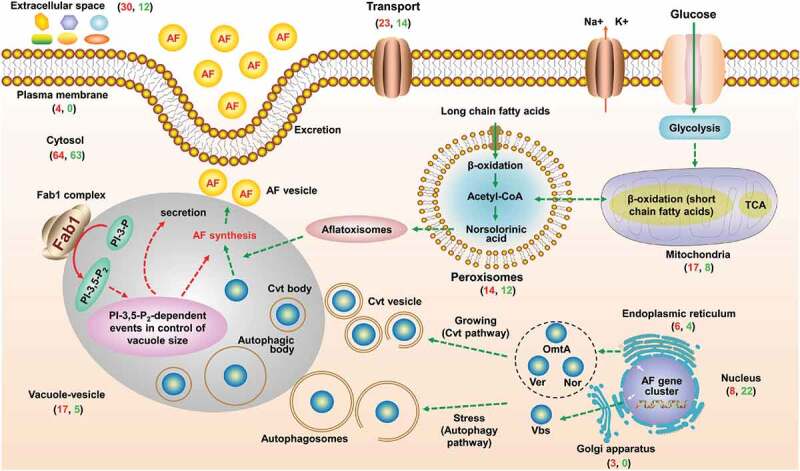
The proposed model for the role of Fab1 in aflatoxin biosynthesis. Fab1 may alter PtdIns-3,5-P2 levels, further disrupting vacuolar homeostasis and impacting the entire metabolic network in A. flavus. Red and green correspond to the number of upregulated and downregulated proteins, respectively
In summary, a novel phosphoinositide kinase (Fab1) was identified in A. flavus. In order to further explore its biological roles, we revealed it to have previously undocumented roles in the regulation of pathogenicity and aflatoxin production. Our results can serve as a valuable resource for future efforts to explore the synthesis, storage, transport, and excretion of secondary metabolites including aflatoxins.
Experimental procedures
Cell culture and protein extraction
All A. flavus strains used in this study are given in Table S8. A. flavus was cultured on liquid yeast extract-sucrose agar (YES) and potato dextrose agar (PDA) and the A. flavus were then grown under several conditions. For the low temperature and pH stresses, 106 conidia were resuspended in fresh YES media at 29°C or YES media (pH 5, with hydrochloride) at 37°C. For the oxidative and hyperosmotic stresses, 106 conidia were resuspended in fresh YES media containing additional H2O2 (0.8 M) or sodium chloride (1 M), respectively.
The cultures were then collected by filtration and washed three times with phosphate-buffered saline (PBS) buffer. The mycelia were ground into powders and reconstituted in buffer [20 mM Tris-Cl (pH 7.5), 150 mM NaCl, 1× phenylmethanesulfonyl fluoride (PMSF)]. The mixtures were shaken at 30°C for 1 h and the debris was removed by centrifugation at 5,000 × g at 4°C for 1 h. Finally, the protein concentration was measured by bicinchonininc acid (BCA) method (Tiangen, Beijing, China).
Real-Time PCR and 3’ rapid amplification of cDNA ends (3’RACE) validations
Total RNA was extracted from the mycelia grown in different conditions (37°C, 29°C, salt and H2O2 stresses) using TRIzol reagent (Invitrogen) as the manufacturer’s protocols. The concentration of total RNA was quantitated using the Nanodrop 2000 spectrophotometer. RT-PCR validation of novel genes was performed using the SYBR Green PCR Master Mix (Applied Biosystems) and the LightCycler 480 Real-Time PCR System (Roche). The actin gene was used as the endogenous control and three independent biological replicates were carried out for duplicate samples. The gene-specific primers were compiled in Table S9.
3’RACE procedures were performed by using 3’-Full RACE Core Set kit (Code No. 6106, Takara) as the manufacturer’s instructions. The first-strand cDNA was synthesized by using 1 μg RNA at the poly(A) tail of the mRNA. Two rounds of nested PCR were performed with the gene-specific forward primers and the RACE adaptor primers. Touch-down PCR protocol was used to confirm the appropriate annealing temperature. Amplicons were detected by electrophoresis on 1% agarose gel to determine the length, and then cloned to T-vector and further sequenced to confirm the specificity of amplification. The gene-specific forward primers were: fab1-RACE1: 5’-GTTTTACGCTGAACAGTTCGATGCTTT-3’ and fab1-RACE2: 5’-CAAGAGTCGGTTCAGGGAAGCAATGGC-3’.
Northern blot analysis
Northern blotting analysis was carried out by using DIG Northern Starter Kit (Roche) following the manufacturer’s protocol. The integral RNA was hybridized with a digoxigenin (DIG)-labeled RNA probe of the fab1 gene overnight at 42°C. The double-stranded DNA probes specific for fab1 was synthesized via PCR using primers 5’-TGAACCTTCCTCGTCCTC-3’ and 5’-CCACATTCGCTAGTCACAG-3’. The actin gene was used as control and the probe was generated via PCR using primers 5’-TGATGAGGCACAGTCCAAGCG-3’ and 5’-CACCGATCCAGACGGAGTATTT-3’. Chemiluminescent signals were detected using a fluorescence scanner (ImageQuant TL, GE Healthcare).
Western blot analysis
The proteins were extracted as mentioned in the protein extraction section and about 50 μg of proteins were used for western blotting analysis as previously described [63].
Construction of the fab1 deletion mutant
A gene deletion mutant was constructed and confirmed by previously described methods [64]. Briefly, the upstream (AP) and downstream (BP) sequences of the fab1 gene, as well as the pyrG gene of A. fumigatus were fused into the interruption fragment (AP-pyrG-BP) using a fusion PCR strategy. The fused fragment was then transformed into A. flavus CA14 PTs protoplasts to construct the fab1 deletion strain (∆fab1). For the construction of the complementary strain, fab1 gene with its upstream promoter region was firstly cloned into the pPTRI vector (Takara) and the recombinant plasmid was then transformed into the ∆fab1 mutant protoplasts to construct the complementary strain (∆fab1::fab1). All mutant and complementary strains were confirmed by PCR and sequencing.
Mycelial growth, conidiation and sclerotia quantification
The YES and PDA media were point inoculated with 105 conidia and kept at 37°C for 5 days in dark to evaluate the growth rate. Then, the colony morphology was recorded and colony diameter was measured. After homogenization and dilution with distilled water, conidia amount was counted using a hemocytometer and a microscope as previously reported [65]. For the sclerotia analysis, 105 conidia were inoculated on Yeast Peptone Dextrose (YPD) media and kept for 9 day at 37°C in the dark and the sclerotial formation was counted. In order to wash away conidia to aid in enumeration of the sclerotia, the plates were washed with 75% ethanol. Each experiment was carried out with three independent biological replicates.
Seed infections
To measure the pathogenicity of A. flavus, mature peanut and maize seeds were washed with 0.05% sodium hypochlorite, 75% ethanol and sterile water, and then inoculated by immersion in a 107 spores suspension for 30 min. The infected seeds were then placed in culture dishes lined with three pieces of moist sterile filter paper to maintain humidity. After 5 days of incubation, the infected seeds were pictured, collected, and mixed with 0.05% Tween 80, followed by vortexing to release the spores. About 100 µl spore suspension was collected, diluted, and counted for further quantification. Aflatoxins were extracted and analyzed by thin layer chromatography (TLC) and mass spectrometer assays according to the protocol described below. Each treatment was performed with three independent biological replicates.
Vesicle-vacuole and non-vesicle-vacuole fractions
A. flavus was cultured in YES media at 37°C for 2 days and the protoplasts were prepared using previously described protocols [64]. The vesicle-vacuole fraction was purified as described [32].
Thin layer chromatography (TLC) and MS quantification of aflatoxins
Aflatoxins from different cultures were extracted by equal volume chloroform with vibration for 30 min and purified by 0.45 u filter. The chloroform layer containing aflatoxins was then dried at 70°C and the precipitates were finally redissolved in 50 ul chloroform for TLC analysis and the fluorescence signal of aflatoxin was analyzed and quantified with Image J suite (http://rsbweb.nih.gov/ij/).
Mass spectrometry was also used to detect aflatoxins. Briefly, the samples were analyzed by an ACQUITY H-Class UPLC system coupled to a Xevo TQ (Waters, Milford, MA). Aflatoxins were separated with an analytical C18-nanocapillary LC column (1.7 μm particle, 2.1 × 50 mm) and eluted with a linear gradient elution program as follows: 0–3 min, 15%-50% solvent B (0.1% formic acid/80% acetonitrile, v/v); 4–5 min, 50%-70%; 6.5–8 min, 70%-100%; 8–10 min, 100%-50%; 10–11 min, 50%-15%; 11–15 min, 15%; at 200 nL/min flow rate. The MS was operated in selected reaction monitoring (SRM) mode. MS source conditions included a cone voltage optimized to 30 V, a capillary voltage set to 3 kV, collision energy set to 35 V, nitrogen gas flow at 50 L/h, a source temperature of 150°C, desolvation temperature at 350°C. Data was acquired using MassLynx 4.0. The precursor ions of aflatoxins (AFB1 and AFB2) were set to 312.92 m/z and 314.78 m/z, respectively.
Microscopic analysis
A. flavus was cultured in YES media at 37°C for 6, 24, 36, 48, or 60 h. After washing three times with PBS to remove the media, hyphal filaments were separated from the mycelia pellet by using forceps, and then stained with neutral red staining solution (C0125, Beyotime, Jiangsu, China) for 5–8 min. In addition, all samples were stained with 50 µm FM4-64 staining solution (N-3-triethy lammoniumpropyl 4-p-diethylamino phenyl-hexa-trienyl pyridinium dibromide) (DE-N4073, BioDee, Beijing, China) for 20–30 min. After washing three times with PBS, the hyphal filaments were placed on a microscope slide, and observed using a Nikon Ti-U microscope. Fluorescent photographs were observed on a Leica TCS SP8 laser scanning confocal microscope (LSCM). Green fluorescent protein (GFP) was excited with a 488 nm laser and FM4-64 with a 558 nm laser. Fluorescence emissions were measured at 507 nm and 734 nm for GFP and FM4-64, respectively.
Measurement of vacuolar and cytosolic pH
The vacuolar and cytosolic pH values were measured as previously described [35,66]. Briefly, all A. flavus strains were cultured in YES media, and then collected and washed three times with PBS buffer to remove the media. The hyphal filaments were then resuspended in PBS buffer containing 50 µM BCECF-AM [2,7-bis(2-carboxyethyl)-5,6-carboxyfluorescein-acetoxymethyl ester] fluorescent dye (GC14603, GLPBIO, Montclair, CA, USA) and incubated at 37°C for 20–30 min. After washing three times with PBS buffer, the fluorescence intensities from different stained hyphal filaments were quantified at the respective excitation/emission wavelengths of 450/535 and 490/535 nm for vacuolar fluorescence, while cytosolic fluorescence intensities were measured at the excitation wavelengths of 405 and 485 nm, with an emission wavelength of 508 nm using an automatic hybrid multi-mode microplate reader (Synergy H1, BioTek, Winooski, VT, USA). The calibration curves for vacuolar and cytosolic pH were established according to previously described [60,61], and the vacuolar and cytosolic pH values were calculated based on the calibration equations: y = −7.1084 + 1.8409x (r2 = 0.9785, for vacuolar pH) and y = −1.8817 + 0.4496x (r2 = 0.9948, for cytosolic pH), where y is the ratio and x is pH value. Each treatment was performed with three independent biological replicates. Statistical comparisons were performed by Student’s t-test.
Measurement of cytosolic Ca2+/Na+/K+ levels
The metal ions in A. flavus were assayed by staining with the Ca2+-specific fluorescence dye Fura-2-AM (Fura-2 acetoxymethyl ester, 40702ES50, Yeasen, Shanghai, China), Na+-specific fluorescence dye SBFI-AM (sodium-binding benzofuran isophthalate acetoxymethyl ester, GC44876, GLPBIO, Montclair, CA, USA) and K+-specific fluorescence dye PBFI-AM (potassium-binding benzofuran isophthalate acetoxymethyl ester, GC18477, GLPBIO, Montclair, CA, USA) as previous reports [36–39]. All A. flavus strains were cultured in YES media, then collected and washed three times with PBS buffer to remove the media. The hyphal filaments were resuspended in PBS buffer containing 10 mM fluorescence dyes (Fura-2-AM, SBFI-AM, or PBFI-AM) and incubated at 37°C for 20–30 min. After washing three times with PBS buffer, the fluorescence intensities from the different stained hyphal filaments were measured at excitation wavelengths of 340/380 nm and an emission wavelength of 505 nm by using a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek, Winooski, VT). The relative metal ions concentrations in different hyphal filaments were calculated based on the ratio of fluorescence intensity after dual-wavelength excitation. The unstained strains were used as controls. Each treatment was performed with three independent biological replicates. Statistical comparisons were performed by Student’s t-test.
Protein extraction, digestion and TMT labeling of wild type (WT) and mutants
Protein lysates from WT and ∆fab1 strains were extracted and quantified as the protocol described in protein extraction section. We precipitated the lysate using 80% ice-cold acetone and then washed three times with ice-cold acetone to remove the pigment and other small molecules. The protein lysates were finally redissolved in 50 mM ammonium bicarbonate. Equal amounts of protein extracts were then in-solution digested by trypsin (Promega) as previously described [63,67] and each sample was prepared with three biological replicates. After trypsin digestion, a total of 100 µg of each sample was further processed as the manufacturer’s protocol for TMT 6plex isobaric label kit (Thermo Fisher Scientific). The labeled peptides were mixed at equal amounts and desalted with C18 (40 μm, 60-Å particle; Agilent Technologies) columns and dried with a vacuum centrifuge.
HPLC fractionation and LC-MS/MS analysis
TMT-labeled peptides were resuspended in 1% (v/v) formic acid and separated into 60 fractions by high pH reverse-phase HPLC using a 4.6 mm × 25 cm C18 column (5 μm particles, Agilent) with a 60 min gradient of 8–32% acetonitrile (10 mM ammonium bicarbonate, pH 9). Finally, the peptides were combined into nine fractions and dried with a vacuum centrifuge.
After dissolution with 0.1% formic acid, the fractioned peptides were directly separated with an online nanoflow EASY-nLC 1200 system (Thermo Fisher Scientific). Peptides were eluted with a gradient of an increase from 6% to 22% of solvent B (0.1% formic acid/90% acetonitrile, v/v) over 38 min, 22%-32% in 4 min, 32%-80% in 4 min, and holding at 80% for the last 4 min at 450 nL/min flow rate. A full MS scan from m/z 350 to 1,600 was acquired at 120,000 resolution. The top 20 most intense precursor ions were selected for following MS/MS fragmentation by HCD with NCE of 28% and analyzed with a resolution of 30,000 in the Orbitrap. The dynamic exclusion was set to 30 s and maximum injection times for both MS and MS/MS were 50 ms and 60 ms, respectively. The isolation width of precursor ion was set to 1.4 m/z and the intensity threshold was set at 8.3E4. AGC for both MS and MS/MS were 3E6 and 1E5, respectively.
Protein identification and quantification
For the identification, the RAW data were searched against the A. flavus protein database concatenated with the protein sequences of common contaminants using MaxQuant software (version 1.6.3.4). Trypsin was used as a protease with two maximum missed cleavages and fixed modification was carbamidomethylation of cysteine. Dynamic modifications were set as deamidation (Asn/Gln) and oxidation (Met). Reporter ion was set as 6plexTMT for quantification. The mass errors for precursor ions and fragment ions were set to 20 ppm and 0.02 Da, respectively. Peptide for quantification was set as unique and razor. The FDR thresholds for peptide and protein identification were specified at maximum 1% and minimum score for peptides was set >40. Proteins identified based upon the presence of at least two unique peptides were accepted and reported.
For the TMT-labeling quantification, the ProteinGroup file from MaxQuant result was loaded to Perseus software (version 1.5.6.0) to facilitate statistical analysis. Only proteins that were identified and quantifiable in three biological replicates were used for relative quantification. The intensity of the TMT reporter ion for each protein was normalized against the total intensity of each sample and the corrected TMT reporter ion intensities in each sample were used to calculate fold changes between samples. A two-sample Student’s t-test was used for the statistical evaluation and the coefficient of variation (CV) was computed from the three biological replicates. The differentially expressed proteins were defined as fold-change ≥1.2 or ≤0.83, and p < 0.05, CV < 20%.
DDA and parallel reaction monitoring (PRM) experiments
The same protein extracts derived from quantitative proteomics were tryptic digested as previously described [63,67]. Each experiment was carried out with three independent biological replicates. The digested peptides were separated on an online nano-flow EASY-nLC 1000 system (Thermo Fisher Scientific) with a gradient of 7–25% solvent B (90% acetonitrile/0.1% formic acid, v/v) over 40 min, 25%-35% in 8 min, 35%-80% in 4 min and holding at 80% for the last 4 min at 350 nL/min flow rate.
For DDA-based experiments, a full MS scan from m/z 350 to 1,800 was acquired at 70,000 resolution. The top 10 most intense precursor ions were selected for following MS/MS fragmentation by HCD with NCE of 28% and analyzed with a resolution of 17,500 in the Orbitrap. The electrospray voltage applied was 2.1 kV and dynamic exclusion was set to 30 s. The maximum injection times for both full MS and MS/MS were 20 ms and 100 ms, respectively. The isolation width of precursor ion was set to 1.6 m/z. AGC for both MS and MS/MS were set at 3E6 and 5E4, respectively.
For PRM experiments, the mass spectrometer was operated in PRM mode with MS1 scan from m/z 400 to 950 at 70,000 mass resolution following by 1 MS/MS acquisitions. The remaining parameters were set as same as DDA-based experiments. PRM data sets were acquired in a time scheduled mode and isolation lists for each performed method can be found in Table S6.
Data analysis of PRM
For the identification of DDA data, raw data files were processed as the protocol described in Protein identification and quantification. For the acquired PRM data, all raw files were imported into the target quantitative software Skyline daily (v.3.5.1.9942). Spectral libraries were built using MS/MS search files from MaxQuant. The precursor of targeted peptide and the 3–10 most intense fragment ions were monitored and used for quantification. Peaks were manually checked for correct integration and the area under the curve of targeted peptide was obtained from the summed AUCs of each transition. The abundance of each peptide was normalized against the average of abundance of each protein. For the protein quantification, the mean of targeted peptide abundance was used to calculate fold changes between samples.
Bioinformatics analysis
The functional annotation of all the identified proteins was performed by using Blast2GO tool and the subcellular localization was analyzed by CELLO web tool [68]. Conservation analysis of the A. flavus proteome was carried out by using reciprocal BLAST [69]. Sequence alignment was also performed using ClustalX2 [70] and visualized by CLC sequence viewer (http://www.qiagenbioinformatics.com/product-downloads/). Domain structures were made by DOG software [71] and a neighbor-joining tree was constructed by using MEGA 5.0 software [72]. The transcripts were retrieved from NCBI (SRX237459, SRX237295, SRX1330586, SRX396791). For RNA-seq data analysis, the retrieved low-quality RNA reads were filtered and then mapped to the A. flavus genome using TopHat (version 2.1.1) [73]. R scripts and Excel were used for the statistical analyses. Statistical significance was analyzed by Student’s t-tests and expressed as a p value. p < 0.05 was considered to be statistically significant.
Statistical analysis
All data represent results from at least three independent experiments. Statistical analysis was performed to assess differences between different experiments. Student’s t-test was used to analyze the statistical significance. p < 0.05 was considered to be statistically significant. One asterisk and two asterisks indicate p < 0.05 and p < 0.01, respectively.
Supplementary Material
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2016YFA0501304), National Natural Science Foundation of China (Grant No. 31772105 and Grant No. 31601069) and CAS Key Technology Talent Program (to M.K.Y). The authors would like to thank the Analysis and Testing Center of Institute of Hydrobiology and the Wuhan Branch, Supercomputing Center, Chinese Academy of Sciences.
Funding Statement
This work was supported by the National Natural Science Foundation of China [31772105]; National Natural Science Foundation of China [31601069];Chinese Academy of Sciences Key Technology Talent Program; National Key Research and Development Program of China [2016YFA0501304].
Authors’ contributions
S.H.W., F.G. and M.K.Y. conceived and designed the study. M.K.Y., Z.Z. performed the proteomics experiments. M.K.Y., Z.Z., Y.H.B. M.Z.L and F.G. analyzed and interpreted the data. The paper was written by M.K.Y., F.G. and S.H.W. and was edited by all authors.
Disclosure statement
The authors have declared that no competing interests exist.
Data availability
All of the raw MS data, TMT-based quantitative proteomic results and the results of PRM validation were uploaded to the public access iProX database (http://www.iprox.org) with the identifier IPX0001753000.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Denning DW, Follansbee SE, Scolaro M, et al. Pulmonary aspergillosis in the acquired immunodeficiency syndrome. N Engl J Med. 1991;324:654–662. [DOI] [PubMed] [Google Scholar]
- [2].Yu J, Cleveland TE, Nierman WC, et al. Aspergillus flavus genomics: gateway to human and animal health, food safety, and crop resistance to diseases. Rev Iberoam Micología. 2005;22:194–202. [DOI] [PubMed] [Google Scholar]
- [3].Banziger M. Workgroup report: public health strategies for reducing aflatoxin exposure in developing countries. Environ Health Perspect. 2006;114:1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Authority EFS . Aflatoxins (sum of B1, B2, G1, G2) in cereals and cereal-derived food products. Efsa Supporting Publications. 2013;10:n/a-n/a. [Google Scholar]
- [5].Roze LV, Hong SY, Linz JE.. Aflatoxin biosynthesis: current frontiers. Annu Rev Food Sci Technol. 2013;4:293–311. [DOI] [PubMed] [Google Scholar]
- [6].Wiseman DW, Buchanan RL.. Determination of glucose level needed to induce aflatoxin production in aspergillus-parasiticus. Can J Microbiol. 1987;33:828–830. [DOI] [PubMed] [Google Scholar]
- [7].Buchanan RL, Stahl HG. Ability of various carbon-sources to induce and support aflatoxin synthesis by aspergillus-parasiticus. J Food Saf. 1984;6:271–279. [Google Scholar]
- [8].Gourama H, Bullerman LB. Inhibition of growth and aflatoxin production of aspergillus-flavus by lactobacillus species. J Food Protect. 1995;58:1249–1256. [DOI] [PubMed] [Google Scholar]
- [9].Gourama H, Bullerman LB. Aspergillus flavus and Aspergillus parasiticus: aflatoxigenic fungi of concern in foods and feeds: a review. J Food Protect. 1995;58:1395–1404. [DOI] [PubMed] [Google Scholar]
- [10].Klich MA. Environmental and developmental factors influencing aflatoxin production by Aspergillus flavus and Aspergillus parasiticus. Mycoscience. 2007;48:71–80. [Google Scholar]
- [11].Veses V, Richards A, Gow NA. Vacuoles and fungal biology. Curr Opin Microbiol. 2008;11:503–510. [DOI] [PubMed] [Google Scholar]
- [12].Yu Q, Wang F, Zhao Q, et al. A novel role of the vacuolar calcium channel Yvc1 in stress response, morphogenesis and pathogenicity of Candida albicans. Int J Med Microbiol. 2014;304:339–350. [DOI] [PubMed] [Google Scholar]
- [13].Rees EM, Thiele DJ. From aging to virulence: forging connections through the study of copper homeostasis in eukaryotic microorganisms. Curr Opin Microbiol. 2004;7:175–184. [DOI] [PubMed] [Google Scholar]
- [14].Hilty J, Smulian AG, Newman SL. The Histoplasma capsulatum vacuolar ATPase is required for iron homeostasis, intracellular replication in macrophages and virulence in a murine model of histoplasmosis. Mol Microbiol. 2008;70:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Veses V, Richards A, Gow NA. Vacuole inheritance regulates cell size and branching frequency of Candida albicans hyphae. Mol Microbiol. 2009;71:505–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Boyce KJ, Kretschmer M, Kronstad JW. The vtc4 gene influences polyphosphate storage, morphogenesis, and virulence in the maize pathogen Ustilago maydis. Eukaryot Cell. 2006;5:1399–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chanda A, Roze LV, Kang S, et al. A key role for vesicles in fungal secondary metabolism. Proc Natl Acad Sci U S A. 2009;106:19533–19538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dove SK, Dong K, Kobayashi T, et al. Phosphatidylinositol 3,5-bisphosphate and Fab1p/PIKfyve underPPIn endo-lysosome function. Biochem J. 2009;419:1–13. [DOI] [PubMed] [Google Scholar]
- [19].Michell RH, Heath VL, Lemmon MA, et al. Phosphatidylinositol 3,5-bisphosphate: metabolism and cellular functions. Trends Biochem Sci. 2006;31:52–63. [DOI] [PubMed] [Google Scholar]
- [20].Nicot AS, Fares H, Payrastre B, et al. The phosphoinositide kinase PIKfyve/Fab1p regulates terminal lysosome maturation in Caenorhabditis elegans. Mol Biol Cell. 2006;17:3062–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ishihara H, Shibasaki Y, Kizuki N, et al. Type I phosphatidylinositol-4-phosphate 5-kinases - Cloning of the third isoform and deletion/substitution analysis of members of this novel lipid kinase family. J Biol Chem. 1998;273:8741–8748. [DOI] [PubMed] [Google Scholar]
- [22].Lang MJ, Strunk BS, Azad N, et al. An intramolecular interaction within the lipid kinase Fab1 regulates cellular phosphatidylinositol 3,5-bisphosphate lipid levels. Mol Biol Cell. 2017;28:858–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Choudhary DK, Bhakt P, Kaur R. Essential role for the phosphatidylinositol 3,5-bisphosphate synthesis complex in caspofungin tolerance and virulence in Candida glabrata. Antimicrob Agents Chemother. 2019;63:e00886–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Adams TH, Boylan MT, Timberlake WE. brlA is necessary and sufficient to direct conidiophore development in Aspergillus nidulans. Cell. 1988;54:353–362. [DOI] [PubMed] [Google Scholar]
- [25].Sewall TC, Mims CW, Timberlake WE. abaA controls phialide differentiation in Aspergillus nidulans. Plant Cell. 1990;2:731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bridges D, Ma JT, Park S, et al. Phosphatidylinositol 3,5-bisphosphate plays a role in the activation and subcellular localization of mechanistic target of rapamycin 1. Mol Biol Cell. 2012;23:2955–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Natsuko J, Kai M, Yui J, et al. Roles for PI(3,5)P2 in nutrient sensing through TORC1. Mol Biol Cell. 2014;25:1171–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dolezal AL, Shu X, OBrian GR, et al. Aspergillus flavus infection induces transcriptional and physical changes in developing maize kernels. Front Microbiol. 2014;5:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Amaike S, Keller NP. Aspergillus flavus. Annu Rev Phytopathol. 2011;49:107–133. [DOI] [PubMed] [Google Scholar]
- [30].Wang JS, Groopman JD. DNA damage by mycotoxins. Mutat Res. 1999;424:167–181. [DOI] [PubMed] [Google Scholar]
- [31].Lee LW, Chiou CH, Klomparens KL, et al. Subcellular localization of aflatoxin biosynthetic enzymes Nor-1, Ver-1, and OmtA in time-dependent fractionated colonies of Aspergillus parasiticus. Arch Microbiol. 2004;181:204–214. [DOI] [PubMed] [Google Scholar]
- [32].Chanda A, Roze LV, Linz JE. Purification of a vesicle-vacuole (V) fraction from Aspergillus. Methods Mol Biol. 2012;944:259–266. [DOI] [PubMed] [Google Scholar]
- [33].Gary JD, Wurmser AE, Bonangelino CJ, et al. Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol. 1998;143:65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Veses V, Richards A, Gow NAR. Vacuoles and fungal biology. Curr Opin Microbiol. 2008;11:503–510. [DOI] [PubMed] [Google Scholar]
- [35].Zhu J, Ying SH, Feng MG. The Na+/H+ antiporter Nhx1 controls vacuolar fusion indispensible for life cycles in vitro and in vivo in a fungal insect pathogen. Environ Microbiol. 2016;18:3884–3895. [DOI] [PubMed] [Google Scholar]
- [36].Mouhoumed AZ, Mou YN, Tong SM, et al. Three proline rotamases involved in calcium homeostasis play differential roles in stress tolerance, virulence and calcineurin regulation of Beauveria bassiana. Cell Microbiol. 2020;22:e13239. [DOI] [PubMed] [Google Scholar]
- [37].Meier SD, Kovalchuk Y, Rose CR. Properties of the new fluorescent Na+ indicator CoroNa Green: comparison with SBFI and confocal Na+ imaging. J Neurosci Meth. 2006;155:251–259. [DOI] [PubMed] [Google Scholar]
- [38].Lindberg S. In-situ determination of intracellular concentrations of K+ in Barley (Hordeum-Vulgare L Cv Kara) Using the K+-binding fluorescent dye benzofuran isophthalate. Planta. 1995;195:525–529. [Google Scholar]
- [39].Kasner SE, Ganz MB. Regulation of intracellular potassium in mesangial cells - a fluorescence analysis using the dye, Pbfi. Am J Physiol. 1992;262:F462–F7. [DOI] [PubMed] [Google Scholar]
- [40].Lam SK, Yu CT, Jiang L, et al. Plant prevacuolar compartments and endocytosis; 2006. [Google Scholar]
- [41].Paris S, Debeaupuis J-P, Crameri R, et al. Conidial hydrophobins of aspergillus fumigatus. Appl Environ Microbiol. 2003;69:1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Etxebeste O, Herrero-García E, Cortese MS, et al. GmcA is a putative glucose-methanol-choline oxidoreductase required for the induction of asexual development in aspergillus nidulans. Plos One. 2012;7:e40292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Navarro RE, Stringer MA, Hansberg W, et al. catA, a new Aspergillus nidulans gene encoding a developmentally regulated catalase. Curr Genet. 1996;29:352–359. [PubMed] [Google Scholar]
- [44].Oh YT, Ahn C-S, Jeong YJ, et al. Aspergillus nidulans translationally controlled tumor protein has a role in the balance between asexual and sexual differentiation and normal hyphal branching. Fems Microbiol Lett. 2013;343:20–25. [DOI] [PubMed] [Google Scholar]
- [45].Paraskevi G, Lockington RA, Kelly JM, et al. The Spt-Ada-Gcn5 acetyltransferase (SAGA) complex in aspergillus nidulans. Plos One. 2013;8:e65221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zeng R, Gao S, Xu L, et al. Prediction of pathogenesis-related secreted proteins from Stemphylium lycopersici. BMC Microbiol. 2018;18:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].van Dongen WD, Niessen WM. LC-MS systems for quantitative bioanalysis. Bioanalysis. 2012;4:2391–2399. [DOI] [PubMed] [Google Scholar]
- [48].Li Z, Li Y, Chen W, et al. Integrating MS1 and MS2 scans in high-resolution parallel reaction monitoring assays for targeted metabolite quantification and dynamic (13)C-labeling metabolism analysis. Anal Chem. 2017;89:877–885. [DOI] [PubMed] [Google Scholar]
- [49].Dove SK, Cooke FT, Douglas MR, et al. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature. 1997;390:187–192. [DOI] [PubMed] [Google Scholar]
- [50].Zolov SN, Dave B, Yanling Z, et al. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci U S A. 2012;109:17472–17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mccartney AJ, Zolov SN, Kauffman EJ, et al. Activity-dependent PI(3,5)P2 synthesis controls AMPA receptor trafficking during synaptic depression. Proc Natl Acad Sci U S A. 2014;111:4896–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bharadwaj R, Cunningham KM, Zhang K, et al. FIG4 regulates lysosome membrane homeostasis independent of phosphatase function. Hum Mol Genet. 2015;25:681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Stein RC, Waterfield MD. PI3-kinase inhibition: a target for drug development? Mol Med Today. 2000;6:347–357. [DOI] [PubMed] [Google Scholar]
- [54].Lang MJ, Strunk BS, Azad N, et al. An intramolecular interaction within the lipid kinase Fab1 regulates cellular phosphatidylinositol 3,5-bisphosphate lipid levels. Mol Biol Cell. 2017;28:858–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hirano T, Sato MH. Diverse physiological functions of FAB1 and phosphatidylinositol 3,5-bisphosphate in plants. Front Plant Sci. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rusten TE, Rodahl LMW, Pattni K, et al. Fab1 phosphatidylinositol 3-Phosphate 5-kinase controls trafficking but not silencing of endocytosed receptors. Mol Biol Cell. 2006;17:3989–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ikonomov OC, Sbrissa D, Delvecchio K, et al. The phosphoinositide kinase PIKfyve is vital in early embryonic development preimplantation lethality of Pikfyve(-/-) embryos but normality of Pikfyve(±) mice. J Biol Chem. 2011;286:13404–13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Takasuga S, Horie Y, Sasaki J, et al. Critical roles of type III phosphatidylinositol phosphate kinase in murine embryonic visceral endoderm and adult intestine. Proc Natl Acad Sci U S A. 2013;110:1726–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Augsten M, Hubner C, Nguyen M, et al. Defective hyphal induction of a Candida albicans phosphatidylinositol 3-phosphate 5-kinase null mutant on solid media does not lead to decreased virulence. Infect Immun. 2002;70:4462–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sung-Yong H, Linz JE. Functional expression and subcellular localization of the aflatoxin pathway enzyme Ver-1 fused to enhanced green fluorescent protein. Appl Environ Microbiol. 2008;74:6385–6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sung-Yong H, Linz JE. Functional expression and sub-cellular localization of the early aflatoxin pathway enzyme Nor-1 in Aspergillus parasiticus. Mycol Res. 2009;113:591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chu ZJ, Sun HH, Zhu XG, et al. Discovery of a new intravacuolar protein required for the autophagy, development and virulence of Beauveria bassiana. Environ Microbiol. 2017;19:2806-2818. [DOI] [PubMed] [Google Scholar]
- [63].Yang MK, Lin XH, Liu X, et al. Genome annotation of a model diatom phaeodactylum tricornutum using an integrated proteogenomic pipeline. Mol Plant. 2018;11:1292–1307. [DOI] [PubMed] [Google Scholar]
- [64].Chang PK, Scharfenstein LL, Wei Q, et al. Development and refinement of a high-efficiency gene-targeting system for Aspergillus flavus. J Microbiol Methods. 2010;81:240–246. [DOI] [PubMed] [Google Scholar]
- [65].Yang K, Qin Q, Liu Y, et al. Adenylate cyclase AcyA regulates development, aflatoxin biosynthesis and fungal virulence in aspergillus flavus. Front Cell Infect Microbiol. 2016;6:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Diakov TT, Tarsio M, Kane PM. Measurement of vacuolar and cytosolic ph in vivo in yeast cell suspensions. Jove-J Vis Exp. 2013;19:50261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yang MK, Yang YH, Chen Z, et al. Proteogenomic analysis and global discovery of posttranslational modifications in prokaryotes. Proc Natl Acad Sci U S A. 2014;111:E5633–E42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yu CS, Lin CJ, Hwang JK. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004;13:1402–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rastogi A, Maheswari U, Dorrell RG, et al. Integrative analysis of large scale transcriptome data draws a comprehensive landscape of Phaeodactylum tricornutum genome and evolutionary origin of diatoms. Sci Rep. 2018;8:4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Larkin MA, Blackshields G, Brown NP, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. [DOI] [PubMed] [Google Scholar]
- [71].Ren J, Wen L, Gao X, et al. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19:271–273. [DOI] [PubMed] [Google Scholar]
- [72].Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the raw MS data, TMT-based quantitative proteomic results and the results of PRM validation were uploaded to the public access iProX database (http://www.iprox.org) with the identifier IPX0001753000.