

ABSTRACT
Immunotherapy using dendritic cells (DCs) is a promising treatment modality for cancer. However, the limited number of functional DCs from peripheral blood has been linked to the unsatisfactory clinical efficacies of current DC-based cancer immunotherapies. We previously generated proliferating antigen-presenting cells (APCs) by genetically engineering myeloid cells derived from induced pluripotent stem cells (iPSC-pMCs), which offer infinite functional APCs for broad applications in cancer therapy. Herein, we aimed to further enhance the antitumor effect of these cells by genetic modification. GM-CSF gene transfer did not affect the morphology, or surface phenotype of the original iPSC-pMCs, however, it did impart good viability to iPSC-pMCs. The resultant cells induced GM-CSF-dependent CD8+ T cell homeostatic proliferation, thereby enhancing antigen-specific T cell priming in vitro. Administration of the tumor antigen-loaded GM-CSF-producing iPSC-pMCs (GM-pMCs) efficiently stimulated antigen-specific T cells and promoted effector cell infiltration of the tumor tissues, leading to an augmented antitumor effect. To address the potential tumorigenicity of iPSC-derived products, irradiation was applied and found to restrict the proliferation of GM-pMCs, while retaining their T cell-stimulatory capacity. Furthermore, the irradiated cells exerted an antitumor effect equivalent to that of bone marrow-derived DCs obtained from immunocompetent mice. Additionally, combination with immune checkpoint inhibitors increased the infiltration of CD8+ or NK1.1+ effector cells and decreased CD11b+/Gr-1+ cells without causing adverse effects. Hence, although GM-pMCs have certain characteristics that differ from endogenous DCs, our findings suggest the applicability of these cells for broad clinical use and will provide an unlimited source of APCs with uniform quality.
KEYWORDS: Cancer immunotherapy, cancer vaccine, dendritic cell, GM-CSF, induced pluripotent stem cell
Introduction
Dendritic cells (DCs) are the most potent antigen-presenting cells (APCs), and play a crucial role in the induction of antitumor immune responses.1 DC-based vaccines, which aim to activate tumor-reactive T cells by administrating tumor antigen-loaded DCs, are effective and well tolerated in many types of cancer.2–5 This method can elicit antitumor responses in at least half of the treated patients and has exhibited a better clinical efficacy than other types of vaccine, such as peptide, viral vector, or inactivated tumor cell-vaccines.6,7 Despite their ability to induce antitumor responses, however, the objective clinical responses have not been satisfied to date, as only 7.1–15.6% of the patients have obtained tumor regression, and the survival benefits have remained insufficient.2,6,8
In experimental mouse models, 1.0 × 105 DCs to 1.0 × 106 DCs are required to suppress cancer.6 To obtain an equivalent effect to treat a human weighing 50 kg, 2.0 × 108 DCs to 2.0 × 109 DCs may be required. However, the differentiation capacities of the DC-progenitors and functions of the differentiated DCs are repressed as disease progresses in cancer patients.9–11 Moreover, the laborious process utilized to generate DCs negatively affects their function and the stability of the manufacturing efficiency, suggesting that, with the current methods using autologous peripheral blood, it may be difficult to obtain a sufficient number of functional DCs to exert the equivalent antitumor effects achieved in the mouse tumor models. In addition, the enormous cost required for personalized medicine hampers the feasibility of this treatment.12 Therefore, the development of an alternate source of cells, of sufficient quality and quantity for treatment, will be required.
Induced pluripotent stem cell (iPSC)-derived immune cells have the potential to be an alternative cell source applicable for cancer therapy.13,14 We previously established a method to generate proliferating myeloid lineage cells (pMCs) from mouse iPSC-derived myeloid cells (iPSC-MCs) by introducing c-Myc gene using a lentiviral vector.15 iPSC-derived proliferating myeloid cells (iPSC-pMCs) can proliferate in a cytokine-dependent manner and differentiate into DC-like cells. Owing to their proliferative ability, direct modification of iPSC-pMCs to impart DC functions may generate an off-the-shelf APC preparation method.
Granulocyte macrophage colony-stimulating factor (GM-CSF), which serves as a growth factor for iPSC-pMCs,15 is a key cytokine essential for the differentiation, proliferation, and recruitment of DCs and promotes their capacity for antigen presentation, co-stimulatory molecule expression, and proinflammatory cytokine production.16,17 Therefore, it has been incorporated as an adjuvant for a variety of cancer vaccines to boost a DC-mediated antitumor immunity.18–21
In this study, we generated GM-CSF-producing APCs by genetically engineering iPSC-pMCs. GM-CSF gene transfer enhanced the viability and proliferative capacity of iPSC-pMCs. Moreover, GM-CSF producing iPSC-pMCs (GM-pMCs) promoted the homeostatic proliferation of the naïve CD8+ T cells, which may amplify the antigen-specific T cell pools at T cell-priming sites. Administration of the cancer antigen-loaded GM-pMCs enhanced the antigen-specific T cell responses and inhibited tumor growth, as with the bone marrow-derived DCs (BM-DCs). Even after irradiation, the GM-pMCs retained their T cell-stimulatory capacity in vivo, while avoiding the tumorigenic risks of iPSC-derived differentiated cells. Although GM-pMCs do not contain all properties of BM-DCs, these cells efficiently stimulated antigen-specific CD8+ T cells, suggesting that they may serve as a cellular platform for an APC-based cancer vaccine, with no need for autologous blood cells.
Results
Generation of GM-CSF-producing myeloid cells
iPSC-pMCs were generated by introducing the c-Myc gene into iPSC-derived myeloid cells and they acquired a GM-CSF-dependent proliferative potential.15 In this study, we constructed GM-CSF-producing iPSC-pMCs (GM-pMCs) by introducing Csf2 gene into iPSC-pMCs (Figure 1a-d and Figures S1a, b). The pMCs proliferated in a GM-CSF-dependent manner, and proliferation was further enhanced by addition of M-CSF. In contrast, GM-pMCs proliferated even in the absence of an exogenous GM-CSF and showed maximal proliferation only with addition of M-CSF (Figure 1e and Figure S1c). No apparent change was observed in morphology compared to pMCs (Figure S1d). GM-pMCs expressed the myeloid lineage markers (CD11b, CD11 c, F4/80, DEC205, Gr-1, and CD33); meanwhile expression of the major histocompatibility complex (MHC)-I/II molecules was lower than those of BM-DCs, and co-stimulatory molecules (CD40, CD80, and CD86) were more highly expressed (Figure 1f and Figure S1e). In the absence of any cytokines, although many pMCs undergo apoptosis after four days, the GM-pMCs showed a tendency to avoid apoptosis, and over 90% survived in vitro (Figure 1g). These data suggest that GM-pMCs possess an APC-like phenotype and that the GM-CSF produced by GM-pMCs was responsible for their high viability and proliferative capacities.
Figure 1.
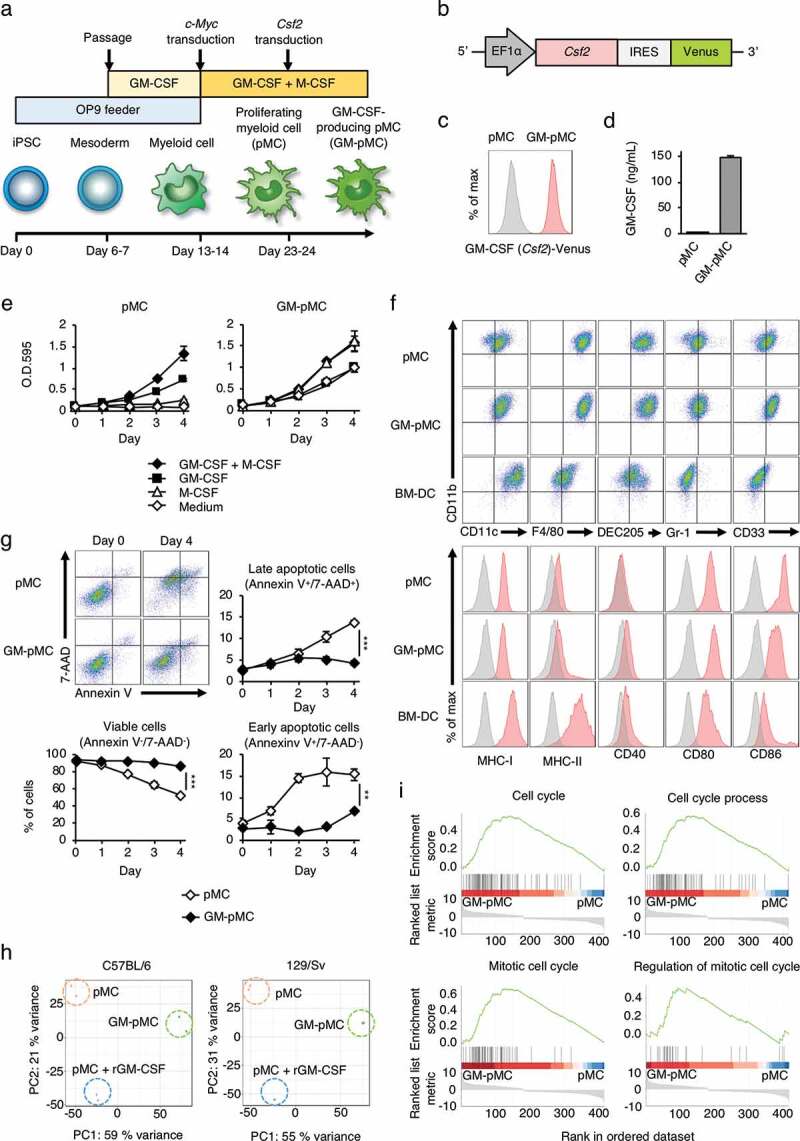
iPSC-derived proliferating myeloid cells were genetically engineered to express GM-CSF.
(a) Protocol for the generation of GM-pMCs. pMCs were established from iPSCs as described.15 After the proliferation of pMCs was stable, the Csf2 gene was introduced using a lentiviral vector. Cells were cultured in α-MEM supplemented with 20% FBS. Other supplements in the medium, and the feeder cells are indicated. (b) Schematic illustration of the lentiviral vector expressing Csf2. (c) The expression of the Csf2 gene was confirmed by flow cytometry using the expression of the Venus gene as an indicator. pMCs; gray, GM-pMCs; red. Additionally, see Figure S1a. (d) GM-CSF production. Cells (5.0 × 105 cells/mL) were cultured for 24 h in 6-well culture plates. GM-CSF levels in the culture supernatants were evaluated using ELISA. Additionally, see Figure S1b. (e) Cell proliferation. Cells (2.0 × 103 cells/well) were seeded in 96-well culture plates in the presence of the indicated cytokines. Proliferation was determined at each time point using the MTT assay. Medium served as a control. Additionally, see Figure S1c. (f) Characterization using flow cytometry. Upper panels, representative flow cytometry profiles of the indicated surface molecules. Lower panels, an expression of surface molecules associated with a T cell stimulation. BM-DC data served as a control. Additionally, see Figure S1e. (g) In vitro viability. Cells (2.0 × 105 cells/well) were cultured in the absence of any cytokine. Cell viability was evaluated by flow cytometry at the indicated time points. Upper left panels, representative flow cytometry profiles. Annexin V−/7-AAD−, Annexin V+/7-AAD−, and Annexin V+/7-AAD+ cells were defined as viable cells, early apoptotic cells, and late apoptotic cells, respectively. Graph data represents the frequency of each cell. (h) Principal component analysis (PCA) of pMCs, pMCs cultured with recombinant GM-CSF, and GM-pMCs. Additionally, see Figure S1f and Tables S1-3. (i) Gene set enrichment analysis (GSEA) of the up-regulated genes in GM-pMCs (overlapping genes between the C57BL/6- and 129/Sv-derived groups). Additionally, see Figure S1g and Table S4. (c–g) Representative data from two independent experiments are presented. (d, e, g) Means ± SD (n = 3) are shown. *p < .05, **p < .01, ***p < .001.
To further characterize the GM-pMCs, we performed an RNA-seq analysis and found that GM-pMCs exhibited distinct gene expression profiles from those of pMCs (Figure 1h). We then identified differentially expressed genes between the GM-pMCs and pMCs, which were derived from the mouse strains C57BL/6 and 129/Sv, respectively (Figure S1f and Tables S1, 2). We then further identified the overlapping up-regulated and down-regulated genes (Figure S1g and Table S3). The RNA-seq data revealed that GM-pMCs were enriched in transcripts associated with “cell cycle”, “cell cycle process”, “mitotic cell cycle”, and “regulation of mitotic cell cycle” (Figure 1i and Table S4); however, these annotation data are attributable to the same set of genes contributing to overlapping pathways. Conceivably, these up-regulated genes may be associated with the proliferative capacity and high viability observed in the GM-pMCs.
GM-pMCs stimulate antigen-specific T cells
DCs secrete proinflammatory cytokines such as IL-12 and tumor necrosis factor-alpha (TNF-α) following stimulation with toll-like receptors.6 In addition, DCs are specialized APCs that acquire, process, and present extracellular antigens in the context of MHC class I molecules, known as cross-presentation, leading to an induction of tumor-reactive CD8+ T cell immune responses.22 We first examined the expression of IL-12 and TNF-α by GM-pMCs. Results show that GM-pMCs did not produce IL-12p40 even when stimulated with OK432 (Figure S2a), whereas they did produce TNF-α in response to OK432; however, the response was lower than that observed for BM-DCs (Figure S2b).
We next evaluated whether GM-pMCs have a DC-like capacity for T cell stimulation and antigen presentation. GM-pMCs induced proliferation of allogeneic T cells, and their activity was markedly higher than that in pMCs, however, was comparable to that in BM-DCs (Figure 2a, b). Further, pMCs and GM-pMCs equally stimulated proliferation of ovalbumin (OVA)-specific CD8+ T cells (OT-1) in the presence of a soluble form of H-2Kb-restricted OVA peptide (OVA257-264 peptide), however, their stimulatory activity was weaker than that of BM-DCs (Figure 2c). Since no difference was observed in the T cell stimulatory activity of pMC and GM-pMC in the experiments using soluble peptides, cells with the antigen washed out 16 h after its addition were used for comparison. When the soluble form OVA257-264 peptide was removed before co-culture, GM-pMCs pre-loaded with OVA257-264 peptide stimulated a proliferative response of OT-1 CD8+ T cells more efficiently than that by pMCs (Figure 2d). Moreover, GM-pMCs pre-loaded with OVA protein also stimulated OT-1 CD8+ T cells more efficiently than pMCs (Figure 2d). The presence of 100 μg/mL OVA protein for four days induced an equivalent T cell proliferative response to that of 10−3 nM OVA257-264 peptide (Figure S2c), however, the level of H-2Kb/OVA peptide complex expression was below the detection limit of the flow cytometry analysis (Figure S2d).
Figure 2.
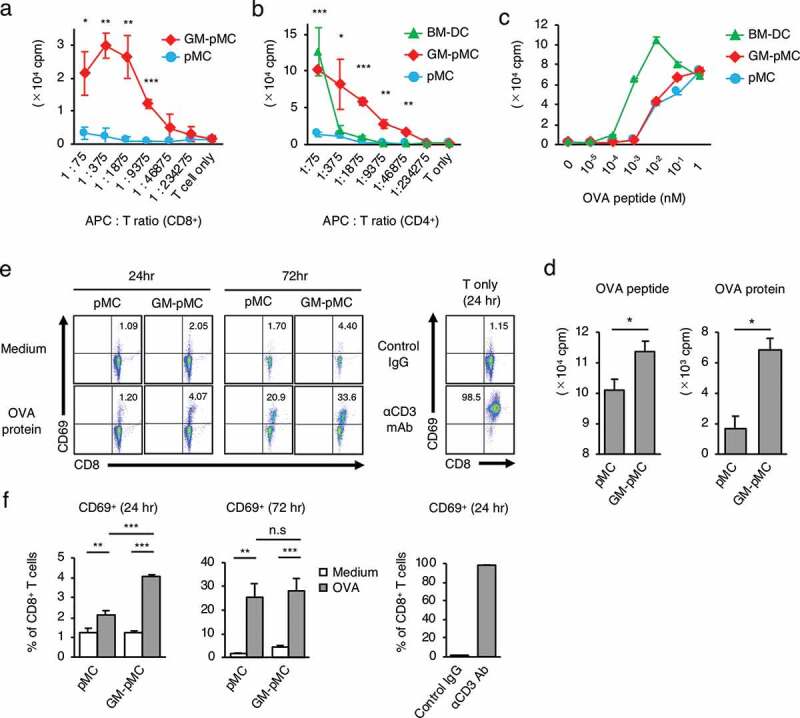
GM-pMCs efficiently stimulate antigen-specific CD8+ T cells.
(a, b) T cell stimulation capacity. MHC-mismatched CD8+ (A) or CD4+ (b) T cells (1.5 × 105 cells/well) from unprimed BALB/c mice were cultured with the graded doses of 85 Gy-irradiated pMCs, GM-pMCs, or BM-DCs (C57BL/6 strain) for six days. (c, d) T cell priming capacity in vitro. (c) Cells (2.5 × 104 cells/well) were irradiated with 85 Gy and co-cultured with the naïve OT-1 CD8+ T cells (5.0 × 104 cells/well) in the presence of OVA257-264 peptide at indicated concentration for four days. (d) Cells (1.0 × 106 cells/mL) were pre-pulsed with 10 μM OVA257-264 peptide or 100 μg/mL OVA protein for 16 h. The cells were then washed twice to remove soluble antigens, seeded in 96-well culture plates (5.0 × 104 cells/well), irradiated with 85 Gy, and co-cultured with the naïve OT-1 CD8+ T cells (1.0 × 104 cells/well) for four days. Also see Figures S2c, d. (e, f) Cells (1.0 × 106 cells/mL) were cultured in the presence or absence of 100 μg/mL OVA protein for 16 h. Then cells were washed twice to remove soluble antigens, seeded in 96-well culture plates (5.0 × 104 cells/well), irradiated with 85 Gy, and co-cultured with naïve OT-1 CD8+ T cells (1.0 × 105 cells/well). The frequency of activated (CD69+) cells in CD8+ T cells was evaluated using flow cytometry at the indicated time points. OT-1 CD8+ T cells stimulated with anti-CD3 mAb or control IgG served as references. (e) Representative flow cytometry profiles. (f) Summary of CD69+ cell frequency. (a–d) Cell proliferation was determined using 3H-thymidine uptake (16 h). (a–d, f) Means ± SD (n = 3) are shown. Representative data from two independent experiments are presented. *p < .05, **p < .01, ***p < .001.
GM-pMCs pre-loaded with OVA protein induced an increase in CD69+ phenotype frequency in co-cultured naïve OT-I T cells, which was higher than with pMCs (Figure 2e, f), suggesting that OVA protein was processed and an H-2Kb-restricted OT-I T cell epitope was efficiently presented by the GM-pMCs. These results collectively indicate that GM-pMCs have a cross-presentation capacity sufficient for stimulating antigen-specific T cells.
GM-CSF produced by GM-pMCs induces CD8+ T cell homeostatic proliferation
Homeostatic proliferation is an essential T cell proliferative response for maintaining the intravital T cell pool and the diversity of the TCR repertoire.23 Interleukin (IL)-7 and IL-15 are known as homeostatic cytokines that induce polyclonal T cell proliferation in an antigen nonspecific manner.24 Since GM-pMCs induced T cell proliferation more strongly than pMCs, they may also induce antigen nonspecific T cell proliferation. We found that recombinant GM-CSF enhanced the spontaneous proliferation of naïve OT-1 CD8+ T cells in a dose-dependent manner (Figure 3a). Furthermore, supernatant from GM-pMC cultures also promoted proliferation, despite the absence of antigenic stimulation, this was attenuated by neutralization of the GM-CSF (Figure 3b, c). These phenomena resemble the T cell homeostatic proliferation induced by IL-7 and IL-15.24 Therefore, we evaluated TCR Vβ usage of naïve CD8+ T cells co-cultured with GM-pMCs and found that GM-pMCs induced polyclonal T cell expansion without affecting TCR clonality (Figure 3d, e, and Figure S3). These findings collectively suggest that GM-CSF produced by GM-pMCs may enhance the homeostatic proliferation of naïve CD8+ T cells.
Figure 3.
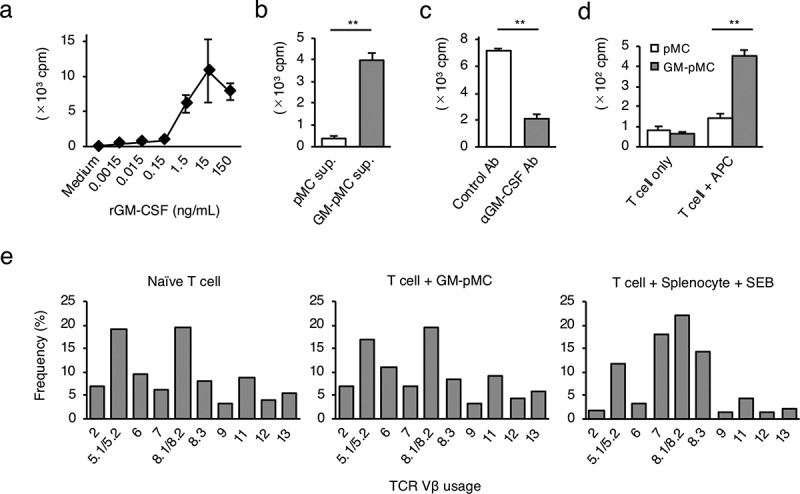
GM-CSF from GM-pMCs induces a CD8+ T cell homeostatic proliferation.
(a) Naïve OT-1 CD8+ T cells (1.0 × 105 cells/well) were cultured for six days in the presence of recombinant GM-CSF at the indicated concentrations. (b) Naïve OT-1 CD8+ T cells were cultured for six days in the presence of cell-free supernatants (1/4 vol.) from the pMCs or the GM-pMCs. (c) Naïve OT-1 CD8+ T cells were cultured for six days in the presence of a cell-free supernatant from GM-pMCs, and anti-GM-CSF or control antibodies. (d) Naïve CD8+ T cells from C57BL/6 mice were cultured with 85 Gy-irradiated pMCs or GM-pMCs (5.0 × 104 cells/well) for four days.(a–d) Cell proliferation was determined using3H-thymidine uptake (16 h). Shown are the results representative of two independent experiments. Means ± SD (n = 3). *p < .05, **p < .01. (e) TCR Vβ usage of naïve CD8+ T cells before, and three days after, coculturing with GM-pMCs. TCR Vβ usage was assessed by flow cytometry. CD8+ T cells co-cultured with syngeneic splenocytes in the presence of 500 ng/mL Staphylococcal enterotoxin B (SEB) served as a control; SEB stimulates the limited T cells expressing specific Vβ usages. Data represents the frequency of cells with a specific TCR Vβ within the CD8+ T cell population. Shown are data representative of three independent experiments.
GM-pMCs efficiently stimulate tumor-reactive T cells to exert antitumor effects
To determine whether GM-pMCs exert antitumor effects, we first performed prophylactic cancer vaccine experiments. The OVA257-264 peptide-loaded GM-pMCs or pMCs (C57BL/6 background) were intraperitoneally (i.p.) administrated to syngeneic mice twice, at seven-day intervals. Seven days after the last treatment, the mice were inoculated subcutaneously (s.c.) with MO4, a mouse melanoma cell line expressing OVA, thereafter tumor growth and survival were evaluated over time (Figure 4a). GM-pMCs significantly inhibited tumor growth and prolonged survival compared to pMCs (Figure 4b-d). In addition, a high frequency of OVA-specific T cells was detected in mice treated with GM-pMCs, which was attributed to promotion of cell division in vivo (Figure 4e, f). We also observed GM-pMC vaccine augmented CD8+ or perforin+ cell infiltration into MO4 tumor tissue, however, it did not affect CD4+ cell infiltration or PD-L1 expression of tumor cells (Figure 4g and Figure S4a).
Figure 4.
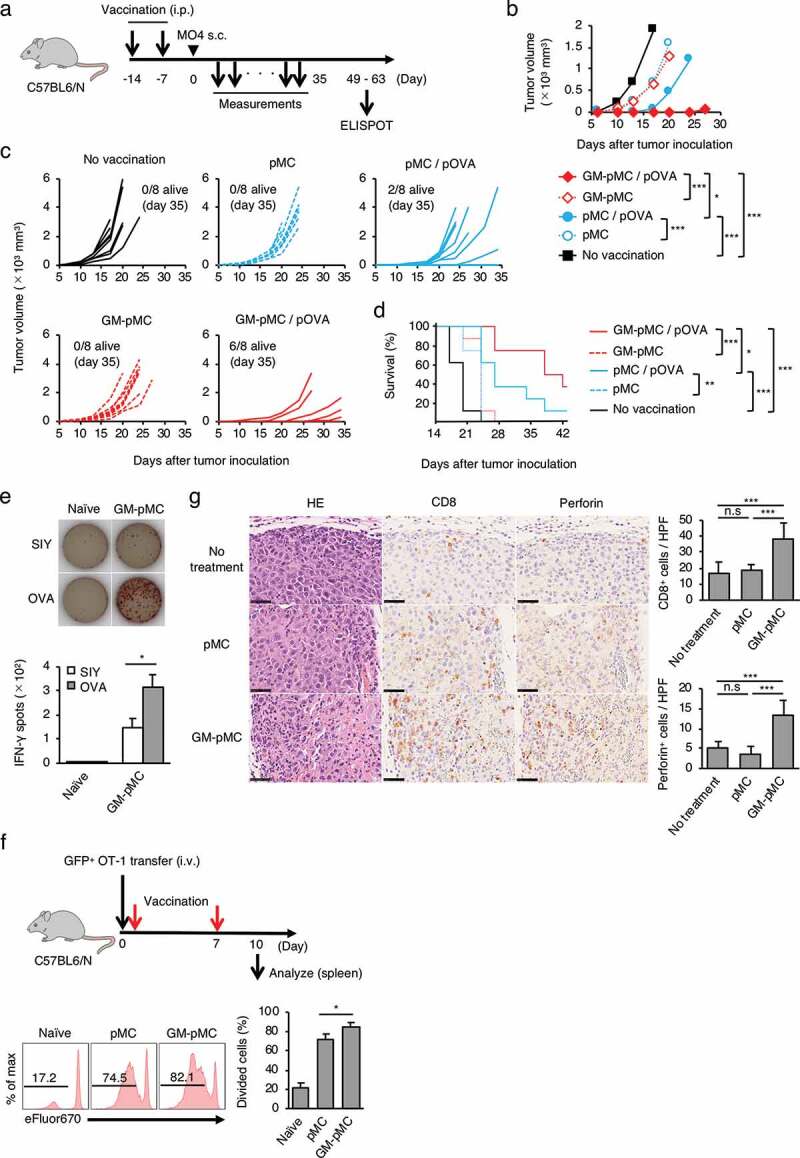
GM-pMCs suppress tumor growth via induction of tumor-reactive CD8+ T cells.
(a) Schematic for vaccination and tumor implantation. C57BL/6 mice were i.p. vaccinated twice followed by s.c. (right flank) inoculation of MO4 tumor cells. Tumor growth and survival were monitored until the mice died or were sacrificed when the tumors exceeded 20 mm in a diameter. (b–e) C57BL/6 mice were i.p. vaccinated with the OVA257-264 peptide-loaded or the unloaded pMCs or the GM-pMCs. (b) Median tumor volume (mm3: length × width2). (c) Tumor growth of individual mice. (d) Kaplan-Meier survival curves. (b–d) Data shown are representative of two independent experiments. (n = 8 mice). (e) IFN-γ ELOSPOT assay ex vivo on day 56 after inoculation. Spleen CD8+ T cells from mice with complete tumor rejection by the OVA257-264 peptide-loaded GM-pMC vaccine or naïve mice were stimulated with the OVA257-264 peptide- or control SIY peptide (SIYRYYGL)-loaded, irradiated RMA-S for 18 h. The number of IFN-γ-producing cells was assessed using the ELISPOT assay. Upper panels, representative samples of each group. Bar graph (lower panel) represents the number of IFN-γ spots. Shown are the mean ± SEM of two independent experiments (n = 7). (f) C57BL/6 mice were intravenously transferred with the eFluor 670-labeled naïve GFP+ OT-1 CD8+ T cells and were left untreated or were vaccinated with the OVA257-264 peptide-loaded pMCs or the GM-pMCs on days 0 and 7 after inoculation. On day 10, GFP+ spleen cell proliferation was assessed based on eFluor 670 dilutions with flow cytometry. Upper left panel, experimental approach. Lower left panels, representative histogram of each group. Bar graph (right panel) shows means ± SD (n = 3). Representative data from two independent experiments are presented. (g) C57BL/6 mice were s.c. inoculated with MO4 tumor cells on day 0. Mice were left untreated or treated with the OVA257-264 peptide-loaded pMCs or the GM-pMCs on days 1 and 8. Immunohistochemical analysis of tumor tissues (day 10). CD8+ T cells (brown in the middle panels) and perforin+ cells (brown in the right panels). Hematoxylin and eosin staining served as references (left panels). Scale bar, 50 μm. Bar graph represents the number of CD8+ cells (upper panel) or perforin+ cells (lower panel) per high power field. Shown are means ± SD (n = 6). Additionally, see Figure S4a. *p < .05, **p < .01, ***p < .001.
As GM-pMCs have a cross-presentation capacity, we next evaluated whether GM-pMCs exert antitumor effects when protein antigens were loaded. The OVA protein-loaded GM-pMCs exhibited a similar antitumor effect as the OVA peptide-loaded GM-pMCs, which was mediated by an increase in OVA-specific T cells (Figure 5a-d).
Figure 5.
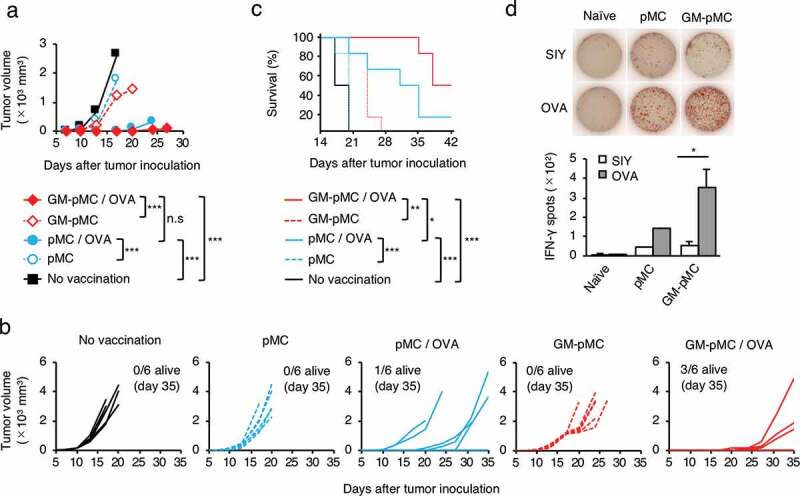
Protein antigen is applicable to GM-pMC-based cancer vaccine.
C57BL/6 mice were i.p. vaccinated twice with the OVA protein-loaded, or unloaded pMCs or GM-pMCs, and s.c. inoculated with MO4 tumor cells and monitored as in Figure 4a. (a) Median tumor volume. (b) Tumor growth of individual mice. (c) Kaplan-Meier survival curves. (a–c) Data shown are representative of two independent experiments. (n = 8 mice). (D) IFN-γ ELOSPOT assay ex vivo on day 56 after inoculation, as in Figure 4e. Shown are the mean ± SEM of two independent experiments (n = 7 except for pMCs). *p < .05, **p < .01, ***p < .001.
Considering the cancer treatment using the iPSC-derived immune cells, it is reasonable to apply allogeneic cells rather than autologous cells. Thus, we performed a vaccine experiment in an MHC-matched-allogeneic setting by administering OVA protein-loaded GM-pMCs from the 129/Sv background into C57BL/6 mice. In the MHC-matched-allogeneic setting, the 129/Sv-derived GM-pMCs exerted a similar antitumor effect in C57BL/6 mice (Figures S4b-e).
As GM-CSF is a proinflammatory cytokine, we investigated whether administration of GM-pMCs triggered adverse events. Administration of GM-pMCs did not affect the leukocyte subset balance, serum GM-CSF levels, or levels of other proinflammatory cytokines, such as IL-6 and TNF-α (Figures S5a, b). Moreover, no other systemic adverse event was observed.
These findings suggest that administration of GM-pMCs loaded with tumor antigens can induce tumor-reactive T cells, leading to antitumor effects without causing adverse effects. Furthermore, our findings suggest that GM-pMCs may be applied to cancer treatment in an MHC-matched-allogeneic setting.
Irradiated GM-pMCs can be used for cancer treatment
As GM-pMCs are generated from iPSCs and have proliferating capacity, there is a concern about their tumorigenic potential, such as induction of teratomas or leukemia.25,26 Therefore, considering the clinical applications, reliable strategies to avoid these adverse events after administration will be required. Irradiation is one strategy for preventing tumor formation of iPSC-derived differentiated cells, however, an excessive dose may attenuate the T cell-stimulation capacity of APCs. Thus, we evaluated whether irradiation could prevent tumorigenic risks while retaining T cell-stimulatory capacity. When irradiated with 85 Gy, most pMCs underwent apoptosis one day after irradiation and disappeared in three days. In contrast, GM-pMCs showed a tendency to avoid apoptosis and survived for three days (Figure 6a and Figures S6a, b). Moreover, irradiated pMCs did not proliferate, while GM-pMCs slightly increased in number for one day and then gradually decreased (Figure 6b). Consistent with the in vitro findings, the irradiated GM-pMCs survived for four days in syngeneic mice, while pMCs disappeared one to two days after irradiation (Figure 6c).
Figure 6.
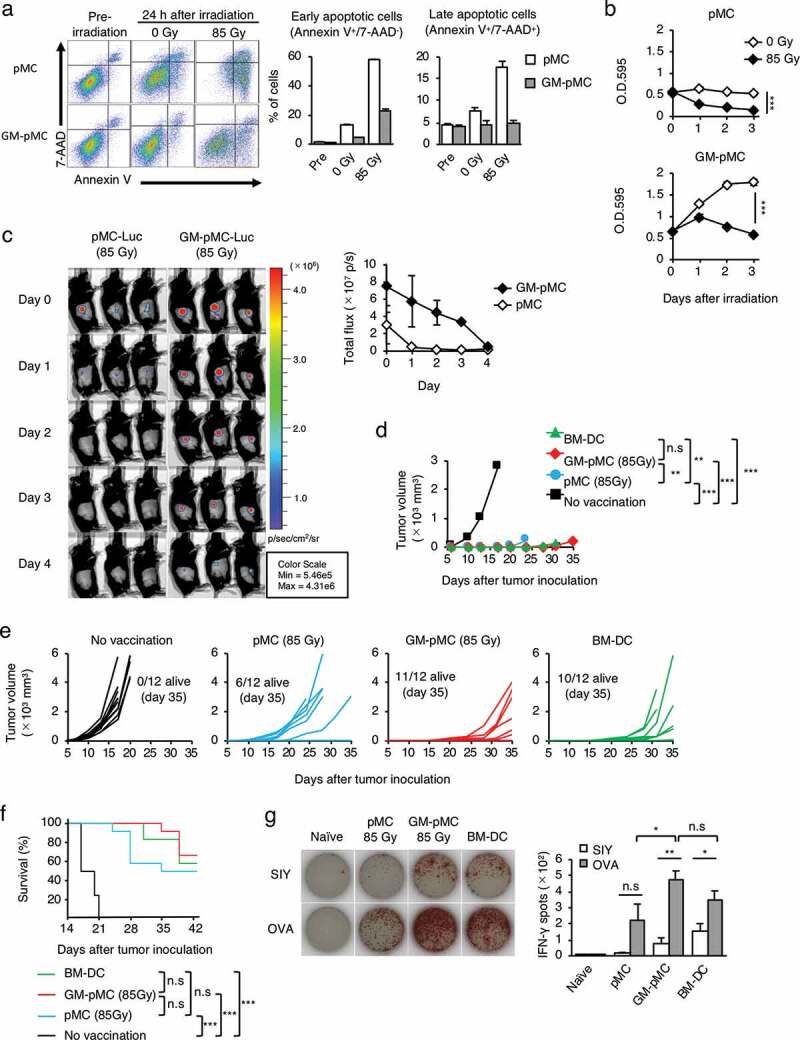
Irradiation controls in vivo GM-pMC survival while retaining antitumor efficacy.
(a) In vitro viability of irradiated pMCs and GM-pMCs. Cells (1.0 × 105 cells/well) were seeded in 96-well culture plates and were 85 Gy-irradiated. The viability of cells (24 h) was evaluated using flow cytometry. Left panels, representative flow cytometry profiles. Bar graph (right panels) represents the frequency of early apoptotic and late apoptotic cells. Additionally, see Figure S6a. (b) Cell proliferation. Cells (2.0 × 104 cells/well) were seeded in 96-well culture plates and left untreated or irradiated with 85 Gy. Proliferation was determined at each time point using the MTT assay. Additionally, see Figures S6a, b. (c) C57BL/6 mice were s.c. inoculated with luciferase-transduced pMCs or GM-pMCs (1.0 × 106 cells) that were irradiated with 85 Gy prior to administration. In vivo cell survival was monitored at the indicated time points using bioluminescence imaging. Left panels, bioluminescence images. Graph (right panel), total flux (photon(s)) at the indicated time points. (a–c) Representative data from two independent experiments are presented. Means ± SD (n = 3) are shown. (d–g) C57BL/6 mice were i.p. vaccinated twice with the OVA257-264 peptide-loaded, the 85 Gy-irradiated pMCs or the GM-pMCs, or non-irradiated BM-DCs, and s.c. inoculated with MO4 tumor cells and monitored as in Figure 4a. Additionally, see Figures S6c-f. (d) Median tumor volume. (e) Tumor growth of individual mice. (f) Kaplan-Meier survival curves. (d–f) Data from two independent experiments are shown. (n = 12 mice). (g) IFN-γ ELOSPOT assay ex vivo on day 56 after inoculation, as in Figure 4e. Shown are the mean ± SEM of two independent experiments (n = 6–8). *p < .05, **p < .01, ***p < .001.
The in vivo survival of APCs correlates to their T cell-stimulatory capacity.27 Thus, to determine whether irradiated GM-pMCs exhibit functional properties of APCs in vivo, we evaluated the antitumor effects in a prophylactic vaccine model with an MO4 tumor. Although the OVA257-264 peptide-loaded pMCs exhibited impaired vaccine effects due to their shortened survival by irradiation, the GM-pMCs retained similar antitumor efficacies as that of non-irradiated cells (Figures S6c-f). Moreover, the effect was similar to that by non-irradiated BM-DCs (Figure 6d-g). These results indicate that in vivo proliferation of GM-pMCs can be appropriately controlled using irradiation. Furthermore, irradiated GM-pMCs can survive for a sufficient period of time to stimulate tumor-reactive T cells in vivo.
GM-pMCs in combination with immune checkpoint inhibitors (ICIs) enhance infiltration of tumor-reactive CD8+ T cells and NK cells
Despite the successful activation of a tumor-reactive T cell response in vivo, it is conceivable that antitumor vaccination may only be effective in a prophylactic setting, and not in a therapeutic setting, due to the established immunosuppressive tumor microenvironment.28 Therefore, additional agents that break such immunosuppressive barriers in combination with cancer vaccines are desirable. Currently, there are two successful ICIs that use different mechanisms for their antitumor effects.29 Cytotoxic T-lymphocyte antigen 4 (CTLA-4), expressed on activated T cells, elicits a suppressive signal by interacting with CD80/CD86 molecules on APCs. In contrast, the programmed death-ligand 1 (PD-L1) is expressed on tumor cells and transmits inhibitory signals via the programmed death-1 (PD-1) molecules on activated T cells. Administration of ICIs in combination with DC-based vaccines is expected to improve the T cell-mediated antitumor effects by removing suppressive signals.30,31 Thus, we assessed the potential of combined treatment with ICIs (anti-CTLA-4 and anti-PD-L1) in two different tumor models that exhibit distinct sensitivity to ICIs (Figure 7a).
Figure 7.
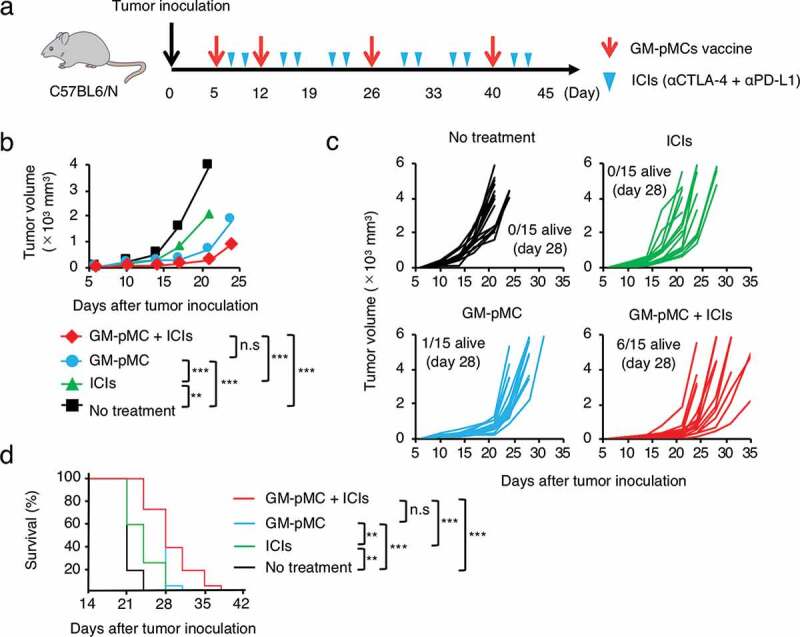
GM-pMC vaccine exhibited a greater therapeutic effect than ICIs in ICI-hyporesponsive tumor.
(a) Schema for tumor implantation and treatment. The mice were s.c. inoculated with tumor cells followed by treatment with GM-pMCs, ICIs i.p. (anti-CTLA-4 Ab + anti-PD-L1 Ab), or in combination. Tumor growth and survival were monitored until the mice died or were sacrificed, when tumors exceeded 20 mm diameter. (b–d) The MO4 tumor-bearing C57BL/6 mice were left untreated or treated with the OVA257-264 peptide-loaded GM-pMCs, the ICIs, or in combination. (b) Median tumor volume. (c) Tumor growth of individual mice. (d) Kaplan-Meier survival curves. Data from two independent experiments are shown (n = 15 mice). *p < .05, **p < .01, ***p < .001.
The MO4 tumor has been reported to be hyporesponsive to ICIs,32 and the GM-pMC vaccine exerted a significantly greater therapeutic efficacy than that exerted by an ICI treatment, as demonstrated by a slower tumor growth and increased survival rate. Although no significant difference was observed, the effect tended to increase when the GM-pMCs were combined with ICIs (Figure 7b-d). We found that intratumoral CD8+ cells and NK1.1+ cells (Figure 8b-d), especially perforin (Pfn) and/or granzyme B (GzmB) expressing cells (Figure 8e-j), were markedly increased by the GM-pMC vaccine, and these observations were more pronounced when combined with ICIs. Indeed, OVA-specific T cells were present in the tumor tissue of mice treated with combination therapy (Figure S7a) and exhibited perforin-dependent cytotoxicity against MO4 cells in vitro (Figure S7b). Moreover, although GM-pMC vaccine alone promoted the infiltration of CD11b+/Gr-1+ myeloid-derived suppressor cells (MDSCs) into the tumor, concomitant ICIs remarkably reduced both the quantity and frequency of MDSCs (Figure 8b-d). These findings indicate that ICIs may confer synergistic antitumor immunity to the GM-pMC vaccine by not only increasing tumor-reactive CD8+ T cells and NK cells, but also by altering the immunosuppressive tumor microenvironment, suggesting the potential of this combination therapy. In terms of safety, there were no apparent adverse events caused by the GM-pMC vaccine or combination therapy, such as an induction of serum proinflammatory cytokines or tissue inflammation (Figure 9a, b).
Figure 8.
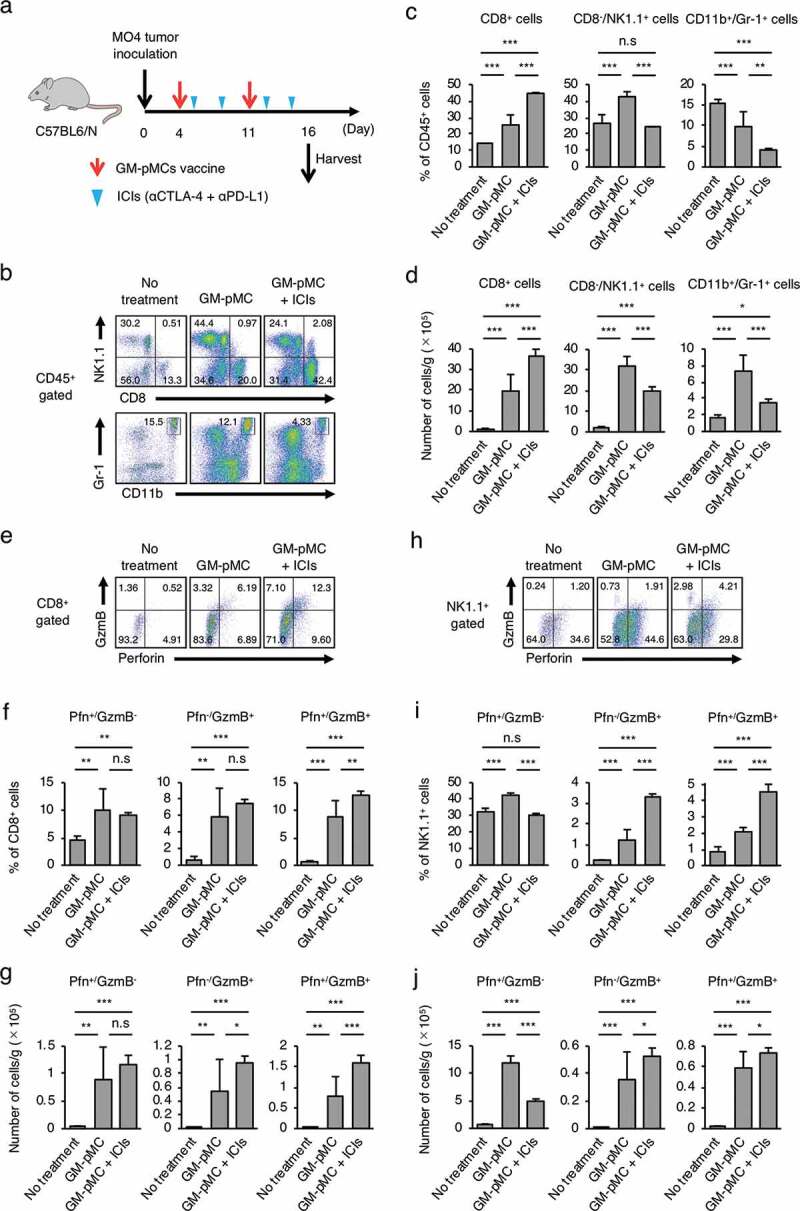
Combined treatment with ICIs enhances antitumor immunity.
(a) Schema for tumor implantation. MO4 tumor-bearing C57BL/6 mice were left untreated or treated with the OVA257-264 peptide-loaded GM-pMCs on days 4 and 11, and ICIs on days 5, 8, 12, and 15. (b–j) Tumors were harvested on day 16, and the intratumoral immune cells were assessed using flow cytometry. (b, e, h) Representative flow cytometry profiles. Graph data represents frequency (c, f, i) and absolute number (per gram of tumor tissue) (d, g, j) of indicated cells. Mean ± SD are shown (n = 5 mice). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Figure 9.
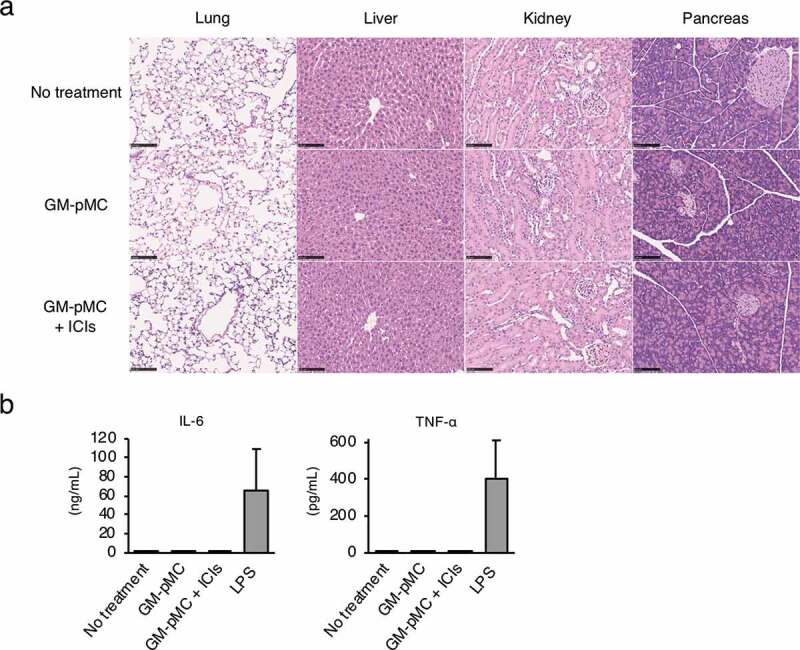
Combination therapy does not induce systemic inflammation.
MO4 tumor-bearing C57BL/6 mice were left untreated or treated as in Figure 7a. (a) Representative hematoxylin and eosin staining of the indicated tissues on day 18. Scale bar, 100 μm. (b) Serum IL-6 and TNF-α were evaluated using ELISA (n = 3). C57BL/6 mice treated with 4.0 mg/kg lipopolysaccharide (LPS) served as a reference (n = 3). Shown are means ± SD. (a, b) Representative data from two independent experiments are presented.
Next, we evaluated the treatment efficacy in an ICI-responsive tumor model (MC38 colon cancer).33 MC38 cells express a mutated ADP-dependent glucokinase (mAdpgk) with a single amino acid mutation.34 This mutation-derived cancer neoantigen is a promising target for cancer immunotherapy as such neoantigens are expressed only in cancer cells and not in normal cells.35,36 Although the wild type Adpgk (wtAdpgk) peptide-loaded GM-pMC vaccine showed no antitumor effect, the mAdpgk peptide-loaded GM-pMC vaccine significantly prolonged survival compared with that observed in the wtAdpgk peptide-loaded GM-pMC vaccine (Figure 10a-c). Moreover, in combination with ICI treatment, survival was significantly prolonged compare to that exhibited by the mAdpgk peptide-loaded GM-pMC vaccine alone. However, the benefit of the combination therapy was not remarkable as ICIs alone sufficiently suppressed this ICIs-responsive tumor.
Figure 10.
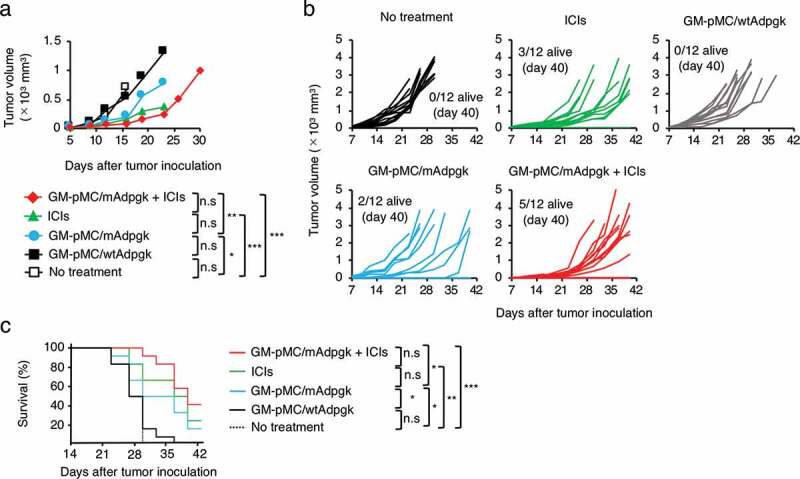
GM-pMC vaccine efficacy does not exceed the effect of ICI on ICI-responsive tumors.
(a–c) MC38 tumor-bearing C57BL/6 mice were left untreated or treated with the wtAdpgk peptide-loaded GM-pMCs, the mAdpgk peptide-loaded GM-pMCs, the ICIs, or a combination of the mAdpgk peptide-loaded GM-pMCs and the ICIs, as in Figure 7a. (a) Median tumor volume. (b) Tumor growth of individual mice. (c) Kaplan-Meier survival curves. Data from two independent experiments are represented (n = 15 mice). *p < .05, **p < .01, ***p < .001.
Recent studies have suggested that stem-like memory CD8+ T cells play a key role in elimination of solid tumors.37,38 Therefore, we evaluated whether GM-CSF is involved in the stemness, or memory function of CD8+ T cells. Results show that even when the CD8+ T cells were stimulated in the presence of GM-CSF, there was no impact on genes associated with stem-like memory or anti-apoptosis, such as the T cell factor 1/7 (Tcf1/7), c-Myb, and Bcl2 in vitro (Figure S8a). In addition, GM-CSF produced by GM-pMCs did not affect the T cell-differentiation state (PD-1/Tcf1), exhausted phenotype (KLRG1/CD62L) (Figures S8b, c), nor did it affect the capacity for cytokine production (Figure S8d) by activated CD8+ T cells.
Discussion
To date, DC-based immunotherapy has been used with autologous DCs generated from peripheral blood-precursor cells.8 However, the stable production of the required quantity and quality of functional DCs with this method has proven challenging. This has been attributed to the variability of individual patient differences or specific clinical conditions, subsequently linked to the limited clinical efficacy of DC-based immunotherapies. Immune cells derived from PSCs are expected to provide an unlimited source of cells for applications in cancer therapies.13,14 However, to generate sufficient numbers of myeloid lineage cells or DCs from PSCs, extensive labor and long periods for cell differentiation are required.39,40 To overcome such issues, we previously developed an iPSC-pMC system that serves as a cellular drug platform for broad applications in cancer therapy, as once a pMC is generated from an iPSC, it can supply an infinite number of homogeneous myeloid cells without requiring their re-preparation.15 However, the system requires two to three days of treatment with IL-4 and GM-CSF for differentiation into functional DCs. Therefore, in the current study we demonstrated that generation of GM-pMCs from iPSCs has the potential to serve as an off-the-shelf APC platform for cancer treatment, allowing for a stable supply of cells with a uniform quality.
GM-pMCs can stimulate antigen-specific T cells by protein-loading due to their cross-presentation capacity. This suggests that GM-pMC vaccines may be useful as a treatment modality for loading antigen proteins, of which the T cell epitopes are unknown. We also found that transduction of the GM-CSF gene imparted a survival advantage to pMCs, which provided a sufficient period of APC/T cell interaction, leading to enhanced T cell priming.27,41 Additionally, GM-CSF produced by GM-pMCs promoted homeostatic proliferation of CD8+ T cells. Although the potency to promote homeostatic proliferation was weaker than that induced by IL-7/IL-15, such amplification of the T cell pool at the T cell-priming site may contribute to an efficient expansion of tumor-reactive T cells. However, systemic administration of recombinant cytokines is often ineffective due to their short half-life. Thus, pMCs in combination with a recombinant GM-CSF may fail to exhibit the equivalent efficacy to GM-pMCs, although these should be clarified in further experiments.
GM-CSF has been combined with various cancer vaccine strategies as an adjuvant to enhance DC functions.20 In contrast to its well-known immune-stimulating functions, GM-CSF often exhibits an immunosuppressive effect via production of prostaglandin E2 from MDSCs.42 A GM-CSF gene-transduced tumor vaccine (GVAX) model suggested that the optimal dose of GM-CSF for eliciting an antitumor immunity may be 36 ng-300 ng/106 tumor cells/24 h.43 In contrast, high dose GM-CSF (> 1,500 ng) induces immunosuppressive effects rather than antitumor efficacy.44 Although a direct comparison is difficult, the amount of GM-CSF produced by GM-pMCs (280 ng/106 cells/24 h) may be optimal for triggering antitumor responses, rather than producing immunosuppressive effects.
By using both the ICI-responsive and -hyporesponsive cancer models, we found that the GM-pMC vaccine exhibited a greater therapeutic effect than that exhibited by ICIs in an ICI-hyporesponsive MO4 cancer. Additionally, combination of ICIs further enhanced GM-pMC-induced effector cell infiltration and showed a tendency to increase effectiveness of the treatment. In contrast, there were no apparent differences in the therapeutic efficacies of ICI and the GM-pMC vaccines, as well as the combination therapy, in an ICI-responsive MC38 cancer. Thus, the GM-pMC vaccine may represent a treatment option for an ICI-hyporesponsive cancer. However, since we examined the efficacy of the therapeutic GM-pMC vaccine only in two tumor models, it is difficult to fully determine the effectiveness of the treatment. Further research is needed to identify the tumors that can serve as effective indicators for combination therapy of GM-pMC and ICIs. A recent study indicated that the neoantigen burden correlated with antitumor T cell responses, particularly with an ICI treatment.36 Nevertheless, although the frequency of the mutant Adpgk allele in the MC38 cancer used in this study was approximately 30%, the transplanted MC38 cancer was completely suppressed in combination therapies in some mice.45 This may be due to the successful elimination of cancer, triggered by an “epitope spreading”.46
Only a single T cell epitope targeting a single cancer antigen was used in this study. This strategy may allow cancer cells to escape an immune surveillance via “cancer immunoediting”;47 therefore, a multiple-antigen targeting strategy is favorable. A recent study suggested that iPSCs and cancer cells may share a number of tumor-associated antigens and that irradiated iPSCs may be promising candidates for cancer vaccines.48 By loading an irradiated iPSC-lysate, our GM-pMCs may efficiently stimulate diverse tumor-reactive T cells, targeting multiple tumor antigens.
Tumorigenicity and histocompatibility are the most critical issues facing iPSC-based cell therapy. GM-pMCs survived for a sufficient period after 85 Gy irradiation, enabling T cell priming in vivo,41 while their proliferative capacity, associated with the risk of tumorigenicity, was appropriately suppressed. Removal of GM-pMCs after fulfilling their immune-stimulatory roles may also avoid the risk of GM-CSF-related adverse events, such as the cytokine-release syndrome. Methods to control the in vivo survival of GM-pMCs by administrating suicide-inducing agents are under investigation.49,50
Considering their clinical use, we recommend using the allogeneic GM-pMCs generated from the HLA-homozygous iPSC stock.51 However, it is difficult to obtain fully HLA haplotype-matched cells. The GM-pMCs expressing mismatched HLA molecules may be eliminated via the host T cell-mediated alloresponse, thereby impairing the antitumor efficacy. Genetic modifications of cell surface HLA molecules may solve the problems associated with histocompatibility. The mismatched HLA allele-specific gene editing may remove the risk of rejection, leading to broader clinical use.52
In conclusion, GM-pMCs generated from iPSCs may serve as an APC cell source for cancer vaccines that replace autologous DCs in the future. Although GM-pMCs harbor different phenotypic features from DCs, these cells can induce an antigen-specific antitumor response equivalent to that induced by DCs. However, it may not be sufficient to determine the APC function of GM-pMCs only in the OVA-expressing tumor model; hence, other intrinsic tumor antigens, such as TRP2 and gp100, should be validated in the future to support the findings herein. Our GM-pMC system, allowing for further genetic manipulations, may result in an excellent cellular platform for cancer therapy. We are currently working on the construction of genetically modified iPSC-pMCs for human use.
Materials and methods
Mice
Female C57BL/6 and BALB/c mice were purchased from Charles River Laboratories (Yokohama, Japan). OT-I CD8+ T cell receptor (TCR)-Tg, and EGFP-Tg mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The EGFP mice were crossed with OT-I mice to generate GFP-expressing OT-1 mice (GFP+ OT-1 mice). In all experiments, animals were randomly assigned to various experimental groups. For experiments designed to evaluate tumor growth, six to ten mice were used for each experiment, which were repeated twice. Animals were sacrificed at the end of the study. All mice were maintained under specific pathogen-free conditions and used when they were 6–12 weeks of age. All animal studies were performed in accordance with the procedures approved by the Animal Research Committee of the National Cancer Center (Tokyo, Japan).
Cells
MO4 is an OVA-expressing B16-F10 melanoma cell line of C57BL/6 origin. MC38 is a colon adenocarcinoma cell line of C57BL/6 origin. RMA-S is a transporter-associated antigen processing (TAP)-defective thymoma cell line of C57BL/6 origin. MO4 and RMA-S cells were cultured in Roswell Park Memorial Institute 1640 medium (RPMI 1640; Sigma-Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco). MC38 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% heat-inactivated FBS. iPSCs of C57BL/6 origin were established as described previously.15 iPSCs of 129/Sv origin were established from peritoneal macrophages using the Cytotune-iPS 2.0 (Medical & Biological Laboratories), according to manufacturer’s instructions. Establishment of mouse iPSC-pMCs was performed as described previously.15 GM-CSF-producing iPSC-pMCs (GM-pMCs) were generated by transducing iPSC-pMCs with a lentivirus vector expressing Csf2 cDNA (GenBank accession number: NM_009969) in the presence of polybrene (Sigma-Aldrich) and were then cultured in alpha-modified Eagle minimum essential medium (α-MEM; Invitrogen) supplemented with 30 ng/mL GM-CSF (Miltenyi Biotec), 50 ng/mL M-CSF (BioLegend), and 20% FBS. All cell lines were tested and found to be free of mycoplasma contamination.
Antibodies and reagents
Anti-mouse GM-CSF antibody (MP1-22E9) for neutralization, and control IgG were purchased from BioLegend. OK-432 (penicillin-killed Streptococcus pyogenes) was purchased from Chugai Pharmaceutical CO., LTD. Staphylococcal enterotoxin B (SEB) was purchased from Toxin Technology Inc. E. coli-derived lipopolysaccharides (LPS O55: B5) was purchased from Sigma-Aldrich. Concanamycin A (CMA) was purchased from AdipoGen Life Sciences.
Generation of recombinant lentivirus
HIV-1-based lentiviral vectors pseudotyped with vesicular stomatitis virus G glycoprotein (VSV-G) were generated by transient transfections with pCAG-HIVgp, pCMV-VSV-G-RSV-Rev, and CSII-EF-Csf2-IRES2-Venus in 293 T cells, using Lipofectamine 2000 (Thermo Fisher Scientific). After 48 h, the vector-containing supernatant was harvested and filtered through 0.45-mm filters. The lentivirus vector was concentrated using Lenti-X Concentrator (Clontech), according to the manufacturer’s instructions.
Generation of DCs from bone marrow cells
DCs were obtained from bone marrow precursors as described previously.15 Briefly, bone marrow cells were cultured in RPMI-1640 supplemented with 10% FBS, 20 ng/mL GM-CSF, 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 μM 2-mercaptoethanol for seven days in petri dishes. GM-CSF was added to the culture on days three and six. On day eight, GM-CSF and 20 ng/mL IL-4 (PeproTech) were added. On day nine, the DCs were collected.
In vivo tumor models
The pMCs, GM-pMCs, and BM-DCs were incubated with OVA257–264 peptides (SIINFEKL, eurofins Genomics), wild type Adpgk peptides (ASMTNRELM, eurofins Genomics), or mutated Adpgk peptides (ASMTNMELM, eurofins Genomics) at a concentration of 10 μM, or OVA protein (100 μg/mL, FUJIFILM Wako Pure Chemical Corporation), for 8–12 h at 37°C. For prophylactic vaccination, mice were treated intraperitoneally with the antigen (OVA257-264 peptide or OVA protein)-loaded or unloaded pMCs, GM-pMCs (1.0 × 105 cells), or BM-DCs (2.0 ×104 cells) twice in a seven-day interval, followed by subcutaneous inoculation with MO4 tumor cells (2.0 × 105 cells) into the right flank, seven days later. For therapeutic vaccination, mice were inoculated subcutaneously (s.c.) with tumor cells (2.0 × 105 MO4 cells or 1 × 105 MC38 cells) in the right flank on day 0. On day five, when the tumors attained a size of 2–3 mm, the mice were randomly assigned to treatment groups and treated i.p. with antigen (OVA257-264 peptide, wild type Adpgk peptide, or mutated Adpgk peptide) loaded GM-pMCs (1.0 × 105 cells) on days 5, 12, 26, and 40. For the checkpoint blockade, mice were i.p. injected with 100 μg anti-PD-L1 monoclonal antibodies (mAb) (10 F.9G2, Bio X cell) and 100 μg anti-CTLA-4 mAb (Armenian Hamster, Bio X cell) twice per week for a total of 12 treatments. The mice were monitored for tumor growth and survival; tumor size was measured twice per week until the mice either died or were sacrificed when the tumors exceeded 20 mm in diameter. Tumor volumes were measured by length (a) and width (b) and calculated as tumor volume = ab2/2.
Cell isolation
Single-cell suspensions of tumor tissues were obtained by combining mechanical dissociation with enzymatic degradation of the extracellular matrix. Tissues were cut into small pieces and suspended in RPMI 1640 medium with 1.0 mg/mL collagenase A (Roche Diagnostics), 0.2 mg/mL hyaluronidase (Nacalai Tesque), and 20 mg/mL DNase I (AppliChem). Tissues were digested using a gentleMACS Dissociator (Miltenyi Biotec) and then passed through a 70 mm filter to generate single-cell suspensions. For intracellular cytokine staining of tumor infiltrating immune cells, the mice were i.p. injected with 10 μg/g GolgiPlug (BD Biosciences) prior to tissue harvesting.
Cytospin
Cell suspensions (1.0 × 105 cells/mL of 0.5% FBS-containing PBS) were loaded onto Superfrost Plus slides using the Cytospin 4 Cytocentrifuge (Thermo Fisher Scientific), spun at 500 rpm for 5 min, and air-dried for 24 h. The dried slides were stained with May-Grünwald/Giemsa (Merck), according to the manufacturer’s instructions.
Flow cytometry
Single-cell suspensions were incubated with the Fc-receptor blocking reagent (Miltenyi Biotec) and then stained with immunofluorescence-conjugated mAbs following the manufacturer’s instructions (Table S5). For transcription factor staining, the cells were first surface stained before fixation and permeabilization using the transcription factor staining kit (eBioscience), followed by intranuclear staining. For intracellular cytokine staining, CD8+ T cells (1.0 × 105 cells) were stimulated in vitro with 50 ng/mL Phorbol-12-myristat-13-acetate (PMA, Sigma-Aldrich) and 1 μg/mL Ionomycin (Sigma-Aldrich) for 5 h in the presence of 5 μg/mL GolgiPlug. Cell surface molecules were stained before fixation and permeabilization, using the fixation/permeabilization kit (BD Biosciences), followed by intracellular staining. Cell viability was determined using the Annexin V apoptosis detection kits (BioLegend), according to manufacturer’s instructions. The cell samples were analyzed on a flow cytometer (FACS Canto II or Accuri C6; BD Biosciences), and data were analyzed using FlowJo software v10.7 (Tree Star Inc.).
Immunohistochemistry
Tissues were fixed with Tissue Fixative (Genostaff), embedded in paraffin, and sectioned at 5 μm. The tissue sections were de-paraffinized with xylene and rehydrated using an ethanol series and Tris-buffered saline (TBS). Antigen retrieval was performed by microwave treatment for 10 min at 500 W in 1 mM ethylenediaminetetraacetic acid (EDTA) buffer, pH 6.0. Endogenous peroxidase was blocked with 0.3% hydrogen peroxide in methanol for 30 min. After washing with TBS, the sections were incubated with G-Block (Genostaff), followed by treatment with an Avidin/Biotin Blocking kit (Vector Laboratories). For CD4, CD8, or perforin single staining, slides were incubated with rat anti-mouse CD4 mAb (eBioscience, 4SM95), rat anti-mouse CD8α mAb (eBioscience, 4SM15), or anti-mouse perforin polyclonal antibody (CB5.4) at 4°C overnight. After washing with TBS, the sections were incubated with biotin-conjugated anti-rat IgG (Vector Laboratories) for 30 min at 25°C, followed by addition of peroxidase-conjugated streptavidin (Nichirei) for 5 min. Peroxidase activity was visualized using diaminobenzidine/hydrogen peroxide. The sections were counterstained with Mayer’s Hematoxylin (Muto Pure Chemicals), dehydrated, and then mounted with G-Mount (Genostaff).
In situ hybridization
The cDNA fragment of murine Cd274 at cDNA positions 59–735 (GenBank accession number: NM_021893.3) was used for generation of sense or anti-sense RNA probes. Digoxigenin (DIG)-labeled RNA probes were prepared using DIG RNA Labeling Mix (Roche Diagnostics). Tissues were fixed with G-Fix (Genostaff), embedded in paraffin on CT-Pro20 (Genostaff) using G-Nox (Genostaff), as a less toxic solvent than xylene, and sectioned at 5 μm. In situ hybridization (ISH) was performed using the ISH Reagent kit (Genostaff) according to the manufacturer’s instructions. Paraffin-embedded sections were hybridized with the DIG-labeled RNA probes at 60°C for 16 h. After hybridization, sections were incubated with anti-DIG alkaline phosphate conjugate (Roche Diagnostics). The bound label was detected using alkaline phosphate color substrates nitro-blue tetrazolium chloride and 5-bromo-4-chloro-3ʹ-indolylphosphatase p-toluidine salt (Sigma-Aldrich). Sections were then counterstained with Kernechtrot Stain Solution (Muto Pure Chemicals, Tokyo, Japan) and mounted with G-Mount (Genostaff).
RNA-seq and analysis
RNA-Seq analysis was performed using the BGISEQ (Hong Kong, China) instrument in the paired-end 2 × 100 bp cycle mode by using library creation kits (BGI original library kit). The sequence reads were mapped to the mouse reference genome (mm10) provided by the UCSC as of October 16, 2016, by using HISAT2 v2.1.0; they were quantified using the HTSeq-count v0.9.1 for read counts and Stringtie v1.3.4 for FPKM, respectively. The data has been deposited in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, accession number GSE136993). Genes differentially expressed between pMCs and GM-pMCs (log fold change ≥ 1 or ≤ −1) were identified using DESeq2.53 Principal component analysis was performed using variable genes. Up-regulated or down-regulated genes shared by C57BL/6 GM-pMCs and 129 Sv GM-pMCs were analyzed for Gene Ontology (GO) biological processes using DAVID.54 Gene set enrichment analysis was performed using gene set enrichment analysis (GSEA) software (http://software.broadinstitute.org/gsea/downloads.jsp).
Real-time PCR
Total RNA was extracted using the RNeasy Mini Kit Plus (QIAGEN), and cDNA was synthesized using the PrimeScript II 1st strand cDNA Synthesis kit (TaKaRa Bio). Transcripts were quantified using real-time quantitative PCR on an ABI PRISM 7500 sequence detector using the TaqMan Gene Expression Assays (Applied Biosystems). The following probes were used: T cell factor 7 (Tcf7; Mm00493445_m1), c-Myb (Myb; Mm00501741_m1), Bcl2 (Bcl2; Mm00477631_m1), Bcl-XL (Bcl2l1; Mm00437783_m1), Bax (Bax; Mm00432051_m1), and glyceraldehyde-3-phosphate dehydrogenase (Gapdh; Mm99999915_g1). mRNA expression levels were calculated using the change-in-cycling-threshold (Ct) method, and the results were normalized to levels of the control gene Gapdh.
Cytokine measurements
Cytokine levels in culture supernatant or serum were measured using an enzyme-linked immunosorbent assay kit (BioLegend), according to the manufacturer’s instructions.
Cytotoxicity
The cytotoxicity of CD8+ T cells was measured using standard 51Cr-release assays. Target cells were labeled with sodium [51Cr]-chromate (5 mCi/mL) for 1 h at 37°C, washed twice, and seeded onto 96-well round-bottomed culture plates (5 × 103 cells/well). Effector cells were added to the target cells at indicated E:T (Effector:Target) ratios and incubated for 4 h at 37°C. The plates were then centrifuged, and supernatants (25 μL/well) were harvested and counted in a liquid scintillation counter (MicroBeta2 LumiJET, PerkinElmer). The percentage of specific lysis was calculated as: 100× [(experimental release – spontaneous release)/(maximal release – spontaneous release)]. Spontaneous release and maximal release were determined in the presence of either medium or 1% Triton X-100, respectively.
Enzyme-linked immunosorbent spot assay
The frequency of IFN-γ-producing cells was detected using an enzyme-linked immunosorbent spot assay kit according to the manufacturer’s instructions (BD Biosciences). Spots were enumerated using an Eliphoto counter (Minervatech).
In vitro cell proliferation
Cell proliferation was evaluated using a standard MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; Sigma-Aldrich) assay and a [3H]-thymidine incorporation assay. For the MTT assay, cells were seeded in 96-well culture plates and labeled with MTT at each time point. After 4 h of labeling, the plate was centrifuged to sediment the cells, and the supernatant was removed by tapping. Subsequently, 200 μL of DMSO was added to each well, and the plate was mixed thoroughly. After 5 min, absorbance at 570 nm was measured. For [3H]-thymidine incorporation assay, cells were cultured for four to six days and pulsed with [3H]-thymidine (PerkinElmer, 1 μCi/well) for the last 16 h. Proliferation was measured as3H-thymidine incorporation by scintillation counting (MicroBeta2 LumiJET, PerkinElmer).
In vivo cell proliferation
GFP-expressing OT-I T cells were isolated from the spleen using the CD8α+ T cell isolation kit (Miltenyi Biotec), labeled with Cell Proliferation Dye eFluor 670 (eBioscience), and intravenously transferred into naïve C57BL/6 mice on day 0. The mice were i.p. injected with OVA257-264 peptide-loaded pMCs or GM-pMCs on days 0 and 7. On day 10, their spleens were harvested, and single-cell suspensions were analyzed on a flow cytometer.
In vivo bioluminescent imaging
Mice were i.p. injected with 200 μL of d-luciferin (15 mg/mL; VivoGlo Luciferin; Promega) under 2% isoflurane as an inhaled anesthesia, and bioluminescence images were obtained using the IVIS Lumina II instrument with the Living Image software version 3.2 (PerkinElmer).
Statistical analysis
Statistical analysis was conducted using JMP Genomics software version 14 (SAS Institute Inc., Cary, NC, USA). Unpaired two-tailed Student’s t-tests were used for comparisons between the two groups. One-way ANOVAs with the Tukey’s test were used for multiple comparisons. The Kaplan-Meier survival analysis was based on reaching the endpoint (when mice died or were sacrificed when the tumors exceeded 20 mm in diameter). Differences between the survival curves were evaluated using log-rank tests. Results were considered significant at the following p-values: *p < .05, **p < .01, ***p < .001.
Supplementary Material
Acknowledgments
We thank Dr. H. Miyoshi (RIKEN) for providing the plasmid vectors (pCSII-EF, pCMV-VSV-G-RSV-Rev, and pCAG-HIVgp). We thank Mrs. M. Ozaki (National Cancer Center) for the secretarial work. We thank Dr. R. Nakaki, Y. Osako, and M. Kawamura (Rhelixa, Inc.) for analyzing the RNA-seq data.
Funding Statement
This research was supported in part by the JSPS KAKENHI under Grant numbers [JP17K16877, JP20K09186, and JP20H03759]; the AMED under Grant numbers [JP19bm0404054 and JP20bm040454]; and the National Cancer Center Research and Development Fund (28-A-8).
Abbreviations
- APC
antigen-presenting cell
- BM-DCs
bone marrow-derived DCs
- DC
dendritic cell
- GM-CSF
granulocyte macrophage colony-stimulating factor
- GM-pMCs
GM-CSF producing iPSC-pMCs
- iPSC
induced pluripotent stem cell
- iPSC-pMCs
iPSC-derived proliferating myeloid cells
- ISH
in situ hybridization
- pMCs
proliferating myeloid lineage cells
Author contributions
Conceptualization: R.Z. S.S., and Y.U.; Methodology: R.Z.; Investigation: H.M., R.Z., Y.H., T.I., M.Y., C. L., N.W., T.Y., and Y.U.; Resources: R.Z., T.L., and T.I.; Writing-Original Draft: H.M., and Y.U.; Writing-Review & Editing: H.M., R.Z., T.K., H.T., R.N., Y.M., S.K., and Y.U.; Administrative support: A.I., H.O., and T.N.; Supervision: Y.U.; Funding Acquisition: R.Z., T.I., S.K., T.N., and Y.U.
Data and code availability
The data have been deposited in NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, accession number GSE136993).
Disclosure statement
Yasushi Uemura received research funding from AGC Inc. Japan. The remaining authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Palucka K, Banchereau J.. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39:38–16. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15:e257–e267. doi: 10.1016/s1470-2045(13)70585-0. [DOI] [PubMed] [Google Scholar]
- 3.Sprooten J, Ceusters J, Coosemans A, Agostinis P, De Vleeschouwer S, Zitvogel L, Kroemer G, Galluzzi L, Garg AD. Trial watch: dendritic cell vaccination for cancer immunotherapy. Oncoimmunology. 2019;8:e1638212. doi: 10.1080/2162402X.2019.1638212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garg AD, Vara Perez M, Schaaf M, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: dendritic cell-based anticancer immunotherapy. Oncoimmunology. 2017;6:e1328341. doi: 10.1080/2162402X.2017.1328341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bloy N, Pol J, Aranda F, Eggermont A, Cremer I, Fridman WH, Fucikova J, Galon J, Tartour E, Spisek R, et al. Trial watch: dendritic cell-based anticancer therapy. Oncoimmunology. 2014;3:e963424. doi: 10.4161/21624011.2014.963424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 7.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: state of the art and beyond. Clin Cancer Res. 2016;22:1897–1906. doi: 10.1158/1078-0432.CCR-15-1399. [DOI] [PubMed] [Google Scholar]
- 9.Failli A, Legitimo A, Orsini G, Romanini A, Consolini R. Numerical defect of circulating dendritic cell subsets and defective dendritic cell generation from monocytes of patients with advanced melanoma. Cancer Lett. 2013;337:184–192. doi: 10.1016/j.canlet.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Orsini G, Legitimo A, Failli A, Ferrari P, Nicolini A, Spisni R, Miccoli P, Consolini R. Defective generation and maturation of dendritic cells from monocytes in colorectal cancer patients during the course of disease. Int J Mol Sci. 2013;14:22022–22041. doi: 10.3390/ijms141122022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verronese E, Delgado A, Valladeau-Guilemond J, Garin G, Guillemaut S, Tredan O, Ray-Coquard I, Bachelot T, N’Kodia A, Bardin-Dit-Courageot C, et al. Immune cell dysfunctions in breast cancer patients detected through whole blood multi-parametric flow cytometry assay. Oncoimmunology. 2016;5:e1100791. doi: 10.1080/2162402X.2015.1100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Y, Lei K, Tang L. Neoantigen vaccine delivery for personalized anticancer immunotherapy. Front Immunol. 2018;9. doi: 10.3389/fimmu.2018.01499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang Z, Han Y, Cao X. Induced pluripotent stem cell (iPSCs) and their application in immunotherapy. Cell Mol Immunol. 2014;11:17–24. doi: 10.1038/cmi.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sachamitr P, Hackett S, Fairchild PJ. Induced pluripotent stem cells: challenges and opportunities for cancer immunotherapy. Front Immunol. 2014;5:176. doi: 10.3389/fimmu.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang R, Liu T-Y, Senju S, Haruta M, Hirosawa N, Suzuki M, Tatsumi M, Ueda N, Maki H, Nakatsuka R, et al. Generation of mouse pluripotent stem cell-derived proliferating myeloid cells as an unlimited source of functional antigen-presenting cells. Cancer Immunol Res. 2015;3:668–677. doi: 10.1158/2326-6066.CIR-14-0117. [DOI] [PubMed] [Google Scholar]
- 16.Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 2013;34:81–89. doi: 10.1016/j.it.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Min L, Mohammad Isa SAB, Shuai W, Piang CB, Nih FW, Kotaka M, Ruedl C. Cutting edge: granulocyte-macrophage colony-stimulating factor is the major CD8+ T cell-derived licensing factor for dendritic cell activation. J Immunol. 2010;184:4625–4629. doi: 10.4049/jimmunol.0903873. [DOI] [PubMed] [Google Scholar]
- 18.Kaufman HL, Ruby CE, Hughes T, Slingluff CL Jr.. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J Immunother Cancer. 2014;2:11. doi: 10.1186/2051-1426-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borrello I, Pardoll D. GM-CSF-based cellular vaccines: a review of the clinical experience. Cytokine Growth Factor Rev. 2002;13:185–193. doi: 10.1016/S1359-6101(01)00034-X. [DOI] [PubMed] [Google Scholar]
- 20.Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, Zhang Y, Zhang L, Yuan ZR, Tan HSW, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res. 2006;16:126–133. doi: 10.1038/sj.cr.7310017. [DOI] [PubMed] [Google Scholar]
- 21.Bezu L, Kepp O, Cerrato G, Pol J, Fucikova J, Spisek R, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: peptide-based vaccines in anticancer therapy. Oncoimmunology. 2018;7:e1511506. doi: 10.1080/2162402X.2018.1511506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe N, Hanabuchi S, Soumelis V, Yuan W, Ho S, de Waal Malefyt R, Liu Y-J. Human thymic stromal lymphopoietin promotes dendritic cell-mediated CD4+ T cell homeostatic expansion. Nat Immunol. 2004;5:426–434. doi: 10.1038/ni1048. [DOI] [PubMed] [Google Scholar]
- 24.Ge Q, Palliser D, Eisen HN, Chen J. Homeostatic T cell proliferation in a T cell-dendritic cell coculture system. Proc Natl Acad Sci U S A. 2002;99:2983–2988. doi: 10.1073/pnas.052714199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutierrez-Aranda I, Ramos-Mejia V, Bueno C, Munoz-Lopez M, Real PJ, Mácia A, Sanchez L, Ligero G, Garcia-Parez JL, Menendez P, et al. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem Cells. 2010;28:1568–1570. doi: 10.1002/stem.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miura K, Okada Y, Aoi T, Okada A, Takahashi K, Okita K, Nakagawa M, Koyanagi M, Tanabe K, Ohnuki M, et al. Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol. 2009;27:743–745. doi: 10.1038/nbt.1554. [DOI] [PubMed] [Google Scholar]
- 27.Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ, Wang J. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006;311:1160–1164. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 28.Johansson-Percival A, He B, Li Z-J, Kjellén A, Russell K, Li J, Larma I, Ganss R. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat Immunol. 2017;18:1207–1217. doi: 10.1038/ni.3836. [DOI] [PubMed] [Google Scholar]
- 29.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 30.Yang P, Song H, Qin Y, Huang P, Zhang C, Kong D, Wang W. Engineering dendritic-cell-based vaccines and PD-1 blockade in self-assembled peptide nanofibrous hydrogel to amplify antitumor T-cell immunity. Nano Lett. 2018;18:4377–4385. doi: 10.1021/acs.nanolett.8b01406. [DOI] [PubMed] [Google Scholar]
- 31.Wilgenhof S, Corthals J, Heirman C, van Baren N, Lucas S, Kvistborg P, Thielemans K, Neyns B. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J Clin Oncol. 2016;34:1330–1338. doi: 10.1200/JCO.2015.63.4121. [DOI] [PubMed] [Google Scholar]
- 32.Chen S, Lee L-F, Fisher TS, Jessen B, Elliott M, Evering W, Logronio K, Tu GH, Tsaparikos K, Li X, et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res. 2015;3:149–160. doi: 10.1158/2326-6066.CIR-14-0118. [DOI] [PubMed] [Google Scholar]
- 33.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, Sharpe AH. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214:895–904. doi: 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu G, Mei L, Vishwasrao HD, Jacobson O, Wang Z, Liu Y, Yung BC, Fu X, Jin A, Niu G, et al. Intertwining DNA-RNA nanocapsules loaded with tumor neoantigens as synergistic nanovaccines for cancer immunotherapy. Nat Commun. 2017;8:1482. doi: 10.1038/s41467-017-01386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, Goedegebuure SP, Gillanders WE. Preclinical and clinical development of neoantigen vaccines. Ann Oncol. 2017;28:xii11–xii17. doi: 10.1093/annonc/mdx681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 37.Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, Hu J, Hocker JD, Hawk NV, Kapoor V, et al. The transcription factor c-Myb regulates CD8+ T cell stemness and antitumor immunity. Nat Immunol. 2019;20:337–349. doi: 10.1038/s41590-018-0311-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50:195–211 e110. doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 39.Sachamitr P, Leishman AJ, Davies TJ, Fairchild PJ. Directed differentiation of human induced pluripotent stem cells into dendritic cells displaying tolerogenic properties and resembling the CD141(+) subset. Front Immunol. 2017;8:1935. doi: 10.3389/fimmu.2017.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi KD, Vodyanik M, Slukvin II. Hematopoietic differentiation and production of mature myeloid cells from human pluripotent stem cells. Nat Protoc. 2011;6:296–313. doi: 10.1038/nprot.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 42.Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569:73–78. doi: 10.1038/s41586-019-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol. 2007;18:226–232. doi: 10.1093/annonc/mdl158. [DOI] [PubMed] [Google Scholar]
- 44.Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64:6337–6343. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- 45.Tsuchiya N, Zhang R, Iwama T, Ueda N, Liu T, Tatsumi M, Sasaki Y, Shimoda R, Osako Y, Sawada Y, et al. Type I interferon delivery by iPSC-derived myeloid cells elicits antitumor immunity via XCR1+ dendritic cells. Cell Rep. 2019;29:162–175.e169. doi: 10.1016/j.celrep.2019.08.086. [DOI] [PubMed] [Google Scholar]
- 46.Heckler M, Dougan SK. Unmasking pancreatic cancer: epitope spreading after single antigen chimeric antigen receptor T-cell therapy in a human Phase I trial. Gastroenterology. 2018;155:11–14. doi: 10.1053/j.gastro.2018.06.023. [DOI] [PubMed] [Google Scholar]
- 47.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16:151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 48.Kooreman NG, Kim Y, de Almeida PE, Termglinchan V, Diecke S, Shao N-Y, Wei -T-T, Yi H, Dey D, Nelakanti R, et al. Autologous iPSC-based vaccines elicit anti-tumor responses in vivo. Cell Stem Cell. 2018;22:501–513 e507. doi: 10.1016/j.stem.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCormick F. Cancer gene therapy: fringe or cutting edge? Nat Rev Cancer. 2001;1:130–141. doi: 10.1038/35101008. [DOI] [PubMed] [Google Scholar]
- 50.Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Umekage M, Sato Y, Takasu N. Overview: an iPS cell stock at CiRA. Inflamm Regen. 2019;39:17. doi: 10.1186/s41232-019-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, Ueda T, Gee P, Nishikawa M, Nomura M, et al. Targeted disruption of HLA genes via CRISPR-cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell. 2019;24:566–578 e567. doi: 10.1016/j.stem.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 53.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data have been deposited in NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, accession number GSE136993).