

ABSTRACT
Infection with human papillomavirus (HPV) is associated with a variety of cancer types and limited therapy options. Therapeutic cancer vaccines targeting the HPV16 oncoproteins E6 and E7 have recently been extensively explored as a promising immunotherapy approach to drive durable antitumor T cell immunity and induce effective tumor control. With the goal to achieve potent and lasting antitumor T cell responses, we generated a novel lymphocytic choriomeningitis virus (LCMV)-based vaccine, TT1-E7E6, targeting HPV16 E6 and E7. This replication-competent vector was stably attenuated using a three-segmented viral genome packaging strategy. Compared to wild-type LCMV, TT1-E7E6 demonstrated significantly reduced viremia and CNS immunopathology. Intravenous vaccination of mice with TT1-E7E6 induced robust expansion of HPV16-specific CD8+ T cells producing IFN-γ, TNF-α and IL-2. In the HPV16 E6 and E7-expressing TC-1 tumor model, mice immunized with TT1-E7E6 showed significantly delayed tumor growth or complete tumor clearance accompanied with prolonged survival. Tumor control by TT1-E7E6 was also achieved in established large-sized tumors in this model. Furthermore, a combination of TT1-E7E6 with anti-PD-1 therapy led to enhanced antitumor efficacy with complete tumor regression in the majority of tumor-bearing mice that were resistant to anti-PD-1 treatment alone. TT1-E7E6 vector itself did not exhibit oncolytic properties in TC-1 cells, while the antitumor effect was associated with the accumulation of HPV16-specific CD8+ T cells with reduced PD-1 expression in the tumor tissues. Together, our results suggest that TT1-E7E6 is a promising therapeutic vaccine for HPV-positive cancers.
KEYWORDS: Cancer vaccine, LCMV, HPV16 E6 E7, HPV16-positive cancer, mouse tumor model
Introduction
Infection with high-risk human papillomavirus (HPV) types is a major cause of head and neck squamous cell carcinoma (HNSCC), cervical and anogenital cancers. HPV16, the most prevalent HPV type, accounts for the majority of these cancers,1,2 in which surgery, chemotherapy and radiotherapy are the traditional standard of care. Recent advances in alternative therapies, such as epidermal growth factor receptor (EGFR) antagonists, tyrosine kinase inhibitors and immune checkpoint inhibitors (ICI), have offered varying degrees of success in clinical trials for HPV-associated cancers.3 However, the standard of care treatment options are limited and often fall short in long-term clinical benefits for patients with HPV-positive, advanced, recurrent, or metastatic HNSCC and other HPV16-positive cancers.
CD8+ cytotoxic T cell (CTL) responses play a pivotal role in antitumor immunity. Several clinical studies have demonstrated that tumor infiltration by CD8+ T cells correlates with improved disease outcome in various cancer types including HPV16-positive HNSCC.4-6 In recent years, ICI targeting programmed cell death 1 (PD-1) or its ligand PD-L1 have been approved for the treatment of recurrent or metastatic HNSCC as well as advanced cervical cancers.7-9
Therapeutic HPV vaccines aiming to induce antitumor CTL responses are emerging as a promising strategy to treat HPV-positive cancers, especially when combined with ICI.10 The HPV oncoproteins E6 and E7 commonly targeted by these therapeutic vaccines are expressed in all HPV-infected cells; persist throughout the viral life cycle and drive the transformation of healthy cells into cancer cells.11,12 In clinical trials and animal studies, HPV E7 and/or E6 vaccine-induced CD8+ T cells not only suppress HPV-positive tumor growth, but also mediate clearance of virus-infected cells.13-15
To date, active immunization has not induced consistent efficacy as cancer immunotherapy in clinical Phase III trials.16-18 The goal of immunotherapy by cancer vaccines is to achieve large numbers of CTLs infiltrating and targeting tumor tissues and to establish memory cells for lasting tumor control. From this perspective, replicating viral vaccines, which can deliver tumor-associated antigens in the context of viral infection, are well suited to fulfill this task. Previously, we have demonstrated the potential of this strategy by generating a stably attenuated, replicating lymphocytic choriomeningitis virus (LCMV)-based vector.19 LCMV, the prototype member of the arenavirus family, was chosen primarily for its ability to elicit potent effector CTL responses, life-long CTL immunity and because LCMV-based vectors have demonstrated the capacity to induce tumor regression in a CD8+ T cell-dependent manner.19,20 In addition, LCMV has low seroprevalence in humans21,22 and the glycosylation of its surface protein renders the host neutralizing Ab response inefficient, which allows for re-administrations of the vector.19,23 The replication-competent, stably attenuated LCMV vector can deliver tumor-associated antigens to professional antigen-presenting cells to induce potent CTL responses and triggers the release of alarmin IL-33 from lymphoid stroma cells resulting in a superior effector CTL response than that induced by replication-deficient LCMV vectors.19
Based on this replicating, attenuated LCMV platform, here, we generated a novel vaccine encoding a non-oncogenic version of the HPV16 oncoproteins E7 and E6 (TT1-E7E6), for which other platforms such as adenovirus vectors have previously been evaluated.24 We evaluated our vector’s potential immunotherapy efficacy in a mouse model of HPV16-positive tumor. We demonstrated vector clearance, HPV 16-specific CD8+ T cell response and tumor control following TT1-E7E6 immunization, which was associated with the accumulation of HPV16-specific CD8 T cells with reduced PD-1 expression in the tumor tissues. Furthermore, TT1-E7E6 synergized with PD-1 checkpoint inhibition to suppress tumor growth. These findings warrant further exploration of this vaccine as immunotherapy for HPV16-positive cancers.
Results
Construction of a replicating LCMV-based vector to target HPV16-positive cancers
A replication-attenuated, therapeutic vaccine vector based on LCMV was engineered as described previously.19 In the natural context, wild-type (wt) LCMV encodes four essential viral proteins on two genomic segments: glycoprotein (GP), nucleoprotein (NP), RNA-directed RNA polymerase (L) and RING finger protein Z (Z). The small (S) genome segment encodes GP and NP, while the large (L) genome segment encodes L and Z (Figure 1(a)). Replicating vectors were generated based on LCMV strain clone 13 (Cl13) with the GP from LCMV strain WE [LCMV (Cl13/WE-GP)] by segregating the viral GP and NP into two artificially duplicated S-segments. Thus, each replication-competent vector contains three genome segments, including one L-segment and two S-segments, which encode either GP or NP but not both. To target HPV16-associated tumors, an inactivated fusion protein of HPV16 antigens E7 and E6, which has been described before, 25 was introduced into both S-segments, yielding the vaccine vector TT1-E7E6 (Figure 1(b)).
Figure 1.
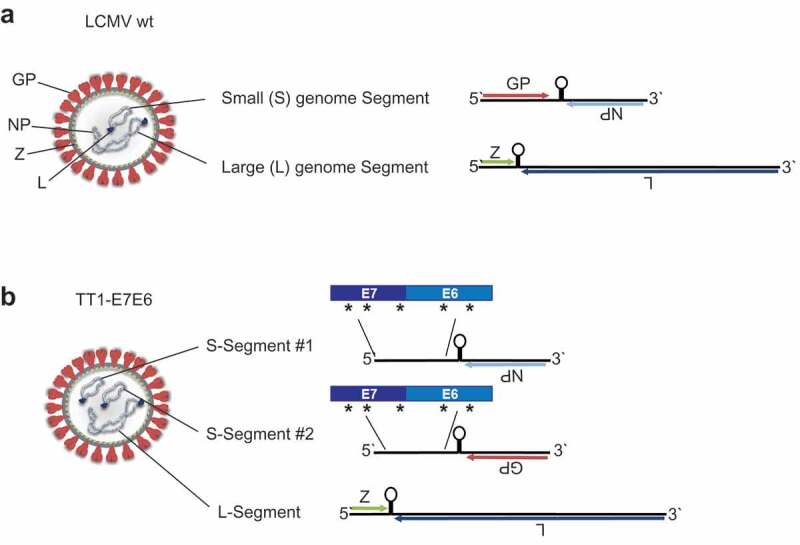
Construction of TT1-E7E6. Schematic view of (a) a LCMV wt particle (left) and its genome (right). The ambisense RNA genome encodes 4 viral proteins: GP (glycoprotein), NP (nucleoprotein), L (RNA-directed RNA polymerase) and Z (RING finger protein Z). (b) The engineered TT1-E7E6 vectors contain artificially duplicated S-segments, encoding either GP or NP but not both, and a fusion protein of HPV16 E7E6, with 5 implemented amino acid mutations as indicated by the asterisks (*), as well as the L-segment.
Co-packaging of the L-Segment and the two different S-Segments is an inefficient process and results in a lower abundance of the replication-competent three-segmented TT1-E7E6 particles (~10%) compared to the two-segmented particles (~90%) (Supplemental Figure S1(a)). Hence, replication of these vaccine vectors was attenuated when compared with parental wt virus. Importantly, two-segmented vector particles, which are formed during vector replication and contained only one S-segment encoding NP and the L-Segment (rLCMV) (Supplemental Figure S1(a)), are active and immunogenic.26 These rLCMV particles can initiate mRNA transcription and genome replication in the target cells, resulting in antigen expression and induction of CTLs, therefore further contributing to immune responses without forming new infectious vector progeny.19
The insertion of E7E6 in the TT1-E7E6 vector genome was verified by PCR and the antigen expression was confirmed by immunoblot of HEK293 cells infected with TT1-E7E6 (Supplemental Figure S1(b and c)). Analysis of vector growth in human cells (HEK293) confirmed that replication of TT1-E7E6 was substantially attenuated in vitro compared to the parental LCMV (Cl13/WE-GP) herein referred to as LCMV wt (Supplemental Figure S1d).
TT1-E7E6 exhibits reduced in vivo replicative capacity and neurovirulence
To examine the replicative capacity of TT1-E7E6 in vivo, immunocompetent mice were treated i.v. with a high dose of replication-competent TT1-E7E6 [2x106 replication-competent virus (RCV) focus forming units (FFU)] or the corresponding parental LCMV wt. Viral load was quantified in the blood post-administration. On Day 4 post-infection, four out of five mice showed low but detectable titers of TT1-E7E6 in the blood, which rapidly decreased below the limit of quantification (LOQ) by Day 8 and remained undetectable at all later time points. In contrast, mice receiving LCMV wt had substantially higher levels of circulating virus early on, and virus persisted in blood throughout the observation period of 20 days (Figure 2(a)). These data recapitulated our previous findings using analogous vectors19 and confirmed that replication of TT1-E7E6 is significantly attenuated compared to LCMV wt in vivo.
Figure 2.
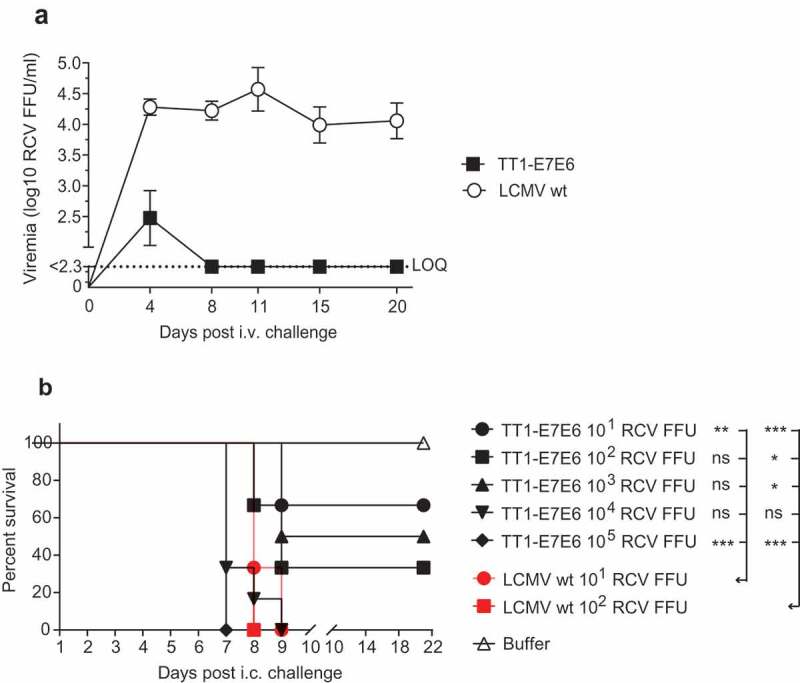
TT1-E7E6 exhibits reduced in vivo replicative capacity and neurovirulence. (a) Immunocompetent mice were i.v. infected with 2 × 106 RCV FFU of TT1-E7E6 or the corresponding LCMV wt. Viral loads in the blood were measured by immunofocus assay on Day 4, 8, 11, 15 and 20 post-administration. Data shown are means ± SD. The dotted line indicates the limit of quantification (LOQ, log10 2.3 RCV FFU). N=5 mice per group. (b) Survival curve of mice inoculated intracranially (i.c.) with TT1-E7E6 or LCMV wt. A tail-spin test was performed on all surviving animals for detection of choriomeningitis, which was defined as the humane endpoint for survival analysis. N=6 mice per group. Mantel-Cox test. Ns, not significant, * p<.05, ** p<.01, *** p<.001.
Although the vast majority of humans accidentally infected with wt LCMV remain completely asymptomatic or develop an unspecific flu-like illness, an estimated 1% to 2% of infected individuals may progress to the second phase of disease, which can manifest as self-limiting aseptic meningitis.27 To assess the potential risk of CNS manifestation post-TT1-E7E6 treatment, neurovirulence was analyzed by injecting the vectors intracranially (i.c.) into mice. Mice receiving LCMV wt rapidly developed progressive lethal choriomeningitis between Day 6 and Day 9, such that all mice in all dose groups (10 and 100 RCV FFU) had to be euthanized by Day 9. In contrast, in the TT1-E7E6 treatment groups, by Day 21, 67%, 33% and 50% of mice survived a challenge dose of 10, 100 and 1000 RCV FFU, respectively (Figure 2(b)). In addition, because the minimal dose required for invariably lethal disease was ≤10 RCV FFU for LCMV wt and ≥1000 RCV FFU for TT1-E7E6, these results suggest that TT1-E7E6 is attenuated several log-fold compared to LCMV wt in terms of neurovirulence.
Immunogenicity of TT1-E7E6
We next investigated the HPV16 E7-specific CD8+ T cell response induced by TT1-E7E6 in doses ranging from 104 to 106 RCV FFU/dose in peripheral blood from mice. TT1-E7E6 was administered i.v. on Day 0 (prime) and Day 21 (boost). While all doses of TT1-E7E6 elicited comparably increased frequencies of E7-specific CD8+ T cells compared to buffer controls on Day 9 post-prime administration, a booster administration augmented the response on Day 30, primarily in mice receiving 105 or 106 RCV FFU (Figure 3(a)). Induction of CD8+ T cells was further analyzed in a kinetic study, in which the prime dose of TT1-E7E6 induced a peak of E7-specific CD8+ T cells on Day 7 and the boost injection led to another peak on Day 26. The peaks were reached 7 and 5 days after the prime and boost injection, respectively, and were followed by a contraction phase after which CD8+ T cell levels evened out at substantially higher levels compared to the levels before administration (Figure 3(b)).
Figure 3.
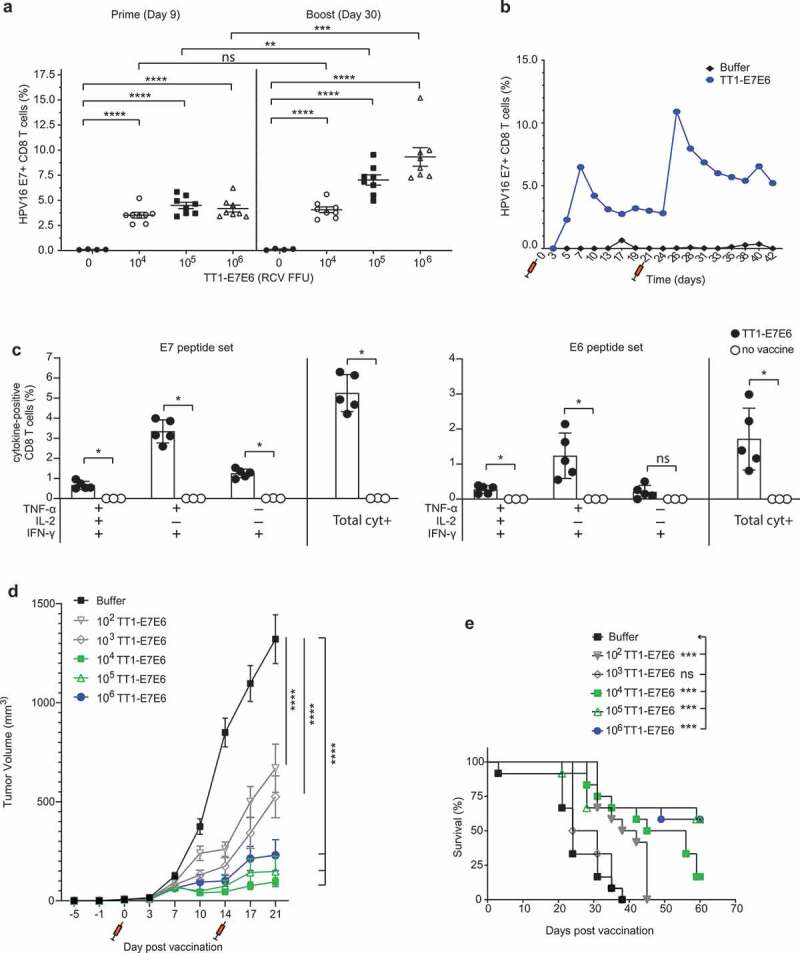
TT1-E7E6 is highly immunogenic and mediates potent antitumor activity. (a) HPV16 E7 responses after immunization. Mice were immunized i.v. with increasing doses of TT1-E7E6 or buffer control on Day 0 and 21. HPV16 E7-specific CD8 (CD8+B220−) T cell frequencies were determined in the blood on Day 9 (prime) and Day 30 (boost). Each dot represents one animal. Shown are means ± SEM. N=8 mice per treatment group; n=4 mice per buffer group. Statistical analysis by unpaired Welch`s t-test. (b) Kinetics study of immunogenicity. Mice were immunized i.v. with 105 RCV FFU of TT1-E7E6 or buffer control on Day 0 and 21. HPV16 E7-specific CD8+ (CD8+B220−) T cell frequencies were determined in the blood up to 42 days. Pooled data from 6 mice per group are shown. (c) Polyfunctional CD8+ T cell responses in immunized mice. Mice were immunized i.v. with 105 RCV FFU of TT1-E7E6. Non-vaccinated naïve mice were used as controls. Frequencies of splenic CD8 T cells producing different combinations of IFN-γ, TNF-α and IL-2 in response to E7 or E6 peptide re-stimulation were determined on Day 9 post-immunization as indicated in the chart. Only cytokine combinations exceeding 0.1% on average are shown. The right-hand bar (Total cyt+) depicts the combined frequency of the three populations of cytokine-secreting cells as depicted on the left. Symbols represent individual mice. N=5 per TT1-E7E6 group, n=3 per no vaccine control group. Bars show means ± SD. Mann Whitney test, ns, not significant, * p<.05. (d) Antitumor efficacy by TT1-E7E6 immunization. Mice were inoculated with 105 TC-1 cells on Day −5. On Day 0 upon appearance of palpable tumors, mice were randomized and immunized i.v. with increasing doses of TT1-E7E6 (prime) or buffer followed by a boost injection on Day 14, as indicated by the syringe symbols. Means ± SEM. N=12 mice per group. Two-way ANOVA was performed for statistical analysis. ns: 102 vs. 103, 104 vs. 105 and 105 vs. 106. p<.01: 104 vs. 106. p<.0001: 102/103 vs. 104/105/106. In the figure, **** p<.0001. (e) Kaplan-Meier survival curves of TC-1 tumor-bearing mice receiving TT1-E7E6 treatment. Mantel-Cox test. ns, not significant, * p<.05, ** p<.01, *** p<.001, **** p<.0001.
To assess whether TT1-E7E6 induced polyfunctional HPV antigen-specific CD8+ T cells, we analyzed cytokine production by these cells using intracellular cytokine staining (ICS). To this end, mice were vaccinated with 105 RCV FFU of TT1-E7E6 i.v. or left untreated (naïve). On Day 9, splenocytes were isolated and re-stimulated with either E6 or E7 peptides ex vivo. As shown in Figure 3(c), HPV16-reactive CD8+ T cells from TT1-E7E6 immunized mice mainly co-produced IFN-γ, TNF-α with or without IL-2, and only a minor population produced IFN-γ alone. CD8+ T cells responding to E6 were less abundant than those specific for E7, but their polyfunctional cytokine production profile was similar to that of E7-specific cells.
Together, these findings suggest that TT1-E7E6 vaccination elicits functional CD8+ T cell responses against the two HPV antigens E7 and E6 in mice.
Therapeutic TT1-E7E6 vaccination mediates potent antitumor activity in a syngeneic TC-1 tumor model
Next, we examined the antitumor efficacy of TT1-E7E6 in vivo. To this end, we employed a syngeneic, HPV16 E7- and E6-expressing TC-1 mouse tumor model.28 In order to establish tumors prior to therapeutic vaccination, mice were engrafted subcutaneously with TC-1 tumor cells (Day −5). Five days later (Day 0), upon the appearance of palpable TC-1 tumors, mice were randomized and immunized i.v. with increasing doses of TT1-E7E6 (102, 103, 104, 105 and 106 RCV FFU) or buffer control. A second immunization with identical doses was administered two weeks post-prime (Day 14).
As shown in Figure 3(d), compared to the buffer control, treatment with TT1-E7E6 significantly suppressed tumor growth at all dose levels, as measured on Day 21, a time point by which all animals in the TT1-E7E6 treatment groups were still alive. Suppression of tumor growth correlated with increasing doses of TT1-E7E6 from 102 to 104 RCV FFU and reached a plateau from 104 to 106 RCV FFU (Figure 3(d)), suggesting that a dose of 104 RCV FFU does induce a maximum effect on tumor suppression in this model with relatively small tumor size. The kinetics of tumor growth in individual animals prior to study termination on Day 59 are shown in Supplemental Figure S2(a). Consistently, survival was significantly prolonged in all TT1-E7E6 dose groups except for the 103 RCV FFU group (Figure 3(e)). Of note, the median survival time (MST) of mice vaccinated with high doses of TT1-E7E6 was >59 d (duration of the study) compared to a 24-d MST in the buffer control group, indicating a significant therapeutic benefit of TT1-E7E6 treatment (Supplemental Figure S2(b)).
We next tested if TT1-E7E6 can treat established tumors of larger size. To this end, TT1-E7E6 treatment was initiated 10 days after TC-1 tumor cell inoculation, a time when tumors reached >300 mm3. Consistent with our findings in the small tumor setting, treatment with a medium dose (105 RCV FFU) of TT1-E7E6 significantly inhibited the growth of these larger tumors and prolonged survival (Supplemental Figure S2(c-d)).
To assess whether TT1-E7E6 treated mice that have cleared their tumors are long term protected from tumor recurrence by established antitumor immune memory, we rechallenged two groups of these mice with TC-1 cells on study Day 143 and 198, respectively. We found that these mice remained tumor-free throughout the follow-up period for over 200 days (Supplemental Figure S3).
Taken together, these results suggest that TT1-E7E6 treatment confers a potent antitumor effect for both small and large tumors in the HPV16 TC-1 tumor model and in mice that succeed in clearing their tumor, affords long-term protection from tumor rechallenge.
TT1-E7E6 in combination with PD-1 checkpoint inhibitor therapy eliminates tumors
PD-1 blockade therapy has recently been approved for the treatment of recurrent or metastatic HNSCC and advanced cervical cancers.7-9 Using an OVA-expressing tumor model, it has previously been demonstrated that the antitumor effect by replicating LCMV vectors, from which TT1-E7E6 is designed, is dependent on CD8+ T cells.19 Here, we sought to evaluate whether the antitumor efficacy of TT1-E7E6 can be enhanced with concomitant anti-PD-1 therapy. To this end, mice were engrafted with TC-1 tumor cells (Day −5) and treated with 106 RCV FFU of TT1-E7E6 on Day 0, 21 and 42. Four doses of anti-PD-1 Abs or isotype control were administered following each TT1-E7E6 immunization (experimental setup see schematic Figure 4(a)).
Figure 4.
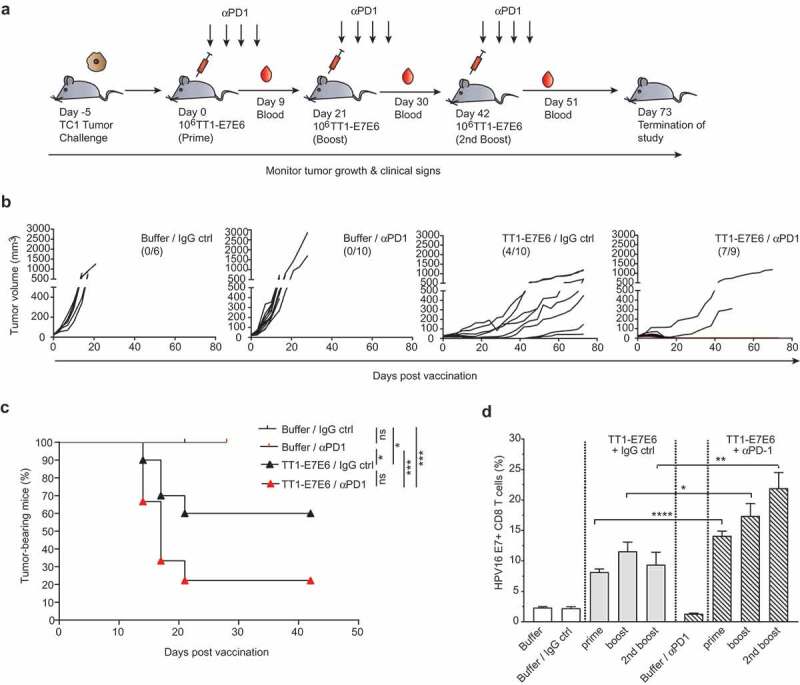
Combination therapy with TT1-E7E6 and PD-1 checkpoint inhibitor eliminates tumors. (a) Schematic design of experiment. All mice were inoculated s.c. with 105 TC-1 tumor cells on Day −5. After tumors became measurable, mice were randomized and treated with 106 RCV FFU of TT1-E7E6 or buffer On Day 0, 21 and 42. Anti-PD-1 antibody (αPD1) or isotype control (IgG ctrl) was administered i.p at a dose of 200 µg/mouse on the same days of TT1-E7E6/buffer treatment, followed by 3 additional doses every 3 days. Tumor growth was monitored. The study was terminated 73 days after start of treatment. (b) Kinetics of tumor growth in individual mice. Numbers in parentheses indicate number of mice with tumor regression/total number of mice in each group. (c) Kaplan-Meier curves showing percentage of TC-1 tumor-bearing mice. Buffer/IgG control, n=6 mice; TT1-E7E6/IgG, n=10 mice; buffer/anti-PD-1, n=10 mice; TT1-E7E6/anti-PD-1, n=9 mice. Mantel-Cox test. ns, not significant, * p<.05, *** p<.001. (d) HPV16 E7-specific CD8+ T cells in tumor-bearing mice were measured 9 days after individual treatment time point (prime, boost and 2nd boost). Means ± SEM. Unpaired Welch`s t-test. * p<.05, ** p<.01, *** p<.001.
Consistent with previous reports, 29,30 anti-PD-1 antibody monotherapy failed to suppress tumor growth in this HPV16-positive TC-1 tumor model with 0 out of 10 mice clearing tumors. TT1-E7E6 alone (with IgG isotype control) led to significant tumor suppression with complete tumor regression in 4 out of 10 animals (40%) by Day 42 with 2 mice staying tumor-free till study termination on Day 73. Strikingly, combined treatment of TT1-E7E6 and anti-PD-1 conferred superior tumor control compared to either of the therapy alone, with complete tumor regression observed in 7 out of 9 animals (78%), all of which stayed tumor-free till study termination. In the two remaining mice of this group, tumor control was comparable to those receiving TT1-E7E6 and isotype control. Moreover, tumor regression was achieved after the first treatment cycle and maintained till the end of the study (Figure 4(b), 4(c) and Supplemental Figure S4).
It is also noted that there was also a synergistic effect by anti-PD-1 antibodies and TT1-E7E6 on the frequencies of E7-specific CD8+ T cells in the tumor-bearing mice (Figure 4(d))
Together, these results demonstrate that TT1-E7E6 is effective in anti-PD-1 refractory tumors and that anti-PD-1 therapy can become effective provided TT1-E7E6 is administered.
Analysis of TT1-E7E6-mediated antitumor effect
To investigate the events underlying antitumor effect mediated by TT1-E7E6 as well as by TT1-E7E6/anti-PD-1 combination therapy, we analyzed immune cells infiltrating tumor tissues in TC-1 tumor-bearing mice by flow cytometry (Figure 5). On Day 16 post-tumor cell inoculation, mice receiving either TT1-E7E6 or TT1-E7E6/anti-PD-1 both showed significant accumulation of HPV16 E7-specific CD8+ T cells in the tumor tissues as compared to the buffer control group, whereas there was no detectable difference between mice treated with TT1-E7E6 and those treated with TT1-E7E6/anti-PD1 (Figure 5(a)). The percentages of PD-1-expressing E7-specific CD8+ T cells were slightly (statistically not significant) decreased upon TT1-E7E6 treatment and were significantly decreased with TT1-E7E6/anti-PD1 treatment compared to the buffer control group (Figure 5(b)). However, PD-1 expression levels on E7-specific CD8+ T cells were significantly lower in mice treated with TT1-E7E6 and were further decreased in the TT1-E7E6/anti-PD1 combination therapy group compared to the buffer control group (Figure 5(c)). In association with this finding, the percentage of PD-L1+ tumor cells as well as their PD-L1 expression levels were substantially and similarly upregulated by both TT1-E7E6 and TT1-E7E6/anti-PD1 treatment, with >90% PD-L1+ tumor cells as compared to 20–30% PD-L1+ cells in the control group (Figure 5(b) and (c), right panels). As for conventional dendritic cells (cDCs), no statistically significant change was detected for the total cell numbers, percentage of PD-L1+ cDCs or the expression levels of PD-L1 among the three groups (Figure 5, middle panels).
Figure 5.
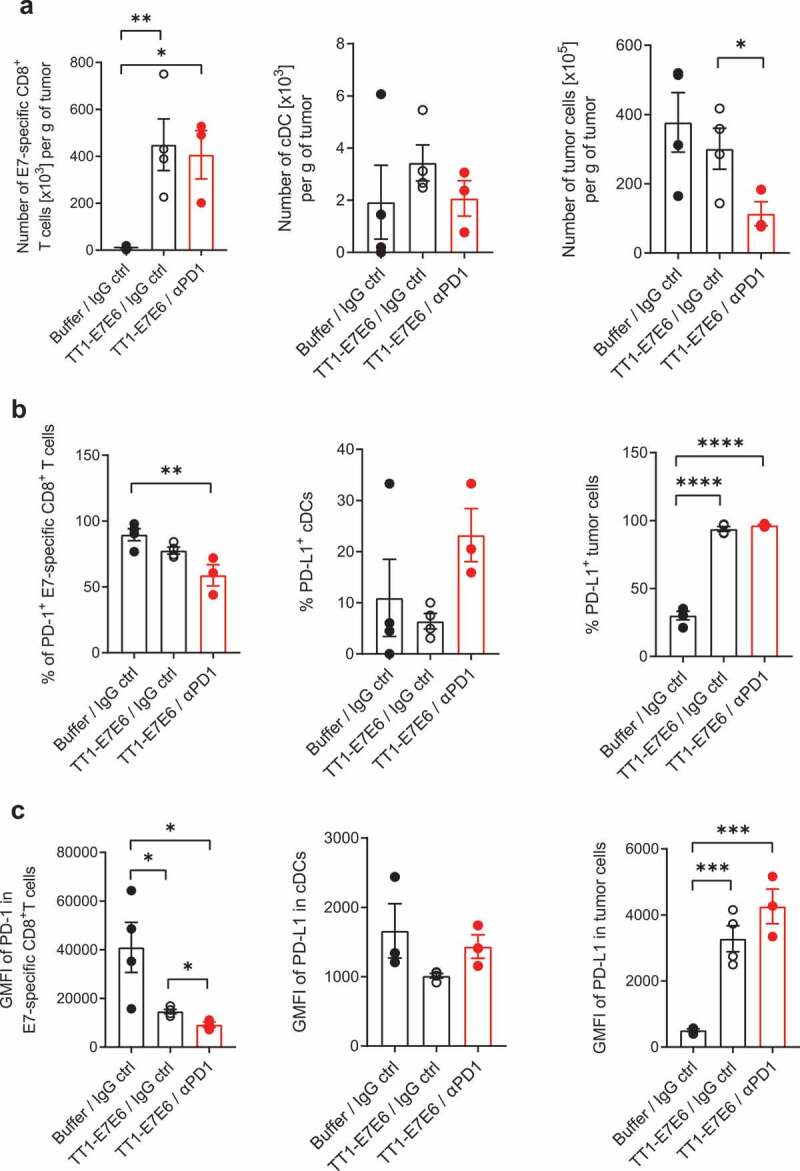
Analysis of tumor-infiltrating immune cells in tumor-bearing mice treated with TT1-E7E6 alone or in combination with anti-PD-1. TC-1 tumor-bearing mice were treated with TT1-E7E6 (1x106 RCV FFU i.v.) on Day 9, with or without anti-PD-1 (clone 29F.1A12, 200 µg/dose i.p.) on Day 9, 12 and 15 after tumor cell implantation. Tumors were extracted on Day 16 and analyzed by flow cytometry. (a) Absolute numbers of HPV16 E7-specific CD8+ T cells (live, CD45+B220−NKp46−CD11b−CD3+CD8+E7+), conventional dendritic cells (cDC) (live, CD45+CD64−F4/80−Lin−I-A/I-E+CD11c+CD26+) and tumor cells (live, CD45−) per gram (g) of tumor tissue. (b) Percentage of PD-1+ E7-specific CD8+ T cells, PD-L1+ cDCs and PD-L1+ tumor cells. (c) Expression of PD-1 (stained with clone RMP1-30) on E7-specific CD8+ T cells and of PD-L1 on cDCs and tumor cells. Geometric mean fluorescence intensity (GMFI). N=4 mice per group for buffer control and TT1-E7E6 groups, N=3 mice for TT1-E7E6/anti-PD1 group. Means ± SEM. One-way ANOVA to compare treatment to buffer groups and unpaired Welch`s t-test to compare both treatment groups. * p<.05, ** p<.01, *** p<.001, **** p<.0001.
Furthermore, to assess whether any antitumor effect was mediated by the vector itself besides the tumor-specific CD8+ T cell response induced by the vectors, we treated TC-1 tumor-bearing mice with an irrelevant TT1-GFP control vector, which devoid of any tumor antigenicity. In comparison with TT1-E7E6, mice treated with TT1-GFP showed a similar increase in total CD8+ T cell levels but no measurable levels of E7-specific CD8+ T cells in the blood (Supplemental Figure S5(a and b)). Accordingly, we observed a minor delay in tumor growth, but no survival benefit in mice treated with TT1-GFP (Supplemental Figure S5(c and d)). These results indicate that induction of HPV-specific immunity by TT1-E7E6 plays a pivotal role in its antitumor effect.
Lastly, we examined the potential oncolytic effect of TT1 vectors by infecting TC-1 tumor cells with TT1-GFP in culture (Supplemental Figure S6(a)). No significant difference in cell viability was observed between cells inoculated with TT1-GFP and control medium up to 72 h (Supplemental Figure S6(b)), with >90% TC-1 tumor cells being infected, as determined by GFP expression after 48 h of incubation (Supplemental Figure S6(c)). These data are in line with the well-document non-cytolytic behavior of LCMV, 31 indicating that the TT1 vectors do not induce lytic cell death of TC-1 tumor cells upon infection.
Discussion
HPV-associated cancers, especially head and neck cancers, remain a significant health concern, as no curative therapies are currently available. With HPV16 being the primary oncogenic virus and causative agent, 1,2 a variety of vaccine platforms including peptide, cell, DNA, RNA and viral vector-based vaccines targeting HPV16 antigens have been explored for HPV-associated cancer therapy.14,15,30,32-36 Here, we describe the generation of a novel replication-attenuated LCMV vector-based vaccine, TT1-E7E6, expressing non-oncogenic variants of the HPV16 oncogenes E7 and E6, for treatment of HPV-associated cancers. We demonstrated that immunization with TT1-E7E6 induced HPV-specific CTL responses and efficiently controlled tumor growth in a murine TC-1 tumor model for HPV16-induced cancers. In addition, combining TT1-E7E6 with anti-PD-1 therapy significantly enhanced its antitumor efficacy. The antitumor effect of TT1-E7E6 was associated with the accumulation of HPV16 E7-specific CD8+ T cells with reduced PD-1 expression in the tumor tissues.
Construction of TT1-E7E6 was based on a previously described replicating, attenuated vector platform based on LCMV.19 Although most wt LCMV infections are asymptomatic, subclinical infections, flu-like illnesses and self-limiting aseptic meningitis have been reported from occasional accidental laboratory infections with wt LCMV.20,37 Interestingly, high doses of unmodified wt LCMV had been administered intravenously to treat patients with advanced cancer in the 1970s, with no evidence of CNS disease (e.g., meningitis or encephalitis) reported.38 To minimize any CNS damaging potential of TT1-E7E6, we applied molecularly defined, robust and stable attenuation mechanism based on an artificial, tri-segmented genome design. As arenaviruses naturally exhibit a bi-segmented genome design, packaging of all three genome segments needed to form replication-competent vector particles is inefficient.19 In the current study, we demonstrated that TT1-E7E6, unlike its parental LCMV wt virus, was unable to establish protracted/chronic infection and was rapidly eliminated from the circulation after systemic administration to mice. Moreover, we showed that the capacity of TT1-E7E6 to cause CNS complications was significantly reduced, as evidenced by several log-fold attenuations in neurovirulence compared to the parental wt LCMV.
Immunization with TT1-E7E6 induced a durable dose-dependent expansion of total and antigen-specific CD8+ T cells, along with potent cytokine (IFN-γ/TNF-α/IL-2) production. These findings are in accordance with previous reports by Kallert and colleagues demonstrating robust CTL responses and generation of long-term CTL immunity by an analogous vaccine based on the same replication-competent LCMV platform.19 The increased frequencies of E7-specific CD8+ T cells after the booster administration of TT1-E7E6 are suggestive of effector T cells generated from the memory pool.
Intravenous TT1-E7E6 therapy significantly inhibited tumor growth and prolonged survival in a TC-1 mouse tumor model. The potent tumor control capacity of TT1-E7E6 was further supported by its efficacy in established tumors with large volumes, where one single dose was sufficient to delay the tumor growth. TT1-E7E6 treated, tumor-free mice rechallenged with TC-1 tumor cells remained tumor-free for over 200 days, suggestive of efficacy in both controlling primary tumors and preventing tumor recurrence39-42.
Analysis of tumor tissue revealed a significant tumor infiltration and accumulation of E7-specific CD8+ T cells in mice treated with TT1-E7E6. These tumor-infiltrating E7-specific CD8+ T cells showed reduced PD-1 expression compared to their counterparts in buffer-treated animals, suggesting recruitment of functional CD8+ T cells from the expanded peripheral T cell pool into the tumor microenvironment. In response to the infiltration of tumor-specific CD8+ T cells upon TT1-E7E6 treatment, the strong upregulation of PD-L1 on tumor cells reflects an adaptive immune resistance mechanism.43
While anti-PD-1 therapy prolonged survival of patients with recurrent/metastatic HNSCC, its efficacy is limited by PD-L1 positivity.44 Multiple studies have shown that when combined with therapeutic HPV vaccines, the efficacy of PD-1 checkpoint inhibitors can be significantly improved.29,30,45-48 In the current study, combined treatment of TT1-E7E6 with anti-PD-1 antibody led to complete tumor regression in 78% of the mice bearing TC-1 tumors, a model known to be resistant to anti-PD-1 treatment.29,30 This superior effect is probably due to the sustained functionality of the CTLs associated with the combination therapy.
Furthermore, using a non-E7E6-expressing control vector, TT1-GFP, we showed that the TT1 vector backbone conferred only marginal tumor control despite a similar increase in total CD8+ T cells in tumor-bearing mice. Failure to induce HPV 16-specific CD8+ T cells by TT1-GFP is likely the reason for its lack of durable antitumor efficacy. In addition, the observation that TT1-GFP did not cause direct cell death of TC-1 cells in vitro indicates that these LCMV-based vectors are non-oncolytic, a mechanism observed for some other viral vectors.49
In conclusion, we demonstrated that a novel cancer therapy targeting HPV16 E7 and E6 based on the replication-attenuated LCMV vector platform was highly immunogenic with robust antitumor efficacy in a murine model of HPV16-positive cancer. The tumor suppression efficacy by TT1-E7E6 is probably attributable primarily, but not exclusively, to the potent tumor antigen-specific CTL response and accumulation of tumor-infiltrating HPV16-specific CD8+ T cells induced by TT1-E7E6. Finally, the addition of a PD-1 checkpoint inhibitor significantly increased the antitumor efficacy of TT1-E7E6, supporting the further investigation into combination immunotherapy for patients with HPV-associated HNSCC, especially those refractory to anti-PD-1 therapy. Based on these preclinical findings, a phase I/II clinical trial of TT1-E7E6 for the treatment of HPV16+ cancer patients has been initiated (ClinicalTrial.gov #NCT04180215).
Materials and methods
Animal studies
Animal studies were carried out at various laboratories. Studies on viremia and cytokine-secreting CD8 T cell responses (ICS) were conducted at the University of Basel, Switzerland, in accordance with the Swiss law for animal protection and with authorizations from the Veterinäramt Basel-Stadt. C57BL/6 J mice were obtained from the laboratory animal service center (LASC) of the University of Zurich, Schlieren, Switzerland. The neurovirulence study (i.c. challenge) was conducted at Meditox (Czech Republic) and was approved by the Institutional Animal Care and Use Committee (IACUC) and the Committee for Animal Protection of the Ministry of Health of the Czech Republic (30/2017). C57BL/6 J mice were obtained from Velaz s.r.o.
For the dose-response study of immunogenicity performed at Preclin Biosystems, Switzerland, female C57BL/6 J mice were obtained from Charles River Laboratories. For tumor studies performed at WuXi Apptec, Shanghai, China, female C57BL/6 J mice were purchased from Shanghai SLAC Laboratory Animal Co., LTD. All the procedures related to animal handling, care and the treatment in the study were performed according to the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of WuXi AppTec following the guidance of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
For tumor studies performed at Hookipa Pharma, C57BL/6 N mice were purchased from Charles River, Sulzfeld, Germany. Studies were approved by the Austrian authorities (Magistrat 58) and were carried out in accordance with the approved guidelines for animal experiments at Hookipa Biotech GmbH. All mice were maintained in specific pathogen-free (SPF) conditions.
Cells and cell lines
TC-1 cells expressing HPV16 E6 and E7 were purchased from Johns Hopkins University.28 Cells were cultured in complete tumor medium containing RPMI 1640 medium (Thermo Fisher) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Corning), Non-essential Amino Acids, 2 mM Glutamine, 1 mM Sodium Pyruvate (all Thermo Fisher), 50 U/ml Penicillin/Streptomycin (Millipore) and 0.4 mg/ml G418 (Thermo Fisher). Cells were maintained as a monolayer culture and were sub-cultured twice weekly by trypsin-EDTA treatment. Cells growing in an exponential growth phase were harvested and resuspended in PBS for tumor inoculation. BHK21 cells constitutively expressing LCMV-GP, and HEK293 cells were obtained from the Institute of Experimental Immunology, University of Zurich. BHK21 cells were cultivated in DMEM (Thermo Fisher) containing 10% FBS, 4 mM Glutamax, 10 mM HEPES (both from Thermo Fisher) and Puromycin (Sigma Aldrich) for selection of cells containing the GP expression cassette encoding the puromycin resistance. HEK293 cells were maintained in CDM4HEK293 medium (GE Healthcare) containing 4 mM Glutamax. All cell lines were kept at 37°C and 5% CO2 in humidified incubators and regularly examined for mycoplasma contamination.
Vector design, propagation and characterization
TT1-E7E6 vectors were generated and titrated as described previously.19,50 Briefly, the coding sequence (cDNA) of the HPV16 vaccine antigen, fusion protein of E7 and E6 (Genbank accession # K02718 with five mutations [three mutations for E7 and 2 mutations for E6] introduced to abrogate oncogenic potential), 25 was synthesized by Genscript and inserted into two plasmids, both encoding the S-Segment of LCMV (based on clone 13), with S-Segment #1 encoding LCMV NP and S-Segment #2 encoding LCMV GP (derived from LCMV strain WE). Vectors were generated by transient transfection of BHK21 cells stably expressing the LCMV GP.26 Briefly, cells were transfected with five plasmids encoding: S-Segment #1, S-Segment #2, L-Segment and two expression plasmids encoding LCMV NP and L, respectively. Three days post-transfection, cells were propagated. On Day 6 post-transfection, virus-containing supernatant was harvested and subsequently titrated by Focus Forming Assays (FFA) on either LCMV GP-complementing cells (FFU) or non-complementing HEK293 cells (RCV FFU) as described before.51 Briefly, monolayers of adherent HEK293-GP or HEK293 seeded in 24-well plates were infected with serial dilutions of the virus, incubated for 48 h, and subsequently fixed and stained with anti-NP antibody (VL-4; Bio X Cell, Lebanon, NH). The number of foci (clusters of infected cells) was determined, and the virus titer was calculated.
To generate fetal calf serum (FCS)-free vector stock, suspension HEK293 cells were infected with TT1-E7E6 at a defined MOI of 0.001 and incubated for 4 days with shaking. Nascent viruses were harvested after low-speed centrifugation and frozen below −65°C. Vector stocks were titrated by FFA and characterized for transgene insertion.
For protein expression, HEK293 cells were infected with TT1-E7E6, followed by proteasome inhibitor treatment with 10 µM MG132 (Selleckchem) or DMSO (solvent control) overnight. Whole-cell lysates were analyzed by western blot. For detection of E7, membranes were stained using a mouse-anti E7 mAb at (Santa Cruz; 1:500) followed by an HRP-coupled anti-mouse Ab (Jackson Immunoresearch; 1:10000). ERK2 (1:2000; Santa Cruz) served as the loading control. The insertion of the transgene (E7E6) on both S-segments was verified by two-step RT-PCR. After viral RNA was isolated from vector containing cell culture supernatant using QIAamp Viral RNA Mini Kit (Qiagen), cDNA from each S-segment was generated by reverse transcription using NP- or GP-specific primers (Superscript III First-Strand Synthesis, Invitrogen). In a second step, transgene-specific fragments were amplified by PCR using transgene-flanking primers. The sizes of the PCR fragments were verified by gel electrophoresis.
For analysis of vector growth kinetics, 3 × 105 HEK293 cells/ml were seeded and subsequently infected with TT1-E7E6 or LCMV wt at an MOI of 0.001. After 2, 24, 48, 72 and 96 h, samples were drawn and subjected to FFU analysis.
Neurovirulence study
Intracranial injection of C57BL/6 mice was performed as described before.19 Briefly, mice were first anesthetized with isoflurane inhalation for approximately 5 min. The inoculum of 30 μl was administered to the immobilized mouse with the needle positioned over the vertex and slightly caudal of the eyes yet cranial of the ears and inserted across the skull into the brain. The needle was prepared to limit and standardize the depth of insertion to approximately 2–3 mm. Mice were challenged with buffer control or increasing doses of TT1-E7E6 (101, 102, 103, 104 and 105 RCV FFU) or the corresponding LCMV wt (101 and 102 RCV FFU). Between Day 5 to 10, animals were monitored twice daily and a tail-spin test was performed to determine the disease condition of the infected mice. Briefly, mice were lifted and spun by the tail, which provoked spasms in diseased animals or convulsions in terminal stages of the infection. Observation of convulsions was defined as the criterion for euthanasia. The study was terminated on day 21 after virus inoculation.
Syngeneic TC-1 tumor model
For all tumor studies, 6–8 week old female C57BL/6 mice were subcutaneously inoculated with 1 × 105 TC-1 cells into their right flank. In each study, clinical signs, body weight, survival and tumor volumes were evaluated. Tumor size was measured at least twice weekly in two dimensions using a caliper, and the tumor volume was expressed in mm3 using the formula: V = 0.5 x a x b2 where a and b are the long and short diameters of the tumor in mm, respectively. Before the start of treatment, mice were assigned to the respective groups by randomized block design based on the tumor volume. For the treatment of small, established tumors, mice were vaccinated i.v. with indicated doses of TT1-E7E6 formulated in PSB-buffer supplemented with 10% D-sorbitol (Fluka analytical, Italy) or buffer as control (PSB, 10 mM HEPES, 150 mM NaCl, 20 mM Glycine, pH 7.4; GE Healthcare) 5 and 19 days post tumor challenge. The two-week prime-boost interval was applied to evaluate antitumor efficacy by different dose levels of TT1-E7E6 (102, 103, 104, 105 and 106 RCV FFU) in the TC-1 tumor model in order to cover possible efficacy by all dose levels given the rapid tumor growth in this model. The study was terminated 60 days after the start of treatment.
For the treatment of large tumors (average 350 mm3), mice were immunized with TT1-E7E6 10 and 31 days after TC-1 tumor inoculation. This study was terminated on Day 40 post tumor transplantation. For combination treatment, mice were i.p. injected with 200 µg of anti-PD-1 antibody (clone 29 F.1A12, BioXcell) or a rat IgG2a isotype control antibody (BioXcell) diluted in 200 µl PBS on each day of the TT1-E7E6 vaccination followed by 3 additional doses every 3 days. The study was terminated 78 days after tumor cell inoculation. One animal receiving TT1-E7E6 and anti-PD-1 Ab died for unknown reasons early in the study (Day 20) with a tumor size of 456 mm3 on the day of death and was excluded from the analysis. The three-week prime-boost interval was used for the treatment of large tumors (105 RCV FFU of TT1-E7E6) as well as in the combination treatment experiments (106 RCV FFU of TT1-E7E6), in which the relatively high doses of TT1-E7E6 were used.
Flow-cytometry
For MHC multimer staining of antigen-specific CD8 T cells, 50–70 µl of blood were collected into BD Falcon tubes with the addition of 1 ml of FACS buffer (PBS supplemented with 2.5% FCS, 10 mM EDTA and sodium azide). Cells were centrifuged at room temperature (RT) and resuspended in FACS buffer containing appropriate fluorescent-conjugated antibodies: anti-B220-FITC, anti-CD8a-PE-Cy7 (both from BD Biosciences) and the MHC Dextramer for HPV16 E7 (H-2 Db/RAHYNIVTF/PE, Immudex, Denmark) for 30 minutes of incubation. After wash, red blood cells were lysed with FACS Lysing solution buffer (BD Biosciences). Finally, cells were washed and resuspended in FACS buffer for analysis on flow-cytometer.
ICS was performed as described before.52 Briefly, mice were sacrificed on Day 9 post-immunization. Single cell suspension was prepared from spleen. Splenocytes were re-stimulated in vitro using overlapping peptide sets for HPV16 E6 and E7 (from JPT). As control, splenocytes were incubated with medium only. These background frequencies were subtracted from those of peptide-stimulated cultures to obtain peptide-specific responses as reported herein. Cells were incubated first with the respective peptide set for 1 h, and then Brefeldin A (Sigma Aldrich) was added for an additional 4 h of incubation. Cells were then stained with anti-mouse anti-CD8a-BV421 (Biolegend) and anti-B220-PerCPCy5.5 (eBioscience) antibodies. Subsequently, cells were fixed using 4% paraformaldehyde and then permeabilized with 0.1% Saponin (Sigma Aldrich). Intracellular staining was performed with anti-mouse IL-2-PE (clone JES6-5H4), anti-mouse TNF-PECy7 (clone MP6-XT22) and anti-mouse IFN-γ-APC (clone XMG1.2) antibodies (all from Biolegend).
For analysis of tumor-infiltrating immune cells, tumors were collected, weighed and cut into small pieces of 2–4 mm. Tumor pieces were transferred into gentleMACS C tubes (Miltenyi Biotec) and 2.5 mL enzyme mix (Miltenyi Biotec) was added. After dissociation on the gentleMACS Dissociator (Miltenyi Biotec), cells were passed through a 70 µm MACS Smart Strainer. After washing cells were passed through a 30 µm MACS Smart Strainer and counted before resuspension in 0.5% MACS BSA Solution. Cells were separated on the autoMACS® Pro Separator using CD45 magnetic beads (Miltenyi Biotec). Positive and negative fractions were counted. CD45+ cells were divided into two parts for staining either CD8+ T cells (live, CD45+B220−NKp46−CD11b−CD3+CD8+E7+) or conventional dendritic cells (cDC) (live, CD45+CD64−F4/80−Lin−I-A/I-E+CD11c+CD26+). The cells were further stained for PD-1 and PD-L1. Fluorochrome conjugated antibodies used for flow cytometry were summarized in the Supplemental Materials and Methods. Staining of PD-1 (clone RMP1-30) was confirmed not being interfered by the anti-PD1 antibody (clone 29 F.1A12) used for i.p. treatment in combination with TT1-E7E6.
Samples were analyzed on BD LSR Fortessa or BD Canto II (Beckton Dickinson, USA). Analysis of flow cytometry data was performed using the FlowJo software (TreeStar, Version 10). For ICS data analysis, background values (medium only) were subtracted. Gating strategy is provided in Supplemental Figure S7.
Statistical analysis
GraphPad Prism 8 was used for data visualization and statistical analysis. If not stated otherwise, graphs show means ± SD or SEM. When comparing differences between two groups, unpaired two-tailed t-tests with Welch’s correction or Mann Whitney test were applied. Single values of multiple groups were compared by two-way analysis of variance (ANOVA). Kaplan–Meier survival curves were assessed using log-rank (Mantel–Cox) test. Results were considered statistically significant if p < .05.
Supplementary Material
Acknowledgments
We are grateful to Johns Hopkins University for providing HPV16 E6 and E7-positive TC-1 tumor cells, WuXi Apptec, Shanghai for performing the mouse tumor studies, Preclin Biosystems, Switzerland for performing the dose-response study of immunogenicity, and Meditox, Czech Republic, for the neurovirulence study.
Funding Statement
Project funded by Österreichische Forschungsförderungsgesellschaft (FFG).
Disclosure of potential conflicts of interests
S.S., F.S., T.K., D.O., U.B., B.K., S.C.S., J.R., M.H., M.S., H.L., X.Q., I.M. and K.K.O are employees of Hookipa Pharma Inc. or its subsidiary Hookipa Biotech GmbH. I.M. is a shareholder of Hookipa Pharma, Inc. D.D.P. is a founder, shareholder and consultant to Hookipa Pharma Inc. and its subsidiary Hookipa Biotech GmbH.
Supplementary material
Supplemental data for this article can be accessed here.
Author contribution
S.S., W.V.B., D.D.P., J.R., M.H., H.L. and K.K.O. conceived and designed the studies, interpreted the results and wrote the manuscript; S.S., W.V.B., F.S., A.R., T.K., A.E.G., D.O., B.K., S.C.S and M.S. performed the experiments; S.S., U.B., T.K., M.H. and W.V.B. performed data analysis; U.B., D.D.P. and I.M. supported the design of studies and interpretation of results. X.Q. supported interpretation of the results and writing of the manuscript.
References
- 1.Gillison ML, D’Souza G, Westra W, Sugar E, Xiao W, Begum S, Viscidi R.. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. 2008;100(6):407–12. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- 2.Saraiya M, Unger ER, Thompson TD, Lynch CF, Hernandez BY, Lyu CW, Steinau M, Watson M, Wilkinson EJ, Hopenhayn C. US assessment of HPV types in cancers: implications for current and 9-valent HPV vaccines. J Natl Cancer Inst. 2015;107(6):djv086. doi: 10.1093/jnci/djv086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frazer IH, Chandra J. Immunotherapy for HPV associated cancer. Papillomavirus Res. 2019;8:100176. doi: 10.1016/j.pvr.2019.100176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balermpas P, Rodel F, Rodel C, Krause M, Linge A, Lohaus F, Baumann M, Tinhofer I, Budach V, Gkika E, et al. CD8+ tumour-infiltrating lymphocytes in relation to HPV status and clinical outcome in patients with head and neck cancer after postoperative chemoradiotherapy: A multicentre study of the German cancer consortium radiation oncology group (DKTK-ROG). Int J Cancer. 2016;138(1):171–181. doi: 10.1002/ijc.29683. [DOI] [PubMed] [Google Scholar]
- 5.Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer. 2017;117(4):451–460. doi: 10.1038/bjc.2017.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward MJ, Thirdborough SM, Mellows T, Riley C, Harris S, Suchak K, Webb A, Hampton C, Patel NN, Randall CJ, et al. Tumour-infiltrating lymphocytes predict for outcome in HPV-positive oropharyngeal cancer. Br J Cancer. 2014;110(2):489–500. doi: 10.1038/bjc.2013.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seiwert TY, Burtness B, Mehra R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–965. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Wu L, Tong R, Yang F, Yin L, Li M, You L, Xue J, Lu Y. PD-1/PD-L1 Inhibitors in Cervical Cancer. Front Pharmacol. 2019;10:65. doi: 10.3389/fphar.2019.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferris RL, Blumenschein G Jr., Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 1856-67;2016:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibata T, Lieblong BJ, Sasagawa T, Nakagawa M. The promise of combining cancer vaccine and checkpoint blockade for treating HPV-related cancer. Cancer Treat Rev. 2019;78:8–16. doi: 10.1016/j.ctrv.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Araldi RP, Assaf SMR, Carvalho RF, et al. Papillomaviruses: a systematic review. Genet Mol Biol. 2017;40:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dadar M, Chakraborty S, Dhama K, et al. Advances in designing and developing vaccines, drugs and therapeutic approaches to counter human papilloma virus. Front Immunol. 2018;9:2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conesa-Zamora P. Immune responses against virus and tumor in cervical carcinogenesis: treatment strategies for avoiding the HPV-induced immune escape. Gynecol Oncol. 2013;131(2):480–488. doi: 10.1016/j.ygyno.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 14.Kenter GG, Welters MJ, Valentijn AR, Lowik MJG, Berends-van der Meer DMA, Vloon APG, Essahsah F, Fathers LM, Offringa R, Drijfhout JW. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361(19):1838–1847. doi: 10.1056/NEJMoa0810097. [DOI] [PubMed] [Google Scholar]
- 15.Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, Edwards L, Parker RL, Denny L, Giffear M, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet. 2015;386(10008):2078–2088. doi: 10.1016/S0140-6736(15)00239-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, Nawrocki S, Ciuleanu T-E, Bosquée L, Trigo JM, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(1):59–68. doi: 10.1016/S1470-2045(13)70510-2. [DOI] [PubMed] [Google Scholar]
- 17.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 18.Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, Jassem J, Yoshimura M, Dahabreh J, Nakayama H, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17(6):822–835. doi: 10.1016/S1470-2045(16)00099-1. [DOI] [PubMed] [Google Scholar]
- 19.Kallert SM, Darbre S, Bonilla WV, et al. Replicating viral vector platform exploits alarmin signals for potent CD8(+) T cell-mediated tumour immunotherapy. Nat Commun. 2017;8:15327. doi: 10.1038/ncomms15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kotturi MF, Swann JA, Peters B, Arlehamn CL, Sidney J, Kolla RV, James EA, Akondy RS, Ahmed R, Kwok WW, et al. Human CD8(+) and CD4(+) T cell memory to lymphocytic choriomeningitis virus infection. J Virol. 2011;85(22):11770–11780. doi: 10.1128/JVI.05477-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Lamballerie X, Fulhorst CF, Charrel RN. Prevalence of antibodies to lymphocytic choriomeningitis virus in blood donors in southeastern France. Transfusion. 2007;47(1):172–173. doi: 10.1111/j.1537-2995.2007.01081.x. [DOI] [PubMed] [Google Scholar]
- 22.Stephensen CB, Blount SR, Lanford RE, Holmes KV, Montali RJ, Fleenor ME, Shaw JFE. Prevalence of serum antibodies against lymphocytic choriomeningitis virus in selected populations from two U.S. Cities. J Med Virol. 1992;38(1):27–31. doi: 10.1002/jmv.1890380107. [DOI] [PubMed] [Google Scholar]
- 23.Sommerstein R, Flatz L, Remy MM, Malinge P, Magistrelli G, Fischer N, Sahin M, Bergthaler A, Igonet S, Ter Meulen J. Arenavirus glycan shield promotes neutralizing antibody evasion and protracted infection. PLoS Pathog. 2015;11(11):e1005276. doi: 10.1371/journal.ppat.1005276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atherton MJ, Stephenson KB, Pol J, Wang F, Lefebvre C, Stojdl DF, Nikota JK, Dvorkin-Gheva A, Nguyen A, Chen L, et al. Customized viral immunotherapy for HPV-associated cancer. Cancer Immunol Res. 2017;5(10):847–859. doi: 10.1158/2326-6066.CIR-17-0102. [DOI] [PubMed] [Google Scholar]
- 25.Cassetti MC, McElhiney SP, Shahabi V, Pullen JK, Le Poole IC, Eiben GL, Smith LR, Kast WM. Antitumor efficacy of Venezuelan equine encephalitis virus replicon particles encoding mutated HPV16 E6 and E7 genes. Vaccine. 2004;22(3–4):520–527. doi: 10.1016/j.vaccine.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Flatz L, Hegazy AN, Bergthaler A, Verschoor A, Claus C, Fernandez M, Gattinoni L, Johnson S, Kreppel F, Kochanek S. Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat Med. 2010;16(3):339–345. doi: 10.1038/nm.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venkatesan A, Murphy OC. Viral encephalitis. Neurol Clin. 2018;36(4):705–724. doi: 10.1016/j.ncl.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 29.Liu Z, Zhou H, Wang W, Fu YX, Zhu M. A novel dendritic cell targeting HPV16 E7 synthetic vaccine in combination with PD-L1 blockade elicits therapeutic antitumor immunity in mice. Oncoimmunology. 2016;5(6):e1147641. doi: 10.1080/2162402X.2016.1147641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grunwitz C, Salomon N, Vascotto F, Selmi A, Bukur T, Diken M, Kreiter S, Türeci Ö, Sahin U. HPV16 RNA-LPX vaccine mediates complete regression of aggressively growing HPV-positive mouse tumors and establishes protective T cell memory. Oncoimmunology. 2019;8(9):e1629259. doi: 10.1080/2162402X.2019.1629259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knipe DM, Howley PM. Chapter 43. Arenavirus. Fields virology. Vol. 1333. 6th ed. Philadelphia (PA): Wolters Kluwer/Lippincott Williams & Wilkins Health; 2013. [Google Scholar]
- 32.Brun JL, Dalstein V, Leveque J, et al. Regression of high-grade cervical intraepithelial neoplasia with TG4001 targeted immunotherapy. Am J Obstet Gynecol. 2011;204:169:e1–8. [DOI] [PubMed] [Google Scholar]
- 33.Garcia F, Petry KU, Muderspach L, Gold MA, Braly P, Crum CP, Magill M, Silverman M, Urban RG, Hedley ML. ZYC101a for treatment of high-grade cervical intraepithelial neoplasia: a randomized controlled trial. Obstet Gynecol. 2004;103(2):317–326. doi: 10.1097/01.AOG.0000110246.93627.17. [DOI] [PubMed] [Google Scholar]
- 34.Hung CF, Cheng WF, Hsu KF, Chai CY, He L, Ling M, Wu TC. Cancer immunotherapy using a DNA vaccine encoding the translocation domain of a bacterial toxin linked to a tumor antigen. Cancer Res. 2001;61:3698–3703. [PubMed] [Google Scholar]
- 35.Melief CJ. Treatment of established lesions caused by high-risk human papilloma virus using a synthetic vaccine. J Immunother. 2012;35(3):215–216. doi: 10.1097/CJI.0b013e318248f17f. [DOI] [PubMed] [Google Scholar]
- 36.Wu A, Zeng Q, Kang TH, Peng S, Roosinovich E, Pai SI, Hung C-F. Innovative DNA vaccine for human papillomavirus (HPV)-associated head and neck cancer. Gene Ther. 2011;18(3):304–312. doi: 10.1038/gt.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aebischer O, Meylan P, Kunz S, Lazor-Blanchet C. Lymphocytic choriomeningitis virus infection induced by percutaneous exposure. Occup Med (Lond). 2016;66(2):171–173. doi: 10.1093/occmed/kqv156. [DOI] [PubMed] [Google Scholar]
- 38.Webb HE, Molomut N, Padnos M, Wetherley-Mein G. The treatment of 18 cases of malignant disease with an arenavirus. Clin Oncol. 1975;1:157–169. [PubMed] [Google Scholar]
- 39.Kostense S, Koudstaal W, Sprangers M, Weverling GJ, Penders G, Helmus N, Vogels R, Bakker M, Berkhout B, Havenga M. Adenovirus types 5 and 35 seroprevalence in AIDS risk groups supports type 35 as a vaccine vector. AIDS. 2004;18(8):1213–1216. doi: 10.1097/00002030-200405210-00019. [DOI] [PubMed] [Google Scholar]
- 40.Roberts DM, Nanda A, Havenga MJ, Abbink P, Lynch DM, Ewald BA, Liu J, Thorner AR, Swanson PE, Gorgone DA, et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature. 2006;441(7090):239–243. doi: 10.1038/nature04721. [DOI] [PubMed] [Google Scholar]
- 41.Pinschewer DD, Perez M, Jeetendra E, Bächi T, Horvath E, Hengartner H, Whitt MA, de la Torre JC, Zinkernagel RM. Kinetics of protective antibodies are determined by the viral surface antigen. J Clin Invest. 2004;114(7):988–993. doi: 10.1172/JCI200422374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalkavan H, Sharma P, Kasper S, Helfrich I, Pandyra AA, Gassa A, Virchow I, Flatz L, Brandenburg T, Namineni S. Spatiotemporally restricted arenavirus replication induces immune surveillance and type I interferon-dependent tumour regression. Nat Commun. 2017;8(1):14447. doi: 10.1038/ncomms14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghanizada M, Jakobsen KK, Gronhoj C, von Buchwald C. The effects of checkpoint inhibition on head and neck squamous cell carcinoma: A systematic review. Oral Oncol. 2019;90:67–73. doi: 10.1016/j.oraloncology.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 45.Lin PL, Cheng YM, Wu DW, Huang Y-J, Lin H-C, Chen C-Y, Lee H. A combination of anti-PD-L1 mAb plus Lm-LLO-E6 vaccine efficiently suppresses tumor growth and metastasis in HPV-infected cancers. Cancer Med. 2017;6(9):2052–2062. doi: 10.1002/cam4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, Feng L, Lee JJ, Tran H, Kim YU. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5(1):67–73. doi: 10.1001/jamaoncol.2018.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moeini S, Saeidi M, Fotouhi F, Mondanizadeh M, Shirian S, Mohebi A, Gorji A, Ghaemi A. Synergistic effect of programmed cell death protein 1 blockade and secondary lymphoid tissue chemokine in the induction of anti-tumor immunity by a therapeutic cancer vaccine. Arch Virol. 2017;162(2):333–346. doi: 10.1007/s00705-016-3091-5. [DOI] [PubMed] [Google Scholar]
- 48.Rice AE, Latchman YE, Balint JP, Lee JH, Gabitzsch ES, Jones FR. An HPV-E6/E7 immunotherapy plus PD-1 checkpoint inhibition results in tumor regression and reduction in PD-L1 expression. Cancer Gene Ther. 2015;22(9):454–462. doi: 10.1038/cgt.2015.40. [DOI] [PubMed] [Google Scholar]
- 49.Eissa IR, Bustos-Villalobos I, Ichinose T, et al. The current status and future prospects of oncolytic viruses in clinical trials against melanoma, glioma, pancreatic, and breast cancers. Cancers (Basel). 2018;10(10):356. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6210336/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ring S, Flatz L. Generation of lymphocytic choriomeningitis virus based vaccine vectors. Methods Mol Biol. 2016;1404:351–364. [DOI] [PubMed] [Google Scholar]
- 51.Cardin RD, Bravo FJ, Pullum DA, Orlinger K, Watson EM, Aspoeck A, Fuhrmann G, Guirakhoo F, Monath T, Bernstein DI. Replication-defective lymphocytic choriomeningitis virus vectors expressing guinea pig cytomegalovirus gB and pp65 homologs are protective against congenital guinea pig cytomegalovirus infection. Vaccine. 2016;34(17):1993–1999. doi: 10.1016/j.vaccine.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 52.Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, Kreutzfeldt M, Hegazy AN, Schrick C, Fallon PG, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science. 2012;335(6071):984–989. doi: 10.1126/science.1215418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.