

Abstract
Abstract
Main protease (Mpro) of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) intervenes in the replication and transcription processes of the virus. Hence, it is a lucrative target for anti-viral drug development. In this study, molecular modeling analyses were performed on the structure activity data of recently reported diverse SARS-CoV-2 Mpro inhibitors to understand the structural requirements for higher inhibitory activity. The classification-based quantitative structure–activity relationship (QSAR) models were generated between SARS-CoV-2 Mpro inhibitory activities and different descriptors. Identification of structural fingerprints to increase or decrease in the inhibitory activity was mapped for possible inclusion/exclusion of these fingerprints in the lead optimization process. Challenges in ADME properties of protease inhibitors were also discussed to overcome the problems of oral bioavailability. Further, depending on the modeling results, we have proposed novel as well as potent SARS-CoV-2 Mpro inhibitors.
Graphic Abstract

Electronic supplementary material
The online version of this article (10.1007/s11030-020-10166-3) contains supplementary material, which is available to authorized users.
Keywords: SARS-CoV-2, Mpro, QSAR, Bayesian model, Ligand-receptor interaction, MD simulation
Introduction
At the end of 2019, a new coronavirus-related infection namely severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) had spread its wings across the globe [1, 2]. For its worldwide impact, this coronavirus disease-2019 (COVID-19) was declared as a global pandemic by the World Health Organization (WHO) and to date over million confirmed cases along with million COVID-19-related mortalities have been reported [1, 3].
Belonging to the betacoronavirus genus, SARS-CoV-2 is responsible for lower respiratory tract infections similar to severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle-East respiratory syndrome coronavirus (MERS-CoV) [1]. Ongoing research highlighted some important druggable targets like spike (S) protein, papain-like protease (PLpro), RNA-dependent RNA polymerase (RdRp) and SARS-CoV-2 main protease/3C-like protease (Mpro/3CLpro). These possess potentiality to become important targets for achieving the most desirable goal that humanity craves in the current situation [1, 2, 4]. The open reading frame 1ab (ORF 1a/b) of coronaviruses translates polyprotein 1a and polyprotein 1ab. The Mpro and PLpro enzymes produce non-structural proteins by processing these polyproteins which in term aids the production of viral structural proteins [5, 6]. Thus, SARS-CoV-2 Mpro enzyme can be a valuable target as it intervenes in the replication and transcription processes of the virus [2]. It possesses high structural similarity (96% sequential resemblance) to SARS-CoV Mpro [5].
Additionally, targeting proteases were successful to provide anti-viral agents for the treatment of viral infections like human immunodeficiency virus (HIV) and hepatitis C virus (HCV) [7, 8]. Thus, small molecule-mediated blocking of Mpro activity is a feasible option for SARS-CoV-2 anti-viral drug development [9–18]. The computer-aided drug design (CADD) and virtual screenings (VS) are viable options. These techniques may be useful to identify promising hit that can aid the design and development of potent anti-viral agents [4]. Meanwhile, drug repurposing was employed as an instant weapon against coronavirus [19]. However, the ongoing rampage of COVID-19 has employed researches in an assignment to discover a permanent solution for this pandemic. In this panorama, the small molecule inhibitors carefully designed by different modeling approaches are one of the most promising tools to achieve success.
Here, we have explored SARS-CoV-2 Mpro inhibitors by different molecular modeling strategies with four main mottos- (i) development of a mathematical relationship between the derivatives and SARS-CoV-2 Mpro enzyme (ii) identification of important fingerprints that module the SARS-CoV-2 Mpro inhibition, (iii) scope of these derivatives to address ADME properties, (iv) design of potent SARS-CoV-2 Mpro inhibitors with significant ADME properties. The current study, a part of our rational drug design and discovery program, [4, 19–21] may offer an initiative to explore the possibility of potent inhibitor design against the Mpro enzyme of SARS-CoV-2.
Methods and materials
Dataset
A number of 33 derivatives, represented by SARS-CoV-2 Mpro inhibitory activity IC50 (µM), were obtained from the published data [5, 6, 9, 14, 15]. The SARS-CoV-2 Mpro inhibitory activity values of the inhibitors are presented in Supplementary Table S1. The pIC50 (i.e., -log IC50) values were used to derive QSAR models [22–24].
Classification-based QSAR
The classification modeling assists to classify the active and inactive molecules in terms of their biological data [25–30]. Here, we employed Bayesian classification approach [31–33].
Bayesian classification study
Performing Bayesian classification study by the aid of Discovery Studio (DS) software [34] enables graphical visualization of critical chemical sub-structural features (fingerprint or fragments) attributed to enhance or decrease the SARS-CoV-2 Mpro inhibitory activity. Additionally, as to conduct this classification-based study, on the basis of their SARS-CoV-2 Mpro inhibitory activity, the dataset molecules were grouped into active (SARS-CoV-2 Mpro pIC50> 5.0) and inactive (SARS-CoV-2 Mpro pIC50< 5.0) molecules (e.g., active = 1, inactive = 0) [23].
The selection of the training and test sets was done by using Generate training and test data tool in DS [34]. The whole data were divided into 20 clusters by maximum dissimilarity approach on the basis of Predefined Set properties including ALogP, Molecular_Weight, Num_H_Donors, Num_H_Acceptors, Num_RotatableBonds, Num_Atoms, Num_Rings, Num_AromaticRings, Num_Fragments, Molecular_PolarSurfaceArea. The whole data set compounds were separated into two groups, a training set SARS-CoV-2 Mpro inhibitors, a test set SARS-CoV-2 Mpro inhibitors (Supplementary Table S1).
Further, to ensure whether the selected test set compounds truly represent the training set or not, principal components analysis (PCA) was performed by Calculate principal components tool in DS [34]. The DS default properties such as ALogP, Molecular_Weight, Num_H_Donors, Num_H_Acceptors, Num_RotatableBonds, Num_Rings, Num_AromaticRings, Molecular_FractionalPolarSurfaceArea were considered for the PCA calculation. The uniform distribution of the test set SARS-CoV-2 Mpro inhibitors in the PCA three-dimensional plot (as given in Supplementary Figure S1) referred a proper division of the training and the test sets.
Finally, the Bayesian classification model was constructed on the training set and was cross-validated by using the test set. Before conducting this Bayesian classification study, several fundamental molecular features namely, ALogP, Molecular_Weight, Num_H_Donors, Num_H_Acceptors, Num_RotatableBonds, Num_Rings, Num_AromaticRings, Molecular_FractionalPolarSurfaceArea of the dataset molecules have been calculated [34]. Alongside those molecular properties, a topological fingerprint descriptor namely extended connectivity fingerprint of diameter 6 (ECFP_6) [35] was also considered for this study. The quality of this classification model was evaluated using the Receiver operating characteristics (ROC) plot [36], sensitivity (Se), specificity (Sp) and accuracy (Acc) for both the training and the test sets [23].
Multiple linear regression analysis
The derivatives with no activity and without definite SARS-CoV-2 Mpro inhibitory activity were not considered for the multiple linear regression (MLR) analysis [23]. Hence, only 25 molecules were recognized for the regression-based QSAR study (Supplementary Table S1).
Meanwhile, a number of 2D and fingerprint descriptors were calculated [37]. Then, the descriptors with constant values were removed from the data matrix [23]. Next, the highly inter-correlated variables were stocked out depending on the specified variance of 0.001 and correlation coefficient cutoff values of 0.99 [38, 39]. Then, several genetic function approximation (GFA) runs were employed to collect a bunch of important descriptors [39]. Finally, stepwise multiple linear regression (S-MLR) model was developed to identify the linear correlation between the structure of SARS-CoV-2 Mpro inhibitors and their respective Mpro inhibitory activities. The robustness of the constructed model was justified by correlation coefficient (R), adjusted R2 (R2A), variance ratio (F) at specified degrees of freedom (df), cross-validated R2 (Q2), standard error of estimate (SEE), and other validation metrics [23]. In addition, Euclidean distance-based applicability domain was also constructed [23, 38] to check the applicability of the MLR model.
Molecular docking & dynamic simulation
For the docking studies, the SARS-CoV-2 Mpro structure was obtained from Protein Data Bank (PDB ID: 6LZE). Subsequently, the compounds were docked in the active site of the Mpro protein using Auto Dock Vina v1.1.2 [40], wherein a grid box of size 16, 14, and 14 with spacing of 1 Å were set around the active site of SARS-CoV-2 Mpro.
Later, the molecular dynamics simulation was performed by the GROMACS 5.1.4 version [41] using the GROMOS43A2 force field and SPC/E water model. To neutralize the charges on each simulating system, an appropriate number of ions (Na+) were added. The energy of each system was minimized by the steepest descent algorithm followed by NVT (at 300 K) and NPT (at 1 bar) ensemble equilibrations for 100 ps. Subsequently, each of the equilibrated system was carried on for the production simulation of 20 ns. The trajectory data of the production simulations were further used for the calculation of root mean square deviation (RMSD), root mean square fluctuations (RMSF), and radius of gyration (Rg) data of each system. The binding energy of each compound in the complex was calculated by g_MMPBSA package of GROMACS, for every 0.1 ns frame of each 20 ns simulation [42].
Result and discussions
SARS-CoV-2 Mpro binding site analyses
SARS-CoV-2 Mpro is a homodimeric protein. Each subunit is termed as protomer. A number of 306 amino acid residue is found in each protomer. It is constructed by three domains [4, 9]. The domain I is 8 to 100 residues long followed by domain II (101 to 184 residues) and domain III (from 199 to 306 residues). Besides, domains II and III are bridged by a long loop (from 185 to 198 residues) [9].
Domains I and II allocated the same fold i.e., an anti-parallel six stranded β-barrel structure, while domain III is semblance by five α-helices arranged into a largely anti-parallel globular cluster. Meanwhile, the domain III helps in the regulation of Mpro dimerization through an inter subunit salt-bridge between E290 from one protomer and R4 from the other protomer. Notably, the substrate-binding site or catalytic site of SARS-CoV-2 Mpro is located at a cleft between domains I and II. The N-terminal amino acid residue of a protomer namely S1 interacts with the E166 of another to form the S1 subsite of the substrate-binding pocket. Hence, the dimerization is essential for protease activity [17].
The research on SARS-CoV-2 Mpro has moved at a much faster after delivering several ligand bound crystal structures. Those have provided useful information for developing inhibitors, but it seems that it is not enough. Analysis of different crystal structures has shown that there is an intrinsic flexibility in the catalytic site. In order to explore the detail binding interactions, few contour maps of the binding site of the SARS-CoV-2 Mpro (PDB: 6WTT) were determined by Display receptor surfaces tool of DS [34]. Six structure-based contour maps for hydrophobic, hydrogen bond, charge, aromatic, ionizability and solvent accessible surface (SAS) are provided in Fig. 1.
Fig. 1.

Six structure-based contour maps for a hydrophobic, b aromatic, c hydrogen bond, d ionizability, e charge, and f solvent accessible surface (SAS)
Figure 1 reveals overall surface topology of SARS-CoV-2 Mpro with its deep binding pocket. The binding site of SARS-CoV-2 Mpro enzyme is a large and wide cavity containing four main hydrophobic sites (Fig. 1a). Analyzing Fig. 1a, b suggests that hydrophobic aromatic substitution may be allowable in the binding pocket. The hydrogen-bond site map (Fig. 1c) shows that acceptor feature exists close to the straight chain amide residues. The S1 cavity is acceptor specific. Near S1 site, H172 endorses acidic ionizability (Fig. 1d), and it is slight negatively charged (Fig. 1e). Significantly, ionizability and interpolated charge contours are more or less consistent. From the SAS contour (Fig. 1f), it may be suggested that a significant part of the catalytic site is solvent exposed. In order to explore the details contribution of fragments/fingerprints of the inhibitors, we moved forward to quantitative structure–activity relationship (QSAR) studies and design of specific SARS-CoV-2 Mpro inhibitors.
Classification-based QSAR
The Bayesian classification modeling is a classification QSAR technique based on the Bayes’ theorem which utilizes data to predict the probability of specific events [43–45]. Additionally, another advantage of this Bayesian classification study with fingerprint descriptor is its capability to recognize important structural fragments of molecules while indicating their positive or negative influence on the activity [23].
In order to describe the statistical quality of the generated Bayesian classification model, different statistical parameters like sensitivity (Se), specificity (Sp), and accuracy (Acc) were calculated. The results were found to be statistically significant as all the parameters were having decent scores to consider the model as robust and predictive as specified in Table 1.
Table 1.
Statistical parameters of the developed model obtained from Bayesian classification study
| Set | ROC | TP | FN | FP | TN | Se | Sp | Acc |
|---|---|---|---|---|---|---|---|---|
| Training (NTrain = 25) | 0.747 | 15 | 0 | 4 | 6 | 1.000 | 0.600 | 0.840 |
| Test (NTest = 6) | 1.000 | 3 | 1 | 0 | 2 | 0.750 | 1.000 | 0.833 |
ROC receiver operating characteristics; TP true positive; FN false negative; FP false positive; TN true negative; Se sensitivity; Sp specificity; Acc accuracy
Further, the ROC (Receiver operating characteristic) curve for training and test sets are found to be 0.747 and 1.000, respectively. This indicates the predictive capability of the model. The ROC curve for training and test set are shown in Supplementary Figure S2.
The mechanistic interpretation of the Bayesian classification study is performed using a fingerprint descriptor ECFP_6. A set of 20 good and 20 bad molecular sub-structural features has been procured with positive and negative influences, respectively, on SARS-CoV-2 Mpro inhibition of the compounds. Twenty good (G1-G20) and twenty bad sub-structural fragments (B1-B20) as constructed from the ECFP_6 fingerprint descriptor are shown in Supplementary Figure S3 and S4, respectively.
Upon observation, the set of 20 good molecular sub-structures can be clustered into four major groups namely: bi-acetyl amine and acetamido group containing 2-oxo pyrrolidine moiety (G1, G7 and G9-G15), cyclohexyl and cyclohexyl methyl groups (G2-G3, G8, G16-G17 and G19-G20), and acetamido methylene (iso-butyl) acetamide moiety (G4-G6). Beside these frequent sub-structures, the oxyanion function (G18) is upheld as positive influencers for the Mpro inhibition as shown in Supplementary Figure S3.
In contrast, among the proposed negatively influencing features, 4-fluoro phenyl and 4-fluoro benzyl moieties are the most commonly displayed bad features (B11-B15 and B17). The branched alkyl (B3-B4, B6, and B8) and amino-alkyl (B1-B2, B5, B10, and B16) groups are indicated to be detrimental for activity. Moreover, oxymethylene carbonyl (B7) and acetate (B9) functions are also suggested as negative regulators of Mpro inhibitory activity (Supplementary Figure S4).
Further analysis of the fragments and the dataset molecules, it is found that the most active M027 having an acetamido methylene (iso-butyl) acetamide function and a negatively charged oxygen ion similar to the sub-structures G4-G6 and G18, respectively (Fig. 2).
Fig. 2.

Structures of some potent SARS-CoV-2 Mpro inhibitors containing good fragments highlighted in deep blue color
From the crystal structure analysis of M027 with SARS-CoV-2 Mpro (PDB: 6WTT), the mentioned fragments are found to involve in several interactions at the enzyme active site. The iso-butyl group of M027 enters into the S2 pocket of the enzyme while the carboxamide function interacts with Q189 and E166 amino acids (Fig. 3) [15].
Fig. 3.

Interaction of compound M027 (PDB: 6WTT) and M013 (PDB: 6Y2F) at the active site of SARS-CoV-2 Mpro
Meanwhile, fragments G2-G3, G8, G16-G17 and G19-G20 exhibit the importance of cyclohexyl moiety. From the SARs, it may be observed that cyclohexyl function is important for the activity. The cyclohexyl methyl moiety is found in active molecules like M009, M011, and M012. The cyclohexyl function embeds itself in the hydrophobic S2 site of SARS-CoV-2 Mpro [6]. Therefore, hydrophobic interactions are essential in these regions. Similarly, the (S)-γ-lactam ring is directed to a hydrophobic S1 pocket (Fig. 3).
The cyclohexyl methyl group of M009 is found to be important for entering into the S2 pocket of the enzyme where its indole ring enters into the S4 pocket (PDB: 6M0K). Meanwhile, the indole moiety of compound M010 also shows identical binding to that of M009 (PDB: 6LZE) [6].
A substituted 2-pyridinone moiety is present in both compound M012 and M013 whereas the 3-amino-Boc substituted 2-pyridinone moiety of M013 forms more than one interaction at the active site of SARS-CoV-2 Mpro (PDB: 6Y2F) as shown in Fig. 3. Also, the 2-carbonyl and the 3-amino functions of the moiety interact with E166 through hydrogen bond formation [5]. The presence of 2-phenyl-4-chromenone moiety can be observed in both M033 and M032. The 6- and 7-hydroxyl group of M033 interacts with L141 and G143 (PDB: 6M2N), respectively [14].
Regarding the bad molecular fragments, compound M021 possessing a 4-fluorophenyl group is inactive (Fig. 4). The acetate function containing M029 exhibits inactivity against Mpro. The oxymethylene carbonyl moiety containing M030 also shows inactivity against Mpro (Fig. 4).
Fig. 4.

Structures of some inactive SARS-CoV-2 Mpro inhibitors containing bad fragments highlighted in deep red color
Challenges in SARS-CoV-2 Mpro inhibitors design
An effective drug candidate/drug-like ligands having promising biological responses should possess the ability to reach its desire domain in sufficient concentration. Drug design and discovery obviously depends on the assessment of absorption, distribution, metabolism and excretion (ADME) characteristics.
In order to check the drug-likeliness of the investigated derivatives Filters ligands using Lipinski and Veber rules protocol of DS was employed [34]. It selects drug-like ligands as per the rules proposed by Lipinski [46] and Veber [47]. The default settings for Lipinski Rule of Five (Hydrogen Bond Donors: 5, Hydrogen Bond Acceptors: 10, Molecular Weight: 500, AlogP: 5, Number of Violations Allowed: 1) and Veber Rule (Rotatable Bonds 10, Polar Surface Area 140, Hydrogen Bond Donors, and Acceptors 12) were considered for this study.
Notably, 19 compounds (Fig. 5) fail to pass the Lipinski and Veber rules. Only 14 compounds (Fig. 5) pass these two rules, therefore, those have a higher probability of good oral bioavailability.
Fig. 5.

Comparison of Lipinski and Vaber rules criteria for the dataset compounds
The protease targeted peptidomimetic inhibitors design is very challenging due to their undesirable pharmacokinetic properties. In contrast, compounds with low molecular weight or non-peptidomimetics exhibit good druglikeliness. However, non-peptidomimetics/low molecular weight derivatives fail to effectively block the proteolytic activity of SARS-CoV-2 Mpro. In these circumstances, the structure of SARS-CoV-2 Mpro in complex with a small molecule baicalein (M033) may be a good option for baicalein-derived lead optimization. The binding mode of baicalein at the active site of SARS-CoV-2 Mpro facilitated a unique protein–ligand interaction pattern.
Since baicalein (M033) possesses a molecular weight of 270.24 Da, it encourages us to anticipate new derivative design by keeping the baicalein core. Lead optimization of baicalein with good molecular fingerprint (as suggested by the Bayesian modeling study) may render new derivatives directed toward S1 and/or S4 site(s). It may effectively block the proteolytic activity of SARS-CoV-2 Mpro.
Taken together these modeling efforts may give rise in a new candidate with broad-spectrum anti-viral properties. However, substitution at the wrong position (as in case of baicalin) resulted in ~ sevenfold loss in SARS-CoV-2 Mpro inhibition (baicalein vs baicalin) [14].
Designing of newer molecules
Considering the finding of the performed QSAR studies, we have designed a set of four chromenone-based molecules (Fig. 6).
Fig. 6.

Designed SARS-CoV-2 Mpro inhibitors (D1–D4)
Bayesian classification model
Primarily, the Bayesian classification model was used to predict the Mpro inhibitory activity of these molecules. The designed compounds (D1–D4) predicted as active. Hence, these compounds may serve as promising molecules against SARS-CoV-2 Mpro.
Multiple linear regression model
To further revalidate prediction credibility, a stepwise multiple linear regression (S-MLR) model has been constructed on the available data. At first, a pool of 2D and fingerprint descriptors for these derivatives was calculated [37]. Then, dataset thinning was introduced followed by several genetic function approximation (GFA) runs [38, 39]. The best model (Eq. 1) through the S-MLR analysis (the stepping criterion of F = 4 for inclusion and F = 3.99 for exclusion) is as follows
| 1 |
Equation (1) explains 94.4% and predicts 92.8% variances of the SARS-CoV-2 Mpro inhibitory activity. The definition and contributions of the descriptors used to develop Eq. (1) are depicted in Table 2. Other details are given in the Supplementary files (Table S2–S4).
Table 2.
The definition and contributions of descriptors used to develop Eq. (1)
| Descriptor | Type | Definition | Contribution |
|---|---|---|---|
| MLFER_A | PaDEL; 2D | Overall or summation solute hydrogen bond acidity | Positive |
| AATS5m | 2D autocorrelations | Average Broto-Moreau autocorrelation—lag 5/weighted by mass | Positive |
| MDEC-33 | PaDEL; 2D | Molecular distance edge between all tertiary carbons | Negative |
| PubchemFP184 | Fingerprint | 1 unsaturated non-aromatic heteroatom-containing ring size 6 | Negative |
| PubchemFP695 | Fingerprint | O=C–C–C–C–C–N | Negative |
Additionally, Euclidean distance-based applicability domain was constructed [23, 38] as illustrated in Fig. 7. It justifies that all the compounds are within the boundary of the hypothetical domain (Fig. 7). Hence, there is no outlier for this dataset.
Fig. 7.
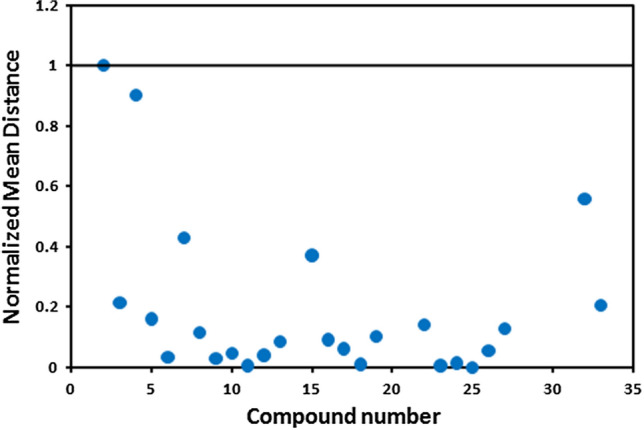
Graphical representation of the applicability domain of Eq. (1) by the Euclidean distance approach
The designed molecules (D1–D4) predicted pIC50 more than 7.523 as depicted in Table 3. This result supports the potential of these designed molecules to become promising Mpro inhibitors.
Table 3.
Drug-like properties and predicted activity of designed SARS-CoV-2 Mpro inhibitors
| Compound | MW | LogP | HBA | HBD | TPSA | nRB | Prediction | Pred pIC50 |
|---|---|---|---|---|---|---|---|---|
| D1 | 493.16 | 2.431 | 8 | 4 | 116.76 | 6 | Active | 8.151 |
| D2 | 473.2 | 2.699 | 8 | 4 | 116.76 | 7 | Active | 7.706 |
| D3 | 489.19 | 3.047 | 9 | 5 | 136.99 | 7 | Active | 8.879 |
| D4 | 507.18 | 2.669 | 8 | 4 | 116.76 | 7 | Active | 8.479 |
Since the drug-likeliness is one of the major challenges in drug discovery. The drug-likeliness of these designed molecules (D1–D4) was investigated using the DruLiTo software [48]. The drug-like properties of the designed molecules are also given in Table 3.
Molecular docking and dynamic simulation
To understand the structural basis of inhibition by compounds (D1–D4), protein–ligand docking studies were performed using AutoDockVina [40]. The outcome of molecular docking shows that all compounds are found to be docked into the active site of SARS-CoV-2 Mpro (Fig. 8).
Fig. 8.

Molecular docking and dynamic simulation analysis: a–d Compounds D1, D2, D3, and D4 are represented as magenta, yellow, gray, and cyan sticks, respectively. The Mpro protein residues showing interaction with compounds are labeled and displayed as stick model in element colors (carbon colored green, nitrogen colored blue, and oxygen colored red), while interactions are represented by black dashed lines. e–g MDS plots are showing RMSD, RMSF, and Rg of the backbone-atoms of the apo Mpro and its complexes
The binding energies of the selected conformation of the compounds are depicted in the Supplementary Table S5, which indicates that the complex with compound D4 has higher binding energy in comparison with the other compounds. Moreover, interacting residues of the docked complexes also reveals similarity with the interacting residues of the reported protein co-crystal structure (PDB: 6LZE).
The respective average RMSD of apo, prt_D1, prt_D2, prt_D3, and prt_D4 are enumerated as 0.284, 0.292, 0.213, 0.258, and 0.276 nm, respectively. Wherein, protein shows lesser deviations in the structure with compound D2 in comparison with the other compounds as well as apo form (Fig. 8). The analysis infers an increase in the stabilization of protein backbone structure after interaction with D2 during the simulation. Simultaneously, the fluctuations in the backbone atom of the protein residues in each system was analyzed by RMSF, which shows apo, prt_D1, prt_D2, prt_D3, and prt_D4 system to possess average RMSF of 0.135, 0.137, 0.117, 0.138, and 0.126 nm, respectively. However, protein residues presented lower fluctuation after interacting with compound D2 than the other complex and apo forms. Decrease in fluctuation of the residues side chain in the presence of D2 indicates the induced stability in rotameric switching of protein residues during dynamic environment.
Further, the comparative analysis of Rg data was performed to determine the protein compactness after interaction with compounds. The respective average Rg value of 2.13, 2.10, 2.13, 2.10, and 2.12 nm are computed for apo, prt_D1, prt_D2, prt_D3, and prt_D4, respectively. The obtained data reveals that all protein complex forms attain level of compactness similar to the apo form, which indicates that each compound interact with protein without disturbing its structural folding in the dynamic environment (Fig. 8). Altogether, our study highlighted that the compound D2 has shown much stable interaction along with the induction of low deviations and fluctuations in the protein structure as compared to apo form and other compounds during MD simulation.
The affinity of the compounds with protein was also analyzed by the binding energy calculation. The average binding energy calculated for each protein–ligand complex is presented in Table 4, which shows that the compound D4 has more binding affinity with Mpro protein during the simulation.
Table 4.
Binding energy calculation of design compounds (D1–D4)
| Compound | van der Waal energy (kJ/mol) | Electrostatic energy (kJ/mol) | Polar solvation energy (kJ/mol) | SASA energy (kJ/mol) | Binding Energy (kJ/mol) |
|---|---|---|---|---|---|
| D1 | − 268.79 ± 2.67 | − 33.40 ± 0.77 | 98.17 ± 1.19 | − 19.61 ± 0.15 | − 223.54 ± 2.31 |
| D2 | − 254.25 ± 1.59 | − 42.77 ± 0.71 | 140.68 ± 1.11 | − 20.26 ± 0.10 | − 176.66 ± 1.49 |
| D3 | − 277.68 ± 1.72 | − 46.58 ± 0.91 | 147.95 ± 1.10 | − 21.52 ± 0.08 | − 197.92 ± 1.71 |
| D4 | − 307.53 ± 2.30 | − 41.80 ± 0.83 | 133.44 ± 1.26 | − 22.45 ± 0.10 | − 238.28 ± 1.88 |
Wherein, the van der Waals energy plays a major role in the binding of compound D4 at the active site of the Mpro in comparison to the other free energies. Moreover, the binding energy analysis shows corroboration with docking studies of the compounds. It infers that the compound D4 has more affinity for the static and dynamic SARS-CoV-2 Mpro structure.
Conclusion
COVID-19 shows worldwide impact as a global pandemic. Till date over million confirmed cases have been reported worldwide. In this communication, QSAR analyses were performed on recently reported structurally diverse Mpro inhibitors to understand structural requirements for higher activity. The study is able to extract the significant molecular attributes of these SARS-CoV-2 Mpro inhibitors.
The main problems for design of SARS-CoV-2 Mpro inhibitors are the perfect binding of susbtituents in putative binding site and the ADME properties. To overcome these problems, we suggest baicalein-derived design as well as lead optimization. Since different ligands induce different conformational changes, the conformation of binding pocket residues could not be easily predicted for different inhibitors. Nonetheless, the S1, S1’, S2, and S4 pockets bear intrinsic flexibility where hydrophobic susbtitutents may trigger the SARS-CoV-2 Mpro inhibition. Our structure-based contours result suggests that Mpro binding pockets should be analyzed carefully to design inhibitors with such flexibilities.
In our previous study, the Monte Carlo optimization-based QSAR, structural and physico-chemical interpretation (SPCI) analysis were successful to deliver several important molecular features from the SARS-CoV Mpro inhibitors [21]. This can be useful to develop effective inhibitors against SARS-CoV-2 Mpro. Additionally, compared to the recent attempts to identify the promising attributes for previous coronavirus inhibitors (Table 5), the current study deals with the existing SARS-CoV-2 Mpro inhibitors.
Table 5.
Comparison of recent QSAR analysis on SARS-CoV and SARS-CoV-2 inhibitors
| Sl | Target | Dataset | QSAR model | References |
|---|---|---|---|---|
| 1 | SARS-CoV Mpro | Diverse set of molecules | Monte Carlo optimization-based QSAR and SPCI analysis | [21] |
| 2 | SARS-CoV Mpro | Aromatic disulfide compounds | Monte Carlo optimization-based QSAR | [49] |
| 3 | SARS-CoV Mpro | Diverse set of molecules | Partial least square study (PLS) | [50] |
| 4 | SARS-CoV Mpro | Carboxamide derivatives | Multiple linear regression (MLR) | [51] |
| 5 | SARS-CoV Mpro | Diverse set of molecules | Multiple linear regression (MLR) | [52] |
| 6 | SARS-CoV-2 Mpro | Diverse set of molecules | Bayesian classification study, Multiple linear regression (MLR) | Current study |
In summary, the modeling results provide useful quantitative and qualitative information about the structural requirements of an effective Mpro inhibitor against SARS-CoV-2.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgement
One of the authors (S. A. Amin) is grateful to the Council of Scientific and Industrial Research (CSIR), New Delhi, India for Senior Research Fellowship (SRF) [FILE NO.: 09/096(0967)/2019-EMR-I, Dated: 01-04-2019]. Suvankar Banerjee and Tarun Jha are also thankful for the financial support from RUSA 2.0 of UGC, New Delhi, India to Jadavpur University, Kolkata, India. The authors acknowledge Center for Modeling Simulation & Design (CMSD), University of Hyderabad, India for computational resources for MD simulation studies. We are very much thankful to the Department of Pharmaceutical Technology, Jadavpur University, Kolkata, India, Department of Pharmaceutical Sciences, Dr. Harisingh Gour University, India and School of Life Sciences, University of Hyderabad, India for providing the research facilities.
Compliance with ethical standards
Conflict of interest
The authors have no conflict of interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Sk. Abdul Amin and Suvankar Banerjee have equal contribution.
Contributor Information
Shovanlal Gayen, Email: shovanlal.gayen@gmail.com.
Tarun Jha, Email: tjupharm@yahoo.com.
References
- 1.Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The Trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20:363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panda PK, Arul MN, Patel P, Verma SK, Luo W, Rubahn H-G, Mishra YK, Suar M, Ahuja R (2020) Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci Adv 6:eabb8097 [DOI] [PMC free article] [PubMed]
- 3.WHO Coronavirus Disease (COVID-19) Dashboard. https://covid19.who.int/. Accessed 29 June 2020
- 4.Amin SA, Jha T. Fight against novel coronavirus: a perspective of medicinal chemists. Eur J Med Chem. 2020;201:112559. doi: 10.1016/j.ejmech.2020.112559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai W, Zhang B, Su H, Li J, Zhao Y, Xie X, Jin Z, Peng J, Liu F, Li C, Li Y, Bai F, Wang H, Cheng X, Cen X, Hu S, Yang X, Wang J, Liu X, Xiao G, Jiang H, Rao Z, Zhang L-K, Xu Y, Yang H, Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 8.Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat Rev Drug Discov. 2010;9:690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang BL, Zhang XL, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Lin D, Kusov Y, Nian Y, Ma Q, Wang J, von Brunn A, Leyssen P, Lanko K, Neyts J, de Wilde A, Snijder EJ, Liu H, Hilgenfeld R. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh AK, Brindis M, Shahabi D, Chapman ME, Mesecar AD. Drug development and medicinal chemistry efforts toward SARS-coronavirus and Covid-19 therapeutics. ChemMedChem. 2020;15:907–932. doi: 10.1002/cmdc.202000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gil C, Ginex T, Maestro I, Nozal V, Barrado-Gil L, Cuesta-Geijo MÁ, Urquiza J, Ramírez D, Alonso C, Campillo NE, Martinez A. COVID-19: drug targets and potential treatments. J Med Chem. 2020 doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- 13.Rut W, Lv Z, Zmudzinski M, Patchett S, Nayak D, Snipas SJ, Oualid FE, Huang TT, Bekes M, Drag M, Olsen SK. Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. BioRxiv. 2020 doi: 10.1101/2020.04.29.068890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su H, Yao S, Zhao W, Li M, Liu J, Shang W, Xie H, Ke C, Gao M, Yu K, Liu H, Shen J, Tang W, Zhang L, Zuo J, Jiang H, Bai F, Wu Y, Ye Y, Xu Y (2020) Discovery of baicalin and baicalein as novel, natural product inhibitors of SARS-CoV-2 3CL protease in vitro. BioRxiv. 10.1101/2020.04.13.038687
- 15.Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, Zhang X, Tarbet B, Marty MT, Chen Y, Wang J (2020) Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. BioRxiv. 10.1101/2020.04.20.051581 [DOI] [PMC free article] [PubMed]
- 16.Goyal B, Goyal D. Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb Sci. 2020;22:297–305. doi: 10.1021/acscombsci.0c00058. [DOI] [PubMed] [Google Scholar]
- 17.Gimeno A, Mestres-Truyol J, Ojeda-Montes MJ, Macip G, Saldivar-Espinoza B, Cereto-Massagué A, Pujadas G, Garcia-Vallvé S. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int J Mol Sci. 2020;21:3793. doi: 10.3390/ijms21113793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang B, He F, Liu D, Fang M, Wu Z, Xu D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. BioRxiv. 2020 doi: 10.1101/2020.03.03.972133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amin SA, Ghosh K, Gayen S, Jha T. Chemical-informatics approach to COVID-19 drug discovery: monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1780946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adhikari N, Baidya SK, Saha A, Jha T. In: Viral proteases and their inhibitors. Gupta SP, editor. USA: Academic Press; 2017. [Google Scholar]
- 21.Ghosh K, Amin SA, Gayen S, Jha T. Chemical-informatics approach to COVID-19 drug discovery: exploration of important fragments and data mining based prediction of some hits from natural origins as main protease (Mpro) inhibitors. J Mol Struct. 2020;1224:129026. doi: 10.1016/j.molstruc.2020.129026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nantasenamat C, Naenna T, Ayudhya CL, Prachayasittikul V. Quantitative prediction of imprinting factor of molecularly imprinted polymers by artificial neural network. J Comp Aided Mol Des. 2005;19:509–524. doi: 10.1007/s10822-005-9004-4. [DOI] [PubMed] [Google Scholar]
- 23.Amin SA, Adhikari N, Gayen S, Jha T. First report on the structural exploration and prediction of new BPTES analogs as glutaminase inhibitors. J Mol Struct. 2017;1143:49–64. doi: 10.1016/j.molstruc.2017.04.020. [DOI] [Google Scholar]
- 24.Abdizadeh R, Hadizadeh F, Abdizadeh T. In silico studies of novel scaffold of thiazolidin-4-one derivatives as anti-Toxoplasma gondii agents by 2D/3D-QSAR, molecular docking, and molecular dynamics simulations. Struct Chem. 2020;31:1149–1182. doi: 10.1007/s11224-019-01458-y. [DOI] [Google Scholar]
- 25.Toropov AA, Toropova AP, Raska I, Benfenati E, Gini G. QSAR modeling of endpoints for peptides which is based on representation of the molecular structure by a sequence of amino acids. Struct Chem. 2012;23:1891–1904. doi: 10.1007/s11224-012-9995-0. [DOI] [Google Scholar]
- 26.dos Santos IM, Agra JPG, de Carvalho TGC, Maia GLA, de Alencar Filho EB. Classical and 3D QSAR studies of larvicidal monoterpenes against Aedes aegypti: new molecular insights for the rational design of more active compounds. Struct Chem. 2018;29:1287–1297. doi: 10.1007/s11224-018-1110-8. [DOI] [Google Scholar]
- 27.Kolodziejczyk W, Kar S, Hill GA, Leszczynski J. A comprehensive computational analysis of cathinone and its metabolites using quantum mechanical approaches and docking studies. Struct Chem. 2016;27:1291–1302. doi: 10.1007/s11224-016-0779-9. [DOI] [Google Scholar]
- 28.Kar S, Roy K. First report on predictive chemometric modeling, 3D-toxicophore mapping and in silico screening of in vitro basal cytotoxicity of diverse organic chemicals. Toxicol In Vitro. 2013;27:597–608. doi: 10.1016/j.tiv.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Serafim MSM, Kronenberger T, Oliveira PR, Poso A, Honório KM, Mota BEF, Maltarollo VG. The application of machine learning techniques to innovative antibacterial discovery and development. Expert Opin Drug Discov. 2020 doi: 10.1080/17460441.2020.1776696. [DOI] [PubMed] [Google Scholar]
- 30.Kar S, Deeb O, Roy K. Development of classification and regression based QSAR models to predict rodent carcinogenic potency using oral slope factor. Ecotoxicol Environ Saf. 2012;82:85–95. doi: 10.1016/j.ecoenv.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Kang Y-L, Zhu Y-Y, Zhao K-X, Liang J-Y, Ding L, Zhang T-G, Zhang J. Novel naïve Bayes classification models for predicting the chemical ames mutagenicity. Toxicol In Vitro. 2017;41:56–63. doi: 10.1016/j.tiv.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 32.Xia X, Maliski EG, Gallant P, Rogers D. Classification of kinase inhibitors using a bayesian model. J Med Chem. 2004;47:4463–4470. doi: 10.1021/jm0303195. [DOI] [PubMed] [Google Scholar]
- 33.Liu L-L, Lu J, Lu Y, Zheng M-Y, Luo X-M, Zhu W-L, Jiang H-L, Chen K-X. Novel Bayesian classification models for predicting compounds blocking hERG potassium channels. Acta Pharmacol Sin. 2014;35:1093–1102. doi: 10.1038/aps.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biovia DS. Discovery studio. San Diego: Biovia; 2016. [Google Scholar]
- 35.David R, Mathew H. Extended-connectivity fingerprints. J Chem Inf Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 36.Fawcett T. An introduction to ROC analysis. Pattern Recog Lett. 2006;27:861–874. doi: 10.1016/j.patrec.2005.10.010. [DOI] [Google Scholar]
- 37.Yap CW. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem. 2011;32:1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- 38.The simple, user-friendly and reliable online standalone tools, http://dtclab.webs.com/software-tools/. Accessed 10 Nov 2020
- 39.Ambure P, Aher RB, Gajewicz A, Puzyn T, Roy K. “NanoBRIDGES” software: open access tools to perform QSAR and nano-QSAR modelling. Chemometr Intell Lab Syst. 2015;147:1–3. doi: 10.1016/j.chemolab.2015.07.007. [DOI] [Google Scholar]
- 40.Trott O, Olson AJ. AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 42.Kumari R, Kumar R. Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 43.Yousefinejad S, Hemmateenejad B. Chemometrics tools in QSAR/QSPR studies: a historical perspective. ChemometrIntell Lab Syst. 2015;149:177–204. doi: 10.1016/j.chemolab.2015.06.016. [DOI] [Google Scholar]
- 44.Kar S, Sanderson H, Roy K, Benfenati E, Leszczynski J. Ecotoxicological assessment of pharmaceuticals and personal care products using predictive toxicology approaches. Green Chem. 2020;22:1458–1516. doi: 10.1039/C9GC03265G. [DOI] [Google Scholar]
- 45.Polishchuk P. Interpretation of quantitative structure-activity relationship models: past, present, and future. J Chem Inf Model. 2017;57:2618–2639. doi: 10.1021/acs.jcim.7b00274. [DOI] [PubMed] [Google Scholar]
- 46.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:2–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 47.Veber DF, Johnson SR, Cheng H-Y, Smith BR, WardKW Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 48.Drug Likeness Tool (DruLiTo), software. http://www.niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_index.html. Accessed 16 Aug 2020
- 49.Toropov AA, Toropova AP, Veselinović AM, Leszczynska D, Leszczynski J. SARS-CoV Mpro inhibitory activity of aromatic disulfide compounds: qSAR model. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1818627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De P, Bhayye S, Kumar V, Roy K. In silico modeling for quick prediction of inhibitory activity against 3CLpro enzyme in SARS CoV diseases. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1821779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan PM, Kumar V, Roy K. In silico modeling of small molecule carboxamides as inhibitors of SARS-CoV 3CL protease: an approach towards combating COVID-19. Comb Chem High Throughput Screen. 2020;23:1–19. doi: 10.2174/1386207323666200120114645. [DOI] [PubMed] [Google Scholar]
- 52.Kumar V, Roy K. Development of a simple, interpretable and easily transferable QSAR model for quick screening antiviral databases in search of novel 3C-like protease (3CLpro) enzyme inhibitors against SARS-CoV diseases. SAR QSAR Environ Res. 2020;31:511–526. doi: 10.1080/1062936X.2020.1776388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.