

Abstract
Small‐cell lung cancer (SCLC) occurs infrequently in never/former light smokers. We sought to study this rare clinical subset through next‐generation sequencing (NGS) and by characterizing a representative patient‐derived model. We performed targeted NGS, as well as comprehensive pathological evaluation, in 11 never/former light smokers with clinically diagnosed SCLC. We established a patient‐derived model from one such patient (DFCI168) harboring an NRAS Q61K mutation and characterized the sensitivity of this model to MEK and TORC1/2 inhibitors. Despite the clinical diagnosis of SCLC, the majority (8/11) of cases were either of nonpulmonary origin or of mixed histology and included atypical carcinoid (n = 1), mixed non‐small‐cell lung carcinoma and SCLC (n = 4), unspecified poorly differentiated carcinoma (n = 1), or small‐cell carcinoma from different origins (n = 2). RB1 and TP53 mutations were found in four and five cases, respectively. Predicted driver mutations were detected in EGFR (n = 2), NRAS (n = 1), KRAS (n = 1), BRCA1 (n = 1), and ATM (n = 1), and one case harbored a TMPRSS2‐ERG fusion. DFCI168 (NRAS Q61K) exhibited marked sensitivity to MEK inhibitors in vitro and in vivo. The combination of MEK and mTORC1/2 inhibitors synergized to prevent compensatory mTOR activation, resulting in prolonged growth inhibition in this model and in three other NRAS mutant lung cancer cell lines. SCLC in never/former light smokers is rare and is potentially a distinct disease entity comprised of oncogenic driver mutation‐harboring carcinomas morphologically and/or clinically mimicking SCLC. Comprehensive pathologic review integrated with genomic profiling is critical in refining the diagnosis and in identifying potential therapeutic options.
Keywords: combination therapy, lung cancers in never smokers, non‐neuroendocrine SCLC, RAS mutation, small‐cell lung cancer
Small‐cell lung cancer occurs infrequently in never/former light smokers. We performed targeted NGS, as well as comprehensive pathological evaluation, in 11 never/former light smokers with clinically diagnosed SCLC. The majority of cases were either of nonpulmonary origin or of mixed histology. Predicted driver mutations were detected in EGFR, NRAS, KRAS, BRCA1, and ATM, and one case harbored a TMPRSS2‐ERG fusion.
Abbreviations
- EGFR
epidermal growth factor receptor
- NE
neuroendocrine
- NSCLC
non‐small‐cell lung cancer
- PDX
patient‐derived xenografts
- SCLC
small‐cell lung cancer
1. Introduction
Small‐cell lung cancer (SCLC) is one of the most challenging cancers to treat, with a 5‐year survival rate of 4–5% (Harris et al., 2012). The standard therapy regimen for SCLC consists of platinum doublet chemotherapy, which has not changed over decades (Rudin et al., 2019). SCLC is known to be strongly correlated with tobacco consumption; however, 2–3% of SCLC patients are reported to be never smokers (Ou et al., 2009; Varghese et al., 2014). Several studies have suggested that SCLC arising in never smokers includes patients with potentially actionable molecular aberrations (Sun et al., 2015; Varghese et al., 2014), yet optimal treatment modalities in this group have not been established due to the rarity of the cases, thus highlighting the urgent need to examine this possible biologically distinct subtype of SCLC to find novel therapeutic approaches.
Small‐cell lung cancer tumors consist of small, round‐shaped cells with a scant cytoplasm, fine granular nuclear chromatin, and frequent nuclear molding (Travis, 2012). The morphological characteristics of SCLC are distinct, and the diagnosis is mainly based on histological features. However, morphologic overlap with other entities, including large‐cell neuroendocrine (NE) carcinoma, basaloid squamous cell carcinoma, and ‘small round blue cell tumors’ including lymphoma, poorly differentiated melanoma, and sarcomas may contribute to diagnostic error and justifies the use of immunohistochemistry (IHC) to confirm the diagnosis. NE markers such as neural cell adhesion molecule (NCAM) (CD56), chromogranin A, synaptophysin, and INSM1 are characteristically expressed in SCLC but are not entirely specific. Recently, several researchers proposed that SCLC without classical NE markers expression can be defined by differential expression of YAP1 (McColl et al., 2017) and POU2F3 (Huang et al., 2018), emphasizing the biological heterogeneity of this morphologic entity and suggesting a need for more comprehensive tumor profiling to better understand this heterogeneous disease.
Concomitant inactivation of RB1 and TP53 is nearly universal in SCLC (George et al., 2015). Known and suspected driver mutations have also been detected in several genes including in PTEN, SLIT2, EPHA7, FGFR1, BRAF, KIT, PIK3CA,CREBBP, EP300, and MLL (George et al., 2015; Peifer et al., 2012). However, the low frequency of clinically actionable driver mutations hinders the successful application of targeted therapies in SCLC. A difference in the mutational profile in SCLC according to the smoking status has also been reported (Cardona et al., 2019; Sun et al., 2015; Varghese et al., 2014). One study by Cardona et al. demonstrated that EGFR, MET, and SMAD4 are more frequently mutated in never smokers, while RB1, CDKN2A, and CEBPA are more frequent in smokers (Cardona et al., 2019).
NRAS, together with KRAS and HRAS, belongs to the RAS oncogene family and encodes a highly conserved small GTPase which regulates cell growth, proliferation, and differentiation (Shimizu et al., 1983). NRAS is mutated in roughly 1% of lung cancers (Ding et al., 2008; Ohashi et al., 2013). To date, no case of SCLC driven by an oncogenic NRAS mutant has been reported on, possibly due to the lack of routine genomic profiling of patients with SCLC. Currently, SW1271 is the only commercially available SCLC cell line with an NRAS‐activating mutation. SCLC harboring HRAS and KRAS mutations is also very uncommon (Rudin et al., 2012; Varghese et al., 2014). The rarity of these SCLC genotypes translates to a poor understanding of its biology and contributes to an ongoing debate on whether SCLC patients with RAF‐MEK‐ERK pathway activation can benefit from targeted therapies (Cristea and Sage, 2016).
In the current study, we evaluate a series of clinically diagnosed pulmonary SCLCs in never/light smokers and demonstrate the potential utility of combined MEK/mTORC1/2 inhibition in an exemplary case with NRAS Q61K mutation. We demonstrate that careful genetic, morphologic, and in vitro characterization highlights the diagnostic ambiguity of clinical SCLC and the essential role for tumor genomic profiling in SCLC arising in this rare clinical context.
2. Materials and Methods
2.1. Patients
Tumor biopsies from 19 treatment naïve SCLC patients who were either never smokers (n = 11) or light former smokers (≤ 10 pack‐years, n = 8) were analyzed for NRAS mutations alone by Sanger sequencing (n = 8) or comprehensively by targeted next‐generation sequencing (n = 11; OncoPanel (Garcia et al., 2017)). All patients provided written informed consent for the analysis of their clinical specimens, and the studies were approved by the Institutional Review Board at Dana‐Farber Cancer Institute (DFCI). The study methodologies conformed to the standards set by the Declaration of Helsinki.
2.2. Cell cultures and Reagents
SW1271, H1299, H2087, H2347, H69, H82, H209, and Glc16 were purchased from American Type Culture Collection (ATCC). All cell lines except SW1271 were maintained in Roswell Park Memorial Institute (RPMI)‐1640 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS, 100 U·mL−1 penicillin, and 100 µg·mL−1 streptomycin (Gibco, Thermo Fisher Scientific). SW1271 was grown in Dulbecco’s modified Eagle medium (Gibco, Thermo Fisher Scientific) with 10% FBS. Trametinib (Yamaguchi et al., 2011), selumetinib (Davis et al., 2011), INK128 (Hsieh et al., 2012), AZD8055 (Chresta et al., 2010), PI103 (Raynaud et al., 2009), BKM120 (Burger et al., 2011), ZSTK474 (Kong and Yamori, 2007), and MK2206 (Hirai et al., 2010) were purchased from Selleck Chemicals (Houston, TX, USA). Torin2 was synthesized using previously published methods (Liu et al., 2013). Stock solutions of all drugs were prepared at 10 mm in DMSO (Sigma‐Aldrich, St. Louis, MO, USA) and stored at −80 °C. Cell lines were authenticated by single tandem repeat analysis at Michigan State University in October 2017 and tested negative for mycoplasma as determined by the Mycoplasma Plus PCR Primer Set (Agilent Technologies, Santa Clara, CA, USA).
2.3. Generation of a patient‐derived cell line (DFCI168)
A pleural effusion was obtained from a patient with SCLC. The sample was subjected to red blood cell lysis using RBC Lysis Buffer (Boston BioProducts, Ashland, MA, USA), and the cells were suspended in HITES medium (Simms et al., 1980) with 10% FBS. We carefully monitored the appearance of the cells during serial passage to minimize the loss of intratumor heterogeneity. Nevertheless, no floating cell aggregates which are the typical morphology of SCLC cell lines were detected throughout the course of cell line creation. Due to the unique cancer cell morphology resembling fibroblasts, the NCAM marker was used to positively select cancer cells using MACS columns (Miltenyi Biotechnology, Auburn, CA, USA). NCAM‐positive cancer cell population was subsequently cloned by limited serial dilution. The established cell line DFCI168 was maintained in RPMI‐1640 media supplemented with 10% FBS.
2.4. Cell proliferation and Growth assays, combination index
Inhibition of growth by targeted kinase inhibitors was evaluated by MTS assay according to the manufacturer’s instructions (Promega, Madison, WI, USA). Cells were plated in 96‐well plates at a density of 2000–8000 per well and treated on the following day. At 72 h after drug addition, cell viability was measured. Combination index (CI) was calculated using compusyn software (Combo Syn, Inc,. Paramus, NJ, USA). For NRAS siRNA knockdown experiments, cell viability was measured using CellTiter‐Glo luminescent assay (Promega).
2.5. Patient‐derived xenograft (PDX) establishment
To establish the DFCI168 PDX model, ~ 5 × 106 cells from pleural effusion were implanted subcutaneously in 8‐week‐old female NSG mice in accordance with the guidelines approved by the DFCI Institutional Animal Care and Use Committee (IACUC) and tumor growth was monitored by caliper measurements. Once tumors grew to a size of 1 cm3, tumors were isolated and cut into pieces of ~ 2 × 2 × 2 mm and transplanted subcutaneously in additional NSG mice. Tumors were passaged for no more than five times. Samples from all passages were viably frozen in liquid nitrogen and used for further experiments. The tumor fidelity from various passages was confirmed by Hematoxylin and eosin (H&E) staining and the existence of the NRAS Q61K mutation.
2.6. Antitumor activity in vivo
All in vivo studies were conducted at DFCI with the approval of the Institutional Animal Care and Use Committee in an AAALAC‐accredited vivarium. The DFCI168 tumor fragments were transplanted subcutaneously on the right flanks of 8‐week‐old female NSG mice. Tumors were allowed to establish to 219 ± 54 mm3 in size before randomization into vehicle‐treated, trametinib‐treated (3 mg·kg−1, PO qd), torin2‐treated (30 mg·kg−1, PO qd), and combo‐treated groups (3 and 30 mg·kg−1, respectively) of 8 mice per group. Trametinib was administered in 0.5% hydroxypropyl methyl cellulose with 0.2% Tween 80. Torin2 was dissolved in captisol (1 : 40) followed by dilution in water to 3 mg·mL−1. Tumor volumes were determined from caliper measurements by using the formula V = (length × width2)/2. Tumor sizes and body weights were measured twice weekly. For pharmacodynamic (PD) study, mice were treated for 2 days and tumor samples were collected at 4 h (n = 3) after the last dose. Tumor samples were analyzed by western blot.
2.7. Flow cytometry experiments
NCAM/epithelial cell adhesion molecule (EpCAM) expression in the DFCI168 cells was evaluated with anti‐human NCAM APC antibody/anti‐human EpCAM‐PE antibody or with fluorophore‐tagged isotype control (eBioscience, Thermo Fisher Scientific). Early and late apoptotic cell death was assessed by Annexin V‐FITC/PI (Life Technologies, Thermo Fisher Scientific) or Annexin V‐PE/7AAD staining (BD Biosciences) according to the manufacturer’s protocol. The samples were analyzed with a BD LSR Fortessa flow cytometer with BD facsdiva software (BD Biosciences, Billerica, MA, USA).
2.8. Immunofluorescence staining
The DFCI168 cells were plated in 8‐well chamber slides at a concentration of 3 × 104 cells per well. Two days after plating, cells were fixed with 4% paraformaldehyde for 10 min, followed by permeabilization with 0.5% Triton X‐100 for 10 min. After blocking with 10% goat serum for 1 h, the cells were stained with Phalloidin Green 488 (1 : 500; BioLegend, San Diego, CA, USA). For vimentin staining, the following antibodies and dilutions were used: rabbit anti‐vimentin (1 : 1000; Abcam, Cambridge, MA, USA) and goat anti‐rabbit Alexa Fluor 594 (1 : 1000; Invitrogen, Thermo Fisher Scientific). Slides were mounted with antifade mounting media containing DAPI. Images were obtained using the Nikon Eclipse 80i microscope (Nikon, Melville, NY, USA).
2.9. Sanger sequence and OncoPanel
The OncoPanel assay surveys exonic DNA sequences of 275 cancer genes and 91 introns across 30 genes for rearrangement detection. DNA was isolated from NRAS mutant cell lines using DNeasy Blood & Tissue Kit (Qiagen, Germantown, MD, USA) and analyzed by massively parallel sequencing using a solution‐phase Agilent SureSelect hybrid capture kit and an Illumina HiSeq 2500 sequencer. Further details were described elsewhere (Sholl et al., 2016).
2.10. Plasmid construction and viral infection
pBabe NRAS Q61K plasmid was a gift from Channing Der (Addgene plasmid # 12543) (Khosravi‐Far et al., 1996). pBabe NRAS Q61K plasmid or pBabe puro empty plasmid and a packaging plasmid pAmpho were co‐transfected into HEK293T cells. Viral supernatants were harvested at 48 h, filtered through 0.45‐µm filter, and spinoculation was performed by spinning at 1000 g for 90 min. After 48 h, cells were selected in puromycin (1 µg·mL−1).
2.11. Antibodies and immunoblotting
Cells were grown and treated as described and lysed with RIPA buffer with Triton X‐100 (Boston BioProducts) supplemented with protease and phosphatase inhibitors. Immunoblotting was performed according to the antibody manufacturers’ recommendations. The following antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA): Anti‐Synaptophysin (SYP), phospho‐ERK1/2 (Thr202/Tyr204), ERK1/2, phospho‐RSK (Thr359/Ser363), RSK, phospho‐S6 (Ser235/236), phospho‐S6 (Ser240/244), S6, phospho‐4EBP (Thr37/46), 4EBP, phospho‐Akt (Ser473), Akt, Hsp90, phospho‐RB (Ser780), phospho‐RB (Ser807/811), RB, Vimentin, ZEB1, Snail, and Lamin B1. Anti‐ASCL1 antibody was purchased from BD Biosciences. Anti‐tubulin antibody is purchased from Sigma‐Aldrich. Anti‐NRAS antibody is purchased from Abcam.
2.12. siRNA experiments
Cells were transfected with 25 nm of control siRNA pool (D‐001206‐13‐05) or with NRAS specific siRNA pool (Dharmacon, Horizon Discovery, Lafayette, CO, USA). After 48 h, cell viability was measured by CellTiter‐Glo. NRAS silencing efficiency was verified by immunoblotting.
2.13. RNA isolation and quantitative real‐time PCR (qPCR)
RNA was extracted using TRIzol (Invitrogen) and column‐purified using RNeasy Kit (Qiagen). cDNA was generated from 1 µg of RNA using QuantiTect Reverse Transcription Kit (Qiagen). qPCR was performed and analyzed on Step One Plus Real‐time PCR System using TaqMan probes (Applied Biosystems, Thermo Fisher Scientific) for E‐cadherin (Hs01023894), vimentin (Hs00958111), SNAI1 (Hs00195591), TWIST (Hs00361186), ZEB1 (Hs00232783), ASCL1 (Hs00269932), and SYP (Hs00300531). GUSB was used as a housekeeping gene.
3. Results
3.1. Pathologic reassessment and genomic testing of clinically diagnosed SCLC patients who are never and light former smokers (≤ 10 pack‐years)
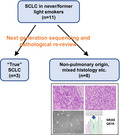
We identified 11 cases of never (n = 7) or light former (n = 4) smokers with clinically diagnosed SCLC treated at DFCI from 2013 to 2018 (Table 1). Mutational analysis was performed using the targeted next‐generation sequencing (NGS) profiling platform, OncoPanel (Garcia et al., 2017) (Tables 1 and S1). Four cases harbored mutations in both RB1 and TP53, and one had a mutation only in TP53. Among RB1 wild‐type (WT) cases (n = 7), two cases were positive for EGFR mutations (case 8: L858R and L861F, case 10: L858R), and the others harbored NRAS Q61K (n = 1), BRCA1 L502Afs*2 (n = 1), and ATM T1953I (n = 1) and high MET amplification (n = 1). In RB1 mutant cases (n = 4), KRAS G12V (n = 1) and TMPRSS2‐ERG fusion (n = 1) were detected. An additional eight patients with SCLC (never smokers (n = 4) or light former smokers (n = 4)) with only limited DNA were specifically queried for an NRAS mutation, but no NRAS mutations were detected (data not shown).
Table 1.
Clinicopathologic characteristics of 11 clinically diagnosed never/light smokers with SCLC. LCNEC, large‐cell neuroendocrine cancer; mt, mutant; ND, not done.
| Patient number | Age | Sex | Smoking (pack‐years) | Pathology diagnosis | RB1/TP53 status | RB protein expression | Myc status |
|---|---|---|---|---|---|---|---|
| 1 | 68 | F | 0 | Combined LCNEC and SCLC | RB1 WT/TP53 WT | ND | |
| 2 | 50 | F | 0 | Combined LCNEC and SCLC | RB1 WT/TP53 WT | Intact | |
| 3 | 60 | M | 0 | Neuroendocrine tumor grade 2/atypical carcinoid, pancreas, or lung primary | RB1 WT/TP53 WT | Intact | |
| 4 | 52 | F | 0 | Small cell of unknown primary hepatobiliary origin suspected | RB1 mt/TP53 mt | ND | Low copy number gain |
| 5 | 70 | M | 0 | SCLC | RB1 mt/TP53 mt | ND | Low copy number gain |
| 6 | 59 | F | 0 | SCLC | RB1 WT/TP53 WT | ND | |
| 7 | 50 | M | 0 | Poorly differentiated carcinoma | RB1 WT/TP53 WT | Intact | |
| 8 | 47 | F | 2 | NSCLC undergoing de novo SCLC transformation | RB1 WT/TP53 WT | ND | |
| 9 | 51 | F | 4 | SCLC | RB1 mt/TP53 mt | ND | Myc, MycL gain |
| 10 | 62 | F | 7 | NSCLC undergoing de novo SCLC transformation | RB1 WT/TP53 mt | ND | |
| 11 | 76 | M | 10 | Metastatic prostate cancer, with small‐cell transformation | RB1 mt/TP53 mt | ND | Myc gain |
Due to the identification of mutations infrequently found in typical SCLC patients, microscopic features were retrospectively re‐evaluated. The review of two cases harboring EGFR mutations (case 8 and case 10) showed admixed non‐small‐cell and small‐cell features, suggestive of de novo small‐cell transformation of EGFR‐mutated adenocarcinomas. Case 1 with an ATM mutation and case 2 with a MET amplification showed features intermediate between large‐cell NE and small‐cell carcinomas. Case 7 with a NRAS mutation was verified to be a keratin‐positive small round blue cell tumor but lacked any specific features to confirm a diagnosis of SCLC. A BRCA1 mutation‐harboring case 3 was reclassified as grade 2 NE tumor/atypical carcinoid of possible pancreas or lung primary in the available clinical and radiographic context. Among the five cases with classic small‐cell carcinoma histology, case 4 with KRAS G12V was suspected to be of possibly of hepatobiliary origin. Case 11 harbored a TMPRSS2‐ERG fusion, an oncogenic fusion common in prostate cancers (Attard et al., 2008; Demichelis et al., 2007) without any reported occurrences in lung cancer. Given the patient’s prior history of prostate carcinoma, this case was reclassified as prostate cancer with small‐cell transformation. Thus, following pathological re‐review, 3/11 (27%) cases remained as SCLC. Of these, two cases had mutations in both TP53 and RB1 (Table 1).
3.2. Identification of NRASQ61K mutation in clinically diagnosed SCLC and establishment of patient‐derived cancer models
We next developed both in vitro and in vivo models from patient 7 (Table 1) harboring a NRAS somatic mutation at codon 61 (Q61K). The patient is a 49‐year‐old male who presented with a large right hilar mass (Fig. S1). Although he had no history of cigarette smoking, he had a history of second‐hand smoking from both parents who were heavy smokers. The histology of the endobronchial biopsy demonstrated morphological features of SCLC, presenting small‐sized cells with a high nuclear‐to‐cytoplasmic ratio, nuclear molding, and frequent single‐cell apoptosis (CK7+, NCAM+, SYP−, Chromogranin−) (Fig. 1A, left, and data not shown). The lack of the expression of some common NE markers (SYP, Chromogranin) together with the unusual genomic features suggests that the tumors of this patient do not possess the features of the classic type of SCLC.
Fig. 1.
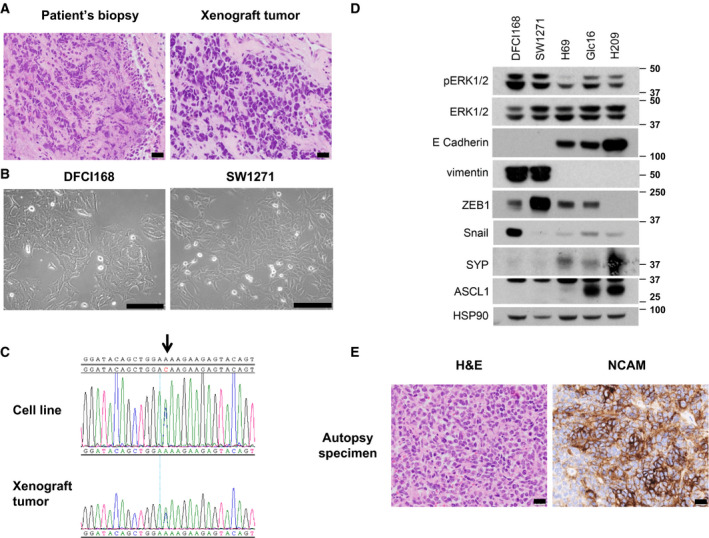
DFCI168 harbors unique non‐NE features. (A) H&E staining (20×) of patient tumor (left) and mouse xenograft tumor derived from pleural effusion from the patient (right). Scale bar, 20 µm (B) Phase‐contrast images of DFCI168 and SW1271. Scale bar, 100 µm (C) Nucleotide sequence tracings showing NRAS Q61K mutation (indicated by arrow) in both the DFCI168 cell line and the xenograft tumor. (D) Western blot analysis of phospho‐ERK1/2, ERK1/2, Vimentin, and NE markers (ASCL1 and SYP) and Hsp90 (loading control). (E) H&E staining (left, 20×) and IHC for NCAM (right, 20×) of autopsy specimens from the patient. Scale bar, 20 µm.
With a clinical diagnosis of limited‐stage disease SCLC, this patient was treated with concurrent chemoradiation therapy, which led to a complete response. Despite the good response, he developed widespread metastatic disease just several months after chemoradiotherapy and could not receive any further systemic treatment due to a declining clinical condition.
A week prior to the patient’s passing, pleural effusion was obtained and established into a cell line, DFCI168. To investigate the characteristics of NRAS mutant SCLC, a PDX model was also established by subcutaneous implantation of cancer cells from the same pleural effusion. The tumor xenografts retained morphological features of SCLC (Fig. 1A, right). The xenograft mice developed spontaneous lung metastases, mimicking the aggressive biology of the patient’s tumor.
Unlike most SCLC cell lines that grow in culture as floating aggregates, DFCI168 grows in a monolayer and displays a spindle‐shaped morphology (Fig. 1B, left). We next assessed SW1271, a commercially available SCLC cell line with a NRAS Q61R mutation. The morphological features of SW1271 were similar and consistent with DFCI168 (Fig. 1B, right). TP53 was mutated (p. C277F), but no RB1 mutation was detected by OncoPanel in SW1271. The NRAS Q61K mutation was confirmed in both the xenograft tumor and the DFCI168 cell line by Sanger sequencing (Fig. 1C). Flow cytometric analysis revealed that DFCI168 was EpCAM‐negative and NCAM‐positive (Fig. S2A). Consistent with the absence of mutations in RB1, RB1 protein expression was maintained in DFCI168 and SW1271 (Fig. S2B). DFCI168 and SW1271 express vimentin and either ZEB1 or Snail, transcription factors known to regulate EMT with concomitant downregulation of E‐cadherin, while the expression of NE markers (SYP and ASCL1) is almost undetectable (Figs 1D and S2D). Due to the genotypic and phenotypic discrepancy between DFCI168 and classic SCLC, we carefully examined the histology of autopsy‐retrieved specimens from this patient. Although the tissue demonstrated patchy‐positive NCAM staining (Fig. 1E, right), the autopsy material showed increased cytoplasm as compared to the original biopsy, striking nuclear pleomorphism and distinct nucleoli (Fig. 1E, left), which do not represent the typical histological characteristics of SCLC. Architectural features of large‐cell NE carcinoma were also lacking. We further investigated the possibility of an alternative diagnosis including either a rhabdomyosarcoma or a melanoma, both of which have recurrent NRAS mutations, but the tissue was negative for all markers specific to these tumor types (SOX10, S100, myf4, and desmin). At autopsy, this tumor was best classified as an undifferentiated carcinoma with NE differentiation by IHC, presenting clinically and being treated as SCLC.
3.3. DFCI168 cells depend on NRAS signaling for survival and show high sensitivity to MEK inhibition
Next, we sought to determine whether the DFCI168 cells depend on the NRAS pathway for their survival using siRNA‐mediated NRAS knockdown (Fig. 2A, left). NRAS downregulation resulted in a significant decrease in cell viability of the DFCI168 cells (P < 0.0001, Fig. 2A, right). We further assessed the sensitivity of DFCI168 and SW1271 to MEK inhibitors and compared them to NRAS WT SCLC cell lines (H82, H209, Glc16). Both trametinib and selumetinib showed selective potency in NRAS mutant cell lines (IC50 in DFCI168: trametinib 2 nm; selumetinib 50 nm), but not in NRAS WT SCLC cell lines (Fig. 2B), despite ERK phosphorylation being significantly suppressed upon MEK inhibition in both SCLC subtypes (Fig. 2C). To verify the apoptotic effects of MEK inhibition on DFCI168 cells, we performed flow cytometry analysis of Annexin V and 7AAD staining. Treatment with 1 µm of selumetinib or trametinib for 48 h significantly increased apoptotic cell population as compared to DMSO. (P = 0.001; one‐way ANOVA) (Fig. 2D).
Fig. 2.
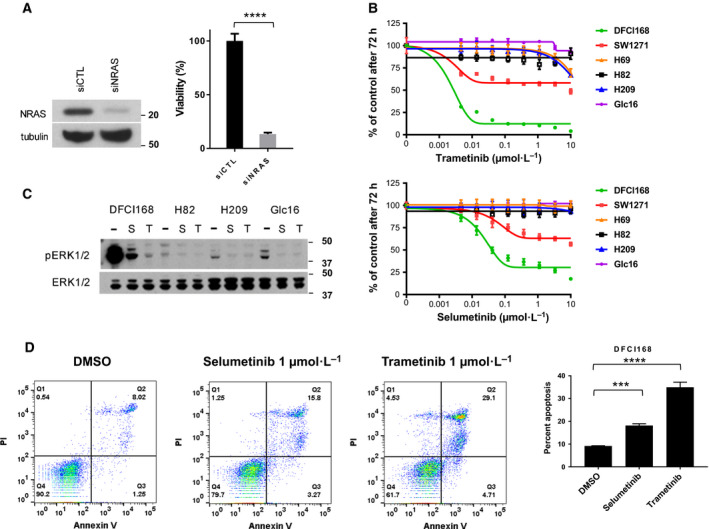
DFCI168 depends on NRAS signaling for survival. (A) DFCI168 cells were transfected with siRNA pool directed against NRAS or with control siRNA pool. The efficiency of NRAS knockdown was confirmed by western blot after 48 h post‐transfection. The cell viability was assessed by CellTiter‐Glo (n = 5, error bars represent the SD of the mean) at 48 h post‐transfection. Significance was assessed by unpaired t‐test. ****P < 0.0001. (B) The SCLC cell lines were treated with increasing doses of MEK inhibitors (trametinib or selumetinib), and the cell viability was assessed by MTS after 72 h (n = 6, mean ± SD). (C) The SCLC cell lines were treated with 1 µm selumetinib (S) or trametinib (T) 100 nm for 6 h, and then, ERK1/2 phosphorylation was evaluated by western blotting. (D) DFCI168 was treated with 1 µm selumetinib or 1 µm trametinib for 48 h. The cells were stained with Annexin V‐PE/7‐AAD (7‐aminoactinomycin D) and analyzed by flow cytometry. The lower right quadrant shows early apoptotic cells, and the upper right quadrant represents late apoptotic cells. The numbers in the graph represent the percentage of cell numbers in each quadrant. Bar graphs represent the mean of percentage from three independent experiments. Significance was assessed by one‐way ANOVA. ***P < 0.001, ****P < 0.0001.
3.4. The diversity in sensitivity to MEK inhibition in NRAS mutant lung cancer cell lines
To further investigate the dependence of lung cancer harboring the NRAS Q61 mutation on MEK signaling, we tested three NRAS mutant non‐small‐cell lung cancer (NSCLC) lines (H1299:NRAS Q61K, large‐cell carcinoma; H2087:NRAS Q61K, adenocarcinoma; H2347:NRAS Q61R, adenocarcinoma) for sensitivity to MEK inhibitors. H1299 showed resistance (IC50 > 10 µm) to trametinib and selumetinib in the cell viability assay (Fig. 3A), which may be attributed to its dependence on PI3K/Akt signaling for survival due to a PTEN promoter methylation (Soria et al., 2002).
Fig. 3.
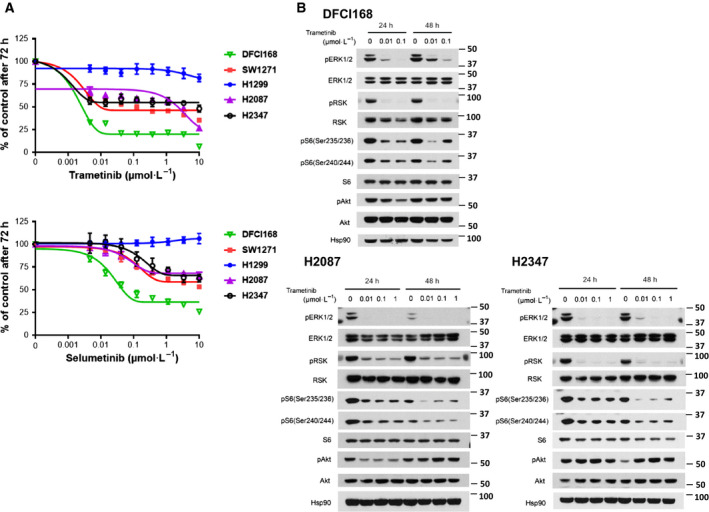
Heterogeneity in response to MEK inhibitors in NRAS mutant lung cancer cell lines. (A) The NRAS mutant lung cancer cell lines were treated with increasing doses of MEK inhibitors (trametinib and selumetinib). The cell viability was assessed by MTS after 72 h. (B) DFCI168, H2087, and H2347 were treated with trametinib at the indicated concentration for the indicated times. The cell extracts were immunoblotted using the indicated antibodies.
H2087 and H2347 showed limited sensitivity to MEK inhibition similar to SW1271, with > 30% of the cells still viable at 10 µm of MEK inhibitor treatment. To elucidate the mechanism underlying the difference in sensitivity to MEK inhibition between the NRAS mutant cell lines, we investigated the downstream effectors of RAS, Akt/mTOR, and ERK. The basal level of p‐ERK varied across the tested NRAS mutant cell lines, exhibiting no obvious trend to predict MEK inhibitor sensitivity. Although high p‐Akt expression is a reported predictor of resistance to MEK inhibition in melanoma patients (Atefi et al., 2011; Catalanotti et al., 2013; Gopal et al., 2010), the p‐Akt level was highest in DFCI168, suggesting that sensitivity to MEK inhibitor treatment does not correlate with the level of p‐Akt in the cell lines we tested (Fig. S2C).
3.5. The combination of MEK and mTORC1/2 inhibitors synergizes to sustain growth inhibition of NRAS mutant lung cancer cells
In order to identify signaling pathways that may compensate for MEK inhibition, and as such explain the diversity of responses to single‐agent MEK inhibition, we analyzed PI3K/Akt and mTOR signaling in the NRAS mutant NSCLC cell lines in the presence of a MEK inhibitor. S6 phosphorylation, especially at residues 240/244, serves as a marker of mTORC1 signaling (Elkabets et al., 2013; Roux et al., 2007). In all five cell lines, Akt and S6 activation persisted during MEK inhibitor treatment (Figs 3B and S3). In DFCI168 and H1299, the levels of phosphorylated S6 increased with higher concentrations of trametinib (100 vs. 10 nm in DFCI168, 1 µm vs. 10 and 100 nm in H1299) (Figs 3B and S3), suggesting feedback regulation between MEK/ERK and mTOR pathways without affecting the p‐Akt levels. These results prompted us to explore whether inhibition of PI3K/Akt/mTOR signaling can enhance cytotoxicity caused by MEK inhibition. To test this, we evaluated the effects of the dual PI3K/mTOR inhibitor (PI103), pan‐class I PI3K inhibitors (BKM120, ZSTK474), Akt inhibitor (MK2206), and mTORC1/2 inhibitors (INK128, torin2, AZD8055) as single agents. Among those tested, mTORC1/2 inhibitors were the most potent against NRAS mutant cell lines (Fig. S4). Importantly, all the NRAS mutant cell lines tested showed substantial sensitivity to single‐agent mTORC1/2 inhibitors (IC50 < 1 µm). We also confirmed that the combination of 100 nm of trametinib and torin2 abrogates the phosphorylation of S6 in DFCI168 and H2087 (Fig. S5).
We further investigated the synergistic effects of the combination of MEK and mTORC1/2 inhibitors. MTS‐based cell viability assays were performed after a 72‐h treatment with trametinib and torin2, either alone or in combination at a constant ratio of 1 : 1 (Fig. 4A). No synergistic or additive effects of the combination treatment were observed in H1299, suggesting that this cell line depends solely on the PI3K/Akt/mTOR pathway for survival due to the known PTEN promoter methylation (Soria et al., 2002). Unexpectedly, single‐agent torin2 showed greater potency than trametinib alone in four out of five cell lines (Fig. 4A). We also performed CI analysis according to the Chou–Talalay equation to determine the synergistic relationship between the two drugs (Fig. 4B). The CI values represent synergistic effects with CI < 1, additive effects with CI = 1, and antagonistic effect with CI > 1. Treated with combinations at a fixed ratio of 1 : 1, CI values were < 1 for H2087, H2347, and SW127 both at ED50 and ED75, and for DFCI168 at ED75. No synergistic effect of the combination was observed in H1299 (Fig. 4B).
Fig. 4.
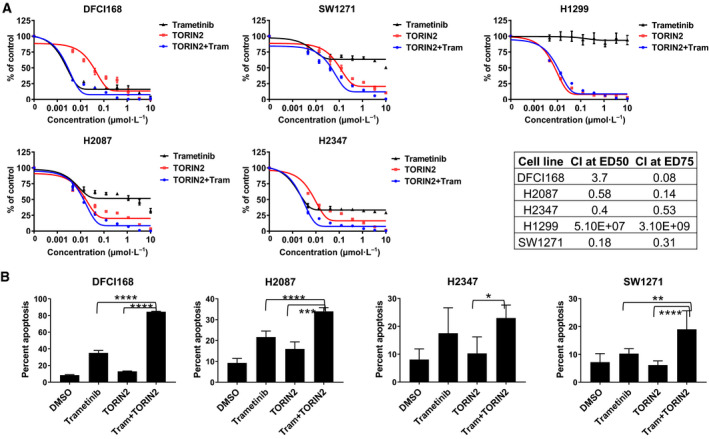
Evaluation of the combination of MEK and mTORC1/2 inhibitors on cell viability in NRAS mutant lung cancer cell lines. (A) The cell lines were treated with increasing doses of combination of trametinib and torin2 at the fixed ratio of 1 : 1. The cell viability was assessed by MTS after 72 h. The CI was calculated for 1 : 1 ratio of combined trametinib and torin2 and presented in the table. The CI values define synergism (CI < 1), additive effect (CI = 1), and antagonism (CI > 1), respectively. (B) The cell lines were treated for 48 h with 1 µm trametinib, 1 µm torin2, or the combination except for DFCI168 which was treated with 50 nm trametinib, 100 nm torin2, or the combination. The treated cells were stained either with Annexin V‐FITC/PI or with Annexin V‐PE/7AAD and analyzed by flow cytometry. Bar graphs represent the mean of percentage from three independent experiments. Significance was assessed by one‐way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To test whether the drugs induce apoptosis, the cell lines were treated with trametinib and torin2 for 48 h and stained with either FITC‐labeled Annexin V and PI‐ or PE‐labeled Annexin V and 7AAD (Fig. 4C). The combination significantly increased the apoptotic cell death compared with the single agents.
3.6. Combined treatment with MEK and mTORC1/2 inhibitors is effective in DFCI168 in vitro and in vivo
Given the good response to single‐agent MEK inhibitors in DFCI168 in vitro, we further evaluated the effect of trametinib in vivo using a PDX model from DFCI 168. The DFCI168 tumor‐bearing mice were treated with trametinib at 3 mg·kg−1 daily for 3 weeks. The tumor initially regressed significantly but grew back within 4 weeks after treatment was stopped, likely from the residual cells not eliminated by drug treatment (Fig. 5A). Next, we tested the in vivo efficacy of the combination of trametinib and torin2 in the DFCI168 PDX model. The mice were treated with trametinib, torin2, or the combination for 28 days (Fig. 5A). Even though all treatments lead to significant tumor regression, all tumors eventually regrew after treatment cessation. The tumor regrowth following the combination treatment withdrawal was significantly slower than the following single‐agent treatment withdrawal (P < 0.01). While trametinib at 3 mg·kg−1 was well‐tolerated, torin2 at 30 mg·kg−1 treatment required a drug holiday in three out of eight mice. The combination treatment resulted in > 15% body weight loss in four out of seven mice, also requiring drug holidays. To confirm on‐target inhibition of the combination treatment in this model, we conducted a 2‐day PD study (Fig. 5B). The combination of trametinib and torin2 significantly suppressed the downstream targets of both MEK and mTOR pathways, p‐ERK1/2 (T202/Y204) and p‐S6 (both S240/244 and S235/235), respectively.
Fig. 5.
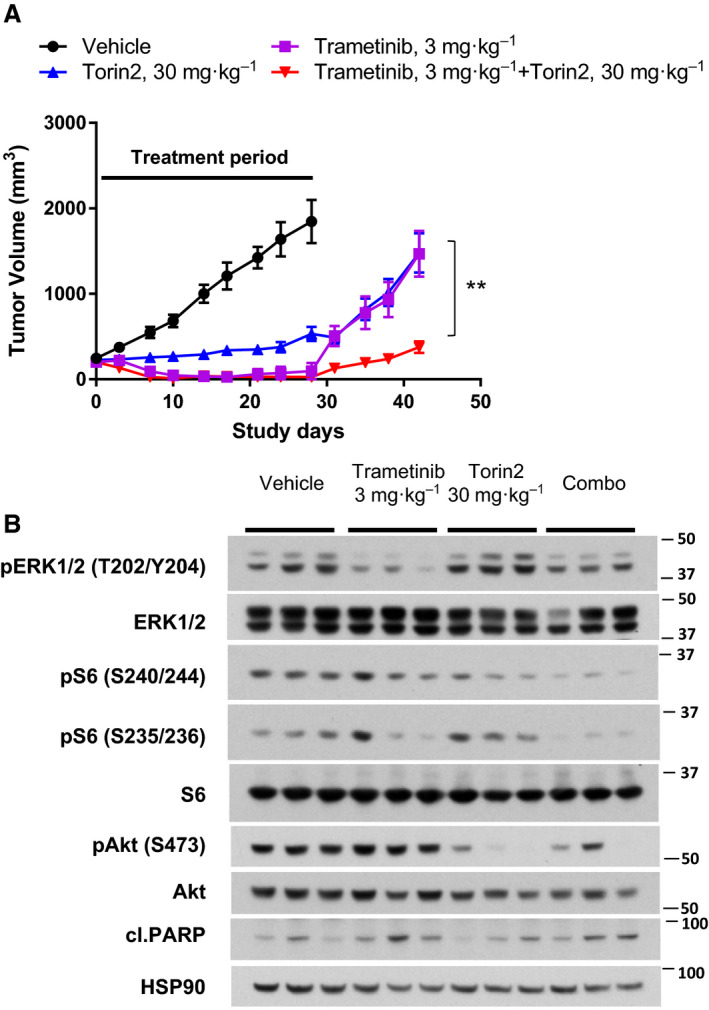
Combined treatment with trametinib and torin2 is effective in DFCI168 PDX tumors. (A) Tumor growth curves treated with vehicle (n = 8), trametinib (3 mg·kg−1, n = 8), torin2 (30 mg·kg−1, n = 8), and combination thereof (n = 7) with indicated 28 days of treatment period. Tumor volumes were recorded biweekly. Significance was assessed by one‐way ANOVA. **P < 0.01 comparing between single‐agent treatment and combination treatment. (B) The PD effect of trametinib and torin2. Mice were treated as indicated for 2 days, and tumor samples were collected at 4 h (n = 3) after the last dose.
3.7. Ectopic expression of NRASQ61K induces non‐NE phenotype
Given the similarity in non‐NE mesenchymal features between DFCI168 and SW1271, we hypothesized that the NRAS‐activating mutation might play a role in the acquisition of this unique phenotype. To address this hypothesis, we retrovirally transduced the classical SCLC cell line, Glc16, with NRAS Q61K. The NRAS Q61K‐expressing Glc16 derivative became adherent, outstretched, and acquired a large elongated morphology similar to that of DFCI168 and SW1271 (Fig. 6A). NRAS mutant protein overexpression and ERK1/2 activation were confirmed by western blotting (Fig. 6B). Quantitative PCR analysis revealed an increase in mRNA expression of vimentin, TWIST, and ZEB1, and a decrease in expression of E‐cadherin in NRAS Q61K transduced cells, consistent with these cells having undergone EMT (Fig. 6C). Furthermore, the NE markers, ASCL1 and SYP, were also shown to have decreased in expression in these cells. Importantly, the NRAS Q61K‐transduced cells acquired sensitivity to MEK inhibitors (Fig. 6D) and showed substantial response to the trametinib/torin2 combination (Fig. 6E).
Fig. 6.
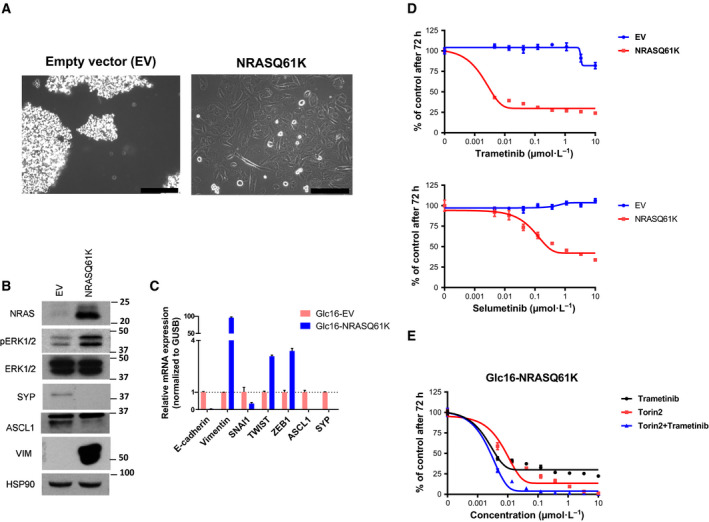
Transition from NE to non‐NE mediated by NRAS Q61K. (A) Phase‐contrast images of Glc16 cells transduced with retrovirus containing NRAS Q61K or the empty vector. Scale bar, 100 µm. (B) The cell extracts were immunoblotted for the detection of indicated protein including NE markers (ASCL1 and SYP). (C) RNA was collected as well, and quantitative PCR was carried out using probes for indicated genes including mesenchymal markers and transcription factors (E‐cadherin, vimentin, SNAI1, TWIST, and ZEB1). (D) The empty vector/NRAS Q61K‐transduced Glc16 cell lines were treated with increasing doses of trametinib or selumetinib, and the cell viability was assessed by MTS after 72 h (n = 6, mean ± SD). (E) The NRAS Q61K transduced Glc16 cell line was treated with increasing doses of trametinib or torin2 alone, or in combination, and the cell viability was assessed by MTS after 72 h (n = 6, mean ± SD).
4. Discussion
Small‐cell lung cancer is an aggressive malignancy most often presenting in patients who are former or current smokers. However, SCLC can occasionally be diagnosed in individuals who are never or light former smokers (Ou et al., 2009; Varghese et al., 2014). Diagnostic specimens in SCLC are generally small hence limiting comprehensive pathological and/or molecular analyses. Furthermore, given no clear targetable alterations in SCLC, these tumors often do not undergo extensive genomic analyses. Recent studies suggest that SCLC can be heterogeneous, consisting of both NE and non‐NE cells, which may warrant an alternative treatment approach (Gazdar et al., 2017; Mollaoglu et al., 2017; Rudin et al., 2019). By both pathological re‐examination and molecular analyses of 11 cases of clinically diagnosed pulmonary SCLC in never/light smokers, our study highlights the biological heterogeneity within this group and that in a subset a nonpulmonary origin should also be considered despite the clinical diagnosis of lung origin. Intriguingly, even after pathological re‐review, not all of the SCLC tumors harbored TP53 and RB1 mutations, suggesting that SCLC in never/light smokers may also be distinct at the genetic level. However, additional studies are needed to support this hypothesis. Our study exemplifies the genomic and phenotypic heterogeneity of SCLC in never smokers and suggests that tumors which clinically and pathologically mimic SCLC can have strikingly different genomic underpinnings and phenotypic plasticity.
We successfully established a cell line and PDX (DFCI168) from a clinically diagnosed SCLC never smoker with a NRAS Q61K mutation (case 7; Table 1). Interestingly, DFCI168 and SW1271 both harbor NRAS‐activating mutations and exhibit characteristics distinct from classic SCLC, such as upregulation of mesenchymal markers and a lack of NE marker expression. Although RB1 inactivation is highly characteristic of SCLC (> 90%) (George et al., 2015), targeted next‐generation sequencing (OncoPanel) analyses revealed no RB1 mutation in either DFCI168 or SW1271. DFCI168 was diagnosed and treated as a SCLC, although detailed molecular and histological analyses, including examination of postmortem specimens, could not definitively classify this as SCLC. SW1271 is listed as a SCLC cell line at the ATCC site (https://www.atcc.org/), but the similarities between SW1271 and DFCI168 raise a question whether SW1271 is in fact a true SCLC cell line. To date, SW1271 has been commonly used as a SCLC cell line in several prior studies (Dabir et al., 2014; Greenberg et al., 2015; Polley et al., 2016) .
This rare subset of pulmonary SCLC in never/light smokers is of particular interest as druggable oncogenic alterations in NSCLC, including EGFR mutations and ALK rearrangements, are more commonly observed in never smokers than in those with a history of smoking (Shigematsu et al., 2005; Wong et al., 2009). We observed two cases of epidermal growth factor receptor (EGFR) inhibitor naïve EGFR mutant SCLC arising in the background of NSCLC (Table 1). Both patients were treated with erlotinib: One had no response, while the other had a sustained clinical response (data not shown). Two independent studies assessed SCLC samples from never smokers by NGS also identified EGFR mutations (6/36 cases), but detected no RAS mutations (Sun et al., 2015; Varghese et al., 2014). We further reviewed data from publicly available SCLC datasets using the cBioPortal website (http://www.cbioportal.org/) and found no NRAS mutations reported in the 210 SCLC cases regardless of smoking status. Considering that NRAS mutations in NSCLC are more frequent in previous/current smokers (Ohashi et al., 2013), comprehensive genomic profiling of SCLC regardless of smoking status will be necessary to determine the true incidence of NRAS mutant SCLC.
We observed heterogeneity in response to MEK inhibitors in five NRAS mutant lung cancer cell lines. Among those tested, DFCI168 was the most sensitive to MEK inhibition. DFCI168 exhibits prominent EMT features, and its high sensitivity to MEK inhibition does not correlate with previous findings where EMT is often associated with drug resistance (Kitai et al., 2016). Several studies using human and mouse cell lines have reported that RAS activation (H‐RAS and K‐RAS) could induce a non‐NE EMT phenotype (Calbo et al., 2011; Falco et al., 1990; Mabry et al., 1988). Here, we show that ectopically expressed NRAS Q61K, too, is sufficient to induce the acquisition of non‐NE characteristics in the classic SCLC cell line, Glc16. Furthermore, the Glc16 NRASQ61K cells acquire sensitivity to MEK inhibition, analogous to DFCI168. However, not all of the NRAS mutant cell lines were sensitive to MEK inhibition. A recent study by Corcoran et al. (2013). revealed the importance of p‐S6, one of the markers of mTORC1 signaling, as a predictor of response to RAF and MEK inhibitors in BRAF mutant melanoma. We previously reported that the mTOR pathway not only serves as a predictive marker for sensitivity to the combination of the EGFR‐TKI inhibitor WZ4002 and trametinib, but also is involved in acquired resistance to this combination treatment in EGFR mutant lung adenocarcinoma (Tricker et al., 2015). Trametinib treatment reduced the p‐S6 levels in the NRAS mutant cell lines we examined, but did not eliminate the activation of S6. In addition, in four out of five cell lines tested, mTORC1/2 inhibitors showed more potency than MEK inhibitors, further emphasizing the importance of inhibiting mTOR signaling in NRAS mutant cell lines. This finding is in agreement with a recent publication by Kiessling et al, who showed that the mTORC1/2 inhibitor, everolimus, alone was sufficient to inhibit cell growth in NRAS mutant neuroblastoma cell lines (Kiessling et al., 2016). In our study, MEK and mTORC1/2 inhibitors exhibited a synergistic effect in 4 out of 5 NRAS mutant lung cancer cell lines. We also demonstrate that the trametinib and torin2 combination is significantly better at delaying the tumor regrowth in vivo than single agent alone, which is consistent with previous studies (Bailey et al., 2014; Kiessling et al., 2015; Vujic et al., 2014). Many signaling pathways converge at mTOR, and the long‐term efficacy of the dual MEK/mTOR inhibition highlights mTOR as an attractive target for combination therapy. While tumor regrowth was slower after the withdrawal of the combination in vivo, it did not completely prevent tumor regrowth. This is possibly due to insufficient drug exposure, as the combination treatment did not completely inhibit p‐ERK1/2 and p‐S6 levels (Fig. 5). Additional studies, including intermittent dosing strategies, are needed to effectively combine these two agents to maximize pathway inhibition.
5. Conclusions
In summary, our findings highlight unique features of SCLC in never/light smokers. Given the aggressive clinical nature of SCLC and the often scant specimens available for pathological and molecular analyses, treatment is commonly based solely on H&E staining with the assistance of IHC and on clinical presentation. However, detailed pathological and molecular analyses reveal that clinically diagnosed SCLC in never/light smokers is in fact more heterogeneous with some cases harboring targetable genomic alterations. The identification and study of such cases may reveal new therapeutic options that are not typically used in classic SCLC. With the increased systematic use of comprehensive genomic sequencing, including in SCLC, more cases of this SCLC subtype may be identified, further underscoring the importance of establishing personalized treatment approaches for these patients.
Conflict of interest
PAJ has received consulting fees from AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Merrimack Pharmaceuticals, Chugai Pharmaceuticals, Takeda Oncology, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, LOXO Oncology, Daiichi‐Sankyo, Voronoi, SFJ Pharmaceuticals, and Biocartis; receives postmarketing royalties from DFCI owned intellectual property on EGFR mutations licensed to Lab Corp; has sponsored research agreements with AstraZeneca, Daiichi‐Sankyo, PUMA, Boehringer Ingelheim, Eli Lilly and Company, Takeda Oncology, and Astellas Pharmaceuticals; and has stock ownership in LOXO Oncology and Gatekeeper Pharmaceuticals. GRO has received consulting fees from AstraZeneca and Inivata and honoraria from Guardant. LMS has received consulting fees from Foghorn Therapeutics and LOXO Oncology and honoraria from AstraZeneca. AC has received honorary/consulting fees from AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Eli Lilly and Company, Novartis, Merck Sharp & Dohme, and Bristol‐Myers Squibb.
All remaining authors have no conflicts of interest.
Author contributions
AO and PAJ conceived and designed the project. AO, JC, MKW, AC, MX, and MC carried out the experiments, and analyzed and interpreted the data. NSG provided the study materials. AO, LMS, and PAJ wrote the paper. ML edited the manuscript. AEA, ESC, MB, and GRO provided the patient data/samples. PCG, SP, PK, and LMS contributed to the study design and data interpretation. PAJ supervised the findings of this work. All authors have read and approved the final version of the manuscript.
Supporting information
Fig. S1. Treatment timeline: Dynamic contrast‐enhanced CT scan and FDG‐PET image are shown.
Fig. S2. Characterization of NRAS mutant lung cancer cell lines. (A) Flow cytometric analysis of NCAM/EpCAM expression on DFCI168. (B) (left) The lysates from nuclear fractions of lung cancer cell lines were examined for RB, pRB (Ser807/811) and lamin B (loading control). (right) IHC image of DFCI168 PDX stained with RB. Scale bar 50 µm. (C) The activation of PI3K/Akt/mTOR and MEK/ERK pathways in the NRAS mutant lung cancer cell lines was assessed by western blot. (D) Immunofluorescence images of DFCI168 stained with phalloidin (green) and VIM (red). Scale bars 100 µm.
Fig. S3. H1299 and SW1271 were treated with trametinib at the indicated concentration for the indicated times. The cell extracts were immunoblotted using the indicated antibodies.
Fig. S4. The comparison of IC50 values of various inhibitors for NRAS mutant lung cancer cell lines after 72 h of treatment. The results were obtained from three independent experiments, and the bar represents the mean ± SD.
Fig. S5. DFCI168 and H2087 were treated with 100 nm of trametinib/torin2 either alone or in combination for the indicated times. The cell extracts were immunoblotted using the indicated antibodies.
Table S1. Mutations and gene amplification found by OncoPanel sequencing in 11 clinically diagnosed never/light smokers with SCLC.
Acknowledgement
The authors thank Mei Zheng for her diligent technical assistance, and Zhengnian Li and Alyssa Verano for helpful discussion. We also thank Magda Bahcall, Brandon P Piel, and Sonal Jhaveri for manuscript preparation.
This work was supported by the American Cancer Society (CRP‐17‐111‐01‐CDD to PAJ), the Mock Family Fund (to PAJ), and the Poduska Family Foundation Research Fund for Thoracic Oncology (to PAJ).
Prior Presentation: This work was previously presented at the 2018 AACR Annual Meeting Chicago, Illinois.
References
- Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin‐Anduix B et al (2011) Reversing melanoma cross‐resistance to BRAF and MEK inhibitors by co‐targeting the AKT/mTOR pathway. PLoS One 6, e28973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, Berney D, Foster CS, Fletcher A, Gerald WL et al (2008) Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 27, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey ST, Zhou B, Damrauer JS, Krishnan B, Wilson HL, Smith AM, Li M, Yeh JJ and Kim WY (2014) mTOR inhibition induces compensatory, therapeutically targetable MEK activation in renal cell carcinoma. PLoS One 9, e104413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S et al (2011) Identification of NVP‐BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med Chem Lett 2, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbo J, van Montfort E, Proost N, van Drunen E, Beverloo HB, Meuwissen R and Berns A (2011) A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 19, 244–256. [DOI] [PubMed] [Google Scholar]
- Cardona AF, Rojas L, Zatarain‐Barron ZL, Ruiz‐Patino A, Ricaurte L, Corrales L, Martin C, Freitas H, Cordeiro de Lima VC, Rodriguez J et al (2019) "Multigene mutation profiling and clinical characteristics of small‐cell lung cancer in never‐smokers vs. heavy smokers (Geno1.3‐CLICaP)." Front Oncol 9, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalanotti F, Solit DB, Pulitzer MP, Berger MF, Scott SN, Iyriboz T, Lacouture ME, Panageas KS, Wolchok JD, Carvajal RD et al (2013) Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY‐142886) in patients with BRAFV600E/K‐mutated melanoma. Clin Cancer Res 19, 2257–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P et al (2010) AZD8055 is a potent, selective, and orally bioavailable ATP‐competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 70, 288–298. [DOI] [PubMed] [Google Scholar]
- Corcoran RB, Rothenberg SM, Hata AN, Faber AC, Piris A, Nazarian RM, Brown RD, Godfrey JT, Winokur D, Walsh J et al (2013) TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF‐mutant melanoma. Sci Transl Med 5, 196ra198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristea S and Sage J (2016) Is the canonical RAF/MEK/ERK signaling pathway a therapeutic target in SCLC? J Thorac Oncol 11, 1233–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabir S, Babakoohi S, Kluge A, Morrow JJ, Kresak A, Yang M, MacPherson D, Wildey G and Dowlati A (2014) RET mutation and expression in small‐cell lung cancer. J Thorac Oncol 9, 1316–1323. [DOI] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK and Zarrinkar PP (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Demichelis F, Fall K, Perner S, Andren O, Schmidt F, Setlur SR, Hoshida Y, Mosquera JM, Pawitan Y, Lee C et al (2007) TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 26, 4596–4599. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB et al (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabets M, Vora S, Juric D, Morse N, Mino‐Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH et al (2013) mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA‐mutant breast cancer. Sci Transl Med 5, 196ra199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falco JP, Baylin SB, Lupu R, Borges M, Nelkin BD, Jasti RK, Davidson NE and Mabry M (1990) v‐rasH induces non‐small cell phenotype, with associated growth factors and receptors, in a small cell lung cancer cell line. J Clin Invest 85, 1740–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, Ligon AH, Sholl LM, Kuo FC, MacConaill LE et al (2017) Validation of oncopanel: a targeted next‐generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med 141, 751–758. [DOI] [PubMed] [Google Scholar]
- Gazdar AF, Bunn PA and Minna JD (2017) Small‐cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer 17, 725–737. [DOI] [PubMed] [Google Scholar]
- George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, Leenders F, Lu X, Fernández‐Cuesta L, Bosco G et al (2015) Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD and Davies MA (2010) Basal and treatment‐induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY‐142886) in Braf‐mutant human cutaneous melanoma cells. Cancer Res 70, 8736–8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg EF, McColl KS, Zhong F, Wildey G, Dowlati A and Distelhorst CW (2015) Synergistic killing of human small cell lung cancer cells by the Bcl‐2‐inositol 1,4,5‐trisphosphate receptor disruptor BIRD‐2 and the BH3‐mimetic ABT‐263. Cell Death Dis 6, e2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K, Khachaturova I, Azab B, Maniatis T, Murukutla S, Chalhoub M, Hatoum H, Kilkenny T, Elsayegh D, Maroun R et al (2012) Small cell lung cancer doubling time and its effect on clinical presentation: a concise review. Clin Med Insights Oncol 6, 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS et al (2010) MK‐2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo . Mol Cancer Ther 9, 1956–1967. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ et al (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Klingbeil O, He XY, Wu XS, Arun G, Lu B, Somerville TDD, Milazzo JP, Wilkinson JE, Demerdash OE et al (2018) POU2F3 is a master regulator of a tuft cell‐like variant of small cell lung cancer. Genes Dev 32, 915–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi‐Far R, White MA, Westwick JK, Solski PA, Chrzanowska‐Wodnicka M, Van Aelst L, Wigler MH and Der CJ (1996) Oncogenic Ras activation of Raf/mitogen‐activated protein kinase‐independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol 16, 3923–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiessling MK, Curioni‐Fontecedro A, Samaras P, Atrott K, Cosin‐Roger J, Lang S, Scharl M and Rogler G (2015) Mutant HRAS as novel target for MEK and mTOR inhibitors. Oncotarget 6, 42183–42196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiessling MK, Curioni‐Fontecedro A, Samaras P, Lang S, Scharl M, Aguzzi A, Oldrige DA, Maris JM and Rogler G (2016) Targeting the mTOR complex by everolimus in NRAS mutant neuroblastoma. PLoS One 11, e0147682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitai H, Ebi H, Tomida S, Floros KV, Kotani H, Adachi Y, Oizumi S, Nishimura M, Faber AC and Yano S (2016) Epithelial‐to‐mesenchymal transition defines feedback activation of receptor tyrosine kinase signaling induced by MEK inhibition in KRAS‐mutant lung cancer. Cancer Discov 6, 754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D and Yamori T (2007) ZSTK474 is an ATP‐competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci 98, 1638–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Xu C, Kirubakaran S, Zhang X, Hur W, Liu Y, Kwiatkowski NP, Wang J, Westover KD, Gao P et al (2013) Characterization of Torin2, an ATP‐competitive inhibitor of mTOR, ATM, and ATR. Cancer Res 73, 2574–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabry M, Nakagawa T, Nelkin BD, McDowell E, Gesell M, Eggleston JC, Casero RA and Baylin SB (1988) v‐Ha‐ras oncogene insertion: a model for tumor progression of human small cell lung cancer. Proc Natl Acad Sci USA 85, 6523–6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl K, Wildey G, Sakre N, Lipka MB, Behtaj M, Kresak A, Chen Y, Yang M, Velcheti V, Fu P et al (2017) Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 8, 73745–73756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD et al (2017) MYC Drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 31, 270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Sequist LV, Arcila ME, Lovly CM, Chen X, Rudin CM, Moran T, Camidge DR, Vnencak‐Jones CL, Berry L et al (2013) Characteristics of lung cancers harboring NRAS mutations. Clin Cancer Res 19, 2584–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou SH, Ziogas A and Zell JA (2009) Prognostic factors for survival in extensive stage small cell lung cancer (ED‐SCLC): the importance of smoking history, socioeconomic and marital statuses, and ethnicity. J Thorac Oncol 4, 37–43. [DOI] [PubMed] [Google Scholar]
- Peifer M, Fernández‐Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T et al (2012) Integrative genome analyses identify key somatic driver mutations of small‐cell lung cancer. Nat Genet 44, 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, Ogle C, Reinhart R, Selby M, Connelly J et al (2016) Small cell lung cancer screen of oncology drugs, investigational agents, and gene and microRNA expression. J Natl Cancer Inst 108, djw122 10.1093/jnci/djw122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud FI, Eccles SA, Patel S, Alix S, Box G, Chuckowree I, Folkes A, Gowan S, De Haven Brandon A, Di Stefano F et al (2009) Biological properties of potent inhibitors of class I phosphatidylinositide 3‐kinases: from PI‐103 through PI‐540, PI‐620 to the oral agent GDC‐0941. Mol Cancer Ther 8, 1725–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N and Blenis J (2007) RAS/ERK signaling promotes site‐specific ribosomal protein S6 phosphorylation via RSK and stimulates cap‐dependent translation. J Biol Chem 282, 14056–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J et al (2012) Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small‐cell lung cancer. Nat Genet 44, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, Heymach JV, Johnson JE, Lehman JM, MacPherson D et al (2019) Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 19, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba KM II, Fong H, Lee S, Toyooka S, Shimizu N et al (2005) Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 97, 339–346. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Goldfarb M, Suard Y, Perucho M, Li Y, Kamata T, Feramisco J, Stavnezer E, Fogh J and Wigler MH (1983) Three human transforming genes are related to the viral ras oncogenes. Proc Natl Acad Sci USA 80, 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, Garcia EP, Jia Y, Davineni P, Abo RP et al (2016) Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, e87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms E, Gazdar AF, Abrams PG and Minna JD (1980) Growth of human small cell (oat cell) carcinoma of the lung in serum‐free growth factor‐supplemented medium. Cancer Res 40, 4356–4363. [PubMed] [Google Scholar]
- Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, Liu DD, Kurie JM, Mao L and Khuri FR (2002) Lack of PTEN expression in non‐small cell lung cancer could be related to promoter methylation. Clin Cancer Res 8, 1178–1184. [PubMed] [Google Scholar]
- Sun JM, Choi YL, Ji JH, Ahn JS, Kim KM, Han J, Ahn MJ and Park K (2015) Small‐cell lung cancer detection in never‐smokers: clinical characteristics and multigene mutation profiling using targeted next‐generation sequencing. Ann Oncol 26, 161–166. [DOI] [PubMed] [Google Scholar]
- Travis WD (2012) Update on small cell carcinoma and its differentiation from squamous cell carcinoma and other non‐small cell carcinomas. Mod Pathol 25(Suppl 1), S18–S30. [DOI] [PubMed] [Google Scholar]
- Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK et al (2015) Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR‐mutant lung cancer. Cancer Discov 5, 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese AM, Zakowski MF, Yu HA, Won HH, Riely GJ, Krug LM, Kris MG, Rekhtman N, Ladanyi M, Wang L et al (2014) Small‐cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol 9, 892–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujic I, Posch C, Sanlorenzo M, Yen AJ, Tsumura A, Kwong A, Feichtenschlager V, Lai K, Arneson DV, Rappersberger K et al (2014) Mutant NRASQ61 shares signaling similarities across various cancer types–potential implications for future therapies. Oncotarget 5, 7936–7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, Ho KK, Au JS, Chung LP, Pik Wong M et al (2009) The EML4‐ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild‐type EGFR and KRAS. Cancer 115, 1723–1733. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Kakefuda R, Tajima N, Sowa Y and Sakai T (2011) Antitumor activities of JTP‐74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo . Int J Oncol 39, 23–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Treatment timeline: Dynamic contrast‐enhanced CT scan and FDG‐PET image are shown.
Fig. S2. Characterization of NRAS mutant lung cancer cell lines. (A) Flow cytometric analysis of NCAM/EpCAM expression on DFCI168. (B) (left) The lysates from nuclear fractions of lung cancer cell lines were examined for RB, pRB (Ser807/811) and lamin B (loading control). (right) IHC image of DFCI168 PDX stained with RB. Scale bar 50 µm. (C) The activation of PI3K/Akt/mTOR and MEK/ERK pathways in the NRAS mutant lung cancer cell lines was assessed by western blot. (D) Immunofluorescence images of DFCI168 stained with phalloidin (green) and VIM (red). Scale bars 100 µm.
Fig. S3. H1299 and SW1271 were treated with trametinib at the indicated concentration for the indicated times. The cell extracts were immunoblotted using the indicated antibodies.
Fig. S4. The comparison of IC50 values of various inhibitors for NRAS mutant lung cancer cell lines after 72 h of treatment. The results were obtained from three independent experiments, and the bar represents the mean ± SD.
Fig. S5. DFCI168 and H2087 were treated with 100 nm of trametinib/torin2 either alone or in combination for the indicated times. The cell extracts were immunoblotted using the indicated antibodies.
Table S1. Mutations and gene amplification found by OncoPanel sequencing in 11 clinically diagnosed never/light smokers with SCLC.