

PTEN protein levels are regulated through ubiquitination. Here, we identify BAP1 as a deubiquitinase that physically binds to and deubiquitinates PTEN, thereby inhibiting PTEN degradation. Stabilized PTEN suppresses prostate cancer progression by inhibiting Akt phosphorylation.

Keywords: BAP1, cancer progression, deubiquitination, prostate cancer, PTEN
Abstract
Deubiquitinase BAP1 is an important tumor suppressor in several malignancies, but its functions and critical substrates in prostate cancer (PCa) remain unclear. Here, we report that the mRNA and protein expression levels of BAP1 are downregulated in clinical PCa specimens. BAP1 can physically bind to and deubiquitinate PTEN, which inhibits the ubiquitination‐mediated degradation of PTEN and thus stabilizes PTEN protein. Ectopically expressed BAP1 in PCa cells increases PTEN protein level and subsequently inhibits the AKT signaling pathway, thus suppressing PCa progression. Conversely, knockdown of BAP1 in PCa cells leads to the decrease in PTEN protein level and the activation of the Akt signaling pathway, therefore promoting malignant transformation and cancer metastasis. However, these can be reversed by the re‐expression of PTEN. More importantly, we found that BAP1 protein level positively correlates with PTEN in a substantial fraction of human cancers. These findings demonstrate that BAP1 is an important deubiquitinase of PTEN for its stability and the BAP1‐PTEN signaling axis plays a crucial role in tumor suppression.
Abbreviations
- BAP1
the BRCA1‐associated protein 1
- DUBs
deubiquitinases
- GEO
Gene Expression Omnibus
- IP
immunoprecipitation
- KEGG
the Kyoto Encyclopedia of Genes and Genomes
- PCa
prostate cancer
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
- qRT–PCR
quantitative real‐time polymerase chain reaction
- TCGA
the Cancer Genome Atlas
- USP
the ubiquitin–proteasome system
- VM
vasculogenic mimicry
1. Introduction
Prostate cancer (PCa) is the most commonly diagnosed cancer and the third leading cause of cancer‐related death in men in the United States [1, 2]. The causes of PCa have been extensively studied but are still poorly understood. Some known alterations of cancer‐related genes/proteins have been described as key drivers and potential biomarkers in PCa progression, and a series of screening and diagnosis markers have recently emerged; however, the sensitivities and specificities of those in detecting high‐grade PCa were limited [3]. Thus, the mechanisms underlying tumorigenesis and cancer progression still need to be deeply explored so as to provide better strategies for diagnosis and treatment.
The ubiquitin–proteasome system (UPS) plays important roles in diverse physiological and pathological processes by mostly controlling the turnover of substrate proteins [4]. Ubiquitination is a dynamic and reversible process, which is deconjugated by a large family of deubiquitinases (DUBs) [5, 6]. DUBs affect the degradation, activation, and localization of target proteins through specifically removing ubiquitin chains, which is implicated in various signaling pathways and cell homeostasis [7, 8, 9]. Either dysfunction or dysregulation of DUBs is closely related to tumorigenesis and the development of multiple cancers [10, 11], and targeting DUBs is a good anticancer therapeutic strategy [12, 13]. BRCA1‐associated protein 1 (BAP1) is a member of the ubiquitin C‐terminal hydrolases (UCH) subfamily of DUBs, which is initially characterized as a deubiquitinase regulating the function of BRCA1 [14]. BAP1 consists of an N‐terminal UCH (ubiquitin carboxyl hydrolase) domain, a HCF (host cell factor)‐binding domain HBM in the middle and two NLS (nuclear localization signal) motifs at the C terminus [15]. BAP1 acts as a deubiquitinase for various proteins including H2A [16, 17, 18], HCF‐1 [19], INO80 [20], KLF5 [21], MCRS1 [22], IP3R3 [23], gamma‐tubulin [24], OGT [25], and BAP1 itself [26], which are involved in diverse cellular processes including chromosome stability, cell proliferation, cell cycle, and apoptosis. Further, BAP1 interacts with several transcription factors such as FOXK1/2 [27] and YY1 [28] to regulate gene expression. BAP1 is emerging as an important tumor suppressor in human cancers [29, 30], which requires its deubiquitinating activity and nuclear localization [31]. Growing evidences show that BAP1 is frequently mutated in many human cancers to suggest BAP1 as a tumor suppressor [32, 33, 34, 35, 36, 37, 38, 39, 40]; however, there are also a few studies reporting that BAP1 plays a role in promoting growth of melanoma [41] and breast cancer [21]. So far, functions and regulations of BAP1 in PCa are largely unexplored.
In this study, we demonstrated that BAP1 acted as a tumor suppressor in PCa progression. More importantly, we characterized its critical role in tumor suppression by deubiquitinating and stabilizing PTEN that is a well‐established tumor suppressor in PCa. Furthermore, relevance analyses of clinical data in PCa and many other cancers revealed a significant positive correlation between BAP1 and PTEN at the protein level, indicating that the BAP1‐PTEN signaling axis might play important roles in tumor suppression.
2. Materials and methods
2.1. Antibodies
Mouse anti‐Akt1 (#2967), anti‐ubiquitin (#3936), anti‐Myc (#2276), rabbit anti‐BAP1 (#13271), anti‐PTEN (#9559), and phospho‐Akt‐S473 (#4060) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit anti‐BAP1 (#10398‐1‐AP) and mouse anti‐beta‐actin (#60008‐1‐Ig) antibodies were purchased from Protein Technology (Rosemont, IL, USA). Mouse anti‐PTEN (ab170941) antibody was purchased from Abcam (Cambridge, UK). Mouse anti‐beta Tubulin (100109‐MM05T) antibody was purchased from Sino Biological (Beijing, China). Anti‐Flag M2 (F1804) antibody was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Anti‐HA (16B12) antibody was purchased from Covance (Berkeley, CA, USA). Anti‐GST (#M0807‐1) antibody was purchased from HuaBio (Hangzhou, Zhejiang, China). Protein G Plus/A agarose suspension (#IP05) was purchased from Calbiochem (San Diego, CA, USA).
2.2. Plasmids
Plasmids Flag‐PTEN, pCDH513B‐PTEN, and GST‐PTEN were described previously [42]. Full‐length BAP1 was inserted into pEF‐HA, pCDH513B, pGEX4T‐1, and pET‐28a vectors. The mutant BAP1C91S was generated by PCR‐directed mutagenesis with KOD‐Plus‐Mutagenesis Kit (TOYOBO, Tokyo, Japan). The shRNAs targeting BAP1 3′‐UTR and AKT1 were cloned into the vector pLKO.1. The truncated PTEN and BAP1 plasmids were obtained by mutagenesis PCR or subclone. All plasmids were checked by sequencing. Primer sequences for plasmids construction, shRNAs, and siRNAs were listed in Table S1.
2.3. Cell culture, transfection, and stable cell line establishment
HEK293T, 293FT, and HeLa cells were cultured in DMEM containing 10% fetal bovine serum (FBS) supplemented with penicillin and streptomycin at 37 °C and 5% CO2. DU145 and PC3 cells were cultured in RPMI‐1640 medium supplemented with 10% FBS. P69 and M12 were cultured in RPMI‐1640 medium containing 5% FBS, supplemented with 10 ng·mL−1 epidermal growth factor (EGF), 0.2 μm dexamethasone, 5 μg·mL−1 insulin, 5 μg·mL−1 transferrin, 5 μg·mL−1 sodium selenite, 50 μg·mL−1 gentamicin, and 100 U·mL−1 penicillin/streptomycin. Plasmid transfection into 293T and 293FT cells was performed using polyethylenimine (PEI) according to manufacturer’s instructions. To establish stable cell lines, the lentiviral vector carrying BAP1, PTEN, or shRNA sequence together with the packaging plasmids (pMD2G and pCMVdR8) was transfected into 293FT cells using PEI. The supernatants were harvested 48 h later and centrifuged at 2500 g for 10 min. DU145, P69, and M12 cells were incubated with viral supernatants in the presence of 5 µg·mL−1 polybrene for 24 h. Stable cell lines were selected with 5–10 μg·mL−1 puromycin for 3–4 days, and the expression levels were analyzed by western blotting.
2.4. Immunoprecipitation (IP) and GST pull‐down assays
293T or HeLa cells transfected with indicated plasmids were lysed in lysis buffer (50 mm Tris/HCl pH7.4, 150 mm NaCl, 0.5% NP‐40, 2 mm EDTA, 0.05% SDS, 0.5 mm DTT, and complete protease inhibitor cocktail) on ice for 1 h. 1 mg of lysates was incubated with protein A/G‐agarose beads and specific antibodies overnight at 4 °C. The complexes bound to agarose beads were washed 5 times in the same lysis buffer and subjected to 8% SDS/polyacrylamide gels for western blotting analysis.
For immunoprecipitation under denaturing conditions, cells were lysed in SDS‐lysis buffer (50 mm Tris/HCl pH7.4, 150 mm NaCl, 1% SDS, and 5 mm DTT) and then boiled for 10 min. The lysates were clarified by centrifugation at 16 000 g for 10 min at 4 °C. The clarified samples were diluted into 0.1% SDS and 0.5 mm DTT with dilution buffer (50 mm Tris/HCl pH7.4, 150 mm NaCl, 0.5% Triton X‐100, 2 mm EDTA, and complete protease inhibitor cocktail). The soluble supernatant fractions were harvested and subjected to immunoprecipitation experiments as described above.
For GST pull‐down assay, purified GST or GST‐PTEN was incubated with GST beads and indicated cell lysates or recombinant proteins at 4 °C. GST beads were then washed three times with lysis buffer. The bound proteins were analyzed by western blotting.
2.5. Immunofluorescence (IF) staining
HeLa cells seeded on coverslips were transfected with the indicated plasmids using Lipofectamine 2000. At 24 h after transfection, cells were fixed with 4% paraformaldehyde. Following incubation in blocking solution, cells were stained with the anti‐HA antibody and then incubated in the second antibody (Alexa Fluor® 568). DU145 stable cells with PTEN overexpression were fixed with 4% paraformaldehyde and then stained with primary antibodies (anti‐PTEN and anti‐BAP1) and subsequently with secondary antibodies (Alexa Fluor® 488 or Alexa Fluor® 568). Nuclei were stained with DAPI. Images were acquired using Leica TSC SP8 confocal microscope.
2.6. qRT–PCR
qRT–PCR was performed according to our previous protocol [43]. Total RNAs were extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the instructions. 1 μg of RNAs was used for reverse transcription by using PrimeScript™ RT–PCR Kit (Takara, Otsu, Shiga, Japan). PTEN mRNA levels were analyzed by using the SYBR‐Green Master PCR Mix (Applied Biosystems) with an ABI StepOne system (Applied Biosystems, Foster City, CA, USA). GAPDH was used for normalization of PTEN mRNA. The primers for qRT–PCR were listed in Table S1.
2.7. Vasculogenic mimicry (VM) formation and three‐dimensional (3D) culture growth assays
The vasculogenic mimicry formation was performed as described before [44]. Briefly, prethawed Matrigel matrix™ (#3445‐005‐01, Trevigen, Gaithersburg, MD, USA) was added into the inner well of µ‐slides (Ibidi Gmbh, Martinsried, Germany) and incubated at 37 °C for 1 h. 50 μL of cells (1 × 105 cells·mL−1) was seeded onto the polymerized matrix. 3D culture growth assay was conducted as described in our previous study [45]. 5 μL of cell solutions (1 × 105 cells·mL−1) mixed with 5 μL of matrix gel was transferred into a well of µ‐slides and incubated at 37 °C for 1 h. The complete cell culture medium was added onto the solidified gel, and the µ‐slides were incubated at 37 °C. Microscopy images were taken after three days.
2.8. Migration assay by wound healing
We used a previously described method to analyze cell migration [46]. Briefly, DU145, P69, or M12 stable cells were seeded into the well of 12‐well plates and cultured until 90% confluence. Scratches were made with sterile pipette tips. The cells were cultured in complete medium. Photographs were captured at the indicated times.
2.9. Migration assay with RTCA‐DP
The protocol for the migration assay with the xCELLigence RTCA‐DP system (Roche) was described previously [45]. Briefly, cells were serum starved for 4–6 h, then detached with trypsin, and resuspended in serum‐free medium with concentration of 1 × 105 cells·mL−1. 100 μL of suspension was seeded into the pre‐equilibrated upper chamber of the CIM plate, and the low chamber was added complete medium for migration. Cell index values were detected every 15 min following procedure. RTCA software v1.2 (Roche Applied Science, Indianapolis, IN, USA) was used to calculate the slopes of the curves at various time points.
2.10. Soft‐agar colony formation assay
The soft‐agar colony formation assay was performed as described before [42]. Briefly, the agar‐medium mixture containing 0.6% low‐melting point agarose (Amresco) and 5% FBS was added in six‐well plate as the base agar gel, and cells resuspended in 0.35% agar (Amresco, Wayne, PA, USA) with 5% FBS were seeded onto top of the base gel as the colony formation gel, and then incubated at 37 °C. After 3–4 weeks, cell colonies were fixed with methanol and stained with 0.005% Crystal Violet. The photographs of the colonies were taken, and the number of colonies was scored.
2.11. Xenograft tumor model
The experiment of xenograft tumor model was conducted as described before [47]. Each of stable DU145 cell lines (at the concentration of 2.5 × 107 cells·mL−1) was injected subcutaneously into 5‐week‐old male BALB/c nude mice (n = 6) on the bilateral backs. The tumor volume was measured at the indicated time. Mice were sacrificed after 28 days. Tumors were dissected, weighted, and photographed. All animal studies were conducted with the approval and guidance of Shanghai Jiao Tong University Medical Animal Ethics Committees.
2.12. Analyses of TCGA and GEO data
The online database Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancerpku.cn/index.html) was used to analyze the RNA sequencing expression of BAP1 in prostate adenocarcinoma (PRAD) patients based on The Cancer Genome Atlas (TCGA) and the Genotype‐Tissue Expression (GTEx) projects.
The reverse‐phase protein array (RPPA) data of BAP1 protein, PTEN protein, and p‐AKT(T308/S473) in TCGA tumor tissue sample were obtained from TCPA (The Cancer Proteome Atlas) which contains cancer proteomic datasets of 8167 tumor samples including all TCGA tumor tissue sample sets. The microarray gene expression profile datasets (accession ID: GSE23035) from Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database were used to analyze in this study. The expression levels were normalized with Limma package by using r software. To identify differentially expressed genes, the fold change cutoff was set to 2 and the adjusted P‐value cutoff was selected as 0.05. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed by the Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/home.jsp).
2.13. Statistical analysis
Statistical analyses were performed with graphpad prism version 7 (GraphPad Software, La Jolla, CA, USA) software package. Mouse xenografts and soft‐agar colony formation are presented as means ± standard error of the mean (SEM). Student’s t‐test (two‐tailed) was used to compare statistical significance between two groups. The correlation between BAP1 protein, PTEN protein, and p‐AKT(T308/S473) levels was analyzed by Pearson correlation test and linear regression. A value P < 0.05 was considered statistically significant. Most of experiments such as IP/WB, GST pull‐down assays, IF, VM, 3D culture, and soft‐agar colony formation were repeated at least 3 times.
3. Results
3.1. BAP1 is downregulated in human PCa
To investigate the expression of BAP1 in prostate cancer, the data were obtained from TCGA (The Cancer Genome Atlas) for analysis. The mRNA levels of BAP1 were downregulated in the prostate adenocarcinoma (PRAD) specimens as compared with those in normal tissue specimens, which datasets were obtained from The Cancer Genome Atlas (TCGA) public PRAD and The Genotype‐Tissue Expression (GTEx) public normal prostate tissues (Fig. 1A). Consistently, BAP1 mRNA levels were lower in tumor tissue than those in normal tissue (Fig. 1B), and higher in early‐stage PCa tissues with Gleason score 6 than those in late‐stage PCa tissue with Gleason score 7/8) (Fig. 1C), which data were obtained from R2 database (https://hgserver1.amc.nl/cgi‐bin/r2/main.cgi). Furthermore, BAP1 protein levels in PCa specimens from The Human Protein Atlas database (http://www.proteinatlas.org/) were analyzed to show that BAP1 protein levels were obviously reduced in the high‐grade specimens compared to those in the low‐ and medium‐grade tissues (Fig. 1D,E). In addition, the translocation of BAP1 was observed during tumor progression. We found that more BAP1 protein located in the cytoplasm or membranous fraction in the low/medium‐grade specimens, whereas BAP1 mainly located in the nucleus fraction in the high‐grade specimens (Fig. 1D), indicating that the localization of BAP1 in the cytoplasm or membranous may be beneficial to its tumor‐suppressive function. Our previous studies demonstrated that P69/M12 cell lines can be used as an experimental model for the study of PCa progression [44, 48]. P69 is a low‐tumorigenic and non‐metastatic prostate epithelial cell line whereas M12 is a highly tumorigenic and metastatic subline derived from P69 by selection in nude mice [45]. To find out the potential role of BAP1 in PCa progression, the protein levels of BAP1 in P69 and M12 cells were detected. The result showed that BAP1 was greatly downregulated in M12 cells compared with that in P69 (Fig. 1F). Moreover, we found a uniform reduced BAP1 protein levels in renal carcinoma cell lines (Fig. S1A), lung cancer cell lines (Fig. S1B), and breast cancer cell lines (Fig. S1C) in comparison with the normal renal cells (HK‐2 cells), lung cells (WI38 and 16HBE cells), and breast cells (MCF10A cells), respectively. Thus, above data suggested that loss or low expression of BAP1 might be an important event in tumorigenesis and progression.
Fig. 1.
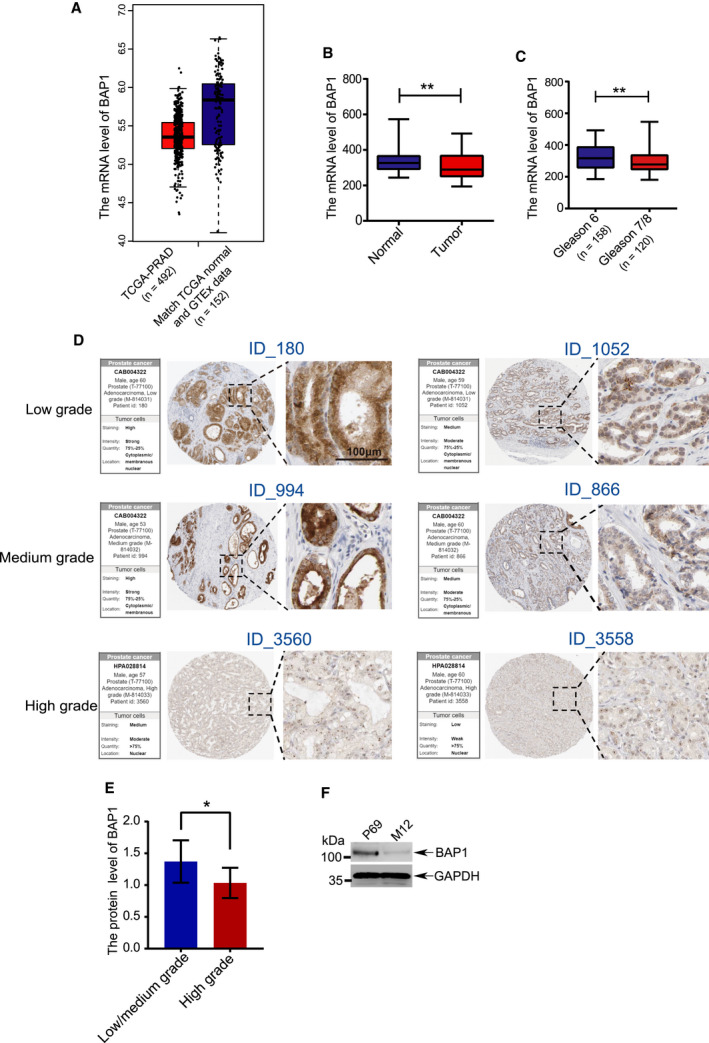
Dysregulation of BAP1 in PCa and cancer cell lines. (A) The mRNA levels of BAP1 were compared between normal tissue specimens (n = 152) and PRAD specimens (n = 492), which derived from The Genotype‐Tissue Expression (GTEx) normal prostate tissue datasets and The Cancer Genome Atlas (TCGA) PRAD datasets. (B)The mRNA levels of BAP1 were compared between prostate normal and tumor tissues derived from R2 database, including normal tissues (n = 70) and tumor tissues (n = 262). (C) The mRNA levels of BAP1 were compared based on PCa gleason scores. Gleason score 6 (n = 158), gleason score 6 and 7 (n = 120). (D) The representative images of IHC (immunohistochemistry) immunoblotted with BAP1 in PCa tissues collected from HPA (human protein atlas) database, including low, medium, and high‐grade PCa tissues. In each grade group, the representative images with strong or weak staining were presented, respectively. The high magnification insets of the images and the detailed information of staining were shown. Scale bars: 100 μm. (E) Quantification of the intensity of IHC by using the software ImageJ. n = 16 (low/medium grade), n = 26 (high grade). (F) The BAP1 protein levels were compared between in low‐tumorigenic P69 and highly tumorigenic M12 cell lines by using western blotting analysis. Error bars in B, C, and E indicated mean ± SD. Data analysis in B, C, and E was conducted by unpaired t‐test (*P < 0.05, **P < 0.01, ***P < 0.001).
3.2. BAP1 suppresses PCa cell progression
To investigate the potential role of BAP1 in PCa progression, BAP1 was silenced by shRNAs targeting the 3′‐terminal untranslated region (UTR) of BAP1 mRNA in DU145 and P69 cells (Fig. S2A), or ectopically expressed in M12 cells by the lentiviral expressing system. Wound‐healing (Fig. 2A and Fig. S2B) and RTCA (real‐time cell analysis) assays (Fig. 2B and Fig. S2C) were conducted to evaluate the effects of BAP1 on migration, showing that cell migratory capacity was enhanced by silencing of BAP1 in DU145 and P69 cells whereas drastically decreased by ectopic expression of BAP1 in M12 cells. The 3D culture growth assay and vasculogenic mimicry (VM) formation assay were performed to assess the effects of BAP1 on cell aggressiveness according to our previous protocol as described [49]. 3D culture growth analysis showed that the knockdown of BAP1 in DU145 and P69 stable cells displayed scattered and invasive morphology compared to the cells with control shRNA (shCtrl), whereas ectopic expression of BAP1 in M12 cells abolished the ability of cell invading into the Matrigel but instead grew into tight colonies (Fig. 2C and Fig. S3A). In consistent with these results, the formation of vascular‐like shape was strongly induced by knockdown of BAP1 in DU145 and P69 cells while reduced by overexpression of BAP1 in M12 cells (Fig. 2D and Fig. S3B). Very similarly, the results of soft‐agar colony formation assays showed that silencing of BAP1 in DU145 and P69 cells massively increased while ectopic expression of BAP1 in M12 cells decreased colony formation both in numbers and sizes (Fig. 2E‐F and Fig. S3C,D). Taken together, above results demonstrated that BAP1 played tumor‐suppressive roles in PCa cell progression through inhibiting cell migration, invasion, and anchorage‐independent growth.
Fig. 2.
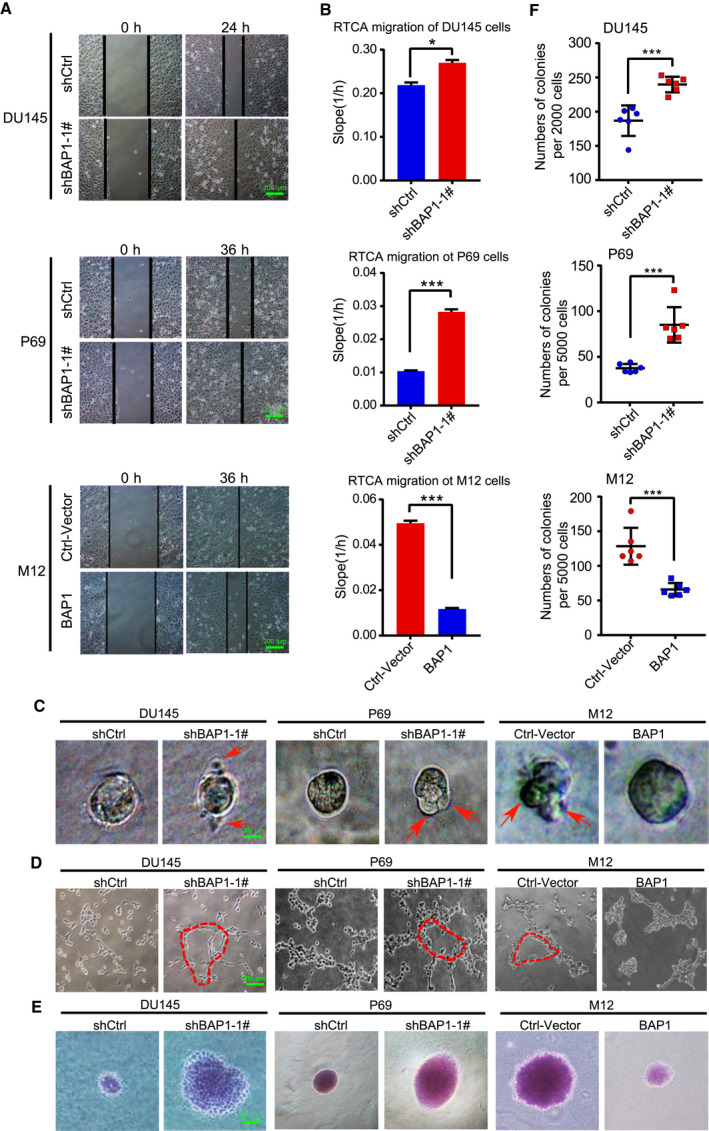
BAP1 suppresses PCa cell progression. (A) Wound‐healing assays for migration analysis of DU145, P69, and M12 stable cell lines. Representative pictures were taken at indicated times. Scale bars: 200 μm. (B) RTCA‐migration assays by using the xCELLigence RTCA‐DP system (n = 3). DU145, P69, and M12 stable cell lines were seeded into a CIM plate and subjected to a dynamic migration assay, respectively. The slope was shown as histogram. (C) 3D cell culture assays for DU145, P69 and M12 stable cell lines. The representative photographs of cell morphology were taken at 4 days. Scale bars: 10 μm. (D) Vasculogenic mimicry assays for DU145, P69 and M12 stable cells. The representative photographs of VM were taken with microscope 20 h later. Scale bars: 200 μm. (E, F) Soft‐agar colony formation assays for DU145, P69, and M12 stable cell lines (n = 6). Scale bars: 10 μm. The representative photographs of colonies were taken (E), and the number of colonies was scored (F). The DU145 and P69 stable cells used in (A–F) were established by using BAP1‐shRNA‐1#. Error bars in B and F indicated mean ± SD. Data analysis in B and F was conducted by unpaired t‐test (*P < 0.05, **P < 0.01, ***P < 0.001).
3.3. BAP1 suppresses PI3K‐Akt pathway by stabilizing PTEN
To investigate the potential mechanism underlying BAP1 in the regulation of tumor suppression, we analyzed the gene expression profile of BAP1‐knockdown U2OS cells, which was obtained from the Gene Expression Omnibus (GEO) public microarray dataset (accession ID: GSE23035) [28]. Differentially expressed genes (fold change ≥ 2, FDR < 0.05) were selected and then subjected to the KEGG pathway enrichment analysis by online tool DAVID (http://david.ncifcrf.gov/home.jsp), which showed that the differentially expressed genes were enriched for tumor‐associated pathways such as PI3K‐Akt signaling pathway and pathways in cancer (Fig. 3A). Among these pathways, BAP1 had the most significant effect on PI3K‐Akt signaling pathway, suggesting that BAP1 suppresses tumorigenesis by regulating key targets involved in the PI3K‐Akt pathway. In order to identify the potential targets of BAP1, lysates from prostate cancer cells P69, M12, DU145, PC3, and BPH1 (a benign prostatic hyperplasia epithelial cell line) as well as HeLa cells were used for western blotting analysis, showing that the protein levels of PTEN, which is a crucial regulator for the PI3K‐Akt pathway, were positively correlated with BAP1, except that in PC3 cells which is lack of PTEN expression [42] (Fig. 3B).
Fig. 3.
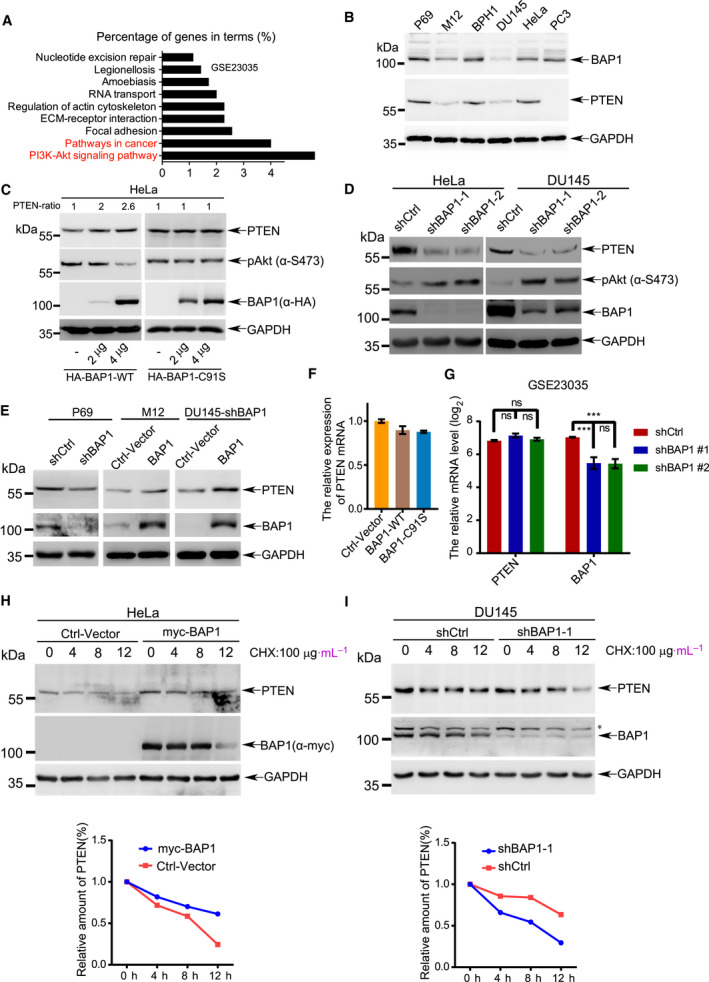
BAP1 suppresses PI3K‐Akt pathway by stabilizing PTEN. (A) The differentially expressed genes (fold change: 2, adjusted P‐value: 0.05) from GEO dataset (accession ID: GSE23035) were subjected to KEGG pathway enrichment analysis by DAVID. (B) The protein levels of BAP1 in cancer cell lines P69, M12, DU145, BPH1, PC3, and HeLa were analyzed by western blotting. (C) Western blotting analysis for endogenous PTEN and p‐Akt (Ser473) in HeLa cells transfected with HA‐BAP1WT or HA‐BAP1C91S. PTEN bands were quantified by ImageJ software. (D) Western blotting analysis for endogenous PTEN, BAP1, and p‐Akt (Ser473) in DU145 and HeLa stable cells with BAP1 knockdown. (E) Western blotting analysis for endogenous PTEN and BAP1 in the indicated PCa stable cell lines. (F) The mRNA levels of PTEN in HeLa cells transfected with BAP1WT or BAP1C91S were analyzed by qRT–PCR. (G) The mRNA levels of PTEN in U2OS cells from GEO database (GSE23035) were analyzed by Limma package. (H, I) The half‐life of endogenous PTEN protein was determined in BAP1 overexpressed HeLa cells (H) and BAP1‐knockdown DU145 cells (I) with treatment of CHX (100 μg·mL−1) for the indicated time points. The asterisk indicated nonspecific band. All above experiments were repeated at least 3 times, and representative images were shown.
PTEN is one of the most important tumor suppressors and plays important roles in tumorigenesis by antagonizing PI3K/Akt signaling [42, 50]. To test whether PTEN protein is a potential target of BAP1, we transiently expressed BAP1‐WT or BAP1‐C91S in HeLa cells. The results showed that PTEN protein levels were significantly increased and sequentially the phosphorylation levels of Akt (pSer473) were decreased (Fig. 3C, left panel), while BAP1 enzyme‐dead mutant (BAP1‐C91S) had no effect (Fig. 3C, right panel). In line with this, decreased protein levels of PTEN and increased phosphorylation levels of Akt were observed in stable knockdown of BAP1 in HeLa and DU145 cells by using the lentiviral shRNA system (Fig. 3D). Furthermore, silencing of endogenous BAP1 in P69 cells downregulated the PTEN protein levels; on the contrary, overexpression of BAP1 in M12 cells upregulated the PTEN protein levels (Fig. 3E). More importantly, re‐expression of BAP1 rescued the effect of PTEN downregulation in DU145‐shBAP1 cells (Fig. 3E, right panel). These data demonstrated that BAP1 increased/stabilized PTEN protein.
Since BAP1 is a deubiquitinating enzyme which is involved in transcriptional regulation through interacting with ASXL1/2, HCF1, YY1, FoxK1/K2, etc. [15]. To exclude the possibility that BAP1 affects PTEN expression on the transcriptional level, total RNAs from HeLa cells transiently expressing wild‐type BAP1 or enzymatically defective mutant BAP1C91S were extracted for quantitative real‐time PCR (qRT–PCR). The results showed there were no significant changes in the PTEN mRNA levels (Fig. 3F), suggesting that BAP1 did not regulate PTEN expression at the transcriptional level. The similar results were also observed in GEO datasets GSE23035 [28] (Fig. 3G). Next to determine whether BAP1 influences the stability of PTEN protein, the half‐life of endogenous PTEN was measured by treatment of cycloheximide (CHX), an inhibitor of protein translation. The result showed that the half‐life of endogenous PTEN protein was prolonged in HeLa cells transfected with Myc‐tagged BAP1 compared with that in the control‐vector transfected cells (Fig. 3H). Conversely, the half‐life of PTEN protein was shortened by knockdown of BAP1 in DU145 cells (Fig. 3I). Collectively, these results suggested that BAP1 stabilized PTEN protein and thus downregulated the PI3K‐Akt signaling pathway.
3.4. BAP1 directly interacts with PTEN
To determine whether BAP1 directly interacts with PTEN, 293T cells were cotransfected with Flag‐tagged PTEN and HA‐tagged BAP1. The reciprocal co‐immunoprecipitation (co‐IP) assays with anti‐Flag or anti‐HA antibody showed that BAP1 interacted with PTEN (Fig. 4A). GST pull‐down assays were performed by using bacterially expressed glutathione S‐transferase (GST)‐tagged PTEN or (GST)‐tagged BAP1 recombinant protein in incubation with lysates from 293T cells overexpressing HA‐tagged BAP1 or HA‐tagged PTEN, respectively, to confirm the interaction between BAP1 and PTEN (Fig. 4B). More confidently, GST‐PTEN was able to directly bind to His‐tagged BAP1 protein which was also purified from E. coli expressing system (Fig. 4C). Furthermore, we validated this direct interaction between endogenous BAP1 and PTEN in HeLa cells by Co‐IP/WB (Fig. 4D).
Fig. 4.
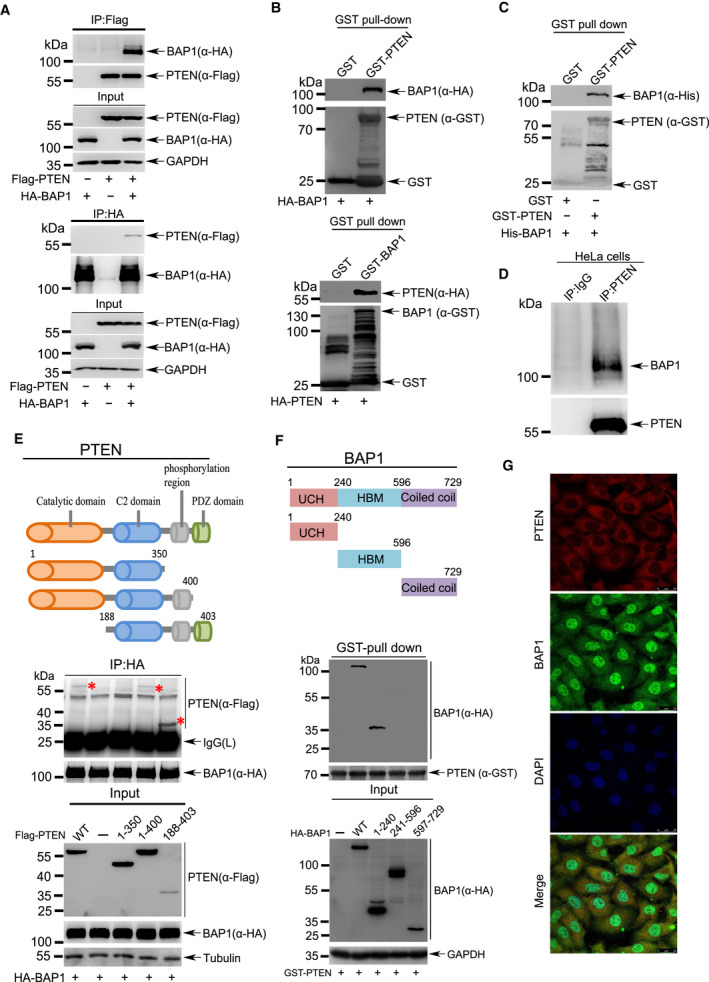
BAP1 directly interacts with PTEN. (A) Lysates from 293T cells transfected with Flag‐PTEN or/and HA‐BAP1 were immunoprecipitated with anti‐Flag antibody, and then immunoblotted with anti‐HA antibody. The reciprocal immunoprecipitation was also conducted to validate the interaction. (B) Purified GST‐PTEN or GST‐BAP1 was incubated with lysates from 293T cells expressing HA‐BAP1 or HA‐PTEN, respectively, for pull‐down assay of protein–protein interaction. (C) GST‐fusion protein pull‐down assay with purified GST‐PTEN and His‐BAP1. (D)The interaction between endogenous BAP1 and PTEN. Lysates from HeLa cells was used for Co‐IP with anti‐PTEN antibody, and followed by western blotting analysis with anti‐BAP1 antibody. Anti‐immunoglobulin (IgG) served as the negative control. (E) Upper panel, a series of truncated forms of Flag‐PTEN were generated based on its domains including a catalytic domain, a C2 domain, a multisite phosphorylation domain, and a PDZ‐binding domain. Middle panel, lysates from 293T cells transfected with HA‐BAP1 full‐length or truncated Flag‐PTEN were co‐immunoprecipitated with anti‐HA antibody, and followed by western blotting analysis with anti‐Flag antibody. Lower panel, immunoblotting was performed for Input. (F) Upper panel, a series of truncated forms of HA‐BAP1 were generated based on its domains including an ubiquitin C‐terminal hydrolase domain (UCH), a HCF‐binding motif (HBM), and a coiled‐coil domain with two nuclear localization sequences (NLS) at its C terminus. Middle panel, lysates from 293T cells transfected with full‐length or truncated HA‐BAP1 were incubated with bacterially produced GST‐PTEN for GST pull‐down assays in vitro. Lower panel, immunoblotting was performed for Input. (G) Colocalization of PTEN and BAP1 in DU145 cells stably expressing PTEN. Scale bars: 25 μm. All above experiments were repeated at least 3 times.
Next to map the BAP1‐binding region of PTEN, we generated a series of truncated forms based on domains of PTEN, which mainly consists of an aminoterminal phosphatase domain, a C2 domain, and a carboxy‐terminal PDZ motif. The truncated PTEN forms as indicated and along with HA‐tagged BAP1 were transfected into 293T cells. The results of co‐IP/WB revealed that BAP1 could efficiently bind to full‐length PTEN, PTEN (1–400), and PTEN (188–403), except the truncated PTEN (1–350) (Fig. 4E), indicating that the BAP1‐binding domain of PTEN might be located in the region of 350–400 amino acid (aa) residues. Moreover, to determine which region of BAP1 is responsible for binding with PTEN, we further generated a series of truncated forms of BAP, which is composed of an ubiquitin C‐terminal hydrolase (UCH) domain, a HBM, and a coiled‐coil domain. GST‐PTEN was incubated with lysates from 293T cells transfected with HA‐BAP1(WT) or three HA‐tagged truncated BAP1 for GST pull‐down assays. The result showed that both the UCH domain (1–240 aa) and the full‐length BAP1 could specifically bind with GST‐PTEN (Fig. 4F), suggesting that the UCH domain of BAP1 was essential for its physical interaction with PTEN. Taken together, these results demonstrated that BAP1 directly interacted with PTEN in vitro and in vivo.
To determine whether BAP1‐PTEN interaction occurs in the cytoplasm or nucleus, we analyzed subcellular fractionations from BPH1, DU145, P69, and M12 cells. The results showed that BAP1 proteins were present both in the nucleus and in the cytoplasm, while PTEN proteins in the cytoplasm were much more than those in the nucleus (Fig. S4A). By using immunofluorescence staining, we confirmed that PTEN colocalized with endogenous or ectopically expressed BAP1 in the cytoplasm of DU145 stable cells (Fig. 4G) and HeLa cells (Fig. S4B), respectively. In addition, the deletion of nuclear localization sequence (ΔNLS) abolished the localization of BAP1 in the nucleus (Fig. S4C), and this form BAP1ΔNLS had same ability as BAPWT in interacting with and stabilizing PTEN (Fig. S4D,E), which further suggested that BAP1 bound to PTEN in the cytoplasm.
3.5. BAP1 deubiquitinates PTEN
Since above results have proven that BAP1 directly interacted with and stabilized PTEN protein, and BAP1 is a member of the ubiquitin C‐terminal hydrolase DUB subfamily, we speculated that BAP1 might remove polyubiquitin chains from PTEN. To validate this assumption, we performed immunoprecipitation assays under natural or denaturing condition to examine PTEN ubiquitination in the presence of BAP1. Plasmids Flag‐PTEN and Myc‐Ub along with an increasing amount of BAP1 were cotransfected into 293T cells. Cell lysates were harvested for IP and western blotting analysis, showing that polyubiquitination of PTEN was gradually reduced by BAP1 in a dose‐dependent manner (Fig. 5A and Fig. S5A). Next, plasmids Flag‐PTEN and Myc‐Ub along with BAP1WT or inactive mutant BAP1C91S were cotransfected into 293T cells. The results showed that polyubiquitination of PTEN was markedly decreased by coexpression of BAP1WT but not BAP1C91S (Fig. 5B and Fig. S5B). Further, only BAP1WT or BAP1C91S was transfected. The similar result that deubiquitination of endogenous PTEN occurred in overexpression of BAP1WT rather than BAP1C91S in HeLa cells was observed (Fig. 5C). These results revealed that ubiquitination of PTEN was specifically reduced by BAP1, whose enzymatic activity was indispensable.
Fig. 5.
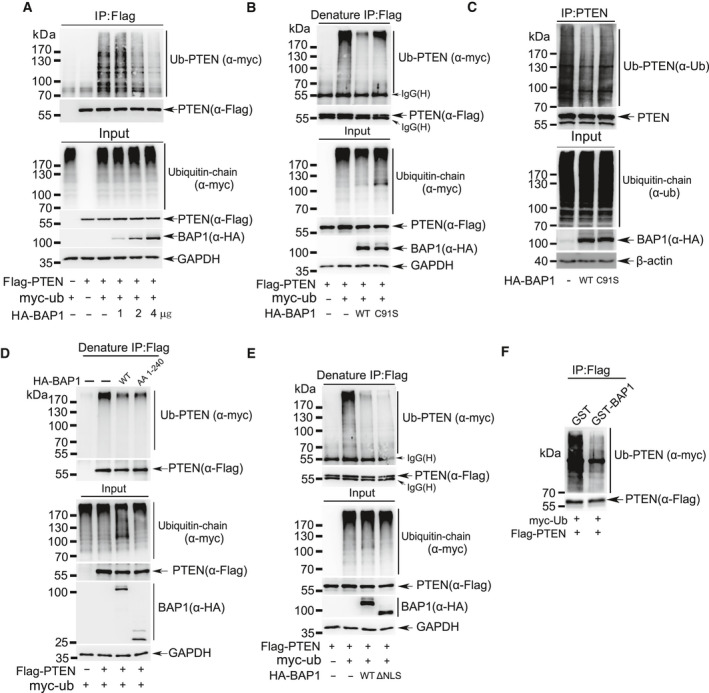
BAP1 deubiquitinates PTEN. (A) Lysates from 293T cells transfected with Flag‐PTEN, Myc‐Ubiquitin together with an increasing amount of BAP1 were immunoprecipitated with anti‐Flag antibody, and followed by western blotting analysis with anti‐Myc antibody. (B) Lysates from 293T cells transfected with Flag‐PTEN, Myc‐Ubiquitin together with BAP1WT or mutant BAP1C91S were immunoprecipitated with anti‐Flag antibody under denaturing condition, and followed by western blotting analysis with anti‐Myc antibody. (C) Lysates from HeLa cells transfected with wild‐type BAP1 or mutant BAP1C91S were immunoprecipitated with anti‐PTEN antibody, and followed by western blotting analysis with anti‐ubiquitin antibody. (D‐E) Lysates from 293T cells transfected with Flag‐PTEN, Myc‐Ubiquitin together with full‐length HA‐BAP1, truncated HA‐BAP11‐240 (D) or HA‐BAP1ΔNLS (E) were immunoprecipitated with anti‐Flag antibody under denaturing condition, and followed by western blotting analysis with anti‐Myc antibody. (F) Ubiquitinated PTEN was purified from 293T cells transfected with Flag‐PTEN and Myc‐Ubiquitin, and then incubated with purified GST‐BAP1 from E. coli. PTEN ubiquitination level was detected by western blotting with anti‐Myc antibody. All above experiments were repeated at least 3 times.
As we have shown that the truncated form BAP11‐240 containing a UCH domain directly interacted with PTEN (Fig. 4F), we wondered that this truncated form of BAP1 is enough and responsible for deubiquitination of PTEN. Flag‐PTEN and Myc‐Ub together with BAP1WT or BAP11‐240 were transfected into 293T cells, respectively. The results of IP/WB analysis revealed that BAP11‐240 was sufficient to deubiquitinate PTEN (Fig. 5D and Fig. S5C). Furthermore, to detected whether recombinant GST‐BAP1 can directly remove polyubiquitination of PTEN in vitro, polyubiquitinated PTEN protein by immunoprecipitation from 293T cells overexpressing Flag‐PTEN and Myc‐Ub was incubated with bacterially produced recombinant protein GST‐BAP1 in a cell‐free system. As expectedly, GST‐BAP1 significantly removed polyubiquitination of PTEN in vitro (Fig. 5E). Collectively, these results suggested that BAP1 was a specific deubiquitinase for PTEN.
3.6. Suppression of tumorigenesis by BAP1 depends on PTEN
As an important tumor suppresser, tiny changes in PTEN protein levels have a big impact on tumorigenesis [42, 51, 52, 53]. To better understand the importance of PTEN in PCa progression, we analyzed the effect of PTEN protein levels on Akt signaling pathway and soft‐agar colony formation. As expectedly, we found that knockdown of PTEN in DU145 cells increased whereas overexpression of PTEN reduced phosphorylation levels of Akt (pSer473) (Fig. S6A). Consistently, the soft‐agar colony formation assay showed that knockdown of PTEN massively increased DU145 cell colony formation, while overexpression of PTEN significantly decreased it (Fig. S6B,C). These results validated that that PTEN suppressed anchorage‐independent cell growth of DU145 by inactivation of Akt pathway.
To verify whether inhibition of tumorigenesis by BAP1 is dependent on PTEN, we stably reintroduced PTEN into a BAP1‐knockdown DU145 cell line (DU145‐shBAP1‐1#) (Fig. 6A) and carried out a series of rescue experiments. Overexpression of PTEN in DU145‐shBAP1‐1# cells abolished the enhanced cell migration (Fig. 6B,C and Fig. S7A), VM formation (Fig. 6D) and cell anchorage‐independent growth (Fig. 6E and Fig. S7B) induced by BAP1 silencing, which suggested that BAP1 repressed tumorigenesis in a PTEN‐dependent way. Furthermore, to investigate the functional correlation between BAP1 and PTEN in vivo, xenografted tumor growth analysis was performed. The same DU145 stable cell lines were subcutaneously injected into the back of male BALB/c nude mice, and tumor growth was monitored. As expectedly, xenografted tumor growth was markedly increased by knockdown of endogenous BAP1, which was reversely rescued by re‐expression of PTEN (Fig. 6F). The expression levels of both BAP1 and PTEN in xenograft tumors were validated by western blotting analysis (Fig. 6G). The photographs were taken, and weight of tumors was analyzed after killing the nude mice at 27 days after injection, showing significant increases in sizes (Fig. 6H) and weights (Fig. 6I) of xenograft tumors of DU145‐shBAP1‐1# cells. Notably, restoring PTEN expression could completely reverse the tumor‐promoting effect of BAP1 knockdown in vivo. To further determine whether BAP1 function of tumor suppression is dependent on inhibiting the Akt signaling pathway, we silenced Akt1 by lenti‐shRNAs in DU145‐shBAP1‐1# cells (Fig. S8A). BAP1/Akt1 double knockdown of DU145 cells led to a reduction on anchorage‐independent cell growth (Fig. S8B,C) and cell aggressiveness (Fig. S8D,E) in compared with only BAP1 knockdown, suggesting that silencing of Akt1 rescued from effects caused by BAP1 depletion. Taken together, BAP1 contributed to tumor suppressor by stabilizing PTEN to suppress Akt activation.
Fig. 6.
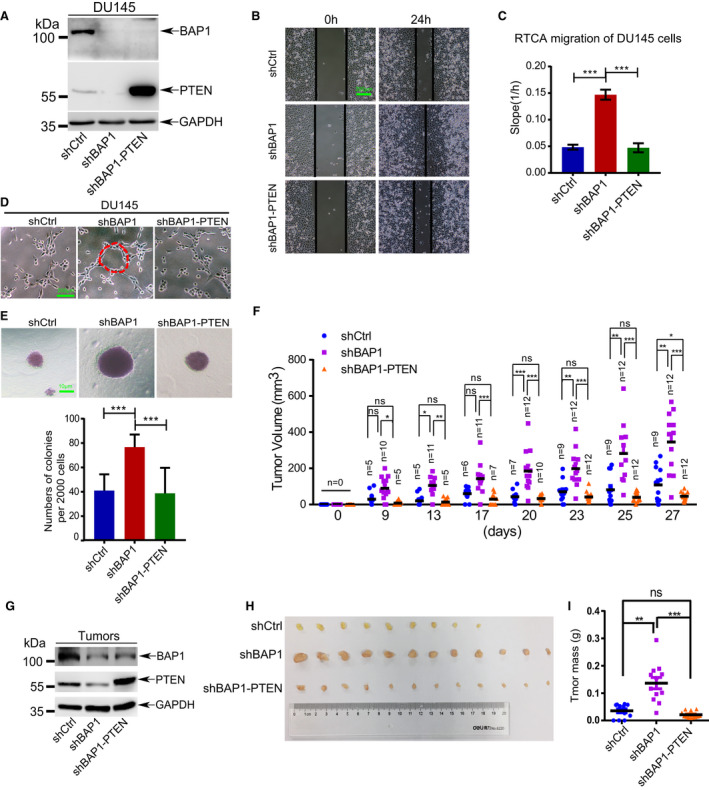
Suppression of tumorigenesis by BAP1 is dependent on PTEN. (A) Western blotting analysis for BAP1 and PTEN in the stable DU145 cell lines, and GAPDH was served as loading control. (B) Wound‐healing assays for migration analysis of the indicated DU145 stable cells. Scale bars: 200 μm. (C) RTCA‐migration assays of DU145 stable cells by using the xCELLigence RTCA‐DP system (n = 3). The slope was shown as histogram. (D) Vasculogenic mimicry assays for the indicated stable cells. The representative photographs were taken. Scale bars: 200 μm. (E) Soft‐agar colony formation assay of DU145 stable cells (n = 6). The representative photographs of colonies were taken (upper panel), and the number of colonies was scored (lower panel). Scale bars: 10 μm. (F) Stable DU145 cells were injected subcutaneously into male BALB/c nude mice, the volume of tumors was measured at the indicated times after injection (n = number of tumors in each group). (G) Western blotting analysis for BAP1 and PTEN in tumor tissues. GAPDH was used as loading control. (H, I) Xenograft tumors were dissected (H), and weight was assessed (I). Error bars in C, E, and I indicated mean ± SD. Data analysis in C, E, and I was conducted by unpaired t‐test (*P < 0.05, **P < 0.01, ***P < 0.001).
3.7. The correlation of BAP1 and PTEN in clinical PCa specimens
Next, we investigated clinical relevance by analyzing the expression levels of BAP1 and PTEN in clinical prostate cancer tissues. We analyzed the protein levels of BAP1 and PTEN in PCa tissues (n = 351) from the TCGA database (The Cancer Genome Atlas) to reveal significant positive correlation between BAP1 and PTEN (linear regression, R 2 = 0.1608, P < 0.0001) (Fig. 7A). However, the positive correlation was not observed between BAP1 mRNAs and PTEN mRNAs (linear regression, R 2 = 0.06118) (Fig. 7B), or between BAP1 proteins and PTEN mRNAs (linear regression, R 2 = 0.001599) (Fig. 7C), which was consistent with our previous results that BAP1 protein stabilized PTEN by direct interaction with each other for deubiquitination of PTEN. Moreover, the BAP1 protein levels were negatively correlated with AKT_pT308 and AKT_pS473 (Fig. 7D). Collectively, BAP1 protein was positively correlated with PTEN protein, whereas both BAP1 and PTEN proteins showed negative associations with AKT_pT308 and AKT_pS473 in clinical PCa specimens (Fig. 7E), revealing that BAP1 made pivotal contribution to the PI3K/Akt signaling pathway through regulating PTEN in PCa progression.
Fig. 7.
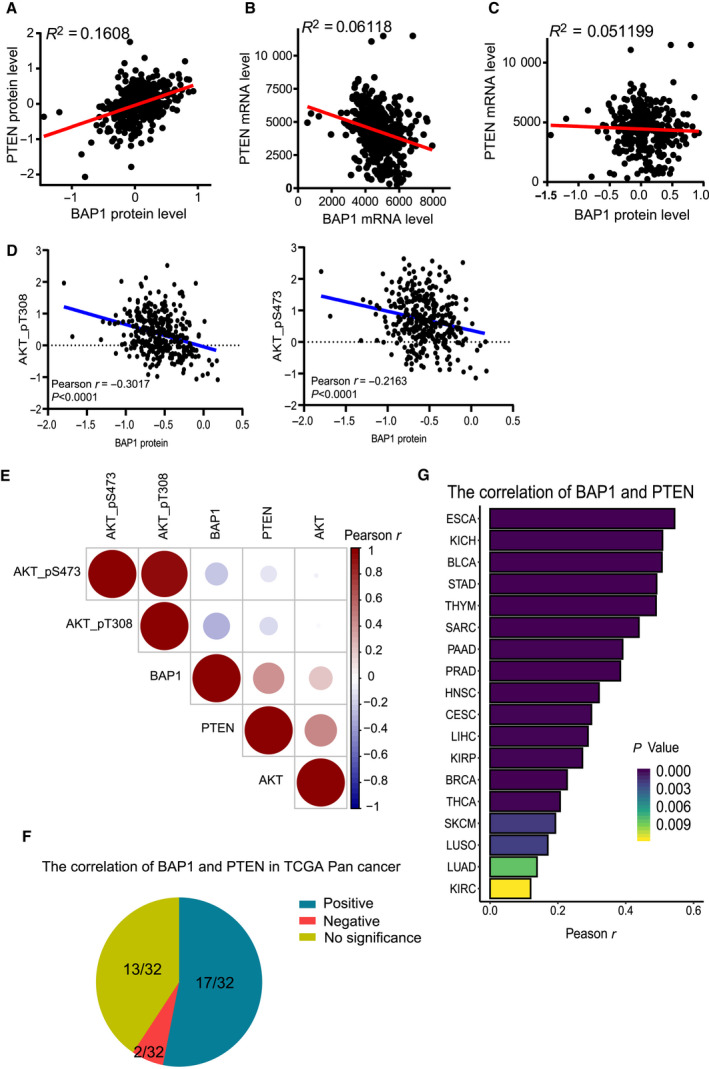
The correlation of BAP1 and PTEN in clinical PCa specimens. (A) The correlation between the protein levels of BAP1 and PTEN in clinical PCa specimens (n = 351) from TCGA database was analyzed by linear regression. (B) The correlation between the mRNA levels of BAP1 and PTEN in clinical specimens from TCGA database was analyzed by linear regression. (C) The correlation between BAP1 protein and PTEN mRNA in clinical specimens from TCGA database was analyzed by linear regression. (D) The correlation between the levels of BAP1 protein and AKT_pT308 (left), or AKT_pS473 (right) in clinical PCa specimens from TCGA database were analyzed by Pearson correlation test and linear regression. (E)The correlation between the levels of BAP1 protein, PTEN protein, AKT protein, AKT_pT308, and AKT_pS473 (right) in clinical PCa specimens from TCGA database was analyzed by Pearson correlation test. (F) The correlation between the protein levels of BAP1 and PTEN in 32 kinds of human cancers from TCGA database. (G) The histogram of human cancers in which the protein levels of BAP1 and PTEN are positively related.
Finally, we also extended our study to a variety of cancers besides PCa. The protein data including 32 kinds of cancers from TCGA (listed in Table S2) were collected for analysis, showing general positive correlations between BAP1 and PTEN in 17 of them, including esophageal carcinoma (ESCA), kidney chromophobe (KICH), bladder urothelial carcinoma (BLCA), and prostate adenocarcinoma (PRAD), whereas slight negative correlations only in brain lower grade glioma (LGG) and testicular germ cell tumors (TGCT) (Fig. 7F,G). These data indicated that BAP1 was an essential regulator of PTEN in many kinds of human cancers, which suggested that the BAP1‐PTEN signaling axis plays important roles in tumor suppression.
4. Discussion
It has been well‐established that BAP1 is an important tumor suppressor and frequently mutated in many human cancers. However, the role of BAP1 in PCa is still unclear. In this study, we revealed that BAP1 suppressed PCa progression through deubiquitinating and stabilizing PTEN. More importantly, large‐scale clinical relevance analyses showed that BAP1 was positively related to PTEN at the protein level in many kinds of human cancers (Fig. 7), suggesting that the positive correlation between BAP1 and PTEN may be ubiquitous.
PTEN is a major tumor suppressor and frequently mutated or deleted in various human cancers. PTEN has been well documented to attenuate cancer cell cycle progression, migration, invasion, and tumorigenesis by antagonizing the PI3K/AKT signaling pathway [50, 54]. Homozygous deletion of Pten in mice results in early embryonic lethality, whereas heterozygous Pten in mice leads to multiple tumors [55, 56, 57]. PTEN alteration is strongly responsible for PCa development. The PTEN prostate‐specific knockout mice show a significant shortened latency of PIN (prostatic intraepithelial neoplasia) formation and metastatic PCa, which are similar with the progression seen in human prostate disease [58]. Dysregulation of PTEN results in tumor initiation and progression with PCa being one of the most sensitive cancers [53].
In addition to PTEN gene mutations and deletions, recent studies have revealed the importance of post‐translational modifications, especially ubiquitination, in the regulation of PTEN stability, activity, and localization. Ubiquitin E3 ligases including NEDD4‐1 [52], XIAP [59], WWP2 [60], CHIP [61], TRIM27 [62], SPOP [63], MKRN1 [64], CRL4B [65], and FBXO22 [66] for PTEN ubiquitination have been identified. On the contrary, the regulation of PTEN deubiquitination remains relatively poorly understood. Several deubiquitinases (DUBs) for PTEN have been reported. USP7/HAUSP can remove the monoubiquitination of PTEN and induce its nuclear export in acute promyelocytic leukemia [67]. Through proteome‐wide screen of DUB members, USP13 [51] and OTUD3 [68] have been identified as deubiquitinases which directly bind to and stabilize PTEN in breast cancer. However, depletion of USP13 or OTUD3 in cells reduces but does not completely abolish the PTEN ubiquitination, suggesting that other deubiquitinases of PTEN may exist. Intriguingly, here we identified that BAP1 served as a novel deubiquitinase of PTEN to control the PI3K‐Akt signaling pathway and cancer progression in PCa. Although it has been reported that somatic mutation of BAP1 is rare in PCa [69], the mRNAs and protein levels of BAP1 were significantly reduced in PCa tissues as compared with adjacent tissues (Fig. 1), which indicates there is a new mechanism underlying PCa progression. Notably, we observed that BAP1 knockdown contributed to PCa cells migration, invasion, transformation, and tumor growth (Fig. 2 and Figs S2 and S3). For the mechanism that BAP1 contributes to PCa suppression, we found that BAP1 deubiquitinated and stabilized PTEN (Fig. 5 and Fig. S5), thereby inhibiting activation of the Akt signaling pathway (Fig. 3). In addition, PTEN overexpression or Akt1 knockdown rescued the aggravated PCa progression caused by BAP1 silencing (Fig. 6 and Fig. S8). Taken together, our findings discovered a novel regulatory mechanism of BAP1‐PTEN‐PI3K/AKT by which BAP1 repressed PCa initiation and progression.
5. Conclusions
In summary, our findings demonstrate that BAP1 is an important deubiquitinase of PTEN for its stability and the BAP1‐PTEN signaling axis plays a crucial role in tumor suppression.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
JY and RD conceived and designed the study. RD, XZ, YG, LL, and JH performed most of the experiments; ZQ, HZ, RC, and YW helped with all experiments; JY, XZ, and RD wrote the manuscript. All authors read and approved the final manuscript.
Supporting information
Fig. S1. The protein level of BAP1 in renal carcinoma, lung cancer and breast cancer cell lines.
Fig. S2. BAP1 suppresses PCa cell migration.
Fig. S3. BAP1 suppresses PCa progression.
Fig. S4. PTEN interacts with BAP1 in the cytoplasm.
Fig. S5. BAP1 deubiquitinates PTEN.
Fig. S6. Downregulated PTEN expression promotes activation of Akt pathway and PCa cell colony formation.
Fig. S7. BAP1 inhibits tumorigenesis through maintenance of PTEN.
Fig. S8. BAP1 suppresses PCa progression in an Akt‐dependent manner.
Table S1. List of DNA or RNA oligonucleotides.
Table S2. The list of clinical cancers used for correlation analysis.
Acknowledgements
This work was supported by grants from NSFC (National Natural Science Foundation of China) 81630075 (to J.Y.), 81702532 (to R.D.), 31671345 (to J.Y.), 81972585 (to X.Z.), and China's National Key R&D Programme (NKP) 2019YFE0110600 (to J.Y.). Jianxiu Yu and Xian Zhao are members of Innovative research team of high‐level local universities in Shanghai.
Rong Deng, Yanmin Guo, Lian Li and Jianfeng He contributed equally to this work.
Contributor Information
Xian Zhao, Email: Zhao20051050134@126.com.
Jianxiu Yu, Email: Jianxiu.Yu@gmail.com.
Data accessibility
All data required to evaluate the conclusions of the paper are present in the main text or the Supplementary Materials of the paper.
References
- 1. Siegel RL, Miller KD & Jemal A (2017) Cancer statistics, 2017. CA Cancer J Clin 67, 7–30. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD & Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70, 7–30. [DOI] [PubMed] [Google Scholar]
- 3. Alford AV, Brito JM, Yadav KK et al, (2017) The use of biomarkers in prostate cancer screening and treatment. Rev Urol 19, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ciechanover A (2006) The ubiquitin proteolytic system: from an idea to the patient bed. Proc Am Thorac Soc 3, 21–31. [DOI] [PubMed] [Google Scholar]
- 5. Sowa ME, Bennett EJ, Gygi SP & Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nijman SM, Luna‐Vargas MP, Velds A, Brummelkamp TR, Dirac AMG, Sixma TK & Bernards R (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786. [DOI] [PubMed] [Google Scholar]
- 7. Sacco JJ, Coulson JM, Clague MJ & Urbe S (2010) Emerging roles of deubiquitinases in cancer‐associated pathways. IUBMB Life 62, 140–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luise C, Capra M, Donzelli M, Mazzarol G, Jodice MG, Nuciforo P, Viale G, Di Fiore PP & Confalonieri S (2011) An atlas of altered expression of deubiquitinating enzymes in human cancer. PLoS One 6, e15891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fraile JM, Quesada V, Rodriguez D, Freije JMP & López‐Otín C (2012) Deubiquitinases in cancer: new functions and therapeutic options. Oncogene 31, 2373–2388. [DOI] [PubMed] [Google Scholar]
- 10. Mofers A, Pellegrini P, Linder S & D'Arcy P (2017) Proteasome‐associated deubiquitinases and cancer. Cancer Metastasis Rev 36, 635–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ciechanover A & Schwartz AL (2004) The ubiquitin system: pathogenesis of human diseases and drug targeting. Biochim Biophys Acta 1695, 3–17. [DOI] [PubMed] [Google Scholar]
- 12. Farshi P, Deshmukh RR, Nwankwo JO, Arkwright RT, Cvek B, Liu J & Dou QP (2015) Deubiquitinases (DUBs) and DUB inhibitors: a patent review. Expert Opin Ther Pat 25, 1191–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen X, Yang Q, Xiao L, Tang D, Dou QP & Liu J (2017) Metal‐based proteasomal deubiquitinase inhibitors as potential anticancer agents. Cancer Metastasis Rev 36, 655–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, Ishov AM, Tommerup N, Vissing H, Sekido Y et al, (1998) BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1‐mediated cell growth suppression. Oncogene 16, 1097–1112. [DOI] [PubMed] [Google Scholar]
- 15. Wang A, Papneja A, Hyrcza M, Al‐Habeeb A & Ghazarian D (2016) Gene of the month: BAP1. J Clin Pathol 69, 750–753. [DOI] [PubMed] [Google Scholar]
- 16. Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly‐Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW & Müller J (2010) Histone H2A deubiquitinase activity of the Polycomb repressive complex PR‐DUB. Nature 465, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sahtoe DD, van Dijk WJ, Ekkebus R, Ovaa H & Sixma TK (2016) BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat Commun 7, 10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Daou S, Hammond‐Martel I, Mashtalir N, Wurtele H, Milot E, Mallette FA, Carbone M & Affar EB (2015) The BAP1/ASXL2 histone H2A deubiquitinase complex regulates cell proliferation and is disrupted in cancer. J Biol Chem 290, 28643–28663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Machida YJ, Machida Y, Vashisht AA, Wohlschlegel JA & Dutta A (2009) The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF‐1. J Biol Chem 284, 34179–34188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee HS, Lee SA, Hur SK, Seo J‐W & Kwon J (2014) Stabilization and targeting of INO80 to replication forks by BAP1 during normal DNA synthesis. Nat Commun 5, 5128. [DOI] [PubMed] [Google Scholar]
- 21. Qin J, Zhou Z, Chen W, Wang C, Zhang H, Ge G, Shao M, You D, Fan Z, Xia H et al, (2015) BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat Commun 6, 8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peng J, Ma J, Li W, Mo R, Zhang P, Gao K, Jin X, Xiao J, Wang C & Fan J (2015) Stabilization of MCRS1 by BAP1 prevents chromosome instability in renal cell carcinoma. Cancer Lett 369, 167–174. [DOI] [PubMed] [Google Scholar]
- 23. Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, Pellegrini L, Signorato V, Olivetto F, Pastorino S et al, (2017) BAP1 regulates IP3R3‐mediated Ca(2+) flux to mitochondria suppressing cell transformation. Nature 546, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zarrizi R, Menard JA, Belting M & Massoumi R (2014) Deubiquitination of gamma‐tubulin by BAP1 prevents chromosome instability in breast cancer cells. Cancer Res 74, 6499–6508. [DOI] [PubMed] [Google Scholar]
- 25. Gambetta MC, Oktaba K & Muller J (2009) Essential role of the glycosyltransferase sxc/Ogt in polycomb repression. Science 325, 93–96. [DOI] [PubMed] [Google Scholar]
- 26. Mashtalir N, Daou S, Barbour H, Sen NN, Gagnon J, Hammond‐Martel I, Dar HH, Therrien M & Affar EB (2014) Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell 54, 392–406. [DOI] [PubMed] [Google Scholar]
- 27. Ji Z, Mohammed H, Webber A, Ridsdale J, Han N, Carroll JS & Sharrocks AD (2014) The forkhead transcription factor FOXK2 acts as a chromatin targeting factor for the BAP1‐containing histone deubiquitinase complex. Nucleic Acids Res 42, 6232–6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu H, Mashtalir N, Daou S, Hammond‐Martel I, Ross J, Sui G, Hart GW, Rauscher FJ, Drobetsky E, Milot E et al, (2010) The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF‐1 and is a critical regulator of gene expression. Mol Cell Biol 30, 5071–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carbone M, Yang H, Pass HI, Krausz T, Testa JR & Gaudino G (2013) BAP1 and cancer. Nat Rev Cancer 13, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Di Nunno V, Frega G, Santoni M, Gatto L, Fiorentino M, Montironi R, Battelli N, Brandi G & Massari F (2019) BAP1 in solid tumors. Future Oncol 15, 2151–2162. [DOI] [PubMed] [Google Scholar]
- 31. Ventii KH, Devi NS, Friedrich KL, Chernova TA, Tighiouart M, Van Meir EG & Wilkinson KD (2008) BRCA1‐associated protein‐1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res 68, 6953–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coupier I, Cousin PY, Hughes D, Legoix‐Né P, Trehin A, Sinilnikova OM & Stoppa‐Lyonnet D (2005) BAP1 and breast cancer risk. Fam Cancer 4, 273–277. [DOI] [PubMed] [Google Scholar]
- 33. Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C & Bowcock AM (2010) Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330, 1410–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goldstein AM (2011) Germline BAP1 mutations and tumor susceptibility. Nat Genet 43, 925–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, Creaney J, Lake RA, Zakowski MF, Reva B et al, (2011) The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet 43, 668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshikawa Y, Sato A, Tsujimura T, Emi M, Morinaga T, Fukuoka K, Yamada S, Murakami A, Kondo N, Matsumoto S et al, (2012) Frequent inactivation of the BAP1 gene in epithelioid‐type malignant mesothelioma. Cancer Sci 103, 868–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wiesner T, Fried I, Ulz P, Stacher E, Popper H, Murali R, Kutzner H, Lax S, Smolle‐Jüttner F, Geigl JB & et al, (2012) Toward an improved definition of the tumor spectrum associated with BAP1 germline mutations. J Clin Oncol 30, e337–e340. [DOI] [PubMed] [Google Scholar]
- 38. Murali R, Wiesner T & Scolyer RA (2013) Tumours associated with BAP1 mutations. Pathology 45, 116–126. [DOI] [PubMed] [Google Scholar]
- 39. Xu J, Kadariya Y, Cheung M, Pei J, Talarchek J, Sementino E, Tan Y, Menges CW, Cai KQ, Litwin S et al, (2014) Germline mutation of Bap1 accelerates development of asbestos‐induced malignant mesothelioma. Cancer Res 74, 4388–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Turunen JA, Markkinen S, Wilska R, Saarinen S, Raivio V, Täll M, Lehesjoki A‐E & Kivelä TT (2016) BAP1 germline mutations in finnish patients with uveal melanoma. Ophthalmology 123, 1112–1117. [DOI] [PubMed] [Google Scholar]
- 41. Kumar R, Taylor M, Miao B, Ji Z, Njauw JC‐N, Jönsson G, Frederick DT & Tsao H (2015) BAP1 has a survival role in cutaneous melanoma. J Invest Dermatol 135, 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang J, Yan J, Zhang J, Zhu S, Wang Y, Shi T, Zhu C, Chen C, Liu X, Cheng J et al, (2012) SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun 3, 911. [DOI] [PubMed] [Google Scholar]
- 43. Liu X, Wang Y, Sun Q, Yan J, Huang J, Zhu S & Yu J (2012) Identification of microRNA transcriptome involved in human natural killer cell activation. Immunol Lett 143, 208–217. [DOI] [PubMed] [Google Scholar]
- 44. Zhao X, Wang Y, Deng R, Zhang H, Dou J, Yuan H, Hou G, Du Y, Chen Q & Yu J (2016) miR186 suppresses prostate cancer progression by targeting Twist1. Oncotarget 7, 33136–33151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu X, Chen Q, Yan J, Wang Y, Zhu C, Chen C, Zhao X, Xu M, Sun Q, Deng R et al, (2013) MiRNA‐296‐3p‐ICAM‐1 axis promotes metastasis of prostate cancer by possible enhancing survival of natural killer cell‐resistant circulating tumour cells. Cell Death Dis 4, e928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu J, Zhang D, Liu J, Li J, Yu Y, Wu X‐R & Huang C (2012) RhoGDI SUMOylation at Lys‐138 increases its binding activity to Rho GTPase and its inhibiting cancer cell motility. J Biol Chem 287, 13752–13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Deng R, Zhao X, Qu Y, Chen C, Zhu C, Zhang H, Yuan H, Jin H, Liu X, Wang Y et al, (2015) Shp2 SUMOylation promotes ERK activation and hepatocellular carcinoma development. Oncotarget 6, 9355–9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Q, Zhao X, Zhang H, Yuan H, Zhu M, Sun Q, Lai X, Wang Y, Huang J, Yan J & et al, (2015) MiR‐130b suppresses prostate cancer metastasis through down‐regulation of MMP2. Mol Carcinog 54, 1292–1300. [DOI] [PubMed] [Google Scholar]
- 49. Xu M, Zhu C, Zhao X, Chen C, Zhang H, Yuan H, Deng R, Dou J, Wang Y, Huang J et al, (2015) Atypical ubiquitin E3 ligase complex Skp1‐Pam‐Fbxo45 controls the core epithelial‐to‐mesenchymal transition‐inducing transcription factors. Oncotarget 6, 979–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Worby CA & Dixon JE (2014) Pten. Annu Rev Biochem 83, 641–669. [DOI] [PubMed] [Google Scholar]
- 51. Zhang J, Zhang P, Wei Y, Piao H‐L, Wang W, Maddika S, Wang M, Chen D, Sun Y, Hung M‐C et al, (2013) Deubiquitylation and stabilization of PTEN by USP13. Nat Cell Biol 15, 1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument‐Bromage H, Tempst P, Cordon‐Cardo C et al, (2007) NEDD4‐1 is a proto‐oncogenic ubiquitin ligase for PTEN. Cell 128, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jamaspishvili T, Berman DM, Ross AE, Scher HI, De Marzo AM, Squire JA & Lotan TL (2018) Clinical implications of PTEN loss in prostate cancer. Nat Rev Urol 15, 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R et al, (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- 55. Di Cristofano A, Pesce B, Cordon‐Cardo C & Pandolfi PP (1998) Pten is essential for embryonic development and tumour suppression. Nat Genet 19, 348–355. [DOI] [PubMed] [Google Scholar]
- 56. Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon‐Cardo C, Catoretti G, Fisher PE & Parsons R (1999) Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA 96, 1563–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, Barrantes IB, Ho A, Wakeham A, ltie A, Khoo W et al, (1998) High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol 8, 1169–1178. [DOI] [PubMed] [Google Scholar]
- 58. Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy‐Burman P, Nelson PS et al, (2003) Prostate‐specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4, 209–221. [DOI] [PubMed] [Google Scholar]
- 59. Van Themsche C, Leblanc V, Parent S & Asselin E (2009) X‐linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J Biol Chem 284, 20462–20466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maddika S, Kavela S, Rani N, Palicharla VR, Pokorny JL, Sarkaria JN & Chen J (2011) WWP2 is an E3 ubiquitin ligase for PTEN. Nat Cell Biol 13, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ahmed SF, Deb S, Paul I, Chatterjee A, Mandal T, Chatterjee U & Ghosh MK (2012) The chaperone‐assisted E3 ligase C terminus of Hsc70‐interacting protein (CHIP) targets PTEN for proteasomal degradation. J Biol Chem 287, 15996–16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee JT, Shan J, Zhong J, Li M, Zhou B, Zhou A, Parsons R & Gu W (2013) RFP‐mediated ubiquitination of PTEN modulates its effect on AKT activation. Cell Res 23, 552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li G, Ci W, Karmakar S, Chen K, Dhar R, Fan Z, Guo Z, Zhang J, Ke Y, Wang L et al, (2014) SPOP promotes tumorigenesis by acting as a key regulatory hub in kidney cancer. Cancer Cell 25, 455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee MS, Jeong MH, Lee HW, Han H‐J, Ko A, Hewitt SM, Kim J‐H, Chun K‐H, Chung J‐Y, Lee C et al, (2015) PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat Commun 6, 7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen Z, Zhang W, Jiang K, Chen B, Wang K, Lao L, Hou C, Wang F, Zhang C & Shen H (2018) MicroRNA‐300 regulates the ubiquitination of PTEN through the CRL4B(DCAF13) E3 ligase in osteosarcoma cells. Mol Ther Nucleic Acids 10, 254–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ge MK, Zhang N, Xia L, Zhang C, Dong S‐S, Li Z‐M, Ji Y, Zheng M‐H, Sun J, Chen G‐Q & et al, (2020) FBXO22 degrades nuclear PTEN to promote tumorigenesis. Nat Commun 11, 1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Song MS, Salmena L, Carracedo A, Egia A, Lo‐Coco F, Teruya‐Feldstein J & Pandolfi PP (2008) The deubiquitinylation and localization of PTEN are regulated by a HAUSP‐PML network. Nature 455, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yuan L, Lv Y, Li H, Gao H, Song S, Zhang Y, Xing G, Kong X, Wang L, Li Y et al, (2015) Deubiquitylase OTUD3 regulates PTEN stability and suppresses tumorigenesis. Nat Cell Biol 17, 1169–1181. [DOI] [PubMed] [Google Scholar]
- 69. Je EM, Lee SH & Yoo NJ (2012) Somatic mutation of a tumor suppressor gene BAP1 is rare in breast, prostate, gastric and colorectal cancers. APMIS 120, 855–856. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The protein level of BAP1 in renal carcinoma, lung cancer and breast cancer cell lines.
Fig. S2. BAP1 suppresses PCa cell migration.
Fig. S3. BAP1 suppresses PCa progression.
Fig. S4. PTEN interacts with BAP1 in the cytoplasm.
Fig. S5. BAP1 deubiquitinates PTEN.
Fig. S6. Downregulated PTEN expression promotes activation of Akt pathway and PCa cell colony formation.
Fig. S7. BAP1 inhibits tumorigenesis through maintenance of PTEN.
Fig. S8. BAP1 suppresses PCa progression in an Akt‐dependent manner.
Table S1. List of DNA or RNA oligonucleotides.
Table S2. The list of clinical cancers used for correlation analysis.
Data Availability Statement
All data required to evaluate the conclusions of the paper are present in the main text or the Supplementary Materials of the paper.