

Abstract
The absence of fucose on asparagine-297 of the human immunoglobulin G (IgG) heavy chain has been shown to enhance antibody-dependent cellular cytotoxicity (ADCC) activity by 10- to 100-fold compared to fucosylated antibody. Our lab is studying the use of adeno-associated virus (AAV) as a vector for the delivery of HIV-specific antibodies for therapeutic purposes. Since the antibody is produced by vector-transduced cells in vivo, current techniques of glycoengineering cannot be utilized. In order to achieve similar enhancement of ADCC with AAV-delivered antibodies, short hairpin RNAs (shRNAs) that target fucosyltransferase-8 (FUT8), were designed, tested, and cloned into AAV vectors used to deliver HIV-specific broadly neutralizing antibodies (bNAbs). Antibodies produced by our glycoengineered-AAV (GE-AAV) vectors were analyzed for fucose content and ADCC. GE-AAV constructs were able to achieve over 80% knockdown of FUT8. Results were confirmed by lectin western blot for α1-6 fucose, which revealed almost a complete absence of fucose on GE-AAV-produced antibodies. GE-AAV-produced antibodies revealed >10-fold enhancement of ADCC, while showing identical neutralization and gp140 trimer binding compared to their fucosylated counterparts. ADCC was enhanced 40- to 60-fold when combined with key Fc mutations known to enhance binding to FcγRIIIA. Our findings define a powerful approach for supercharging AAV-delivered anti-HIV antibodies.
Keywords: fucosyltransferase 8, antibody-dependent cellular cytotoxicity, broadly neutralizing antibodies, glycoengineering, adeno-associated virus, viral reservoir, gene therapy, immunotherapy
Graphical Abstract
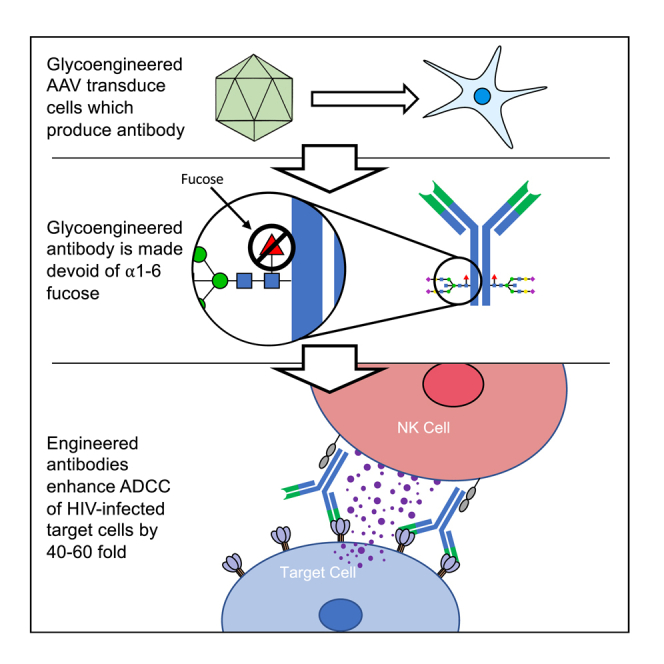
Termini et al. describe the creation and validation of a novel glycoengineered-AAV vector for the delivery of anti-HIV antibodies. These AAV are capable of knocking down fucosyltransferase-8 following transduction and, when combined with antibody-Fc mutations, produce broadly neutralizing antibodies with 40- to 60-fold higher ADCC.
Introduction
While continuous antiviral drug therapy can suppress HIV replication for many years, even lifelong treatment does not represent a cure. The vast majority of effectively-suppressed individuals quickly show viral rebound when treatment is removed.1,2 The cause of this rebound is the large HIV reservoir of latently infected cells where virus persists and can reactivate at any time.3 In recent years, this latent viral reservoir has received much attention as it has been suggested to be the last critical hurdle in the search for a cure. Although viral reactivation does occur at very low levels, the HIV reservoir is typically transcriptionally silent.3,4 HIV can reactivate from latently infected cells spontaneously or upon activation, leading to viral rebound in the absence of anti-retroviral therapy (ART). Because these reservoirs are long-lived, one proposed strategy to target the viral reservoir is to reverse latency. By using latency reversing agents, one can reactivate HIV transcription, protein expression, and virion production. This approach, commonly referred to as shock and kill, aims at reducing or potentially eliminating the reservoir by causing latently infected cells to undergo cytolysis and/or immune-mediated clearance.5,6 This approach is typically performed while the patient is on ART to prevent infection of new cells and the potential establishment of a new reservoir. Although latency reversing agents have been shown to reactivate latent HIV in the clinic, many have failed to demonstrate a reduction of the viral reservoir or have exhibited a limited clearance of reactivated cells.7, 8, 9
Antibody-based approaches to therapy have been made possible by an incredible array of monoclonal antibodies (mAbs) with potent neutralizing activity against a broad range of HIV isolates. These broadly neutralizing antibodies (bNAbs) are typically capable of neutralizing the majority of circulating HIV strains. Passive administration of combinations of these potent broadly-neutralizing anti-HIV antibodies has been shown to dramatically reduce levels of replicating virus as impressively as the drug therapies currently in routine use.10, 11, 12, 13, 14, 15, 16, 17, 18 However, in contrast to most drug therapies that block viral replication in the downstream cell, antibody-based therapies can not only neutralize newly produced virus, but their effector functions can also target and kill the latently infected reservoir cells that are reactivated to express virus.19 Although passive administration of bNAbs is an attractive approach for the treatment of HIV, regular injections are required to maintain serum concentrations that are high enough for therapeutic efficacy.10, 11, 12, 13 Also, large amounts of protein are required for regular injections, making passive immunization a costly therapy that is prone to adherence issues. For these reasons, many in the field have turned their attention to adeno-associated virus (AAV) as a vector to deliver mAbs. AAV delivery will allow for a single administration to provide long-term virologic suppression. Our lab has previously demonstrated that rhesus macaques can maintain high levels of delivered bNAbs following a single AAV intramuscular injection.20,21 In some monkeys such as macaque 84-05, these levels can reach 300–400 μg/mL20 and are capable of persisting indefinitely post AAV administration.22 Additionally, long-term virologic suppression has been exhibited in macaques such as the Miami Monkey.21,23 While the suppression exhibited in the Miami Monkey represents a functional cure, this cannot be considered an actual sterilizing cure; viral reservoirs remain present and SHIV was recovered from the Miami Monkey after 84 weeks of undetectable viral loads.21 These studies beg the question: might there be ways of more effectively targeting the viral reservoirs with such antibody-based therapies?
One way to potentially improve the targeting of the viral reservoir by antibody-based therapies can be taken from the cancer therapeutics field. To maximize the efficiency of cancer treatment, many groups have explored modification to the Fc sequence and glycan content of immunoglobulin G (IgG), which can increase antibody Fc binding and effector functions. Of these methods, glycoengineering is one of the most effective. IgG contains a single N-linked oligosaccharide at asparagine 297 (Asn-297) with an optional α1-6 fucose residue on the first N-acetylglucosamine. Removal of the α1-6 fucose at Asn-297 can drastically enhance antibody-dependent cellular cytotoxicity (ADCC).24, 25, 26 When anti-CD20 IgG1 (Rituximab) was made in a non-fucosylated form, higher affinity for FcγRIIIA and a 100-fold increase in B cell depleting activity was observed.27 Much lower concentrations of antibody were necessary to achieve identical clinical efficacy. Glycoengineering also holds enormous potential for the treatment of HIV. High levels of ADCC have been associated with slowed progression, better viral control, and lower viral set points.28, 29, 30, 31, 32, 33 Glycoengineering has also been successful at enhancing the potency of anti-HIV mAbs; when b12 was produced devoid of fucoslyation, 10-fold higher levels of viral inhibition were observed when compared to wild-type b12.34 ADCC has also been suggested to be effective in the clearance of reactivated latent HIV-1 reservoirs.35 These findings suggest that glycoengineering may be an important avenue to pursue in attempts to maximize the effectiveness of AAV-delivered mAbs.
In the current study, we set out to develop an AAV viral vector capable of glycoengineering in vivo. In vivo glycoengineering via AAV transduction has not previously been demonstrated. By using short hairpin RNA (shRNA) expressed by our AAV vector to knock down fucosyltransferase-8 (FUT8, Gene ID: 2530), the only glycosyltransferase capable of α1-6 fucoslyation at Asn-297,36 we were able to glycoengineer AAV-delivered IgG to enhance ADCC activity. By increasing the ADCC potential of AAV-delivered, HIV-specific antibodies 10- to 100-fold, we seek to increase the antibodies’ ability to impact, and perhaps even destroy, the competent viral reservoir.
Results
Generation and validation of FUT8 knockout (KO) cell lines
FUT8 KO cell lines were generated using a well-established protocol from our lab previously used to generate cell lines deficient in O-linked glycosylation.37 CRISPR-Cas9 was used to target and disrupt the human FUT8 gene. To achieve this, gRNA expression vectors were cloned to include a green fluorescent protein (GFP) tag. HEK293T cells were transfected with three gRNA expression vectors all targeting FUT8 to ensure complete disruption of the gene. Cells within the top 20% of GFP-expression were sorted (Figure 1A) into 96-well plates with one cell per well to establish clonal lines. Cells were allowed to grow to confluence before confirming that all copies of the FUT8 gene were disrupted.
Figure 1.

Generation of a FUT8 KO cell line
HEK293T cells were transfected with three gRNA targeting FUT8 and a CAS9/GFP expression plasmid. (A) After 24 h, cells were sorted and the 20% highest GFP expressing cells were individually sorted into a 96-well plate and cultured until confluent. (B) Four of the FUT8 KO clones isolated from cell sorting and a HEK293T wild-type control were transfected with a 10-1074 expression plasmid. After an additional 4 days, supernatant was harvested and filtered to remove cell debris. IgG was purified by protein A column and 3 μg were run on 4%–12% bris-tris gels in duplicate. After the transfer, one membrane was stained with anti-IgG-HRP and the other was probed with AAL-HRP lectin to visualize the presence of α1-6 fucose.
Four HEK293T-FUT8 KO clones were analyzed for their ability to fucosylate human IgG. Expression vectors encoding the human anti-HIV mAb 10-1074 were transiently transfected into FUT8 KO clones, as well as the HEK293T parental cell line. To avoid bovine serum IgG contamination in our purified antibody preparations, we continuously cultured cell lines in ultra-low IgG FBS followed by a PBS wash and media change to serum-free media 1 day post-transfection. 5 days post-transfection, secreted 10-1074 was affinity-purified using protein A columns. Purified antibodies were analyzed by western blot using an IgG probe to ensure equal amounts of protein were loaded into each lane and by lectin western blot using a Aleuria aurantia lectin-horseradish peroxidase (AAL-HRP) lectin to detect α1-6 fucose presence on the IgG (Figure 1B). Aleuria aurantia lectin (AAL) is known to bind specifically to α1-6 fucose, which can only be added to a growing N-glycan chain by FUT8. Therefore, lack of α1-6 fucose would confirm a lack of FUT8 enzymatic activity. Although equal amounts of IgG were loaded in each lane, there was no detectable α1-6 fucose present on IgG produced in the four FUT8 KO cell lines (Figure 1B).
Design and development of GE-AAV vectors
Due to the high similarity between human and rhesus FUT8 genes, we designed five shRNAs capable of targeting both human and rhesus FUT8. By selecting shRNAs that can target both human and rhesus FUT8, GE-AAV vectors can be used for both in vitro work with human cell lines and macaque animal experiments. Candidate human shRNAs were aligned to rhesus macaque FUT8 to demonstrate the homology (Figure 2A). Only shRNA 61 had a one base-pair difference when compared to the rhesus FUT8 sequence.
Figure 2.

FUT8 shRNA and glycoengineering AAV design
Five candidate shRNAs were selected to regions of FUT8 with >99% homology between human and rhesus macaque FUT8. (A) Candidate shRNAs (in caps) were aligned to rhesus macaque FUT8 (lowercase) to demonstrate the homology. Only shRNA 61 had a one base pair difference when compared to the rhesus FUT8 sequence. (B) HEK293T cells were transfected with pLKO.1 expression vector with each candidate shRNA. After 24 h, cells were harvested and analyzed for FUT8 mRNA expression by real-time PCR in triplicate. Data are presented as percentage knockdown compared to wild-type HEK293T cells. (C) Diagram depicting the design of the GE-AAV knockdown constructs. The poly(A) depicted is the poly(A) tail of the IgG being expressed by the AAV. U6, H1, and 7SK promoters were used to drive the expression of individual shRNAs. Spacer size was adjusted between constructs to maximize knockdown while maintaining maximal IgG expression. (D) Diagram depicting the cloning of the FUT8 knockdown constructs into the AAV vector. The construct was inserted downstream of the poly(A) tail and upstream of the 3′ ITR.
Candidate shRNAs were cloned into the pLKO.1 expression vector under the control of a U6 promoter. Expression vectors were transfected into HEK293T cells and levels of FUT8 mRNA were measured by real-time PCR. Although all clones exhibited high levels of knockdown, shRNAs 52, 53, and 59 exhibited the highest levels (Figure 2B). All three clones exhibited greater than 60% knockdown, with clone 59 achieving the highest knockdown approaching 80%. These three shRNAs were chosen for the development of our GE-AAV vectors.
GE-AAV constructs were designed such that the shRNA insert region did not exceed 1,000 base pairs due to packaging limitations of the single-stranded AAV (ssAAV) vector. Constructs with varying spacer lengths and number of shRNAs were tested (Figure 2C). All shRNAs were designed to have independent Pol III promoters. U6, H1, and 7SK promoters were used to drive the expression of each individual shRNA. Care was taken to use the strongest promoter to drive the shRNA that exhibited the highest levels of FUT8 knockdown. shRNA constructs were cloned into our ssAAV vector downstream of the IgG poly(A) tail and upstream of the 3′ ITR using an existing SalI restriction site and screening for proper orientation in the final constructs (Figure 2D).
Spacer length was quickly identified as an important variable for construct design. Early versions of constructs #1 and #2 were designed with the U6 promoter directly after the SalI restriction site downstream of the IgG poly(A) tail. These constructs were found to have reduced antibody production (34% and 47%, respectively) when compared to ssAAV-antibody constructs without shRNA constructs. Once an 83 bp spacer was added between the SalI restriction site and the beginning of the U6 promoter, we observed antibody production comparable to that of the wild-type ssAAV-antibody vectors. Similarly, if the individual shRNA and the start of the next Pol III promoter were in close proximity, a decrease in FUT8 knockdown was observed (data presented in the GE-AAV validation section below). Spacer lengths were optimized to allow for maximal FUT8 knockdown without inhibiting IgG expression. The five constructs presented here are a result of this optimization.
Validation of GE-AAV vectors
AAV plasmid DNA encoding 4L6 IgG and the various FUT8 shRNA constructs was transfected into HEK293T cells. After 24 h, cells were harvested and real-time PCR was performed on the transfected cells in order to measure levels of human FUT8 mRNA. Percent knockdown was calculated compared to FUT8 mRNA levels present in non-transfected HEK293T control cells. Constructs #5, #6, and #7 demonstrated relative knockdown of approximately 80% (Figure 3A). Surprisingly, construct #5, while only containing one shRNA, demonstrated similar levels of knockdown to construct #7, which contained three different shRNAs targeting FUT8.
Figure 3.

FUT8 knockdown validation by GE-AAV constructs
HEK293T and FRhK-4 cells were transfected with plasmid DNA of the AAV vector plasmids containing the shRNA constructs outlined in Figure 2. (A and B) After 24 h, (A) HEK293T cells and (B) FRhK-4 were harvested and analyzed for FUT8 mRNA by real-time PCR. Samples were analyzed in triplicate. Data are presented as percentage knockdown as compared to wild-type HEK293T cells. (C) Wild-type HEK293T cells were transfected with AAV vector plasmids expressing 4L6 with or without a FUT8 shRNA construct. After 18 h, cells were washed and transferred to serum-free media for an additional 3 days. Media was aspirated and replaced with fresh serum-free media to eliminate IgG produced before full knockdown of FUT8. On day 7, supernatant was harvested and filtered to remove cell debris. IgG was purified using a protein A column and 3 μg were loaded onto 4%–12% bris-tris gels in duplicate. After transfer, one membrane was stained with anti-IgG-HRP and the other was probed with AAL-HRP lectin to visualize the presence of α1-6 fucose.
To confirm that our shRNA constructs were equally effective in rhesus cells as they are in human HEK293T cells, we also assessed FUT8 knockdown in FRhK-4 cells, a rhesus epithelial cell line derived from Macaca mulatta. Although FRhK-4 cells are more difficult to transfect than HEK-239T, transfection conditions were optimized for this particular cell line to obtain similar levels of transfection efficiency. Similar to what was observed in HEK293T cells, our FUT8 shRNA constructs produced high levels of knockdown of rhesus FUT8 in the FRhK-4 cells (Figure 3B). In the FRhK-4 cells, construct 7 achieved the highest levels of knockdown (∼80%). These levels are similar to the levels achieved in human HEK293T cells (Figure 3A). These data suggest that the FUT8 shRNA constructs are effective at knocking down rhesus FUT8.
AAV plasmid DNA encoding 4L6 IgG and the various FUT8 shRNA constructs was transfected into HEK293T cells. After 3 days, cells were washed and media replaced to remove any antibody secreted before optimal FUT8 knockdown. After an additional 4 days, supernatants were harvested and 4L6 antibody present in the supernatant was purified. To analyze the presence of fucose on the secreted 4L6, we analyzed purified antibody by western and lectin western blots. 4L6 antibody produced in wild-type HEK293T cells was loaded as a control. One blot was probed with anti-rhesus IgG-HRP and the other membrane was probed with AAL-HRP lectin to visualize the presence of α1-6 fucose (Figure 3C).
Although the IgG probe indicated that equal amounts of 4L6 antibody were indeed loaded per lane, when probed with AAL lectin, varying amounts of α1-6 fucose were observed. Constructs #5, #6, and #7 had the lowest levels of α1-6 fucose (Figure 3C). This is consistent with knockdown observed in the real-time PCR (Figure 3A). Although constructs #1 and #6, as well as constructs #2 and #7, contain the same promoters and shRNA sequences (Figure 2C), the difference in α1-6 fucose levels present on purified 4L6 was likely a result of increased spacer lengths between shRNAs (83 bp and 50 bp increase, respectively). Levels of α1-6 fucose present on 4L6 generated from constructs #6 and #7 were almost undetectable by western blot. These data suggest that knockdown levels would be sufficient to glycoengineer cells in vivo and that the majority of secreted antibody from a cell containing the engineered vector would lack α1-6 fucose.
ADCC of 10-1074 expressed by GE-AAV stable cell lines
Construct shRNA expression typically required around 3 days before achieving FUT8 knockdown levels sufficient to produce antibodies with low levels of a1-6 fucose. However, in vivo, following intramuscular injection of AAV, muscle cells will be able to continuously express IgG and our shRNA constructs indefinitely.22 Due to the high rate of transduction of muscle cells following intramuscular injection of AAV and the long-lived nature of this transduction, we set out to more accurately model this scenario in vitro. Constructs #2, #5, and #6 were chosen to make stable cell lines on the basis of the variety of shRNAs in each construct.
shRNA constructs were cloned into a lentiviral vector with a puromycin resistant selectable marker and a GFP tag. HEK293T cells were incubated with high titer lentiviruses for 48 h before adding 1 μg/mL puromycin. Puromycin dosage was escalated to 2 μg/mL after week one and to 4 μg/mL after week 2 to select for well-transduced cells. shRNA construct expression was monitored by flow cytometry for GFP expression (Figure 4A). High levels of GFP were observed in all constructs indicating high shRNA construct expression.
Figure 4.

shRNA construct and ADCC activity validation
HEK293T cells were transduced with lentivirus expressing constructs #2, #5, and #6. Lentiviruses also expressed a GFP tag along with a selectable puromycin resistance gene. 48 h after transduction, puromycin was added to the cell media and allowed to select for positively transduced cells. For an additional 2 weeks, puromycin concentration was gradually increased to select a population of high expressing cells. (A) Flow cytometry was performed on lentivirus-transduced cells to monitor the levels of GFP expression. These levels were expected to correlate with levels of shRNA construct expression. The GFP+ gate was set on HEK293T cells not expressing GFP. (B) 10-1074 IgG expressing plasmids were transfected into wild-type HEK293T cells, FUT8 KO cell lines, and lenti-transduced HEK293T cell lines expressing constructs #2, #5, or #6. 10-1074 was purified by protein A column and quantified. ADCC activity was assessed in triplicate. The dashed line indicates 50% RLU or 50% ADCC activity against HIV-1 NL(AD8)-infected target cells. The loss of RLU indicates the loss of virus-infected cells during the 8-h incubation period and represents higher ADCC activity. 10-1074 FUT8 was included as a positive control due to the complete lack of fucose on the purified IgG. Samples were analyzed for statistical significance compared to controls using a two-tailed paired t test.
HEK293T cells, FUT8 KO cell lines, and our FUT8 knockdown stable cell lines were transfected with an expression vector for 10-1074 IgG. Antibody was purified from the supernatant and ADCC activity was measured to quantify NK cell activity toward HIV-1 NL(AD8)-infected target cells.38 Effector cells were combined with infected target cells before the addition of serially diluted antibodies. ADCC activity was measured as a loss of luciferase activity at the end of an 8-h incubation.
As predicted, we observed a >10-fold enhancement of ADCC activity when 10-1074 was generated in the FUT8 KO cell line as compared to the wild-type (Figure 4B). Interestingly, 10-1074 produced in all three FUT8 knockdown stable cell lines also displayed ∼10-fold enhancement in ADCC activity, with only slightly less ADCC activity than observed in the FUT8 KO cell lines. There was no statistical different between ADCC of 10-1074 produced in FUT8 knockdown stable cell lines compared to ADCC of 10-1074 produced in FUT8 KO cell lines. ADCC activity is consistent with minimal amounts of fucose observed on purified 4L6 IgG in Figure 3C. These data suggest that antibody produced by AAV-transduced muscle cells in vivo should demonstrate a similar enhancement of ADCC activity.
ADCC of 3BNC117 following AAV2 transduction
To further confirm our hypothesis that GE-AAV transduced cells would produce monoclonal antibody (mAb) with enhanced ADCC, we produced AAV2 expressing constructs #6 and #7. For this experiment, constructs #6 and #7 were chosen due to the higher level of FUT8 knockdown as confirmed by real-time PCR and AAL lectin western blot (Figures 3A–3C). HEK293T culture supernatant was changed 72 h post-transduction to remove any antibody that may have been produced before optimal knockdown of FUT8.
AAV2-produced 3BNC117 IgG was compared to GE-AAV2-produced 3BNC117 from vectors containing FUT8 shRNA constructs #6 and #7. Both construct #6 and #7 produced 3BNC117 with significantly higher ADCC when compared to AAV2-produced 3BNC117 (p = 0.0003 and p = 0.0004) and 3BNC117 generated by HEK293T transfection (p = 0.0023 and p = 0.0103). Similar to the results observed in our stable cell line experiments (Figure 4B), IgG produced by AAV2-3BNC117-construct #6 exhibited similar levels of ADCC compared to 3BNC117 produced in the FUT8 KO cell line (Figure 5A). Although construct #7 displayed similar levels of knockdown as measured by real-time PCR (Figure 3A), levels of ADCC observed by AAV2 transduction were slightly less than that achieved by construct #6. Consistent with this finding, 3BNC117 produced by construct #7 had slightly higher levels of fucose when examined by AAL lectin western blot (Figure 3C). There was no significant difference in ADCC of AAV2-3BNC117 produced by construct #6 or construct #7 when compared to FUT8 KO-produced 3BNC117 (p = 0.6477 and p = 0.1556, respectively).
Figure 5.

GE-AAV production and ADCC activity validation
(A) 3BNC117 was produced in HEK293T cells by AAV2 transduction at an MOI of 100,000. 72 h after transduction, cells were washed and transferred to serum-free media. After an additional 4 days of culture, supernatants were harvested, clarified, and analyzed for 3BNC117 concentration by protein A/anti-rhesus IgG ELISA. Antibody concentrations were normalized and analyzed for ADCC activity. ADCC levels were compared to 3BNC117 produced in HEK293T cells, as well as FUT8 KO cells as a representation of ADCC in IgG devoid of fucose. Both control antibodies were produced by plasmid transfection. ADCC activity was measured against HIV-1 NL(AD8)-infected target cells in triplicate. Samples were analyzed for statistical significance using a two-tailed paired t test. (B) Genome copies per AAV preparation was calculated by taking the total genome copies divided by the number of preparations performed.
AAV2 yields appear to have been impacted by the size of the insert. Of the three AAV2 preparations, construct #7 had the lowest genome copies per AAV preparation with a yield of 5.8 times less than that of AAV2-3BNC117 (Figure 5B). When compared to construct #6, the reduction in yield was only 1.3 times lower when compared to AAV2-3BNC117. These data are consistent with the observation that as the size of shRNA insert approaches 1,000 base pairs, AAV yield may suffer.
Fc mutations in combination with glycoengineering
Another common way to increase antibody effector functions such as ADCC is through Fc mutations that increase affinity for the Fc receptor. However, little is known about the effects of combining Fc mutations with glycoengineering. In order to determine whether there is any added value in combining the two approaches for AAV vector-based delivery of anti-HIV antibodies, we also explored common Fc mutations used in antibody therapies. The first two Fc mutations tested were the LS (M428L/N434S)39 and LALA (L234A/L235A)40 mutations. The LS mutation, while having no effect on ADCC, is known to increase binding to the neonatal Fc receptor, consequently increasing serum half-life of an antibody.39 This mutation has been used for antibodies in passive transfer experiments, as well as in AAV gene transfer experiments.21,41 The second Fc mutation, LALA, has been shown to abrogate ADCC activity by disrupting the binding to FcγRIIIA.40,42 These mutants were also tested in combination and annotated LALA-LS in our experiments.
Both 10-1074 and 3BNC117 and their respective Fc mutants were produced in both HEK293T cells and FUT8 KO cells. Antibodies were purified and analyzed by Coomassie Blue to visualize total protein and AAL lectin blot to visualize the presence of α1-6 fucose (Figures 6A and 7A). Although equal amounts of antibody were loaded per lane, both 10-1074 and 3BNC117 produced in FUT8 KO cells were devoid of α1-6 fucose.
Figure 6.

ADCC of common 10-1074 Fc variants lacking fucose
10-1074 was produced in both HEK293T cells and FUT8 KO cell lines as wild-type IgG, LS mutant, LALA mutant, or a combination of LALA and LS mutants. Antibodies were purified by protein A column. (A) 3 μg IgG were loaded onto a 4%–12% bis-tris gel in duplicate. One was processed for Coomassie staining, the second was probed with AAL lectin to monitor the presence of α1-6 fucose. (B) SF162 gp140 trimer binding ELISA. Starting at an antibody concentration of 1 μg/mL followed by 3-fold serial dilutions, 10-1074 variants were incubated for ELISA measurement against SF162 gp140 trimer. High absorbance indicates high binding. (C) Neutralization curve of HIV-1 NL(AD8) with 10-1074 IgG1 starting at 0.625 μg/mL. The dashed line indicates 50% RLU representing 50% neutralization activity against HIV-1 NL(AD8). Lowest RLU indicates highest neutralization. (D) 10-1074 variants were tested for ADCC activity in triplicate. The dashed line indicates 50% RLU or 50% ADCC activity against HIV-1 NL(AD8)-infected target cells. ADCC was measured by the luciferase activity in HIV-infected cells after an 8-h incubation in the presence of a human CD16+ NK cell line and a serial dilution of antibodies. The loss of RLU indicates the loss of virus-infected cells during the 8-h incubation period and represents higher ADCC activity. Samples were analyzed for statistical significance compared to controls in (B)–(D) using a two-tailed paired t test.
Figure 7.

ADCC of common 3BNC117 Fc variants lacking fucose
3BNC117 was made in both HEK293T cells and FUT8 KO cell lines as wild-type IgG, LS mutant, LALA mutant, or a combination of LALA and LS mutants. Antibodies were purified by protein A column. (A) 3 μg IgG were loaded onto a 4%–12% bis-tris gel in duplicate. One gel was processed for Coomassie staining, the second was transferred and probed with AAL lectin to monitor the presence of α1-6 fucose. (B) SF162 gp140 trimer binding ELISA. Starting at an antibody concentration of 1 μg/mL followed by 3-fold serial dilutions, 3BNC117 variants were incubated for ELISA measurement against SF162 gp140 trimer. High absorbance indicates high binding. (C) Neutralization curve of HIV-1 NL(AD8) with 3BNC117 IgG1 starting at 0.625 μg/mL. The dashed line indicates 50% RLU representing 50% neutralization activity against HIV-1 NL(AD8). Lowest RLU indicates highest neutralization. (D) 3BNC117 variants were tested for ADCC activity in triplicate. The dashed line indicates 50% RLU or 50% ADCC activity against HIV-1 NL(AD8)-infected target cells. ADCC was measured by the luciferase activity in HIV-infected cells after an 8-h incubation in the presence of a human CD16+ NK cell line and a serial dilution of antibodies. The loss of RLU indicates the loss of virus-infected cells during the 8-h incubation period and represents higher ADCC activity. Samples were analyzed for statistical significance compared to controls in (B)–(D) using a two-tailed paired t test.
Next, the antibodies were analyzed for their ability to bind gp140 using an SF162 gp140 trimer binding ELISA. 10-1074 and 3BNC117 variants were serially diluted and incubated with plate-bound gp140 trimer. As expected, neither glycoengineering nor Fc mutations had any impact on gp140 binding (Figures 6B and 7B). There was no significant difference in gp140 binding between any of the samples tested. Importantly, the consistency of the assay when comparing different antibodies suggests little to no variability in protein quantification of the IgG. These data suggest that any differences observed in ADCC activity are truly due to the antibody modifications and not due to sample to sample variability.
To determine whether glycoengineering in combination with Fc receptor mutations had any impact on neutralization capacity, we performed neutralization assays with 10-1074, 3BNC117, and their variants (Figures 6C and7C). Neutralization assays were carried out using HIV-1 NL(AD8). Lowest relative light units (RLU) indicates highest neutralization. As expected, neither glycoengineering nor Fc receptor mutations, individually or in combination, impacted the ability of 10-1074 and 3BNC117 to neutralize HIV-1 NL(AD8). There was no significant difference in neutralization between any of the samples tested.
10-0174 and 3BNC117 variants were also tested for ADCC activity (Figures 6D and 7D). Consistent with what was observed with 4L6, 10-1074 and 3BNC117 both display >10-fold enhancement of ADCC when produced in the FUT8 KO cell line as compared to wild-type HEK293T cells (p = 0.0015 and p = 0.0187, respectively; Figures 6D and 7D). As expected, the LS mutation did not impact ADCC activity even in combination with FUT8 KO. However, the LALA mutation produced some unexpected results. The LALA mutation has been shown to abrogate ADCC activity.42 This can be observed in both the 10-1074-LALA and 3BNC117-LALA. ADCC activity for the LALA mutants is almost non-existent even at the highest antibody concentrations. However, when 10-1074-LALA and 3BNC117-LALA IgG were produced in FUT8 KO cells, ADCC activity was enhanced to levels significantly greater than wild-type 10-1074 and 3BNC117 (p = 0.0002 and p = 0.0392, respectively). Both antibodies exhibited a ∼5-fold enhancement of ADCC activity compared to wildtype 10-1074 and 3BNC117 (Figures 6D and 7D).
Due to the observation that the ADCC activity of LALA mutant IgG can not only be restored but enhanced by the removal of α1-6 fucose, we set out to determine whether increased ADCC levels associated with other Fc mutations could be further enhanced by removal of α1-6 fucose. If so, Fc mutations to enhance ADCC would be an ideal approach to combine with our GE-AAV vectors. For these experiments, we selected the well documented DEL mutation (S239D/I332E/A330L)43. Both 10-1074-DEL and 3BNC117-DEL were cloned and then produced in HEK293T and FUT8 KO cells. ADCC activity was determined from purified IgG (Figures 8A and 8B). DEL mutations significantly enhanced ADCC activity by ∼10 fold when compared to wild-type 10-1074 and 3BNC117 (p = 0.0061 and p = 0.0385). These enhancements in ADCC were almost identical to those observed by fucose removal with no significant difference between the two groups. However, when combined, 3BNC117 FUT8 KO DEL and 10-1074 FUT8 KO DEL displayed an additive effect on ADCC activity with a ∼40- to 60-fold enhancement when compared to the wild-type antibodies, respectively (p = 0.0071 and p = 0.0159; Figures 8A and 8B).
Figure 8.

10-1074 and 3BNC117 ADCC enhancement by combining FUT8 removal with Fc mutation
10-1074 and 3BNC117 were produced in HEK293T cells and FUT8 KO cells. Antibodies were also made with the DEL mutations (S239D/I332E/A330L). Variants were tested for ADCC activity in triplicate. The dashed line indicates 50% RLU or 50% ADCC activity against HIV-1 NL(AD8)-infected target cells. ADCC was measured by the luciferase activity in HIV-infected cells after an 8-h incubation in the presence of a human CD16+ NK cell line and a serial dilution of antibodies. The loss of RLU indicates the loss of virus-infected cells during the 8-h incubation period and represents higher ADCC activity. (A) 10-1074 IgG1 variant ADCC assay. (B) 3BNC117 IgG1 variant ADCC assay. Samples were analyzed for statistical significance compared to controls using a two-tailed paired t test.
Discussion
While AAV delivery of bNAbs has shown great promise for impressive long-term virologic suppression,21 our results suggest that there may be ways to improve its efficacy even further. Due to the fact that AAV-delivered antibodies are produced in muscle cells in vivo and not in engineered cell lines, normal methods of glycoengineering cannot be utilized. To our knowledge, our study is the first to demonstrate glycoengineering of AAV-produced proteins. In this study, we were able to clone shRNA constructs capable of knocking down FUT8 mRNA expression by ∼80% in the AAV vector backbone (Figures 3A and 3B). These vectors were able to produce 4L6 antibody with drastically reduced fucose content (Figure 3C). shRNA constructs were able to knock down expression of both human and macaque FUT8 (Figures 3A and 3B), allowing for future experiments in our SIV- and SHIV-infection models in rhesus macaques. These constructs were small enough to allow for successful packaging into the AAV2 capsid (Figure 5B). These data predict that antibody produced by GE-AAV-transduced human and macaque muscle cells will produce IgG with reduced α1-6 fucose and enhanced ADCC activity.
To further demonstrate this concept, we engineered HEK293T cells to stably express candidate FUT8 shRNA constructs. Due to the high level of transduction of muscle cells by AAV and the long-lived expression of the delivered construct and transgene, we felt that this would more accurately simulate the expected results of in vivo administration of our AAV constructs. As expected, antibody produced in stable cell lines expressing FUT8 shRNA constructs #2, #5, and #6 exhibited ∼10-fold higher ADCC activity as compared to wild-type 10-1074 (Figure 4B). More importantly, these antibodies displayed similar levels of ADCC activity to those of 10-1074 produced in a CRISPR-Cas9 generated FUT8 KO cell line. These data suggest that our FUT8 shRNA constructs are capable of producing high levels of FUT8 knockdown and that prolonged expression in an in vivo model will likely result in bNAbs with little to no α1-6 fucose content.
Most importantly, when HEK293T cells were transduced with our GE-AAV2 expressing both 3BNC117 and our shRNA constructs, there was a similar enhancement in ADCC (Figure 5A). Construct #6 displayed levels of ADCC that were almost identical to those of 3BNC117 produced in the FUT8 KO cell line. Due to the low transduction efficiency of AAV in vitro we found these results promising as they would suggest similar if not better performance in vivo. Although the ADCC activity of construct #7 was slightly lower than expected, we still observed a significant enhancement in ADCC activity as compared to that of wild-type AAV2-delivered 3BNC117. This finding is consistent with the observed fucose content in Figure 3C. Although construct #7 contains 3 shRNAs compared to 2 shRNAs in construct #6, less fucose was observed on antibodies produced by construct #6. It is also important to note that we observed a large decrease in AAV yield from construct #7 due to the size approaching the packaging limit (Figure 5B). These data suggest that smaller constructs with fewer shRNAs can be equally effective as compared to larger constructs and can lead to enhanced packaging of viral vectors with higher virus yields.
Because α1-6 fucose and the Fc mutations both affect Fc receptor binding and downstream effector functions, we felt it was important to determine whether common Fc mutations could impact the ADCC enhancements realized by FUT8 removal. We started off testing the LS (M428L/N434S)39 and LALA (L234A/L235A)40 mutations. Although there was no impact to the gp140 binding and HIV neutralization (Figures 6B, 6C, 7B, and 7C), α1-6 fucose removal was able to enhance the ADCC of both the LS and LALA mutant antibodies (Figures 6D and 7D). Interestingly, the LALA mutation was previously believed to abrogate all ADCC activity. However, when produced in a FUT8 KO cell line, ADCC activity of 10-1074-LALA and 3BNC117-LALA antibodies was significantly improved relative to the wild-type HEK293T-produced antibodies. This finding is very important for the therapeutic antibody field. If an AAV-delivered LALA antibody were to transduce a cell type with low rates of fucoslyation, the resulting antibody could have high levels of ADCC, negating the safety aspects of the LALA mutation.
Because high levels of ADCC have been associated with slowed HIV progression, better viral control, and lower viral set points,20,28,29,31, 32, 33,44 we attempted to maximize the ADCC activity of AAV-delivered IgG by combining Fc mutations known to enhance ADCC with α1-6 fucose removal. Could the combination possibly be additive? If so, this increased ADCC activity could be beneficial to target and destroy viral reservoirs. As suggested by our data with the DEL mutant IgG (Figures 8A and 8B), Fc mutations and reduced fucose content can additively enhance ADCC activity. For instance, only 4.2 ng/mL of 10-1074 FUT8 KO DEL was required to induce the same levels of ADCC as 240.6 ng/mL of wild-type 10-1074. This 57-fold enhancement in ADCC could have a dramatic impact on the efficacy of AAV-delivered bNAbs. Since both FUT8 shRNA knockdown and Fc receptor mutations can easily be combined in a single AAV vector, examination of their performance in monkey trials is clearly warranted. Whether such supercharged mAbs continuously expressed from AAV vector can take the infectious viral reservoir to undetectable levels remains to be seen.
Materials and methods
gRNA generation
Guide RNAs (gRNAs) (SantaCruz Biotechnology, Dallas, TX, USA) were generated to our gene of interest: FUT8 (Gene ID: 2530). Target sequences were determined using the GeCKO v2 human library. Three gRNAs to FUT8 were used, targeting both strands of DNA, to ensure full KO of the gene of interest. gRNAs were cloned into an expression vector with a GFP tag to allow for single cell GFP sort.
FUT8 CRISPR-Cas9 KO gRNAs
1: 5′-ACGCGTACTCTTCCTATAGC-3′
2: 5′-ATTGATCAGGGGCCAGCTAT-3′
3: 5′-TACTACCTCAGTCAGACAGA-3′
Cell line generation
HEK293T cells (ATCC, Manassas, VA, USA) were transfected with three gRNAs for human FUT8 using JetPrime transfection reagent (Polyplus-Transfection, New York, NY, USA). Cells were examined by GFP fluorescent microscopy at 24 h post-transfection using a Zeiss Axio Observer A1 Microscope to gauge sufficient levels of expression necessary for downstream flow cytometric analysis and cell sorting. Following microscopy, cells were harvested, washed in PBS, and resuspended in DMEM with 1 mM EDTA to prevent the formation of cell aggregates. Cells were sorted on a 5-laser 17-color BD FACS SORP Aria-IIu with an Automatic Cell Deposition Unit (ACDU). The top 20% GFP-expressing cells were individually sorted into a 96-well plate. FSC-W by FSC-A and SSC-W by SSC-A were used to reduce the rate of duplets. 4 h post-sort, cells were inspected to ensure that all wells contained only one cell. Any wells that contained duplets were excluded from further processing. Once clones reached confluence in a 6-well plate, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific, Waltham, MA, USA) and used for western blot analysis.
Upon acceptance of this manuscript, FUT8 KO cell line clone 4 will be licensed for distribution through the University of Miami Office of Technology Transfer and will be commercially available for purchase.
Western blots to confirm FUT8 CRISPR KO
Four FUT8 KO clones were selected for further screening. HEK293T wild-type control and four FUT8 KO clones were transfected with a 10-1074 monoclonal antibody expression plasmid using JetPrime transfection reagent (Polyplus-Transfection). After 18 h, cells were washed with and transferred to BIO-MPM-1 serum-free media (Biological Industries, Kibbutz Beit-Haemek, Israel). After an additional 4 days, supernatant was harvested and filtered through a 0.45 μm asymmetric polyethersulfone (aPES) filter (Thermo Fisher Scientific) to remove cell debris. IgG was purified using HiTrap protein A column (GE Healthcare) and 3 μg was run on 4%–12% bris-tris gels (Thermo Fisher Scientific) in duplicate. 10-1074 antibody produced in wild-type HEK293T cells was loaded in the first lane as a control. Protein was transferred to a polyvinylidene fluoride (PVDF) membrane using the iBlot Dry Blotting System (Thermo Fisher Scientific). After transfer, one membrane was probed with anti-human IgG-HRP (SouthernBiotech, Birmingham, AL, USA) using the iBind western system (Thermo Fisher Scientific) and the other blot was probed with Aleuria aurantia lectin-HRP (AAL-HRP) (BioWorld, Dublin, OH, USA) to visualize the presence of α1-6 fucose. Membranes were developed using the SuperSignal Pico Substrate (Thermo Fisher Scientific) and images were captured on an ImageQuant LAS 4000 mini Luminescent Image Analyzer (GE Healthcare).
FUT8 shRNA design
Five candidate shRNAs were selected to regions of human FUT8 with near identical homology between human and rhesus macaque FUT8. Candidate shRNAs were aligned to rhesus macaque FUT8 to demonstrate the homology using Serial Cloner 2-6-1. Candidate shRNAs were cloned into the pLKO.1 expression vector to allow for transfection experiments.
FUT8 real-time PCR
Wild-type HEK293T cells and FRhK-4 cells, a rhesus epithelial cell line from Macaca mulatta, were transfected with pLKO.1 expression vector with each candidate shRNA, GE-AAV vector plasmids, and GFP expression vector where indicated using JetPrime transfection reagent (Polyplus-Transfection). After 24 h, cells were harvested and washed with PBS. FUT8 shRNA expression was analyzed by real-time PCR using the TaqMan Gene Expression Assay HS00189535_m1 (Thermo Fisher Scientific) and the Cells-to-CT 1-Step TaqMan kit (Thermo Fisher Scientific) according to the manufacturer’s specified protocol. Data are presented as percentage knockdown compared to wild-type HEK293T cells.
GE-AAV cloning
Coding sequences for antibodies 4L6 and 10-1074 were cloned into a single-stranded AAV (ssAAV) vector as previously described45 using a bicistronic expression cassette containing F2A peptide and a furin peptide. All antibody sequences were codon-optimized and synthesized by Genscript.
shRNA constructs were constructed to include one, two, or three shRNAs targeting various regions of FUT8 and all under the control of individual Pol III promoters. Pol III promoters included were the U6, 7SK, and H1. The strongest promoters were used to drive expression of the shRNA that exhibited the highest levels of knockdown by real-time PCR. shRNA constructs were cloned into the ssAAV vectors containing 4L6 and 10-1074 antibody sequences. shRNA constructs were inserted downstream of the poly(A) tail and upstream of the 3′ ITR.
All GE-AAV vectors were tested for levels of knockdown by real-time PCR as described above. Full sequences of all shRNA constructs are included in Table S1.
Lectin western blots to confirm FUT8 knockdown
GE-AAV vectors expressing 4L6 and FUT8 shRNA knockdown constructs were transiently transfected into HEK293T cells using JetPrime transfection reagent (Polyplus-Transfection). After 18 h, cells were washed with and media was replaced with BIO-MPM-1 serum-free media (Biological Industries). After an additional 4 days, supernatant was harvested and filtered through a 0.45 μm aPES filter (Thermo Fisher Scientific) to remove cell debris. IgG was purified using HiTrap protein A column (GE Healthcare) and 3 μg was run on 4%–12% bris-tris gels (Thermo Fisher Scientific) in duplicate. 4L6 antibody produced in wild-type HEK293T cells was loaded in the first lane as a control. Protein was transferred to a PVDF membrane using the iBlot Dry Blotting System (Thermo Fisher Scientific). After transfer, one membrane was probed with anti-human-IgG-HRP (SouthernBiotech) using the iBind western system (Thermo Fisher Scientific) and the other blot was probed with AAL-HRP lectin (BioWorld) to visualize the presence of α1-6 fucose. Lectin blots were blocked, probed, and washed using RIPA buffer (Thermo Fisher Scientific). Membranes were developed using the SuperSignal Pico Substrate (Thermo Fisher Scientific) and images were captured on an ImageQuant LAS 4000 mini Luminescent Image Analyzer (GE Healthcare).
shRNA knockdown cell lines
shRNA constructs #2, #5, and #6 were cloned into the pGFP-C-shLenti lentiviral vector. Lentivirus was packaged using the Lenti-vpak packaging kit (Origene, Rockville, MD, USA) according to the manufacturer’s specified protocol. HEK293T cells were plated into a 6 well plate the day before transduction at a density in which cells would reach ∼70% confluence on the day of transduction. 1 mL of harvested viral supernatant was incubated with HEK293T cells for 48 h before adding 1 μg/mL puromycin (Thermo Fisher Scientific). Puromycin dosage was escalated to 2 μg/mL after week one and to 4 μg/mL after week two to select for well transduced cells. shRNA construct expression was monitored by flow cytometry for GFP expression in comparison to HEK293T wild-type control cells.
gp140 ELISA
10-1074 and 3BNC117 variants were tested for their ability to bind SF162 gp140 trimer (NIH AIDS Reagent Program) by ELISA. High binding ELISA plates were coated with recombinant SF162 gp140 overnight at 4°C in PBS. Plates were washed using PBS-Tween20 (Sigma-Aldrich) and subsequently blocked with 5% nonfat dry milk in PBS (Bio-Rad, Hercules, CA, USA). 10-1074 and 3BNC117 variants were serially diluted 1:3 in blocking buffer and added to the test plate. After 1 h of incubation at 37°C, the plates were washed again and an HRP-conjugated goat anti-human IgG H+L (SouthernBiotech) was then added for detection. After 1 h at 37°C, plates were washed 10 times to remove unbound secondary antibody. Subsequently, plates were developed with TMB substrate (EMD Biosciences) and the reaction was stopped with stop solution (SouthernBiotech). Absorbance at 450 nm was measured in a Wallac Victor3 plate reader (PerkinElmer, Waltham, MA, USA).
Viral neutralization assay
Neutralization assays against HIV-1 NL(AD8) were performed in TZM-bl cells as previously described,38 using 2 ng HIV-1 p24 per well. 5,000 TZM-bl cells per well were plated in flat-bottom 96-well cellBIND plates the day before the neutralization assay. Antibody dilutions and viruses were incubated for 1 h at 37°C before being combined with the TZM-bl reporter cells. Luciferase activity in TZM-bl cells was measured after 3 days using BriteLite Plus luciferase substrate (PerkinElmer) on a Wallac Victor3 plate reader (PerkinElmer). The antibody titers required to neutralize 50% of the viral infection were calculated.
ADCC assay
ADCC activity was measured by a previously established assay to quantify natural killer (NK) cell activity toward virus-infected target cells expressing luciferase as previously described38 with slight modifications outlined below. A CD4+, CCR5+ T cell line derived from CEM.NKR-CCR5 CD4+ cells and modified to express firefly luciferase under the control of the LTR promoter was used as the target cells. Infection of the target cells was carried out by spinoculation in round-bottom 12 × 75 mm tubes using 200 ng p24 HIV-1 NL(AD8). Virus and target cells were centrifuged for 2 h at 1,200 × g at 25°C. Effector NK cells were derived from the KHYG-1 cell line modified to express human CD16.
ADCC assays were performed in round-bottom, 96-well plates, with each well containing 104 target cells and 105 NK effector cells in a 200 μL final volume. Effector cells were combined with washed target cells immediately before addition to assay plates. 4-fold serial dilutions of antibodies were performed in triplicate. Once target cells, effector cells, and serially diluted antibody were combined, assay plates were incubated for 8 h at 37°C. After the 8-h incubation, plates were spun down and 100 μL of media was removed from the top. 100 μL of BriteLite Plus (Perkin Elmer) was added to each well and mixed by pipetting. 150 μL of the mixture was transferred to a white 96-well plate. Luciferase activity was read using a Wallac Victor3 plate reader (PerkinElmer). 50% ADCC activity was calculated as the dilution at which a line connecting the values above and below 50% RLU would intercept the 50% RLU line.
AAV production
Production of rAAVs was conducted as described previously.46 HEK293 cells were transfected with a rAAV vector plasmid and two helper plasmids to allow generation of infectious AAV particles. After harvesting transfected cells and cell culture supernatant, rAAV was purified by three sequential CsCl centrifugation steps. Vector genome number was assessed by real-time PCR, and the purity of the preparation was verified by electron microscopy and silver-stained SDS-PAGE.
AAV in vitro transduction
HEK293T cells were seeded in R10 media in a T225 cellBIND flask (Corning, Corning, NY, USA) 1 day prior to transduction. On the day of AAV transduction, cells reached a confluence of ∼50%–70% and were infected with a total of 1 × 105 rAAV particles per cell. Cells were transduced with AAVs expressing 3BNC117 with or without shRNA targeting FUT8. After 72 h, cells were washed and cell culture media was changed to BIO-MPM-1 serum-free media (Biological Industries). Supernatant was clarified by centrifugation at 16,000 RCF and 4°C for 10 min. Concentration of secreted 3BNC117 IgG1 in cell culture supernatant was measured by Protein A/anti-rhesus IgG ELISA using purified rhesus IgG as a standard, as previously described.45 Equal amounts of 3BNC117 were analyzed for ADCC activity as described above.
Graphing and statistical analysis
All graphs were generated using GraphPad Prism 8. All statistical analysis was performed using Prism 8. For ADCC assays, 50% ADCC activity was calculated as the dilution at which a line connecting the values above and below 50% RLU would intercept the 50% RLU line. All ADCC samples were performed in triplicate and treatment groups were compared to the control antibodies using a two-tailed paired t test. p values were reported in the text.
Acknowledgments
We would like to acknowledge the assistance of the Flow Cytometry Core Facility of the Sylvester Comprehensive Cancer Center at the University of Miami, Miller School of Medicine, for the provision of fluorescence analysis and cell sorting service. We want to thank Michael Alpert for assistance with statistical analysis of our ADCC assays. We want to thank Leydi Machado for administrative assistance. We also want to thank Anabetsy Rivero and William Lauer for proofreading the manuscript. This work was made possible through funding support from the Florida Department of Health Pilot Award program (contract CODMR), as well as grants R01AI098446 and U19AI149646 from the NIH.
Author contributions
Conceptualization, J.M.T.; Methodology, J.M.T., J.M.M.-N., S.P.F., and R.C.D.; Investigation, J.M.T. and G.G.; Validation, J.M.T., and R.C.D.; Formal Analysis, J.M.T.; Writing-Original Draft, J.M.T., and R.C.D.; Writing-Review and Editing J.M.T., R.C.D., S.P.F., and J.M.M.-N.; Funding Acquisition, J.M.T. and R.C.D.; Supervision, R.C.D.; Project Administration, J.M.T. and R.C.D.
Declaration of interests
G.G. is a scientific co-founder of Voyager Therapeutics and holds equity in the company. G.G. is an inventor on patents with potential royalties licensed to Voyager Therapeutics and other biopharmaceutical companies. G.G. is a scientific co-founder of Aspa Therapeutics and holds equity in the company. G.G. is an inventor on patents with potential royalties licensed to Aspa Therapeutics and other bio-pharmaceutical companies. The remaining authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.11.001.
Supplemental information
References
- 1.Chun T.W., Davey R.T., Jr., Engel D., Lane H.C., Fauci A.S. Re-emergence of HIV after stopping therapy. Nature. 1999;401:874–875. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- 2.Hill A.L., Rosenbloom D.I., Goldstein E., Hanhauser E., Kuritzkes D.R., Siliciano R.F., Henrich T.J. Real-Time Predictions of Reservoir Size and Rebound Time during Antiretroviral Therapy Interruption Trials for HIV. PLoS Pathog. 2016;12:e1005535. doi: 10.1371/journal.ppat.1005535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joel N. Blankson, Deborah Persaud, a. & Siliciano, R. F. The Challenge of Viral Reservoirs in HIV-1 Infection. Annu. Rev. Med. 2002;53:557–593. doi: 10.1146/annurev.med.53.082901.104024. [DOI] [PubMed] [Google Scholar]
- 4.Chun T.W., Finzi D., Margolick J., Chadwick K., Schwartz D., Siliciano R.F. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1995;1:1284–1290. doi: 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- 5.Kim Y., Anderson J.L., Lewin S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe. 2018;23:14–26. doi: 10.1016/j.chom.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abner E., Jordan A. HIV “shock and kill” therapy: In need of revision. Antiviral Res. 2019;166:19–34. doi: 10.1016/j.antiviral.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Archin N.M., Kirchherr J.L., Sung J.A., Clutton G., Sholtis K., Xu Y., Allard B., Stuelke E., Kashuba A.D., Kuruc J.D. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Invest. 2017;127:3126–3135. doi: 10.1172/JCI92684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elliott J.H., McMahon J.H., Chang C.C., Lee S.A., Hartogensis W., Bumpus N., Savic R., Roney J., Hoh R., Solomon A. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet HIV. 2015;2:e520–e529. doi: 10.1016/S2352-3018(15)00226-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen T.A., Tolstrup M., Brinkmann C.R., Olesen R., Erikstrup C., Solomon A., Winckelmann A., Palmer S., Dinarello C., Buzon M. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. 2014;1:e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 10.Gautam R., Nishimura Y., Pegu A., Nason M.C., Klein F., Gazumyan A., Golijanin J., Buckler-White A., Sadjadpour R., Wang K. A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature. 2016;533:105–109. doi: 10.1038/nature17677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garber D.A., Adams D.R., Guenthner P., Mitchell J., Kelley K., Schoofs T., Gazumyan A., Nason M., Seaman M.S., McNicholl J. Durable protection against repeated penile exposures to simian-human immunodeficiency virus by broadly neutralizing antibodies. Nat. Commun. 2020;11:3195. doi: 10.1038/s41467-020-16928-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Julg B., Tartaglia L.J., Keele B.F., Wagh K., Pegu A., Sok D., Abbink P., Schmidt S.D., Wang K., Chen X. Broadly neutralizing antibodies targeting the HIV-1 envelope V2 apex confer protection against a clade C SHIV challenge. Sci. Transl. Med. 2017;9:eaal1321. doi: 10.1126/scitranslmed.aal1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moldt B., Rakasz E.G., Schultz N., Chan-Hui P.Y., Swiderek K., Weisgrau K.L., Piaskowski S.M., Bergman Z., Watkins D.I., Poignard P., Burton D.R. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc. Natl. Acad. Sci. USA. 2012;109:18921–18925. doi: 10.1073/pnas.1214785109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bar-On Y., Gruell H., Schoofs T., Pai J.A., Nogueira L., Butler A.L., Millard K., Lehmann C., Suárez I., Oliveira T.Y. Safety and antiviral activity of combination HIV-1 broadly neutralizing antibodies in viremic individuals. Nat. Med. 2018;24:1701–1707. doi: 10.1038/s41591-018-0186-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caskey M., Schoofs T., Gruell H., Settler A., Karagounis T., Kreider E.F., Murrell B., Pfeifer N., Nogueira L., Oliveira T.Y. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 2017;23:185–191. doi: 10.1038/nm.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caskey M., Klein F., Lorenzi J.C., Seaman M.S., West A.P., Jr., Buckley N., Kremer G., Nogueira L., Braunschweig M., Scheid J.F. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. 2015;522:487–491. doi: 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen Y.Z., Lorenzi J.C.C., Krassnig L., Barton J.P., Burke L., Pai J., Lu C.L., Mendoza P., Oliveira T.Y., Sleckman C. Relationship between latent and rebound viruses in a clinical trial of anti-HIV-1 antibody 3BNC117. J. Exp. Med. 2018;215:2311–2324. doi: 10.1084/jem.20180936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheid J.F., Horwitz J.A., Bar-On Y., Kreider E.F., Lu C.L., Lorenzi J.C., Feldmann A., Braunschweig M., Nogueira L., Oliveira T. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature. 2016;535:556–560. doi: 10.1038/nature18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grobben M., Stuart R.A., van Gils M.J. The potential of engineered antibodies for HIV-1 therapy and cure. Curr. Opin. Virol. 2019;38:70–80. doi: 10.1016/j.coviro.2019.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Fuchs S.P., Martinez-Navio J.M., Piatak M., Jr., Lifson J.D., Gao G., Desrosiers R.C. AAV-Delivered Antibody Mediates Significant Protective Effects against SIVmac239 Challenge in the Absence of Neutralizing Activity. PLoS Pathog. 2015;11:e1005090. doi: 10.1371/journal.ppat.1005090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Navio J.M., Fuchs S.P., Pantry S.N., Lauer W.A., Duggan N.N., Keele B.F., Rakasz E.G., Gao G., Lifson J.D., Desrosiers R.C. Adeno-Associated Virus Delivery of Anti-HIV Monoclonal Antibodies Can Drive Long-Term Virologic Suppression. Immunity. 2019;50:567–575. doi: 10.1016/j.immuni.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Navio J.M., Fuchs S.P., Mendes D.E., Rakasz E.G., Gao G., Lifson J.D., Desrosiers R.C. Long-Term Delivery of an Anti-SIV Monoclonal Antibody With AAV. Front. Immunol. 2020;11:449. doi: 10.3389/fimmu.2020.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liberatore R.A., Ho D.D. The Miami Monkey: A Sunny Alternative to the Berlin Patient. Immunity. 2019;50:537–539. doi: 10.1016/j.immuni.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Thomann M., Schlothauer T., Dashivets T., Malik S., Avenal C., Bulau P., Rüger P., Reusch D. In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and ADCC activity. PLoS ONE. 2015;10:e0134949. doi: 10.1371/journal.pone.0134949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamane-Ohnuki N., Satoh M. Production of therapeutic antibodies with controlled fucosylation. MAbs. 2009;1:230–236. doi: 10.4161/mabs.1.3.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peipp M., Lammerts van Bueren J.J., Schneider-Merck T., Bleeker W.W., Dechant M., Beyer T., Repp R., van Berkel P.H., Vink T., van de Winkel J.G. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood. 2008;112:2390–2399. doi: 10.1182/blood-2008-03-144600. [DOI] [PubMed] [Google Scholar]
- 27.Iida S., Kuni-Kamochi R., Mori K., Misaka H., Inoue M., Okazaki A., Shitara K., Satoh M. Two mechanisms of the enhanced antibody-dependent cellular cytotoxicity (ADCC) efficacy of non-fucosylated therapeutic antibodies in human blood. BMC Cancer. 2009;9:58. doi: 10.1186/1471-2407-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wren L.H., Chung A.W., Isitman G., Kelleher A.D., Parsons M.S., Amin J., Cooper D.A., Stratov I., Navis M., Kent S.J., ADCC study collaboration investigators Specific antibody-dependent cellular cytotoxicity responses associated with slow progression of HIV infection. Immunology. 2013;138:116–123. doi: 10.1111/imm.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baum L.L., Cassutt K.J., Knigge K., Khattri R., Margolick J., Rinaldo C., Kleeberger C.A., Nishanian P., Henrard D.R., Phair J. HIV-1 gp120-specific antibody-dependent cell-mediated cytotoxicity correlates with rate of disease progression. J. Immunol. 1996;157:2168–2173. [PubMed] [Google Scholar]
- 30.Lambotte O., Ferrari G., Moog C., Yates N.L., Liao H.X., Parks R.J., Hicks C.B., Owzar K., Tomaras G.D., Montefiori D.C. Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. AIDS. 2009;23:897–906. doi: 10.1097/QAD.0b013e328329f97d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang P., Gajjar M.R., Yu J., Padte N.N., Gettie A., Blanchard J.L., Russell-Lodrigue K., Liao L.E., Perelson A.S., Huang Y., Ho D.D. Quantifying the contribution of Fc-mediated effector functions to the antiviral activity of anti-HIV-1 IgG1 antibodies in vivo. Proc. Natl. Acad. Sci. USA. 2020;117:18002–18009. doi: 10.1073/pnas.2008190117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asokan M., Dias J., Liu C., Maximova A., Ernste K., Pegu A., McKee K., Shi W., Chen X., Almasri C. Fc-mediated effector function contributes to the in vivo antiviral effect of an HIV neutralizing antibody. Proc. Natl. Acad. Sci. 2020;117:18754–18763. doi: 10.1073/pnas.2008236117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bournazos S., Klein F., Pietzsch J., Seaman M.S., Nussenzweig M.C., Ravetch J.V. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell. 2014;158:1243–1253. doi: 10.1016/j.cell.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moldt B., Shibata-Koyama M., Rakasz E.G., Schultz N., Kanda Y., Dunlop D.C., Finstad S.L., Jin C., Landucci G., Alpert M.D. A nonfucosylated variant of the anti-HIV-1 monoclonal antibody b12 has enhanced FcγRIIIa-mediated antiviral activity in vitro but does not improve protection against mucosal SHIV challenge in macaques. J. Virol. 2012;86:6189–6196. doi: 10.1128/JVI.00491-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee W.S., Parsons M.S., Kent S.J., Lichtfuss M. Can HIV-1-Specific ADCC Assist the Clearance of Reactivated Latently Infected Cells? Front. Immunol. 2015;6:265. doi: 10.3389/fimmu.2015.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Becker D.J., Lowe J.B. Fucose: biosynthesis and biological function in mammals. Glycobiology. 2003;13:41R–53R. doi: 10.1093/glycob/cwg054. [DOI] [PubMed] [Google Scholar]
- 37.Termini J.M., Silver Z.A., Connor B., Antonopoulos A., Haslam S.M., Dell A., Desrosiers R.C. HEK293T cell lines defective for O-linked glycosylation. PLoS ONE. 2017;12:e0179949. doi: 10.1371/journal.pone.0179949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alpert M.D., Heyer L.N., Williams D.E., Harvey J.D., Greenough T., Allhorn M., Evans D.T. A novel assay for antibody-dependent cell-mediated cytotoxicity against HIV-1- or SIV-infected cells reveals incomplete overlap with antibodies measured by neutralization and binding assays. J. Virol. 2012;86:12039–12052. doi: 10.1128/JVI.01650-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zalevsky J., Chamberlain A.K., Horton H.M., Karki S., Leung I.W., Sproule T.J., Lazar G.A., Roopenian D.C., Desjarlais J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010;28:157–159. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu D., Alegre M.L., Varga S.S., Rothermel A.L., Collins A.M., Pulito V.L., Hanna L.S., Dolan K.P., Parren P.W., Bluestone J.A. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell. Immunol. 2000;200:16–26. doi: 10.1006/cimm.2000.1617. [DOI] [PubMed] [Google Scholar]
- 41.Gaudinski M.R., Coates E.E., Houser K.V., Chen G.L., Yamshchikov G., Saunders J.G., Holman L.A., Gordon I., Plummer S., Hendel C.S., VRC 606 Study Team Safety and pharmacokinetics of the Fc-modified HIV-1 human monoclonal antibody VRC01LS: A Phase 1 open-label clinical trial in healthy adults. PLoS Med. 2018;15:e1002493. doi: 10.1371/journal.pmed.1002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hezareh M., Hessell A.J., Jensen R.C., van de Winkel J.G., Parren P.W. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J. Virol. 2001;75:12161–12168. doi: 10.1128/JVI.75.24.12161-12168.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lazar G.A., Dang W., Karki S., Vafa O., Peng J.S., Hyun L., Chan C., Chung H.S., Eivazi A., Yoder S.C. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA. 2006;103:4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gómez-Román V.R., Patterson L.J., Venzon D., Liewehr D., Aldrich K., Florese R., Robert-Guroff M. Vaccine-elicited antibodies mediate antibody-dependent cellular cytotoxicity correlated with significantly reduced acute viremia in rhesus macaques challenged with SIVmac251. J. Immunol. 2005;174:2185–2189. doi: 10.4049/jimmunol.174.4.2185. [DOI] [PubMed] [Google Scholar]
- 45.Fuchs S.P., Martinez-Navio J.M., Gao G., Desrosiers R.C. Recombinant AAV Vectors for Enhanced Expression of Authentic IgG. PLoS ONE. 2016;11:e0158009. doi: 10.1371/journal.pone.0158009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mueller C., Ratner D., Zhong L., Esteves-Sena M., Gao G. 2012. Production and discovery of novel recombinant adeno-associated viral vectors. Curr Protoc Microbiol. Chapter 14, Unit14D.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.