

Abstract
The myotonic dystrophies are the commonest cause of adult-onset muscular dystrophy. Phenotypes of DM1 and DM2 are similar, but there are some important differences, including the presence or absence of congenital form, muscles primarily affected (distal vs proximal), involved muscle fiber types (type 1 vs type 2 fibers), and some associated multisystemic phenotypes. There is currently no cure for the myotonic dystrophies but effective management significantly reduces the morbidity and mortality of patients. For the enormous understanding of the molecular pathogenesis of myotonic dystrophy type 1 and myotonic dystrophy type 2, these diseases are now called “spliceopathies” and are mediated by a primary disorder of RNA rather than proteins. Despite clinical and genetic similarities, myotonic dystrophy type 1 and type 2 are distinct disorders requiring different diagnostic and management strategies. Gene therapy for myotonic dystrophy type 1 and myotonic dystrophy type 2 appears to be very close and the near future is an exciting time for clinicians and patients.
Key words: myotonic dystrophy type 2, DM2, proximal myotonic myopathy, PROMM, DMPK, CNBP
Introduction
The myotonic dystrophies are the more frequent muscle disorders in adulthood. So far 2 distinct entities have been described: myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2).
In this article I review the discovery of the gene, the clinical features, pathogenetic and management of more recently described DM2. All findings related mainly to clinical aspects, pathomolecular mechanisms, new guidelines of management have been updates to 2020.
Discovery of the genes
Myotonic dystrophies represent a group of dominantly inherited, multisystem (eye, heart, brain, endocrine, gastrointestinal tract, uterus, skin) diseases that share the core features of myotonia, muscle weakness, and early onset cataracts (before 50 years of age). The gene defect responsible for myotonic dystrophy described by Steinert on 1908, was discovered in 1992 and was found to be caused by expansion of a CTG repeat in the 3’ untranslated region of myotonic dystrophy protein kinase gene (DMPK), a gene located on chromosome 19q13.3, encoding a protein kinase 1-3. After the discovery of this gene defect, DNA testing revealed a group of patients with dominantly inherited myotonia, proximal more than distal weakness, and cataracts; these patients were previously diagnosed as having myotonic dystrophy of Steinert but lacked the gene defect responsible for this disease. Subsequent clinical studies of kindreds with patients having these characteristics led to new diagnostic labels for these patients: myotonic dystrophy type 2 4, proximal myotonic myopathy (PROMM) 5,6, or proximal myotonic dystrophy (PDM) 7. Later studies demonstrated that many of the families identified as having myotonic dystrophy type 2, PROMM, or PDM had a single disorder that results from an unstable tetranucleotide CCTG repeat expansion in intron 1 of the nucleic acid-binding protein (CNBP) gene (previously known as zinc finger 9 gene, ZNF9) on chromosome 3q21 8,9.
Myotonic dystrophy of Steinert, the classical form of myotonic dystrophy that results from an unstable trinucleotide repeat expansion on chromosome 19q13.3, was termed myotonic dystrophy type 1-DM1. Patients with the clinical picture of myotonic dystrophy type 2, PROMM, or PDM who have positive DNA testing for the unstable tetranucleotide repeat expansion on chromosome 3q21 were classified as having myotonic dystrophy type 2 (DM2). Reliability of DNA testing to establish or to exclude the diagnosis of myotonic dystrophy type 1 is close to 100% 10. However, caution is necessary in the diagnosis of myotonic dystrophy type 2. At present, much more information is available on the natural history of DM1 than DM2, but knowledge of myotonic dystrophy type 2 will increase at a rapid pace over the next several years.
Biological basis: pathomolecular mechanisms
Myotonic dystrophy type 2 results from an unstable tetranucleotide repeat expansion, CCTG in intron 1 of the nucleic acid-binding protein (CNBP) gene (previously known as zinc finger 9 gene, ZNF9) on chromosome 3q21 8,9,11,12. The cause for the unstable expansion is unknown. In contrast to the (CTG)n repeat in myotonic dystrophy type 1, in myotonic dystrophy type 2/proximal myotonic myopathy the (CCTG)n repeat is a part of the complex repetitive motif (TG)n(TCTG)n(CCTG)n, and the (CCTG)n repeat tract is generally interrupted in healthy range alleles by 1 or more GCTG, TCTG, or ACTG motifs, whereas it is typically uninterrupted in the expanded alleles 9,11,13.
The size of the (CCTG)n repeat is below 30 repeats in normal individuals, whereas the range of expansion sizes in myotonic dystrophy type 2 patients is huge 13. The smallest reported mutations vary between 55 and 75 CCTG 9,13 and the largest expansions have been measured to be about 11,000 repeats 9. The expanded myotonic dystrophy type 2 alleles show marked somatic instability, with significant increase in length over time 9,14, thus the threshold size of the disease-causing mutation remains to be determined. The size of the CCTG repeat appears to increase over time in the same individual, and, like myotonic dystrophy type 1, this is a dynamic gene defect 14. These 2 genetic findings complicate the correlation between genotype and phenotype (Tab. I). The gene mutation responsible for myotonic dystrophy type 2 appears to have arisen from a Northern European founder 11,12, but single-kindred Afghan 15 and Japanese 16 cases have been described. Both mutations are believed to have occurred after migration out of Africa, between 120,000 and 60,000 years ago. The age of the myotonic dystrophy type 2 founder mutation has been estimated at 4000 to 12,000 years (about 200 to 540 generations) 11. The molecular pathomechanism leading to the manifestations of myotonic dystrophy type 2 is felt to be similar to that in myotonic dystrophy type 1 and relates to a toxic effect of the abnormally expanded RNA that accumulates in the muscle nuclei 17-21.
Table I.
Etiology of DM1 and DM2.
| DM1 | DM2 | |
|---|---|---|
| Chromosomal locus | 19q 13.3 | 3q 21.3 |
| Gene | DMPK | ZNF9/CNBP |
| Inheritance | Autosomal dominant | Autosomal dominant |
| Mechanism | CTG repeat expansion | CCTG repeat expansion |
| Normal repeat size | < 37 | < 27 |
| Pathologic repeat size | > 50 | > 75? |
| Expanded repeat range | 50-4000 | 75-5000 -> 11000 |
| Anticipation | Yes | ----- |
The fact that 2 repeat sequences located in entirely different genes can cause such similar disease features implies a common pathogenic mechanism. The clinical and molecular parallels between myotonic dystrophy type 1 and type 2 strongly suggest that the mutant RNAs containing the repeat expansions that accumulate in the cell nuclei as foci are responsible for the pathological features common to both disorders. It is now clear that the gain-of-function RNA mechanism is the predominant cause of myotonic dystrophy pathogenesis in which the CUG and CCUG repeats alter cellular function of several RNA-binding proteins. It has been demonstrated that mutant CUG and CCUG RNAs are very stable due to a deficiency of RNA helicase p68 22. The expanded CUG and CCUG RNA form hairpins, imperfect double-stranded structures that lead to dysregulation of 2 important RNA-binding proteins: muscleblind like 1 (MBNL1) and CUG-binding protein 1 (CUGBP1), which are antagonist regulators of alternative splicing of various genes 23,24. Data demonstrate that MBNL1-containing foci in myotonic dystrophy type 2 cells also sequester snRNPs and hnRNPs, splicing factors involved in the early phases of transcript processing 25,26, thus strengthening the hypothesis that a general alteration of pre-mRNA posttranscriptional pathway could be at the basis of the multifactorial phenotype of myotonic dystrophy type 2 patients. In myotonic dystrophies, the downregulation of MBNL1, due to its sequestration in mutant RNA foci, and the upregulation of CUGBP1 result in abnormal expression of embryonic isoforms in adult tissues. The alteration of pre-mRNA processing strengthens the hypothesis of a spliceopathy that leads to an expression of isoforms inadequate for a particular tissue or developmental stage 27,28. In both myotonic dystrophy type 1 and type 2, missplicing of insulin receptor gene (INSR) was associated with insulin resistance. However, Renna and colleagues reported that post-receptor insulin signal transduction via both IRS1-Akt/PKB and Ras-ERK pathway is impaired in myotonic dystrophy skeletal muscle, thus contributing to insulin resistance observable in myotonic dystrophy type 1 and type 2 patients 29. Moreover, myotonic dystrophy skeletal muscle exhibits a lower expression of the insulin receptor in type 1 fibers, contributing to the defective activation of the insulin pathway 30. It is now clear that the molecular pathomechanism of myotonic dystrophies is more complex than actually suggested 31.
miRNAs are small, noncoding RNA modulating gene expression at the posttranscriptional level, and their expression and intracellular distribution are deregulated in many human diseases, including muscular dystrophies 32-36. Both in myotonic dystrophy type 1 and in myotonic dystrophy type 2 it has been demonstrated that the highly regulated pathways of miRNA are altered in skeletal muscle, potentially contributing to myotonic dystrophy pathogenetic mechanisms 34-36. A deregulation of microRNA in skeletal muscle and plasma from myotonic dystrophy type 2 patients has been also reported 36,37. The identification of minimally invasive analytical biomarkers for myotonic dystrophies and the established potential of circulating miRNAs as prognostic and diagnostic biomarkers are particularly important to monitor myotonic dystrophies progression and the effectiveness of new drug treatments.
A novel molecular mechanism that may contribute to the pathogenesis of myotonic dystrophies has been described by Zu and collaborators 38. RNA transcripts containing expanded CAG or CUG repeats can be translated in the absence of a starting ATG, and this noncanonical translation, called repeat associated non-ATG translation (RAN-translation), occurs across expanded repeats in all reading frames to produce potentially toxic homopolymeric proteins 38,39. It has been demonstrated that RAN-translation also occurs across transcripts containing the myotonic dystrophy type 2 CCUG or CAGG expansion mutation, producing tetra-repeat expansion proteins with a repeating Leu-Pro-Ala-Cys (LPAC) or Gln-Ala-Gly-Arg (QAGR) motif 40. Both LPAC and QAGR RAN proteins accumulate in myotonic dystrophy type 2 human autopsy brains in distinct patterns. For LPAC, cytoplasmic aggregates are found in neurons, astrocytes, and glia in the gray matter regions of the brain. In contrast, QAGR RAN protein accumulation, which is nuclear, is found primarily in oligodendrocytes located in white matter regions of the brain. Moreover, it has been evidenced that RAN protein accumulation can be modulated by MBNL1 levels and that nuclear sequestration of CCUG, CUG, or CAG RNAs decrease steady-state levels of RAN proteins 40. These data suggest that RAN-translation may be common to both myotonic dystrophy type 1 and type 2 and that RAN proteins may be responsible for some of the CNS features of myotonic dystrophies.
Another open question in the field of myotonic dystrophies is to clarify the pathomechanisms underlying the phenotypic differences between myotonic dystrophy type 1 and type 2. Clinical signs in myotonic dystrophy type 1 and type 2 are similar, but there are some distinguishing features: myotonic dystrophy type 2 is generally less severe and lacks a prevalent congenital form. This suggests that other cellular and molecular pathways are involved besides the shared toxic-RNA gain of function hypothesized. An important step forward in understanding the differences between myotonic dystrophy type 1 and type 2 has been made. Indeed, rbFOX1 has been reported as a novel RNA binding protein that specifically binds to expanded CCUG repeats, but not to expanded CUG repeats. rbFOX1 is enriched in skeletal muscle, heart, and brain and is involved in the regulation of various aspects of mRNA metabolism. In the study, it has been demonstrated that rbFOX1 co-localizes with CCUG RNA foci in muscle cells and skeletal muscle tissues of individuals with myotonic dystrophy type 2, but not with CUG RNA foci in myotonic dystrophy type 1 samples 41. Interestingly, rbFOX1 competes with MBNL1 for binding to CCUG expanded repeats, and its overexpression partly releases MBNL1 from sequestration within CCUG RNA foci in muscle cells. Furthermore, expression of rbFOX1 corrects alternative splicing alterations and rescues muscle atrophy, climbing, and flying defects caused by expression of expanded CCUG repeats in a Drosophila model of myotonic dystrophy type 2 41.
Several studies have revealed a role for CNBP in myotonic dystrophy type 2. CNBP deletion in several animal models results in severe brain and muscle phenotypes and other abnormalities similar to those seen in myotonic dystrophy type 2 42-45. These defects can be rescued by reintroduction of wild-type levels of CNBP, suggesting that a loss of CNBP function likely contributes to myotonic dystrophy type 2. Two reports using cell models describe a reduction of the rate of protein translation in myotonic dystrophy type 2 muscle cells due to a decrease of CNBP protein levels in myotonic dystrophy type 2 myoblasts and adult muscle 46 and due to the interaction of CCUG repeats with cytoplasmic multiprotein complexes, which dysregulate translation and degradation of proteins in patients 47. Sammons and colleagues report that CNBP activity is reduced in myotonic dystrophy type 2 human myoblasts leading to a decrease in CNBP activation of IRES-mediated translation of the human ODC and suggest that CNBP activity may contribute to myotonic dystrophy type 2 phenotype 48. Moreover, the reduction of CNBP expression has been reported in myotonic dystrophy type 2 muscle biopsies but not in myotonic dystrophy type 1, thus explaining some of the phenotypic disparities between both types of myotonic dystrophies 49. Taken together, these data suggest that myotonic dystrophy type 2 pathology may be due to a combination of an RNA gain of function and CNBP loss of function.
The role of CUGBP1 in myotonic dystrophy type 2 is particularly intriguing, with contradictory results being reported 47,49-51. Cardani and colleagues demonstrated that this protein is overexpressed in muscle biopsies from patients affected by the adult classical form of myotonic dystrophy type 1 but not in muscle from myotonic dystrophy type 2 patients, suggesting that sequestration of MBNL1 evidently has a central role in splicing misregulation in both types of myotonic dystrophies, whereas CUGBP1 overexpression might be an additional pathogenic mechanism in myotonic dystrophy type 1 not shared by myotonic dystrophy type 2 49. However, it has been shown that MBNL1 overexpression in a mouse model of RNA toxicity (DM200) is not effective in reversing myotonic dystrophy type 1 phenotypes such as myotonia and cardiac conduction abnormalities. Also, the mice do not show improvement in function assays such as grip strength or treadmill running, and MBNL1 overexpression notably increases muscle histopathology and results in variable rescue of a number of splicing targets 52.
Vihola and collaborators investigated the molecular basis of muscle weakness and wasting and the differences in muscle phenotype between myotonic dystrophy type 1 and type 2. They identified differences in muscle gene expression and splicing between myotonic dystrophy type 1 and type 2 patients. In particular, the aberrant splicing isoform of TNNT3 is twice as frequent in myotonic dystrophy type 2 compared to myotonic dystrophy type 1. Moreover, in myotonic dystrophy type 1 and type 2, a different protein expression pattern has been found in the highly atrophic fibers 53. Concerning myotonic dystrophy type 2, skeletal muscle phenotype has been studied in heterozygous Cnbp KO mice and in human muscle samples 54. The study demonstrates that CNBP protein expression is reduced in cytoplasm of myotonic dystrophy type 2 muscle fibers, and it is predominantly localized at membrane level where its interaction with α-dystroglycan is increased compared to controls. These findings suggest that alterations of CNBP in myotonic dystrophy type 2 might cause muscle atrophy, not only via misregulation of mRNA but also via protein-protein interactions with membrane proteins affecting myofiber membrane function 54.
Epidemiology
Myotonic dystrophy type 2 appears to have a lower prevalence than myotonic dystrophy type 1 and primarily affects populations with a Northern European heritage 12. For myotonic dystrophy type 2, there are currently no established prevalence estimates; myotonic dystrophy type 2 is generally thought to be rarer than myotonic dystrophy type 1, but large-scale population studies to confirm this have not been performed. In Germany, 267 mutation-verified molecular diagnoses were made between 2003 and 2005 compared with 277 myotonic dystrophy type 1 diagnoses within the same period. These data suggest that myotonic dystrophy type 2 appears to be more frequent than previously thought, with most myotonic dystrophy type 2 patients currently undiagnosed with symptoms frequently occurring in the elderly population 55. However, many patients in older generations with myotonic dystrophy type 1 or type 2 with milder symptoms are clearly undiagnosed. It is noteworthy that recessive mutations in the chloride channel gene CLCN1, which have a high frequency in the general population, can act as modifiers in patients with myotonic dystrophy type 2 disease by amplification of their myotonia 56-58. Meola’s group has identified myotonic dystrophy type 2 patients presenting an atypical phenotype characterized by early and severe myotonia without mutation on the CLCN1 gene but with mutations on SCN4A gene 59-61. Thus, both CLCN1 and SCN4A mutations may contribute to exaggerate the myotonia in myotonic dystrophy type 2 60.
Clinical manifestation
There are no distinct clinical subgroups in DM2, and clinical presentation comprises a continuum ranging from early adult-onset severe forms to very late-onset mild forms that are difficult to differentiate from normal aging. Only 2 cases of neonatal forms have been reported so far in the literature: 1 of these patients had reduced intrauterine movements and muscle hypotonia after birth 62, and the second had only congenital talipes equinovarus without any other clinical sign 63. At present, there is no evidence of a congenital or childhood form of myotonic dystrophy type 2 14. The main difference in DM2 in comparison to DM1 is the absence of congenital form. Myotonic dystrophy type 2 typically presents in adulthood and has variable manifestations such as early onset cataracts (less than 50 years of age), various grip myotonias, thigh muscle stiffness, muscle pain, and weakness (in hip flexors, hip extensors, or long flexors of the fingers) 4-6,14,64-67. These complaints often appear between 20 and 50 years of age. Posterior subscapular cataract before 50 years of age is a characteristic feature of myotonic dystrophy type 2, and early onset cataract can be a presenting feature of the disease, preceding all other symptoms 68. Pain is a common as well as a highly relevant problem for many patients with myotonic dystrophy type 2, with an estimated lifetime prevalence of 76% and a negative effect on quality of life 69. Patients and their care providers ascribe the symptoms to overuse of muscles, “pinched nerves”, “sciatica”, arthritis, or fibromyalgia. In comparison to other chronic muscle disorder patients, myotonic dystrophy type 2 patients more frequently describe a pain that is sometimes reported to be exercise-related, temperature-modulated, and palpation-induced (Tab. II) 70. Younger patients may complain of stiffness or weakness when running up steps, whereas they infrequently complain of cramps. The muscle pain in myotonic dystrophy type 2 has no consistent relationship to exercise or to the severity of myotonia found on clinical examination. The pain, which tends to come and go without obvious cause, usually fluctuates in intensity and distribution over the limbs. It can last for days to weeks. This pain seems qualitatively different from the muscle and musculoskeletal pain that occurs in patients with myotonic dystrophy type 1. In a study on qualitative as well as quantitative aspects of pain in patients with myotonic dystrophy type 2, it has been observed that mechanical hyperalgesia is the main finding present in the rectus femoris, trapezius, and thenar, suggestive of at least a peripheral mechanism of pain 69. Pain appears to be most often located symmetrically in the proximal limbs 69. Myotonic dystrophy type 2 scored significantly lower than myotonic dystrophy type 1 on the bodily pain scale, indicating more body pain in myotonic dystrophy type 2. This finding has a high disease impact on physical as well as on mental health functioning 71, and on professional performance 72. A transcriptomic analysis performed on 12 muscle biopsy specimens obtained from myotonic dystrophy type 2 patients has identified 14 muscle genes significantly up- or down-regulated in myalgic patients compared to nonmyalgic myotonic dystrophy type 2 patients. These data support the idea that molecular changes in the muscles of myotonic dystrophy type 2 patients are associated with muscle pain and suggest that muscle-specific molecular pathways might play a significant role in myalgia 73.
Table II.
Multisystemic aspects of adult onset DM2.
| Brain |
|
| Heart |
|
| Respiratory |
|
| Anesthesia |
|
| Hypersomnia and fatigue |
|
| Endocrine |
|
| Pregnancy |
|
| Muscle pain |
|
Early in the presentation of myotonic dystrophy type 2, there is only mild weakness of hip extension, thigh flexion, and finger flexion. Myotonia of grip and thigh muscle stiffness varies from minimal to moderate severity over days to weeks. Direct percussion of forearm extensor and thenar muscles is the most sensitive clinical test for myotonia in myotonic dystrophy type 2. Myotonia may appear only on electromyographic testing after examination of several muscles 14,64. Facial weakness is mild in myotonic dystrophy type 2 as is muscle wasting in the face and limbs (Fig. 1). Weakness of neck flexors is frequent. Trouble arising from a squat is common, especially as the disease progresses (Fig. 2). Calf muscle hypertrophy occasionally is prominent (Fig. 3). Other manifestations, such as excessive sweating, hypogonadism, glucose intolerance, cardiac conductions disturbances, cognitive alterations, and neuropsychological alterations, may also occur and worsen over time 6,14,65,74. Sleep complaints and breathing disorders are also frequent in myotonic dystrophy type 2 75.
Figure 1.
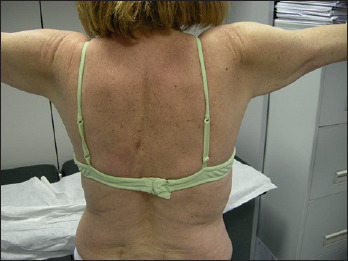
Mild atrophy, grade 4 MRC proximal muscle weakness in upper limbs in a patient affected by DM2.
Figure 2.
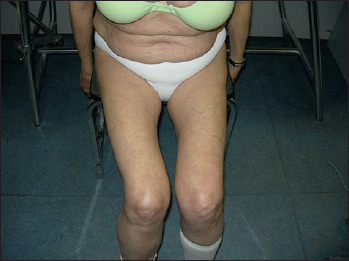
Moderate atrophy and weakness of proximal lower limbs (grade 3 MRC) with difficulty in arising from a chair in a patient affected by DM2.
Figure 3.
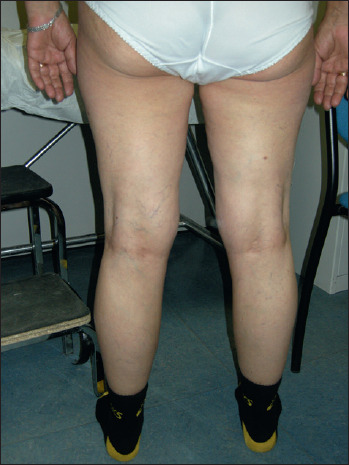
Calf hypertrophy in a patient affected by DM2.
A study on frequency and progression of cardiac and muscle involvement in a large cohort of patients with myotonic dystrophy type 2 demonstrated that the frequency and severity of cardiac involvement and muscle weakness are reduced in myotonic dystrophy type 2 compared to myotonic dystrophy type 1 and that progression is slower and less severe 76. Nevertheless, careful cardiac evaluation is recommended to identify patients at risk for potential cardiac major arrhythmia. A retrospective study comprised of 62 adult patients with myotonic dystrophy type 2 showed that cardiac conduction and rhythm defects are relatively rare in myotonic dystrophy type 2, although diastolic dysfunction is common, suggesting that regular ECG and echocardiography screening is needed in these patients 77. Cardiovascular magnetic resonance demonstrates that in myotonic dystrophy type 2 patients subclinical myocardial injury was already detectable in preserved left ventricular ejection fraction. Moreover, extracellular volume was also increased in regions with no focal fibrosis and myocardial fibrosis was related to conduction abnormalities 78.
Patients with both myotonic dystrophy type 1 and type 2 have lower scores on frontal lobe functioning tests compared to controls and have an increased prevalence of avoidant personality disorders 6. In a study aimed to analyze personality patterns in a cohort of myotonic dystrophy type 1 and type 2 patients, no significant personality impairments have been observed in patients with myotonic dystrophy type 2, and the most common clinical symptoms observed in these patients were anxiety and somatization 79. In patients with type 2 disease, conventional brain MRI findings can be entirely normal. However, in advanced stages or more severe cases, diffuse white-matter changes can be present although be less pronounced than or different to that in myotonic dystrophy type 1 80,81. It has been reported that the main transcranial sonography finding in myotonic dystrophy type 2 patients is brainstem raphe hypoechogenicity, which is associated with fatigue and excessive daytime sleepiness. In addition, substantia nigra hyperechogenicity and increased diameter of the third ventricle has been observed 82. The type of cognitive impairment that occurs in myotonic dystrophy type 2 is similar to but less severe than that of myotonic dystrophy type 1. A specific type of “avoidant” personality and a significant impairment in frontal lobe function (especially limited ability to perform executive functions) have been observed in myotonic dystrophy type 1 and type 2 patients, although these abnormalities were milder in myotonic dystrophy type 2 patients 82. Similar observations have been reported in a study performed in a larger cohort of myotonic dystrophy type 2 patients 84. In conclusion there are clinical, neuropsychological, and neuroimaging data that support the hypothesis of central nervous system involvement also in myotonic dystrophy type 2 85.
Gastrointestinal manifestations are common in myotonic dystrophy type 2 patients, affecting their quality of life. A study on progression of gastrointestinal manifestations in these patients reports that during the 5 years of follow-up, the most common changes are the development of trouble swallowing and constipation and that female patients demonstrate a greater risk of a gastrointestinal manifestation 86. A relatively high frequency of cholecystectomy on average before 45 years of age is also reported 86.
It has been reported that hearing impairment is a frequent symptom in myotonic dystrophy type 2 patients and that the sensorineural hearing impairment is located in the cochlea 87. This suggests it is important to perform audiometry when hearing impairment is suspected in order to propose early hearing rehabilitation with hearing aids when indicated.
In a study conducted on a large cohort of 307 genetically-confirmed myotonic dystrophy type 2 patients, a profound gender and age influence on the phenotype has emerged, emphasizing that female gender and aging may be associated with a higher disease burden 88. Indeed, it appears that with aging, there is a tendency towards the worsening of weakness, whereas myalgia and myotonia tend to decrease. Females seem to be more severely affected than men as they show more frequently muscle weakness, multisystem involvement, and need of using walking aids. This study suggests that these age- and gender-specific differences should be considered in diagnostics, management, and future clinical studies of myotonic dystrophy type 2.
It has been observed that metabolic syndrome is common in myotonic dystrophy type 2 patients but not more frequent than in healthy subjects. However, treatment of metabolic disturbances may reduce cardiovascular complications and improve quality of life in patients with myotonic dystrophy type 2 89.
Body composition assessed by DEXA (dual-energy x-ray absorptiometry) reveals that patients with myotonic dystrophy type 1 and type 2 have similar total body mass, bone mineral content, fat mass, and lean tissue mass. Patients with myotonic dystrophy type 2 have less visceral fat deposition than those affected by myotonic dystrophy type 1. Also, right rib bone mineral density was lower in myotonic dystrophy type 2 patients 90.
Overall the prognosis for patients with myotonic dystrophy type 2 is more favorable than for individuals with myotonic dystrophy type 1. Patients usually have a slower, less severe, and less widespread progression of muscle weakness and less muscle wasting. Does not seem to be a more severe phenotype associated with the homozygotic form of this disease 15. As in myotonic dystrophy type 1, patients with myotonic dystrophy type 2 who have an earlier onset of symptoms have an earlier onset of myotonia and weakness 91. The natural history of myotonic dystrophy type 2 remains to be fully defined, but present information indicates that most patients have a normal lifespan. Respiratory failure, hypersomnia, and recurrent aspiration or pneumonia are not common in myotonic dystrophy type 2 72. Cardiac conduction disturbances occur 67, but they are less frequent compared to myotonic dystrophy type 1 92,93. An investigation using a variety of standard tests of autonomic function (response to Valsalva maneuver, deep breathing, change in posture, grip, analysis of heart rate variability) reveals no major abnormalities in patients with myotonic dystrophy type 2 94.
Diagnosis
The gold standard for establishing the diagnosis of myotonic dystrophy type 2/proximal myotonic myopathy is to demonstrate the presence of abnormal CCTG repeats in the 3q21 zinc finger protein 9 (ZNF9/CNBP) gene involved with myotonic dystrophy type 2.
Leucocyte DNA testing is also available for myotonic dystrophy type 2, but previous DNA analysis for diagnosing myotonic dystrophy type 2 and proximal myotonic myopathy may have missed as many as 20% of affected individuals 14. As for myotonic dystrophy type 1, a new ready to use genetic test has been validated to identify the myotonic dystrophy type 2 disease, with the advantage to reduce errors that can be introduced using homemade reagents 95. However, the myotonic dystrophy type 2 diagnostic odyssey is complicated by the difficulties to develop an accurate, robust, and cost-effective method for a routine molecular assay 60.
A more practical tool for myotonic dystrophy type 2 diagnosis than the complex genotyping procedure is via in situ hybridization detection of nuclear accumulations of CCUG-containing RNA in myotonic dystrophy type 2 muscle biopsy using specific probes 21,96. Moreover, because MBNL1 is sequestered by mutant RNA foci, it is possible to visualize the nuclear accumulation of MBNL1 by immunofluorescence on muscle sections. However, although MBNL1 represents a histopathological marker of myotonic dystrophies, it does not allow one to distinguish between myotonic dystrophy type 1 and myotonic dystrophy type 2 96. Another tool to investigate muscle weakness and wasting is muscle imaging with MRI. In type 2 disease, early muscular changes develop in the anterior vastus group of thigh muscles, with relative sparing of the rectus femoris 98. The main aspects of multisystemic involvement are summarized in the Table II.
Management
In general, the management of myotonic dystrophy type 2 is similar to myotonic dystrophy type 1, but there is less need for supportive care like bracing, scooters, or wheelchairs. Cataracts require monitoring, and serial monitoring of ECG is necessary to check for covert arrhythmia. Disturbances in cardiac rhythm are less frequent in myotonic dystrophy type 2, but abnormalities do occur 14,67. Hypogonadism and insulin resistance need monitoring as in myotonic dystrophy type 1. Myotonia tends to be less marked and less troublesome in myotonic dystrophy type 2, but in specific circumstances, especially if muscle stiffness is frequent and persistent, anti-myotonia therapy with mexiletine is helpful. Cognitive difficulties also occur in myotonic dystrophy type 2, as in myotonic dystrophy type 1, and appear to be associated with decreased cerebral blood flow to frontal and anterior temporal lobes 74 and decreased brain volume 94,99,100. The changes are less severe than in myotonic dystrophy type 1. Their etiology is unknown but may relate to the toxic effect of intranuclear accumulations of abnormally expanded RNA. Management of these brain symptoms is similar to that for myotonic dystrophy type 1.
A frequent and difficult problem in myotonic dystrophy type 2 is the peculiar muscle pain described earlier. The exact mechanism underlying the pain is unknown, and there is no well-established effective treatment. Carbamazepine or mexiletine along with nonsteroidal anti-inflammatory medications ameliorate this pain in some patients. However, others with severe pain may require opiates on a regular basis to obtain relief. Fortunately, this peculiar muscle pain is not typical in myotonic dystrophy type 1. Guidelines on diagnosis and management have been published 98. Care considerations and management issues on the wide spectrum of disease manifestations in DM2 have been published recently by a Consortium of international Experts 101.
For pregnancy and anesthesia there are some special considerations.
Pregnancy
Studies of prenatal diagnosis using sensitive DNA testing for myotonic dystrophy type 2 myopathy 14 are theoretically possible and more information is likely to become available in near future. If a mother has myotonic dystrophy type 2 with only minimal symptoms at the time of her pregnancy, she may have an increased risk of developing myotonia and weakness in the later stages of the pregnancy 14,102. In 1 study of 96 pregnancies in 42 myotonic dystrophy type 2 women, it was found that 21% of the women had their first myotonic symptoms during their pregnancy. Additionally, 17% of their pregnancies ended in miscarriages, and 27% ended in preterm labor 103. Two reports suggest that the symptoms that develop during pregnancy reverse after delivery 14,102, but more information is necessary to make such a prediction with certainty.
Anesthesia
One study of a large number of individuals with myotonic dystrophy type 2 has found no significant problems with the ability of patients to tolerate general anesthesia 14. In a report of a large German patient cohort, the overall frequency of severe complications was 0.6% (2 of 340). The overall lower risk seems to be predominantly related to the minor respiratory involvement in myotonic dystrophy type 2 than in myotonic dystrophy type 1 104.
Conclusions
Twenty-eight years have passed since the (CTG)n repeat expansion mutation was discovered in patients with myotonic dystrophy type 1, and 19 years ago the (CCTG)n mutation was identified in type 2 disease. Emerging data indicate that molecular pathomechanisms are much more complex than could have been envisioned when the respective mutations were just identified. RNA toxicity clearly has a major role, yet spliceopathy alone does not seem to fully account for all aspects of the multisystemic phenotype in myotonic dystrophies. Other pathomechanisms consistent with the toxic RNA model probably entail regulation of gene expression and translation and various cellular stress pathways and extend beyond the nucleus to the cytoplasm. Nevertheless, it is important to emphasize that despite clinical and genetic similarities, myotonic dystrophy type 1 and type 2 are distinct disorders requiring different diagnostic and management strategies.
Although treatment of myotonic dystrophy type 1 and myotonic dystrophy type 2 is currently limited to supportive therapies, new therapeutic approaches based on pathogenic mechanisms may become feasible in near future.
The future holds great promise for advances in translational research in DM2. The teamwork will expedite the development of targeted therapies and improve the lives of patients and their families 105.
Figures and tables
Acknowledgments
This study was mainly supported by grants from Fondazione Malattie Miotoniche-FMM, Milan, Italy.
References
- 1.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 1992;68:799-808. https://doi.org/10.1016/0092-8674(92)90154-5 10.1016/0092-8674(92)90154-5 [DOI] [PubMed] [Google Scholar]
- 2.Fu YH, Pizzuti A, Fenwick RG, Jr, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992;255:1256-8. https://doi.org/10.1126/science.1546326 10.1126/science.1546326 [DOI] [PubMed] [Google Scholar]
- 3.Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science 1992;255:1253-5. https://doi.org/10.1126/science.1546325 10.1126/science.1546325 [DOI] [PubMed] [Google Scholar]
- 4.Thornton CA, Griggs RC, Moxley RT., 3rd Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol 1994;35:269-72. https://doi.org/10.1002/ana.410350305 10.1002/ana.410350305 [DOI] [PubMed] [Google Scholar]
- 5.Ricker K, Koch MC, Lehmann-Horn F, et al. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology 1994;44:1448-52. https://doi.org/10.1212/wnl.44.8.1448 10.1212/wnl.44.8.1448 [DOI] [PubMed] [Google Scholar]
- 6.Meola G, Sansone V, Radice S, et al. A family with an unusual myotonic and myopathic phenotype and no CTG expansion (proximal myotonic myopathy syndrome): a challenge for future molecular studies. Neuromuscul Disord 1996;6:143-50. https://doi.org/10.1016/0960-8966(95)00040-2 10.1016/0960-8966(95)00040-2 [DOI] [PubMed] [Google Scholar]
- 7.Udd B, Krahe R, Wallgren-Pettersson C, et al. Proximal myotonic dystrophy – a family with autosomal dominant muscular dystrophy, cataracts, hearing loss and hypogonadism: heterogeneity of proximal myotonic syndromes. Neuromuscul Disord 1997:217-28. https://doi.org/10.1016/s0960-8966(97)00041-2 10.1016/s0960-8966(97)00041-2 [DOI] [PubMed] [Google Scholar]
- 8.Ranum LP, Rasmussen PF, Benzow KA, et al. Genetic mapping of a second myotonic dystrophy locus. Nature Genetics 1998;19:196-8. https://doi.org/10.1038/570 10.1038/570 [DOI] [PubMed] [Google Scholar]
- 9.Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001;293:864-7. https://doi.org/10.1126/science.1062125 10.1126/science.1062125 [DOI] [PubMed] [Google Scholar]
- 10.Valaperta R, Sansone V, Lombardi F, et al. Identification and characterization of DM1 patients by a new diagnostic certified assay: neuromuscular and cardiac assessments. Biomed Res Int 2013;2013:958510 https://doi.org/10.1155/2013/958510 10.1155/2013/958510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bachinski LL, Udd B, Meola G, et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet 2003;73:835-48. https://doi.org/10.1086/378566 10.1086/378566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liquori CL, Ikeda Y, Weatherspoon M, et al. Myotonic dystrophy type 2: human founder haplotype and evolutionary conservation of the repeat tract. Am J Hum Genet 2003;73:849-62. https://doi.org/10.1086/378720 10.1086/378720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bachinski LL, Czernuszewicz T, Ramagli LS, et al. Premutation allele pool in myotonic dystrophy type 2. Neurology 2009;72:490-7. https://doi.org/10.1212/01.wnl.0000333665.01888.33 10.1212/01.wnl.0000333665.01888.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 2003;60:657-64. https://doi.org/10.1212/01.wnl.0000054481.84978.f9 10.1212/01.wnl.0000054481.84978.f9 [DOI] [PubMed] [Google Scholar]
- 15.Schoser BG, Kress W, Walter MC, et al. Homozygosity for CCTG mutation in myotonic dystrophy type 2. Brain 2004a;127(Pt 8):1868-77. https://doi.org/10.1093/brain/awh210 10.1093/brain/awh210 [DOI] [PubMed] [Google Scholar]
- 16.Saito T, Amakusa Y, Kimura T, et al. Myotonic dystrophy type 2 in Japan: ancestral origin distinct from Caucasian families. Neurogenetics 2008;9:61-3. https://doi.org/10.1007/s10048-007-0110-4 10.1007/s10048-007-0110-4 [DOI] [PubMed] [Google Scholar]
- 17.Mankodi A, Thornton CA. Myotonic syndromes. Curr Opin Neurol 2002;15:545-52. https://doi.org/10.1097/00019052-200210000-00005 10.1097/00019052-200210000-00005 [DOI] [PubMed] [Google Scholar]
- 18.Ranum LP, Day JW. Myotonic dystrophy: clinical and molecular parallels between myotonic dystrophy type 1 and type 2. Curr Neurol Neurosci Rep 2002;2:465-70. https://doi.org/10.1007/s11910-002-0074-6 10.1007/s11910-002-0074-6 [DOI] [PubMed] [Google Scholar]
- 19.Timchenko LT, Tapscott SJ, Cooper TA, et al. Myotonic dystrophy: discussion of molecular basis. Adv Exp Med Biol 2002;516:27-45. https://doi.org/10.1007/978-1-4615-0117-6_2 10.1007/978-1-4615-0117-6_2 [DOI] [PubMed] [Google Scholar]
- 20.Mankodi A, Teng-Umnuay P, Krym M, et al. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann Neurol 2003;54:760-8. https://doi.org/10.1002/ana.10763 10.1002/ana.10763 [DOI] [PubMed] [Google Scholar]
- 21.Cardani R, Mancinelli E, Sansone V, et al. Biomolecular identification of (CCTG)n mutation in myotonic dystrophy type 2 (DM2) by FISH on muscle biopsy. Eur J Histochem 2004;48:437-42. https://doi.org/10.4081/918 10.4081/918 [DOI] [PubMed] [Google Scholar]
- 22.Jones K, Wei C, Schoser B, et al. Reduction of toxic RNAs in myotonic dystrophies type 1 and type 2 by the RNA helicase p68/DDX5. Proc Natl Acad Sci U S A 2015;112:8041-5. https://doi.org/10.1073/pnas.1422273112 10.1073/pnas.1422273112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tapscott SJ, Thornton CA. Biomedicine. Reconstructing myotonic dystrophy. Science 2001;293:816-7. https://doi.org/10.1126/science.1063517 10.1126/science.1063517 [DOI] [PubMed] [Google Scholar]
- 24.Day JW, Ranum LP. RNA phatogenesis of the myotonic dystrophies. Neuromuscl Disord 2005;15:5-16. https://doi.org/10.1016/j.nmd.2004.09.012 10.1016/j.nmd.2004.09.012 [DOI] [PubMed] [Google Scholar]
- 25.Fakan S. Perichromatin fibrils are in situ forms of nascent transcriptions. Trend Cell Biol 1994;4:86-90. https://doi.org/10.1016/0962-8924(94)90180-5 10.1016/0962-8924(94)90180-5 [DOI] [PubMed] [Google Scholar]
- 26.Perdoni F, Malatesta M, Cardani R, et al. RNA/MBNL1-containing foci in myoblast nuclei from patients affected by myotonic dystrophy type 2: an immunocytochemical study. Eur J Histochem 2009;53:151-8. https://doi.org/10.4081/ejh.2009.151 10.4081/ejh.2009.151 [DOI] [PubMed] [Google Scholar]
- 27.Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet 2006;15:r162-9. https://doi.org/10.1093/hmg/ddl181 10.1093/hmg/ddl181 [DOI] [PubMed] [Google Scholar]
- 28.Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta 2015;1852:594-606. https://doi.org/10.1016/j.bbadis.2014.05.019 10.1016/j.bbadis.2014.05.019 [DOI] [PubMed] [Google Scholar]
- 29.Renna LV, Bosè F, Iachettini S, et al. Receptor and post-receptor abnormalities contribute to insulin resistance in myotonic dystrophy type 1 and type 2 skeletal muscle. PLoS One 2017;12:e0184987 https://doi.org/10.1371/journal.pone.0184987 10.1371/journal.pone.0184987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Renna LV, Bosè F, Brigonzi E, et al. Aberrant insulin receptor expression is associated with insulin resistance and skeletal muscle atrophy in myotonic dystrophies. PLoS One 2019;14:e0214254 https://doi.org/10.1371/journal.pone.0214254 10.1371/journal.pone.0214254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sznajder ŁJ, Swanson MS. Short tandem repeat expansions and RNA-mediated pathogenesis in myotonic dystrophy. Int J Mol Sci 2019;20:E3365 https://doi.org/10.3390/ijms20133365 10.3390/ijms20133365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eisenberg I, Alexander MS, Kunkel LM. miRNAS in normal and diseased skeletal muscle. J Cell Mol Med 2009;13:2-11. https://doi.org/10.1111/j.1582-4934.2008.00524.x 10.1111/j.1582-4934.2008.00524.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greco S, De Simone M, Colussi C, et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J 2009;23:3335-46. https://doi.org/10.1096/fj.08-128579 10.1096/fj.08-128579 [DOI] [PubMed] [Google Scholar]
- 34.Gambardella S, Rinaldi F, Lepore SM, et al. Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J Transl Med 2010;8:48 https://doi.org/10.1186/1479-5876-8-48 10.1186/1479-5876-8-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perbellini R, Greco S, Sarra-Ferraris G, et al. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul Disord 2011;21:81-8. https://doi.org/10.1016/j.nmd.2010.11.012 10.1016/j.nmd.2010.11.012 [DOI] [PubMed] [Google Scholar]
- 36.Greco S, Perfetti A, Fasanaro P, et al. Deregulated microRNAs in myotonic dystrophy type 2. PLoS One 2012;7:e39732 https://doi.org/10.1371/journal.pone.0039732 10.1371/journal.pone.0039732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perfetti A, Greco S, Cardani R, et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci Rep 2016;6:38174 https://doi.org/10.1038/srep38174 10.1038/srep38174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zu T, Gibbens B, Doty NS, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 2011;108:260-5. https://doi.org/10.1073/pnas.1013343108 10.1073/pnas.1013343108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pearson CE. Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet 2011;7:e1002018 https://doi.org/10.1371/journal.pgen.1002018 10.1371/journal.pgen.1002018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zu T, Cleary JD, Liu Y, et al. RAN translation regulated by Muscleblind proteins in myotonic dystrophy type 2. Neuron 2017;95:1292-305. https://doi.org/10.1016/j.neuron.2017.08.039 10.1016/j.neuron.2017.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sellier C, Cerro-Herreros E, Blatter M, et al. rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat Commun 2018;9:2009 https://doi.org/10.1038/s41467-018-04370-x 10.1038/s41467-018-04370-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen W, Liang Y, Deng W, et al. The zinc-finger protein CNBP is required for forebrain formation in the mouse. Development 2003;130:1367-79. https://doi.org/10.1242/dev.00349 10.1242/dev.00349 [DOI] [PubMed] [Google Scholar]
- 43.Abe Y, Chen W, Huang W, et al. CNBP regulates forebrain formation at organogenesis stage in chick embryos. Dev Biol 2006;295:116-27. https://doi.org/10.1016/j.ydbio.2006.03.012 10.1016/j.ydbio.2006.03.012 [DOI] [PubMed] [Google Scholar]
- 44.Chen W, Wang Y, Abe Y, et al. Haploinsufficiency for Znf9 in Znf9+/2 mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J Mol Biol 2007;368:8-17. https://doi.org/10.1016/j.jmb.2007.01.088 10.1016/j.jmb.2007.01.088 [DOI] [PubMed] [Google Scholar]
- 45.Weiner AM, Allende ML, Becker TS, et al. CNBP mediates neural crest cell expansion by controlling cell proliferation and cell survival during rostral head development. J Cell Biochem 2007;102:1553-70. https://doi.org/10.1002/jcb.21380 10.1002/jcb.21380 [DOI] [PubMed] [Google Scholar]
- 46.Huichalaf C, Schoser B, Schneider-Gold C, et al. Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J Neurosci 2009;29:9042-9. https://doi.org/10.1523/JNEUROSCI.1983-09.2009 10.1523/JNEUROSCI.1983-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salisbury E, Schoser B, Schneider-Gold C, et al. Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am J Pathol 2009;175:748-62. https://doi.org/10.2353/ajpath.2009.090047 10.2353/ajpath.2009.090047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sammons MA, Antons AK, Bendjennat M, et al. ZNF9 activation of IRES-mediated translation of the human ODC mRNA is decreased in myotonic dystrophy type 2. PLoS One 2010;5:e9301 https://doi.org/10.1371/journal.pone.0009301 10.1371/journal.pone.0009301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardani R, Bugiardini E, Renna LV, et al. Overexpression of CUGBP1 in skeletal muscle from adult classic myotonic dystrophy type 1 but not from myotonic dystrophy type 2. PLoS One 2013;8:e83777 https://doi.org/10.1371/journal.pone.0083777 10.1371/journal.pone.0083777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 2006;15:2087-97. https://doi.org/10.1093/hmg/ddl132 10.1093/hmg/ddl132 [DOI] [PubMed] [Google Scholar]
- 51.Pelletier R, Hamel F, Beaulieu D, et al. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis 2009;36:181-90. https://doi.org/10.1016/j.nbd.2009.07.009 10.1016/j.nbd.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 52.Yadava RS, Kim YK, Mandal M, et al. MBNL1 overexpression is not sufficient to rescue the phenotypes in a mouse model of RNA toxicity. Hum Mol Genet 2019;28:2330-8. https://doi.org/10.1093/hmg/ddz065 10.1093/hmg/ddz065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vihola A, Bachinski LL, Sirito M, et al. Differences in aberrant expression and splicing of sarcomeric proteins in the myotonic dystrophies DM1 and DM2. Acta Neuropathol 2010;119:465-79. https://doi.org/10.1007/s00401-010-0637-6 10.1007/s00401-010-0637-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei C, Stock L, Schneider-Gold C, et al. Reduction of cellular nucleic acid binding protein encoded by a myotonic dystrophy type 2 gene causes muscle atrophy. Mol Cell Biol 2018;38:e00649-17. https://doi.org/10.1128/MCB.00649-17 10.1128/MCB.00649-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suominen T, Bachinski LL, Auvinen S, et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet 2011;19:776-82. https://doi.org/10.1038/ejhg.2011.23 10.1038/ejhg.2011.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suominen T, Schoser B, Raheem O, et al. High frequency of co-segregating CLCN1 mutations among myotonic dystrophy type 2 patients from Finland and Germany. J Neurol 2008;255:1731-6. https://doi.org/10.1007/s00415-008-0010-z 10.1007/s00415-008-0010-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cardani R, Giagnacovo M, Botta A, et al. Co-segregation of DM2 with a recessive CLCN1 mutation in juvenile onset of myotonic dystrophy type 2. J Neurol 2012:2090-9. https://doi.org/10.1007/s00415-012-6462-1 10.1007/s00415-012-6462-1 [DOI] [PubMed] [Google Scholar]
- 58.Peddareddygari LR, Grewal AS, Grewal RP. Focal seizures in a patient with myotonic disorder type 2 co-segregating with a chloride voltage-gated channel 1 gene mutation: a case report. J Med Case Rep 2016;10:167 https://doi.org/10.1186/s13256-016-0958-8 10.1186/s13256-016-0958-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bugiardini E, Rivolta I, Binda A, et al. SCN4A mutation as modifying factor of myotonic dystrophy type 2 phenotype. Neuromuscul Disord 2015;25:301-7. https://doi.org/10.1016/j.nmd.2015.01.006 10.1016/j.nmd.2015.01.006 [DOI] [PubMed] [Google Scholar]
- 60.Meola G, Cardani R. Myotonic dystrophy type 2 and modifier genes: an update on clinical and pathomolecular aspects. Neurol Sci 2017;38:535-46. https://doi.org/10.1007/s10072-016-2805-5 10.1007/s10072-016-2805-5 [DOI] [PubMed] [Google Scholar]
- 61.Binda A, Renna LV, Bosè F, et al. SCN4A as modifier gene in patients with myotonic dystrophy type 2. Sci Rep 2018;8:11058 https://doi.org/10.1038/s41598-018-29302-z 10.1038/s41598-018-29302-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kruse B, Wöhrle D, Steinbach P, et al. Does proximal myotonic myopathy show anticipation. Hum Mutat 2008;29:e100-2. https://doi.org/10.1002/humu.20791 10.1002/humu.20791 [DOI] [PubMed] [Google Scholar]
- 63.Renard D, Rivier F, Dimeglio A, et al. Congenital talipes equinovarus associated with myotonic dystrophy type 2. Muscle Nerve 2010;42:457 https://doi.org/10.1002/mus.21738 10.1002/mus.21738 [DOI] [PubMed] [Google Scholar]
- 64.Ricker K, Koch MC, Lehmann-Horn F, et al. Proximal myotonic myopathy. Clinical features of a multisystem disorder similar to myotonic dystrophy. Arch Neurol 1995;52:25-31. https://doi.org/10.1001/archneur.1995.00540250029009 10.1001/archneur.1995.00540250029009 [DOI] [PubMed] [Google Scholar]
- 65.Day JW, Roelofs R, Leroy B, et al. Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2). Neuromuscul Disord 1999;9:19-27. https://doi.org/10.1016/s0960-8966(98)00094-7 10.1016/s0960-8966(98)00094-7 [DOI] [PubMed] [Google Scholar]
- 66.Meola G. Clinical and genetic heterogeneity in myotonic dystrophies. Muscle Nerve 2000;23:1789-99. https://doi.org/10.1002/1097-4598(200012)23:12<1789::aid-mus2>3.0.co;2-4 [DOI] [PubMed] [Google Scholar]
- 67.Moxley RT, 3rd, Meola G, Udd B, et al. Report of the 84th ENMC workshop: PROMM (proximal myotonic myopathy) and other myotonic dystrophy-like syndromes: 2nd workshop. 13-15th October 2000. Loosdrecht, The Netherlands. Neuromuscul Disord 2002;12:306-17. https://doi.org/10.1016/s0960-8966(01)00284-x 10.1016/s0960-8966(01)00284-x [DOI] [PubMed] [Google Scholar]
- 68.Papadopoulos C, Kekou K, Xirou S, et al. Early onset posterior subscapular cataract in a series of myotonic dystrophy type 2 patients. Eye (Lond) 2018;32:622-25. https://doi.org/10.1038/eye.2017.280 10.1038/eye.2017.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Vliet J, Tieleman AA, Verrips A, et al. Qualitative and quantitative aspects of pain in patients with myotonic dystrophy type 2. J Pain 2018b;19:920-30. https://doi.org/10.1016/j.jpain.2018.03.006 10.1016/j.jpain.2018.03.006 [DOI] [PubMed] [Google Scholar]
- 70.George A, Schneider-Gold C, Zier S, et al. Musculoskeletal pain in patients with myotonic dystrophy type 2. Arch Neurol 2004:61:1938-42. https://doi.org/10.1001/archneur.61.12.1938 10.1001/archneur.61.12.1938 [DOI] [PubMed] [Google Scholar]
- 71.Tieleman AA, Knoop H, van de Logt AE, et al. Poor sleep quality and fatigue but no excessive daytime sleepiness in myotonic dystrophy type 2. J Neurol Neurosurg Psychiatry 2010;81:963-7. https://doi.org/10.1136/jnnp.2009.192591 10.1136/jnnp.2009.192591 [DOI] [PubMed] [Google Scholar]
- 72.Suokas KI, Haanpää M, Kautiainen H, et al. Pain in patients with myotonic dystrophy type 2: a postal survey in Finland. Muscle Nerve 2012;45:70-4. https://doi.org/10.1002/mus.22249 10.1002/mus.22249 [DOI] [PubMed] [Google Scholar]
- 73.Moshourab R, Palada V, Grunwald S, et al. A molecular signature of myalgia in myotonic dystrophy 2. EBioMedicine 2016;7:205-11. https://doi.org/10.1016/j.ebiom.2016.03.017 10.1016/j.ebiom.2016.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meola G, Sansone V, Perani D, et al. Reduced cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathy. Neurology 1999;5:1042-50. https://doi.org/10.1212/wnl.53.5.1042 10.1212/wnl.53.5.1042 [DOI] [PubMed] [Google Scholar]
- 75.Romigi A, Maestri M, Nicoletta C, et al. Sleep complaints, sleep and breathing disorders in myotonic dystrophy type 2. Curr Neurol Neurosci Rep 2019;19:9 https://doi.org/10.1007/s11910-019-0924-0 10.1007/s11910-019-0924-0 [DOI] [PubMed] [Google Scholar]
- 76.Sansone V, Brigonzi E, Schoser B, et al. The frequency and severity of cardiac involvement in myotonic dystrophy type 2 (DM2): long-term outcomes. Int J Cardiol 2013;168:1147-53. https://doi.org/10.1016/j.ijcard.2012.11.076 10.1016/j.ijcard.2012.11.076 [DOI] [PubMed] [Google Scholar]
- 77.Peric S, Bjelica B, Aleksic K, et al. Heart involvement in patients with myotonic dystrophy type 2. Acta Neurol Belg 2019a;119:77-82. https://doi.org/10.1007/s13760-018-1052-3 10.1007/s13760-018-1052-3 [DOI] [PubMed] [Google Scholar]
- 78.Schmacht L, Traber J, Grieben U, et al. Cardiac involvement in myotonic dystrophy Type 2 Patients with preserved ejection fraction: detection by cardiovascular magnetic resonance. Circ Cardiovasc Imaging 2016;9:e004615 https://doi.org/10.1161/CIRCIMAGING.115.004615 10.1161/CIRCIMAGING.115.004615 [DOI] [PubMed] [Google Scholar]
- 79.Paunic T, Peric S, Parojcic A, et al. Personality traits in patients with myotonic dystrophy type 2. Acta Myol 2017;36:14-8. PMID [PMC free article] [PubMed] [Google Scholar]
- 80.Romeo V, Pegoraro E, Ferrati C, et al. Brain involvement in myotonic dystrophies: neuroimaging and neuropsychological comparative study in DM1 and DM2. J Neurol 2010;257:1246-55. https://doi.org/10.1007/s00415-010-5498-3 10.1007/s00415-010-5498-3 [DOI] [PubMed] [Google Scholar]
- 81.Minnerop M, Weber B, Schoene-Bake JC, et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 2011;134(Pt 12):3530-46. https://doi.org/10.1093/brain/awr299 10.1093/brain/awr299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rakocevic-Stojanovic V, Peric S, Savic-Pavicevic D, et al. Brain sonography insight into the midbrain in myotonic dystrophy type 2. Muscle Nerve 2016;53:700-4. https://doi.org/10.1002/mus.24927 10.1002/mus.24927 [DOI] [PubMed] [Google Scholar]
- 83.Meola G, Sansone V, Perani D, et al. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM1) and in proximal myotonic myopathy (DM2/PROMM). Neuromuscul Disord 2003;13:813-21. https://doi.org/10.1016/s0960-8966(03)00137-8 10.1016/s0960-8966(03)00137-8 [DOI] [PubMed] [Google Scholar]
- 84.Peric S, Mandic-Stojmenovic G, Stefanova E, et al. Frontostriatal dysexecutive syndrome: a core cognitive feature of myotonic dystrophy type 2. J Neurol 2015;262:142-8. https://doi.org/10.1007/s00415-014-7545-y 10.1007/s00415-014-7545-y [DOI] [PubMed] [Google Scholar]
- 85.Meola G, Sansone V. Cerebral involvement in myotonic dystrophies. Muscle Nerve 2007;36(3):294-306. https://doi.org/10.1002/mus.20800. 10.1002/mus.20800 [DOI] [PubMed] [Google Scholar]
- 86.Hilbert JE, Barohn RJ, Clemens PR, et al. High frequency of gastrointestinal manifestations in myotonic dystrophy type 1 and type 2. Neurology 2017;89:1348-54. https://doi.org/10.1212/WNL.0000000000004420 10.1212/WNL.0000000000004420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van Vliet J, Tieleman AA, van Engelen BGM, et al. Hearing impairment in patients with myotonic dystrophy type 2. Neurology 2018a;90:e615-22. https://doi.org/10.1212/WNL.0000000000004963 10.1212/WNL.0000000000004963 [DOI] [PubMed] [Google Scholar]
- 88.Montagnese F, Mondello S, Wenninger S, et al. Assessing the influence of age and gender on the phenotype of myotonic dystrophy type 2. J Neurol 2017;264:2472-80. https://doi.org/10.1007/s00415-017-8653-2 10.1007/s00415-017-8653-2 [DOI] [PubMed] [Google Scholar]
- 89.Vujnic M, Peric S, Calic Z, et al. Metabolic impairments in patients with myotonic dystrophy type 2. Acta Myol 2018;37:252-6. PMID [PMC free article] [PubMed] [Google Scholar]
- 90.Peric S, Bozovic I, Nisic T, et al. Body composition analysis in patients with myotonic dystrophy types 1 and 2. Neurol Sci 2019b;40:1035-40. https://doi.org/10.1007/s10072-019-03763-0 10.1007/s10072-019-03763-0 [DOI] [PubMed] [Google Scholar]
- 91.Schneider C, Ziegler A, Ricker K, et al. Proximal myotonic myopathy: evidence for anticipation in families with linkage to chromosome 3q13. Neurology 2000;55:383-8. https://doi.org/10.1212/wnl.55.3.383 10.1212/wnl.55.3.383 [DOI] [PubMed] [Google Scholar]
- 92.Meola G, Sansone V, Marinou K, et al. Proximal myotonic myopathy: a syndrome with a favourable prognosis. J Neurol Sci 2002;193:89-96. https://doi.org/10.1016/s0022-510x(01)00649-9 10.1016/s0022-510x(01)00649-9 [DOI] [PubMed] [Google Scholar]
- 93.Schoser BG, Ricker K, Schneider-Gold C, et al. Sudden cardiac death in myotonic dystrophy type 2. Neurology 2004b;63:2402-4. https://doi.org/10.1212/01.wnl.0000147335.10783.e4 10.1212/01.wnl.0000147335.10783.e4 [DOI] [PubMed] [Google Scholar]
- 94.Flachenecker P, Schneider C, Cursiefen S, et al. Assessment of cardiovascular autonomic function in myotonic dystrophy type 2 (DM2/PROMM). Neuromuscul Disord 2003;13:289-93. https://doi.org/10.1016/s0960-8966(02)00277-8 10.1016/s0960-8966(02)00277-8 [DOI] [PubMed] [Google Scholar]
- 95.Valaperta R, Lombardi F, Cardani R, et al. Development and validation of a new molecular diagnostic assay for detection of myotonic dystrophy type 2. Genet Test Mol Biomarkers 2015;19:703-9. https://doi.org/10.1089/gtmb.2015.0135 10.1089/gtmb.2015.0135 [DOI] [PubMed] [Google Scholar]
- 96.Sallinen R, Vihola A, Bachinski LL, et al. New methods for molecular diagnosis and demonstration of the (CCTG)n mutation in myotonic dystrophy type 2 (DM2). Neuromuscul Disord 2004;14:274-83. PMID [DOI] [PubMed] [Google Scholar]
- 97.Cardani R, Mancinelli E, Rotondo G, et al. Muscleblind-like protein 1 nuclear sequestration is a molecular pathology marker of DM1 and DM2. Eur J Histochem 2006;50:177-82. PMID [PubMed] [Google Scholar]
- 98.Udd B, Meola G, Krahe R, et al. Myotonic dystrophy type 2 (DM2) and related disorders report of the 180th ENMC workshop including guidelines on diagnostics and management 3-5 December 2010, Naarden, The Netherlands. Neuromuscul Disord 2011;21:443-50. https://doi.org/10.1016/j.nmd.2011.03.013 10.1016/j.nmd.2011.03.013 [DOI] [PubMed] [Google Scholar]
- 99.Chang L, Ernst T, Osborn D, et al. Proton spectroscopy in myotonic dystrophy: correlations with CTG repeats. Arch Neurol 1998;55:305-11. https://doi.org/10.1001/archneur.55.3.305 10.1001/archneur.55.3.305 [DOI] [PubMed] [Google Scholar]
- 100.Akiguchi I, Nakano S, Shiino A, et al. Brain proton magnetic resonance spectroscopy and brain atrophy in myotonic dystrophy. Arch Neurol 1999;56:325-30. https://doi.org/10.1001/archneur.56.3.325 10.1001/archneur.56.3.325 [DOI] [PubMed] [Google Scholar]
- 101.Schoser BG, Montagnese F, Bassez G, et al. Consensus-based care recommandations for adults with myotonic dystrophy type 2. Neurol Clin Pract 2019;9:343-53. https://doi.org/10.1212/CPJ.0000000000000645 10.1212/CPJ.0000000000000645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Newman B, Meola G, O’Donovan DG, et al. Proximal myotonic myopathy (PROMM) presenting as myotonia during pregnancy. Neuromuscular Disord 1999;3:144-9. https://doi.org/10.1016/s0960-8966(98)00118-7 10.1016/s0960-8966(98)00118-7 [DOI] [PubMed] [Google Scholar]
- 103.Rudnik-Schoneborn S, Schneider-Gold C, Raabe U, et al. Outcome and effect of pregnancy in myotonic dystrophy type 2. Neurology 2006;66:579-80. https://doi.org/10.1212/01.wnl.0000198227.91131.1e 10.1212/01.wnl.0000198227.91131.1e [DOI] [PubMed] [Google Scholar]
- 104.Kirzinger L, Schmidt A, Kornblum C, et al. Side effects of anesthesia in DM2 as compared to DM1: a comparative retrospective study. Eur J Neurol 2010;17:842-5. https://doi.org/10.1111/j.1468-1331.2009.02942.x 10.1111/j.1468-1331.2009.02942.x [DOI] [PubMed] [Google Scholar]
- 105.Moxley RT, Hilbert JE, Meola G. The myotonic dystrophies. Rosenberg RN, Pascual JM, Eds. Rosenberg’s molecular and genetic basis of neurological and psychiatric disease. 6th edition. Vol. 2, San Diego (CA): Elsevier; 2020. [Google Scholar]