

Abstract
The term ‘limb girdle muscular dystrophy’ (LGMD) was first used in the seminal paper by Walton and Nattrass in 1954, were they identified LGMD as a separate clinical entity In LGMD description it is pointed out that the category of LGMD most likely comprises a heterogeneous group of disorders. After that the clinical entity was discussed but the LMGD nosography reached a permanent classification during two ENMC workshops held in 1995 and 2017, in the last one an operating definition of LGMD was agreed. This last classification included dystrophies with proximal or distal-proximal presentation with evidence at biopsy of fibre degeneration and splitting, high CK, MRI imaging consistent with degenerative changes, fibro-fatty infiltration present in individuals that reached independent walking ability. To be considered in this group at least two unrelated families should be identified.
A review is done of the first genetic characterisation of a number of LGMDs during the late twentieth century and a historical summary is given regarding how these conditions were clinically described and identified, the progresses done from identification of genetic loci, to protein and gene discoveries are reported. The LGMD described on which such historical progresses were done are the recessive calpainopathy (LGMD 2A/R1), dysferlinopathy (LGMD 2B/R2), sarcoglycanopathy (LGMD 2C-2F/R3-R6) types and the dominant type due to TPNO3 variants named transportinopathy (LGMD 1F/D2). Because of new diagnostic techniques such as exome and genome sequencing, it is likely that many other subtypes of LGMD might be identified in the future, however the lesson from the past discoveries can be useful for scientists and clinicians.
Key words: limb girdle dystrophy, calpain-3, dysferlin, sarcoglycans, transportin-3
Introduction
At the end of 19th century, G.B. Duchenne and W. Gowers described patients of both genders whose clinical symptoms and course were overlapping those of the Duchenne (DMD) type. In 1876-1880, a clinical form of atrophic pelvic muscle disease different from DMD was recognized by E. Leyden and P. Möbius (later referred as atrophic, pelvic girdle or pelvi-femoral type of Leyden-Möbius).
In 1954 J.N. Walton (Fig. 1) and F.J. Nattrass introduced the expression limb girdle muscular dystrophy (LGMD) to identify patients of both genders who presented onset of muscle weakness within the third decade of life, with weakness and atrophy of proximal muscles in the limb girdles, with sparing of facial muscles, and with moderately rapid course. They identified LGMD as a separate clinical entity from the more common, X-linked recessive DMD, and pointed out that LGMD most likely comprises a heterogeneous group of disorders 1.
Figure 1.

Lord John Walton of Detchant (the founder of Muscular Dystrophy Association in UK) and Giovanni Nigro (President and organizer) (A), and Alan E.H. Emery and Corrado Angelini (B) at the open ceremony of the 12th ICNMD congress held in Naples in 2010, taking place in the suggestive location of the San Carlo Royal Theatre.
The autosomal recessive LGMD of childhood, which was reported to be common in Brazil and North Africa, was initially referred as Severe Childhood-onset Recessive Muscular Dystrophy (SCARMD), with clinical features indistinguishable from DMD 2. The definition of LGMD soon emphasized its heterogeneity at both clinical and genetic level, and also the difficulty in discriminating between the different entities.
Identification of genetic loci of LGMDs
In the pre-molecular era, the diagnosis of LGMD was based only on clinical phenotype and by exclusion of other diagnoses, such as X-linked recessive DMD or its milder BMD variant (also using dystrophin gene and protein testing, available from 1987). In the following years, a molecular diagnosis of LGMD became possible using linkage analysis in large inbred families with many affected individuals.
In 1991 the first locus to be mapped in a recessive LGMD was to chromosome 15q15 (LGMD2A/R1) 3, probably due to the easy availability of large families, an observation that indirectly implies a high population frequency of the disease.
In the following years, many additional loci in inbred SCARMD or recessive LGMD families have been mapped to chromosome region 13q12 (LGMD2C/R5) 4, to 17q21 (LGMD2D/R3)5, to 4q12 (LGMD2E/R4) 6, to 5q33 (LGMD2F/R6 7,8, to 2p (LGMD2B/R2) 9.
Although possible in some instances, the genetic diagnosis of LGMD using linkage analysis was available only on research basis and unfeasible in the majority of patients who are isolated cases.
Such huge limitations in defining the genetic and molecular basis the LGMD patients, had for many years, important consequences on genetic counselling on family relatives because the mode of inheritance remained unclear in isolated cases.
In 2020, the large majority of LGMD patients achieve a genetic diagnosis, due to the recent development of advanced genome sequencing techniques, and the research is open to the future development of specific therapies. Patients’ management is so far limited to physical rehabilitation, clinical follow up of cardiologic and respiratory complications, but several therapies have been tested (including corticosteroids 10 and myostatin inhibitors) with variable success 11.
Classification of LGMDs
Due to the genetic characterisation of a number of LGMDs in the late 20th century, in 1995 the first consortium meeting, under the auspices of the European Neuromuscular Center (ENMC), reached a consensus on a classification of LGMD subtypes based on molecular and genetic criteria 12,13 the autosomal dominant loci were designated as LGMD type 1, and the autosomal recessive loci were designated as LGMD type 2. LGMD nomenclature adopted a progressive alphabetical letter indicating the order of gene mapping identification (Tab. I), also avoiding imprecise and often lengthy nomenclature in use (e.g. SCARMD). During the following 10 years, the number of LGMD loci/genes identified increased rapidly, and occupied all the alphabetic letters (from LGMD2A to LGMD2Z); furthermore, the classification system applied by the Online Mendelian Inheritance in Man (OMIM) catalogue was no longer used by the clinical community, because it included also inherited myopathies with normal CK levels which should be more properly classified between categories other than LGMD (e.g. congenital myopathies, scapulo-peroneal myopathies, metabolic myopathies, etc.). Because of this dilemma, it was time to update and modify the LGMD nomenclature (Tab. I).
Table I.
Genetic classification of LGMD.
| LGMD type | Disease OMIM# | Cytogenetic locus | Gene OMIM# | Gene symbol | Protein |
|---|---|---|---|---|---|
| 1A | 159000 | 5q31.2 | 604103 | MYOT | Myotilin |
| 1B | 159001 | 1q22 | 150330 | LMNA | Lamin A/C |
| 1C | 607801 | 3p25.3 | 601253 | CAV3 | Caveolin-3 |
| 1D/D1 | 603511 | 7q36.3 | 611332 | DNAJB6 | DNAJ/HSP40 homolog |
| 1F/D2 | 608423 | 7q32.1 | 610032 | TPNO3 | Transportin-3 |
| D3 | 609115 | 4p21.22 | - | - | Heterogeneous molecular ribonucleic D-like protein |
| D4 | 613530 | 15q15.1 | - | CAPN3 | Calpain-3 |
| 2A/R1 | 253600 | 15q15.1 | 114240 | CAPN3 | Calpain-3 |
| 2B/R2 | 253601 | 2p13.2 | 603009 | DYSF | Dysferlin |
| 2C/R5 | 253700 | 13q12.12 | 608896 | SGCG | γ-sarcoglycan |
| 2D/R3 | 608099 | 17q21.33 | 600119 | SGCA | α-sarcoglycan |
| 2E/R4 | 604286 | 4q12 | 600900 | SGCB | β-sarcoglycan |
| 2F/R6 | 601287 | 5q33.3 | 601411 | SGCD | δ-sarcoglycan |
| 2G/R7 | 601954 | 17q12 | 604488 | TCAP | Telethonin |
| 2H/R8 | 254110 | 9q33.1 | 602290 | TRIM32 | Tripartite motif containing protein-32 |
| 2I/R9 | 607155 | 19q13.32 | 606596 | FKRP | Fukutin-related-protein |
| 2J/R10 | 608807 | 2q31.2 | 188840 | TTN | Titin |
| 2K/R11 | 609308 | 9q34.13 | 607423 | POMT1 | Protein O-mannosyl transferase-1 |
| 2L/R12 | 611307 | 11p14.3 | 608662 | ANO5 | Anoctamin-5 |
| 2M/R13 | 611588 | 9q31.2 | 607440 | FKTN | Fukutin |
| 2N/R14 | 613158 | 14q24.3 | 607439 | POMT2 | Protein O-mannosyl transferase-2 |
| 2O/R15 | 613157 | 1p34.1 | 606822 | POMGnT1 | Protein O-mannose N-acetyl-glucosaminyl Transferase-1 |
| 2P/R16 | 613818 | 3p21 | 128239 | DAG1 | Dystroglycan |
| 2Q/R17 | 613723 | 8q24.3 | 601282 | PLEC1 | Plectin |
| 2R/R18 | 615325 | 2q35 | 125660 | DES | Desmin |
| 2S/R19 | 615356 | 4q35.1 | 614138 | TRAPPC11 | Transport-protein-particle-complex-11 |
| 2T/R20 | 615352 | 3p21.31 | 615320 | GMPPB | GDP-mannose-pyrophosphorylase B |
The 2nd ENMC workshop, which was held in March 2017 in Naarden, the Netherlands, on the classification and nomenclature of the LGMDs, was aimed to reach consensus of an updated definition of LGMD, and to review and evaluate suggestions of potential new classifications of LGMD subtypes 14. At this meeting, the classification was revised naming the autosomal dominant LGMDs as D and numbering them from 1 to 5, and the recessive forms as R with numbers from 1 to 23. The classification of LGMD included dystrophies with proximal or disto-proximal presentation with evidence at biopsy of fiber degeneration and splitting, high CK, muscle MRI imaging consistent with degenerative change and fibro-fatty infiltration.
As of today, more than 30 different genetic subtypes of LGMD have been identified. Like for other inherited conditions that display huge genetic heterogeneity, nomenclature has become a significant problem with the increased speed in which new disease genes are discovered. Because of new diagnostic techniques 15, such as exome and genome sequencing, it is likely that many more subtypes of LGMD will be identified in the future.
With the exception of autosomal inheritance, the different LGMD subtypes listed in the current classification system have little in common. The broad, original definition has led to potential inaccuracies over what is considered a form of LGMD. Over the past sixty years, the original clinical definition of LGMD has been useful, but our increased molecular and pathogenetic understanding of LGMD subtypes is beginning to call into question this definition and subsequent classification only by phenotype. Phenotypically LGMD subtypes are highly variable in their age of onset, speed of disease progression and overall severity; however, they do not share a common pathological mechanism that would distinguish them from other forms of muscular dystrophy and progressive limb girdle weakness is not always the leading clinical feature. Advances in genetic medicine and the identification of new genetic loci made the nomenclature increasingly difficult for LGMD, as mutations in a number of genes that have been assigned to subtypes of LGMD can also cause allelic conditions presenting with a different phenotype and be more commonly known under a different name. Mutations in the TTN or DYSF gene have e.g. also been associated with distal myopathic phenotypes. Several of the LGMDs are dystroglycanopathies that are also associated with a group of congenital muscular dystrophy syndromes, including Fukuyama congenital muscular dystrophy, Muscle-Eye-Brain disease, and Walker-Warburg-Syndrome. All dystroglycanopathies were grouped together due to the recognition that mutations in at least 18 different genes all interfere with the glycosylation of a-dystroglycan, and thus dystroglycan’s function as a matrix receptor. Understanding the role of dystroglycan and its carbohydrate moieties as a basement membrane receptor will therefore be relevant for therapy development for a large number of diseases, beyond the boundaries of traditional classification systems. Because industry is starting to show increased interest in LGMD, it will be important to have clarity about the classification of diseases caused by mutations in the same gene, as this will affect feasibility studies, inclusion criteria for clinical trials and recruitment strategies.
Nomenclature of disease is an important educational topic for both clinicians and patients and serves as a critical nosological reference point. Widely accepted nomenclature should therefore only be changed with caution and by consulting key clinical opinion leaders and patient advocacy groups. Any change of the classification also needs to take more general reforms of nosology into account (e.g. International Classification of Diseases (ICD)). Patients and their families will often have an emotional link with their diagnosis, identifying with the name of their disease. Changes to nomenclature therefore can have distressing effects for patients.
Description of LGMD
The term LGMD defines a progressive weakness with onset in the proximal limb girdle muscles, with age at onset of symptoms varying from early childhood (not congenital) to late adulthood. The progression of muscle weakness is usually symmetrical and variable among individuals and genetic type. The term LGMD used to molecularly classify the disease, is however sometime inappropriate for some patients when it is utilized to describe the clinical severity. Indeed, these disorders present a wide spectrum of muscle involvement and wasting, spanning from very severe forms, such as those with childhood onset and rapid progression, to relatively benign forms with late onset.
The clinical phenotypes due to mutation in the LGMD genes include severe childhood-onset forms, distal and proximal myopathies, pseudo-metabolic myopathies, eosinophilic myositis, and hyperCKemia. Because there is a spectrum of phenotypes under the same genetic entity, and a wide genetic heterogeneity under the same phenotype 16, it is crucial to identify suitable selection criteria to be used when screening patients for the proteins and genes responsible for LGMD.
LGMD is a genetically inherited condition that primarily affects skeletal muscle leading to progressive, predominantly proximal muscle weakness at presentation caused by a loss of muscle fibres. To be considered a form of LGMD the condition must be described in patients achieving independent walking, must have an elevated serum creatine kinase (CK) activity, must demonstrate degenerative changes on muscle imaging over the course of the disease, and have dystrophic changes on muscle histology ultimately leading to end-stage pathology for the most affected muscles.
Proximal muscle weakness and genetic inheritance were kept from the original definition as important factors of LGMD. To distinguish LGMD from congenital muscular dystrophies, patients must achieve independent walking, this criterium was felt to be superior to setting a defined age limit (i.e. two years). An elevated CK activity is seen in almost all LGMD patients in early stages of the disease process and is related to muscle fibre breakdown, which is considered a hallmark of muscular dystrophy. Degenerative changes on muscle MRI are defined as the replacement of skeletal muscle with adipose tissue as detected on standard T1 weighted axial images. Dystrophic changes on muscle histology include fibre necrosis and regeneration together with an increase in fibrosis and adipose tissue. The term ‘end-stage pathology’ refers to progressive replacement with fibro-adipose tissue seen on histological examination.
Calpainopathy identification (LGMD2A/R1)
Following the identification of the disease locus to chromosome 15 in 1991 3, the first mutations in the calpain-3 gene have been identified in 1995 by Isabelle Richard17] and the pioneer work of Michel Fardeau in 1996 (Fig. 2) analysed clinically a group of LGMD2A patients in a small community in the Reunion Island 18. The discovery of this isolate was followed by the identification of other genetic clusters of LGMD2A/R1 throughout the world, also in other specific isolates, e.g. in the venetian lagoon 19 or in the Alps 20.
Figure 2.

George Karpati and Michel Fardeau at the Myology Institute amphitheater, Hôpital Pitié-Salpetrière in Paris.
LGMD2A is due to mutations in the gene encoding calpain-3 (CAPN3), a neutral protease 17. This is the first muscular dystrophy to be recognized to be due to an enzyme defect rather than a structural protein defect 21. The disease is typically characterized by a selective and progressive involvement of proximal limb-girdle muscles 22. Two phenotypes have been identified based on the distribution of muscle weakness at onset: the pelvi-femoral form of Leiden-Möbius, which is the most frequently observed, in which muscle weakness is first evident in the pelvic girdle and later evident in the shoulder girdle, and the scapulo-humeral form of Erb, which is usually a milder phenotype with infrequent early onset, in which muscle weakness is first evident in the shoulder girdle and later evident in the pelvic girdle 23. Another early and transient feature in LGMD2A/R1 is eosinophilic myositis, which has been reported in patients presenting with increased CK levels. CK levels are always elevated since infancy (5-80 times the normal). The age at onset of muscle weakness ranges between 2 and 40 years (in average 15 years). The first clinical symptoms are usually difficulty in running, the tendency to walk on tiptoes, and scapular winging caused by weakness of scapular girdle muscles. Weakness and wasting of the hip adductors/extensors and the posterior thigh muscles (gluteus maximus, semimembranosus, biceps femoris) are evident on both clinical examination and on muscle CT and MRI imaging scan. Joint contractures are typically seen and may be early present. As the disease progresses, waddling gait, difficulty in climbing stairs, lifting weights, and getting up from the floor or a chair are evident. Muscle involvement is mainly symmetrical and atrophic. Facial and neck muscles are usually spared. LGMD2A/R1 is the most prevalent form of LGMD in most European countries, where it would account for about 40-50% of total LGMD cases 24. Its high prevalence, which was estimated to be about 1:15,000-1:150,000, is due to a high heterozygote frequency in the general population (about 1:100-120) 19. Calpainopathy phenotype with Erb presentation shares some similarities with facio-scapulo-humeral muscular dystrophy (FSHD) and may be confused with this latter disorder especially in isolated cases: muscle weakness at onset in the shoulder girdle, scapular winging, elevated CK levels, and nonspecific myopathic changes on muscle biopsy; however, facial weakness and asymmetrical muscle involvement, which are frequent in FSHD, are uncommon in LGMD2A/R1. The diagnosis of BMD should be also excluded in isolated male patients who have weakness in the lower girdle muscles in adolescence or adulthood and high CK levels. Some patients with LGMD2A/R1 have been reported with pseudo-metabolic myopathy, presenting asthenia, myalgia, exercise intolerance, proximal muscle weakness, and excessive lactate production.
The lack of therapy is still distressing in a disease that have been described from 20 years. Attempts to replace the missing protein in an animal model has led to a cardiac phenotype.
A variety of clinical presentation and disease course may be due to primary calpainopathy including cases with respiratory insufficiency. Symptoms caused by respiratory failure may be specific, such as breathlessness, or more general, such as fatigue, lethargy, poor appetite, weight loss and impaired concentration. Patients may describe breathlessness at rest or on exertion, depending on the severity of the muscle weakness. With the association of the diaphragm, symptoms of orthopnoea or breathlessness may be apparent when bending over. Breathlessness experienced when a person is immersed in water above the waist is a rare but classical symptom of diaphragm weakness.
When the upper airway musculature is affected, speech and swallowing difficulties start to develop. Snoring, apnoeic episodes and daytime somnolence point to the possibility of obstructive sleep apnoea. If patients under-ventilate at night, the resultant hypercapnia may cause early-morning headaches, reduced concentration, or clouded consciousness. Blurring of vision from papillo-edema has been described but is rare and only seen in severe hypercapnia. A history of recurrent chest infections may indicate an ineffective cough. Coughing requires activation of the inspiratory muscles, closure of the glottis and then contraction of the expiratory muscles, particularly those of the abdominal wall; finally, the expulsive phase is initiated by opening the glottis. A poor cough can result from weakness or in-coordinated contraction of the inspiratory, glottic or expiratory muscles.
A patient that suffered from calpainopathy with respiratory insufficiency was Federico Milcovich, the founder of Italian Muscular Dystrophy Association (UILDM).
Sarcoglycanopathies identification (LGMD2C/R5, 2D/R3, 2E/R4, 2F/R6)
LGMD named SCARMD: Adhal (γ-sarcoglycanopathy, LGMD2C/R5) and Hetairosin (β-sarcoglycanopathy, LGMD2E/R4) proteins and genes discovery
Severe childhood-onset autosomal recessive muscular dystrophy (SCARMD) was first described by Ben Hamida and Fardeau in a symposium in Venice in 1980. In 1983 they reported 93 patients belonging to Tunisian inbred families with a severe form of muscular dystrophy, with childhood onset and early loss of ambulation (20-30 years of age) 2; the CK was markedly raised in the early stages of disease, muscle wasting affected mainly limb-girdle and trunk muscles, and calf muscle hypertrophy was usually present.
In 1992 the gene segregating in Tunisian, Moroccan, Algerian, Egyptian SCARMD families was mapped to chromosome region 13q12 4. In the same year, the deficiency of the 50k dystrophin-associated glycoprotein (DAG) (now called α-sarcoglycan, or α-SG) was identified in the muscle from 13q12-linked SCARMD patients, initially suggesting that the defect of this protein was the primary cause of the disease 25. In 1994, Romero et al. 26 proposed to refer to the 50k DAG with the term “adhalin” from the arabic word for muscle, and consequently the disorder originating from this protein deficiency was originally called “adhalinopathy”. This disorder was considered typical of Northern Africa, Arabian peninsula, but similar cases originated from Japan, Europe, and United States.
It was speculated that the gene encoding 50k DAG protein might be localized in 13q12, and that mutations in this gene may be responsible for SCARMD. However, SCARMD has been shown to be genetically heterogeneous, as families of Brazilian and European descent did not demonstrate linkage to 13q12 27,28.
In 1995 Noguchi 29 discovered that the causative gene in Maghrebian SCARMD patients was that encoding for γ-SG, reported the first mutations in γ-sarcoglycanopathy (LGMD2C/R5), and introduced the term “sarcoglycanopathies” to refer to this group of disorders.
LGMD2C/R5 is often the most severe of autosomal muscular dystrophies 30 and since its first genetic characterization in 1995, a number of patients have been identified worldwide, with a high frequency in gypsy population or other genetic clusters 31-33. Using linkage analysis in SCARMD patients in the inbred Amish population of North America, a second locus was mapped to chromosome 4q216. Since the secondary deficiency of 50k DAG was common to all disorders due to a component of the sarcoglycan protein complex, the term “adhalinopathy” was initially used also for this second sarcoglycan gene mutated.
The gene was that encoding α-sarcoglycan protein, which was initially termed “hetairosin” (which means “accompanying”), an equivocal meaning according to Michel Fardeau, and therefore both terms adhalinopathy and hetairosin have now only an historical role. The first mutations in β-sarcoglycanopathy (LGMD2E/R4) were described contemporarily in patients from the Amish community [6] and in an Italian girl with SCARMD34. The clinical phenotype in this disorder was variable, including relatively mild LGMD cases (Amish patients) and patients with severe SCARMD phenotype.
α-sarcoglycanopathy (LGMD2D/R3) and δ-sarcoglycanopathy (LGMD2F/R6) identification
Linkage analysis in SCARMD patients with 50k DAG protein deficiency have excluded the 13q12 locus 26-28. In 1993 another locus was mapped to 17q21 5 and the first mutations in the gene encoding 50k DAG protein (or adalin or α-sarcoglycan) were identified in French patients 35. The phenotype of LGMD2D/R3 is the most variable among all sarcoglycanopathies, including severe SCARMD patients and very mild myopathic patients 10,30.
In 1996 a locus for LGMD in brazilian families was mapped to 5q33-34 7, and in the same year it was identified the gene and the encoded protein δ-sarcoglycan was discovered by Vincenzo Nigro in a brasilian family 8,36. The phenotype of δ-sarcoglycanopathy (LGMD2F/R6) is that of a severe SCARMD phenotype associated with dilated cardiomyopathy.
Frequency of sarcoglycanopathies
Among total LGMD patients, sarcoglycanopathies constitute about 10-25% of cases in most countries, with higher frequency among inbred populations 24.
LGMD2D/R3 is the most frequent form of sarcoglycanopathies in most countries, followed by LGMD2C/R5 (which, however, is the most frequent form in Maghreb, India and Europe) and by LGMD2E/R4 and LGMD2F/R6. Severe childhood-onset LGMD patients have a higher probability of obtaining a molecular diagnosis than adult-onset LGMD patients. Among patients with the severe SCARMD phenotype, the frequency of sarcoglycanopathies ranges in various populations from between 22 to 69% of cases, whereas among adult-onset LGMD it is only 4-8% 24.
A few genetic epidemiological studies have estimated the prevalence of total sarcoglycanopathies to be about 1:178,000 and 1:370,000 inhabitants 37.
Clinical features of sarcoglycanopathies
The phenotypes of sarcoglycanopathies are rather, similar to those of dystrophinopathies (DMD, BMD), except for the absence of cognitive dysfunction 30 and more frequent occurrence of scapular winging. The most common presentation is a DMD-like phenotype with onset of weakness in childhood (especially in LGMD2C/R5, LGMD2E/R4, LGMD2F/R6), and the disease is more severe and rapid than in other LGMDs. Most patients have a severe and rapid course, leading to loss of independent walking ability before age 30-40 years. On average, the earlier the onset, the more rapid the progression, but in some cases the progression is not linear. Tiptoe walking in early childhood is often present before muscle weakness is detected. Adult-onset patients may be seen especially in LGMD2D/R3 and LGMD2C/R5. The ability to rise from the floor (presence of Gowers’ sign), and to run, jump, and hop, as well as sporting ability may be affected in childhood or may be normal even until middle age. Muscle hypertrophy, especially of the calves and the tongue, is common 30,38.
Clinical variability in the phenotype and in severity has been observed between unrelated patients sharing the same mutation and even between patients belonging to the same family 10, suggesting that different factors of both genetic (intragenic polymorphisms, modulating genes) and non-genetic origin (nutrition, sport activity, body mass index, drugs, infections, inflammatory process) might have a role in determining the clinical phenotype and disease progression.
Cardiac, respiratory involvement and management in sarcoglycanopathies
Dilated cardiomyopathy may occur in all forms, but it is frequent and severe (sometimes fatal) in LGMD2E/R4 and LGMD2F/R6, while in LGMD2C/R5 and LGMD2D/R3 it is mild or occasional 39-42. The subtypes mostly associated with cardiac involvement (manifest as conduction disorders and/or myocardial disease) are those associated with a defect in the genes encoding for the β-SG, or δ-SG proteins, which are expressed in heart and skeletal muscle,
Preclinical cardiomyopathy (44% of cases), and initial cardiomyopathy (19% of cases) are frequent, as well as arrhythmias and dilated cardiomyopathy 39,40. Signs of hypoxic myocardial damage may occur in LGMD2E/R4, LGMD2F/R6, LGMD2C/R5. Abnormal coronary smooth muscle function has been suggested to be involved in the development of cardiomyopathy in LGMD2E and LGMD2F, since β-SG and δ-SG are also expressed in the coronary arteries 43.
Impaired vasoregulation occurs via marked reduction in membrane-associated neuronal nitric oxide synthase (nNOS) in both cardiac and skeletal muscle. Without dystrophin, nNOS mislocalizes to the cytosol; this greater distance between nNOS and the sarcolemma may impair NO diffusion through the myocyte membrane to the microvasculature. As a consequence, insufficient NO release follows muscle contraction resulting in muscle ischemia. Unopposed vasoconstriction may explain the necrosis observed in skeletal and cardiac muscle of dystrophinopathy patients. Microvasculature abnormalities have also been shown to result primarily from absence of dystrophin or sarcoglycan components in cardiomyocytes. The sarcolemmal nNOS expression correlated with the clinical severity 44 and muscle fatigue: absence or severe reduction of sarcolemmal nNOS expression was associated with a severe and childhood-onset form of muscular dystrophy and in most cases also with dilated cardiomyocytes.
Mice lacking either γ-SG or δ-SG display progressive focal cardiomyocyte degeneration that leads to reduced cardiac function and death. This model of cardiomyopathy closely parallels what is seen in humans with SG and dystrophin gene mutations. Null mice for β-SG and δ-SG (but not for αγ-SG) presented a disruption of the vascular smooth muscle SG complex. The perturbed vascular function induces ischemic injury in cardiac and skeletal muscle, suggesting that this mechanism could contribute to the development of cardiomyopathy and exacerbate skeletal myopathy.
It is well known that vascular spasm is an important contributor to cardiac pathology. Elevated levels of intracellular calcium, disturbances of the NOS pathway, and increased activity of protein kinase C, have been implicated in increased contractility and/or spasm of the microvasculature.
Therefore, the observation that sarcolemmal nNOS can be absent or mislocalized in sarcoglycanopathy muscle 45 provides a possible link between this pathogenetic mechanism and the development of cardiomyopathy in sarcoglycanopathies, offering further insights for therapeutic interventions.
NO stimulates soluble guanylate cyclase (sGC) to produce cyclic guanosine monophosphate (cGMP), and in the absence of dystrophin the NO-sGC-cGMP pathway is disrupted. The nucleotide phosphodiesterases (PDEs) hydrolize the cGMP and regulate their downstream signaling. PDE5 expression in cardiomyocytes is low at baseline and increases in response to ischemia or pressure overload from heart failure. Impaired blood flow in muscle and heart in mdx dystrophin-deficient and NOS deficient mice was rescued by inhibition of PDE5. Unfortunately, in DMD and BMD patients a clinical trial with PDE5 inhibitor (Sidenafil) did not improve cardiomyopathy, since 30% of patients progressed to ventricular dilatation.
Long term dietary supplementation of L-arginine (a NOS substrate) was not a viable therapy for dystrophinopathy, but the use of antioxidants that attenuate the superoxide attack and restore the bioactive NO level, might be useful approaches for the treatment of these disorders.
Most patients present respiratory involvement of variable severity, which is especially relevant in the advanced stage of the disease 40, and sometimes results in respiratory failure while patients are still ambulant. Reduced respiratory function can be determined by measurements of forced vital capacity in both sitting and supine position. Respiratory insufficiency can be treated by non-invasive intermittent positive-pressure ventilation with nasal masks. Symptoms that suggest nocturnal hypoventilation are sleep disturbances, early morning headache, and daytime drowsiness. The demonstration of night-time hypoventilation by overnight pulse oximetry should lead to non-invasive nocturnal ventilation. Early detection of cardiomyopathy is important, since institution of cardio-protective medical therapies may slow adverse cardiac remodeling and attenuate heart failure symptoms in these patients. Although electrocardiography and echocardiography are typically advocated for screening, cardiovascular magnetic resonance has shown promise in revealing early cardiac involvement when standard cardiac evaluation is unremarkable.
Joint range of movement necessitates physiotherapy and sometimes orthopaedic intervention. Scoliosis may be a problem in more severely affected patients, and they might need spinal surgery.
Dysferlinopathy identification (LGMD2B/R2)
A number of families of Palestinian and Italian origin suffered from a proximo-distal myopathy mapped to chromosome 2p9 (Fig. 3). Limb-girdle muscular dystrophy type 2B and the distal muscular dystrophy of Miyoshi (MM) are caused by mutations in the DYSF gene encoding the protein dysferlin 46,47. Although the clinical features of LGMD2B/R2 and MM are different, both phenotypes can be detected among patients belonging to the same family 48. The clinical heterogeneity might be attributed to additional epigenetic factors. Dysferlin immunolocalizes to the sarcolemma and has a central role in membrane fusion and repair of the plasmalemma lesions generated by eccentric muscle contraction, as demonstrated by the presence of many crowded vesicles just beneath the sarcolemma 49. Several studies have reported a prominent inflammatory response in dysferlinopathy muscle 50 and increased ubiquitin-proteasomal and autophagic degradation secondary due to high levels of regeneration and inflammation 51. The detection of dysferlin deficiency in muscle offers an important diagnostic tool 46,52.
Figure 3.
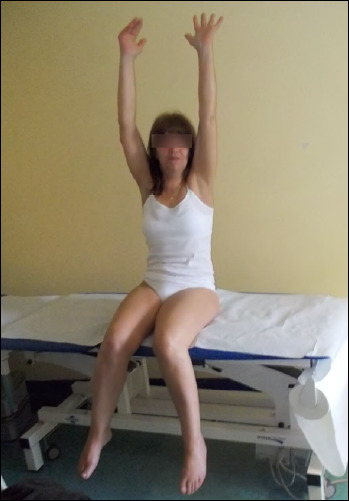
Dysferlinopathy-LGMD phenotype in a patient belonging to a family with affected individuals presenting various clinical phenotypes.
Transportinopathy identification (LGMD1F/D2)
The locus for one form of LGMD with autosomal dominant inheritance has been identified on chromosome 7 in 2003 thorough clinical and genetic analysis of a large Spanish family 53,54.
The identification of the gene and its encoded protein (transportin-3) was obtained in 2013 55,56.
A large three generation family with several branches in Spain and Italy was previously examined and described in detail 54. The clinical history in 29 patients was collected. There were early onset patients who became wheelchair bound around 30 years of age, while in milder cases, walking ability was preserved up to 65 years of age. Some patients had an early onset weakness, but others had the adult onset of the disease, with onset as late as 58 years. The severity appearance of phenotype does not always increase in successive generations 57, at difference to what originally reported by Gamez et al. 54. In fact, anticipation phenomenon is generally seen in triplet expansion disorders, such as myotonic dystrophy or Huntington Chorea but not in this disorder. In this LGMD, the observed main features were dysphagia, dysarthria with bulbar, distal and axial weakness, variably occurring in the family members. The most involved muscles were at onset the lower limb muscles and then weakness spread to upper girdle muscles especially involving the triceps. Abnormal long fingers (arachnodactyly) characterize also the clinical phenotype 57. There was a prominent neck axial weakness (flexor more than extensor). A main clinical sign was observed when patients were lying in bed in fact they were able to raise arm horizontally, but in standing position they were not able to fully lift arms over the head, because of scapular winging.
Transportin-3 protein is involved in HIV virus and other proteins transport in nucleus. The genetic mutations that cause transportinopathy make patients immune to AIDS, holding promise for research in this field and reassuring the patients regarding a possible HIV infection risk. The mutation could block the activity of the HIV-1 intasome and makes it unable to interact with cargo protein causing a block of nuclear import of proteins involved in lentiviral replication: CD3,CD28 peripheral blood cells from transportinopathy patients of the italo-spanish family show in fact lower production of viral proteins in patients than control 58.
Figures and tables
References
- 1.Walton JN, Nattrass FJ. On the classification, natural history and treatment of the myopathies. Brain 1954; 77:169-231. https://doi.org/10.1093/brain/77.2.169 10.1093/brain/77.2.169 [DOI] [PubMed] [Google Scholar]
- 2.Ben Hamida M, Fardeau M, Attia N. Severe childhood muscular dystrophy affecting both sexes and frequent in Tunisia. Muscle Nerve 1983;6:469-80. https://doi.org/10.1002/mus.880060702 10.1002/mus.880060702 [DOI] [PubMed] [Google Scholar]
- 3.Beckmann JS, Richard I, Hillaire D, et al. A gene for limb-girdle muscular dystrophy maps to chromosome 15 by linkage. C R Acad Sci III 1991;312:141-8. PMID 1901754 [PubMed] [Google Scholar]
- 4.Ben Othmane K, Ben Hamida M, Pericak-Vance MA, et al. Linkage of Tunisian autosomal recessive Duchenne-like muscular dystrophy to the pericentromeric region of chromosome 13q. Nat Genet 1992;2:315-7. https://doi.org/10.1038/ng1292-315 10.1038/ng1292-315 [DOI] [PubMed] [Google Scholar]
- 5.McNally EM, Yoshida M, Mizuno Y, et al. Human adhalin is alternatively spliced and the gene is located on chromosome 17q21. Proc Natl Acad Sci USA 1994;91:9690-4. https://doi.org/10.1073/pnas.91.21.9690 10.1073/pnas.91.21.9690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim LE, Duclos F, Broux O, et al. Beta-sarcoglycan: characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet 1995;11:257-65. https://doi.org/10.1038/ng1195-257 10.1038/ng1195-257 [DOI] [PubMed] [Google Scholar]
- 7.Passos-Bueno MR, Moreira ES, Vainzof M, et al. Linkage analysis in autosomal recessive limb-girdle muscular dystrophy (ARLGMD) maps a sixth form to 5q33-34 (LGMD2F) and indicates that there is at least one more subtype of ARLGMD. Hum Molec Genet 1996;5:815-20. https://doi.org/10.1093/hmg/5.6.815 10.1093/hmg/5.6.815 [DOI] [PubMed] [Google Scholar]
- 8.Nigro V, Piluso G, Belsito A, et al. Identification of a novel sarcoglycan gene at 5q33 encoding a sarcolemmal 35 kDa glycoprotein. Hum Molec Genet 1996;5:1179-86. https://doi.org/10.1093/hmg/5.8.1179 10.1093/hmg/5.8.1179 [DOI] [PubMed] [Google Scholar]
- 9.Bashir R, Strachan T, Keers S, et al. A gene for autosomal recessive limb-girdle muscular dystrophy maps to chromosome 2p. Hum Molec Genet 1994;3:455-7. https://doi.org/10.1093/hmg/3.3.455 10.1093/hmg/3.3.455 [DOI] [PubMed] [Google Scholar]
- 10.Angelini C, Fanin M, Menegazzo E, et al. Homozygous a-sarcoglycan mutation in two siblings: one asymptomatic and one steroid responsive mild limb-girdle muscular dystrophy patient. Muscle Nerve 1998;21:769-75. [DOI] [PubMed] [Google Scholar]
- 11.Angelini C, Fanin M. Limb girdle muscular dystrophies: clinical-genetical diagnostic update and prospects for therapy. Exp Opin Orphan Drugs 2017;5:769-84. https://doi.org/10.1002/(sici)1097-4598(199806)21:6<769::aid-mus9>3.0.co;2-5 [DOI] [Google Scholar]
- 12.Bushby KM. Diagnostic criteria for the limb-girdle muscular dystrophies: report of the ENMC Consortium on Limb-Girdle Dystrophies. Neuromusc Disord 1995;5:71-4. https://doi.org/10.1016/0960-8966(93)e0006-g 10.1016/0960-8966(93)e0006-g [DOI] [PubMed] [Google Scholar]
- 13.Bushby KMD, Beckmann JS. The limb girdle muscular dystrophies. Proposal for a new nomenclature. Neuromusc Disord 1995;5:337-43. https://doi.org/10.1016/0960-8966(95)00005-8 10.1016/0960-8966(95)00005-8 [DOI] [PubMed] [Google Scholar]
- 14.Straub V, Murphy A, Udd B, et al. 229th ENMC international workshop: limb girdle muscular dystrophies. Nomenclature and reformed classification. Neuromusc Disord 2018;28:702-10. [DOI] [PubMed] [Google Scholar]
- 15.Nigro V, Piluso G. Next generation sequencing (NGS) strategies for the genetic testing of myopathies. Acta Myol 2012;33:1-12. PMID 23620651 [PMC free article] [PubMed] [Google Scholar]
- 16.Bushby KM. The limb-girdle muscular dystrophies-multiple genes, multiple mechanisms. Hum Mol Genet 1999;8:1875-82. https://doi.org/10.1093/hmg/8.10.1875 10.1093/hmg/8.10.1875 [DOI] [PubMed] [Google Scholar]
- 17.Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995;81:27-40. https://doi.org/10.1016/0092-8674(95)90368-2 10.1016/0092-8674(95)90368-2 [DOI] [PubMed] [Google Scholar]
- 18.Fardeau M, Hillaire D, Mignard C, et al. Juvenile limb girdle muscular dystrophy. Clinical, histopathological, and genetic data from a small community living in the Reunion Island. Brain 1996;119:295-308. https://doi.org/10.1093/brain/119.1.295 10.1093/brain/119.1.295 [DOI] [PubMed] [Google Scholar]
- 19.Fanin M, Nascimbeni AC, Fulizio L, et al. The frequency of limb girdle muscular dystrophy 2A in northeastern Italy. Neuromusc Disord 2005;15:218-24. https://doi.org/10.1016/j.nmd.2004.11.003 10.1016/j.nmd.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 20.Fanin M, Benedicenti F, Fritegotto C, et al. An intronic mutation causes severe LGMD2A in a large inbred family belonging to a genetic isolate in the Alps. Clin Genet 2012;82:601-2. https://doi.org/10.1111/j.1399-0004.2012.01873.x 10.1111/j.1399-0004.2012.01873.x [DOI] [PubMed] [Google Scholar]
- 21.Fanin M, Nascimbeni A, Fulizio L, et al. Loss of calpain-3 autocatalytic activity in LGMD2A patients with normal protein expression. Am J Pathol 2003;163:1929-36. https://doi.org/10.1016/S0002-9440(10)63551-1 10.1016/S0002-9440(10)63551-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angelini C, Nardetto L, Borsato C, et al. The clinical course of calpainopathy (LGMD2A) and dysferlinopathy (LGMD2B). Neurol Res 2010;32:41-6. https://doi.org/10.1179/174313209X380847 10.1179/174313209X380847 [DOI] [PubMed] [Google Scholar]
- 23.Fanin M, Angelini C. Protein and genetic diagnosis of limb girdle muscular dystrophy type 2A: the yield and the pitfalls. Muscle Nerve 2015; 52:163-73. https://doi.org/10.1002/mus.24682 10.1002/mus.24682 [DOI] [PubMed] [Google Scholar]
- 24.Fanin M, Nascimbeni AC, Aurino S, et al. Frequency of LGMD gene mutations in Italian patients with distinct clinical phenotypes. Neurology 2009; 72:1432-5. https://doi.org/10.1212/WNL.0b013e3181a1885e 10.1212/WNL.0b013e3181a1885e [DOI] [PubMed] [Google Scholar]
- 25.Matsumura K, Tomè FMS, Collin H, et al. Deficiency of the 50k dystrophin-associated glycoprotein in severe childhood autosomal recessive muscular dystrophy. Nature 1992;359:320-2. https://doi.org/10.1038/359320a0 10.1038/359320a0 [DOI] [PubMed] [Google Scholar]
- 26.Romero NB, Tomè FMS, Leturcq F, et al. Genetic heterogeneity of SCARMD with adhalin (50 kDa dystrophin-associated glycoprotein) deficiency. CR Acad Sci Paris 1994;317:70-6. PMID 7987694 [PubMed] [Google Scholar]
- 27.Passos-Bueno MR, Oliveira JR, Bakker E, et al. Genetic heterogeneity for Duchenne-like muscular dystrophy (DLMD) based on linkage and 50 DAG analysis. Hum Molec Genet 1993;2:1945-7. https://doi.org/10.1093/hmg/2.11.1945 10.1093/hmg/2.11.1945 [DOI] [PubMed] [Google Scholar]
- 28.Fardeau M, Matsumura K, Tomè FMS, et al. Deficiency of the 50 kDa dystrophin associated glycoprotein (adhalin) in severe autosomal recessive muscular dystrophies in children native from European countries. CR Acad Sci Paris 1993;316:799-804. PMID 8044705 [PubMed] [Google Scholar]
- 29.Noguchi S, McNally EM, Ben Othmane K, et al. Mutations in the dystrophin-associated protein γ-sarcoglycan in chromosome 13 muscular dystrophy. Science 1995; 270:819-22. https://doi.org/10.1126/science.270.5237.819 10.1126/science.270.5237.819 [DOI] [PubMed] [Google Scholar]
- 30.Angelini C, Fanin M, Freda MP, et al. The clinical spectrum of sarcoglycanopathies. Neurology 1999;52:176-9. https://doi.org/10.1212/wnl.52.1.176 10.1212/wnl.52.1.176 [DOI] [PubMed] [Google Scholar]
- 31.Duggan DJ, Gorospe JRM, Fanin M, et al. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med 1997;336:618-24. https://doi.org/10.1212/wnl.52.1.176 10.1212/wnl.52.1.176 [DOI] [PubMed] [Google Scholar]
- 32.Merlini L, Kaplan JC, Navarro C, et al. Homogeneous phenotype of the gypsy limb-girdle MD with the gamma-sarcoglycan C283Y mutation. Neurology 2000;54:1075-9. https://doi.org/10.1212/wnl.54.5.1075 10.1212/wnl.54.5.1075 [DOI] [PubMed] [Google Scholar]
- 33.Fanin M, Hoffman EP, Angelini C, et al. Private beta- and gamma-sarcoglycan gene gene mutations: evidence of a founder effect in northern Italy. Hum Mut 2000;16:13-7. https://doi.org/10.1002/1098-1004(200007)16:1<13::AID-HUMU3>3.0.CO;2-V [DOI] [PubMed] [Google Scholar]
- 34.Bonnemann CG, Modi R, Noguchi S, et al. β-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet 1995;11:266-73. https://doi.org/10.1038/ng1195-266 10.1038/ng1195-266 [DOI] [PubMed] [Google Scholar]
- 35.Roberts SL, Leturcq F, Allamand V, et al. Missense mutations in the adhalin gene linked to autosoml recessive muscular dystrophy. Cell 1994;78:625-33. https://doi.org/10.1016/0092-8674(94)90527-4 10.1016/0092-8674(94)90527-4 [DOI] [PubMed] [Google Scholar]
- 36.Nigro V, Moreira E, Piluso G, et al. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the δ-sarcoglycan gene. Nat Genet 1996;14:195-8. https://doi.org/10.1038/ng1096-195 10.1038/ng1096-195 [DOI] [PubMed] [Google Scholar]
- 37.Fanin M, Duggan DJ, Mostacciuolo ML, et al. Genetic epidemiology of muscular dystrophies resulting from sarcoglycan gene mutations. J Med Genet 1997;34:973-7. https://doi.org/10.1136/jmg.34.12.973 10.1136/jmg.34.12.973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Angelini C, Fanin M. Pathogenesis, clinical features and diagnosis of sarcoglycanopathies. Exp Opin Orphan Drugs 2016;4:1239-51. [Google Scholar]
- 39.Melacini P, Fanin M, Duggan DJ, et al. Heart involvement in muscular dystrophies due to sarcoglycan gene mutations. Muscle Nerve 1999;22:473-9. https://doi.org/10.1002/(sici)1097-4598(199904)22:4<473::aid-mus8>3.0.co;2-5 [DOI] [PubMed] [Google Scholar]
- 40.Politano L, Nigro V, Passamano L, et al. Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromusc Disord 2001;11:178-85. https://doi.org/10.1016/s0960-8966(00)00174-7 10.1016/s0960-8966(00)00174-7 [DOI] [PubMed] [Google Scholar]
- 41.Fanin M, Melacini P, Boito C, et al. LGMD2E patients risk developing dilated cardiomyopathy. Neuromusc Disord 2003;13:303-9. https://doi.org/10.1016/s0960-8966(02)00280-8 10.1016/s0960-8966(02)00280-8 [DOI] [PubMed] [Google Scholar]
- 42.Barresi R, Di Blasi C, Negri T, et al. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by beta sarcoglycan mutations. J Med Genet 2000;37:102-7. https://doi.org/10.1136/jmg.37.2.102 10.1136/jmg.37.2.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coral-Vasquez R, Cohn RD, Moore SA, et al. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell 1999;98:465-74. https://doi.org/10.1016/s0092-8674(00)81975-3 10.1016/s0092-8674(00)81975-3 [DOI] [PubMed] [Google Scholar]
- 44.Fanin M, Tasca E, Nascimbeni AC, et al. Sarcolemmal neuronal nitric oxide synthase defect in limb-girdle muscular dystrophy: an adverse modulating factor in the disease course? J Neuropathol Exp Neurol 2009;68:383-90. https://doi.org/10.1097/NEN.0b013e31819cd612 10.1097/NEN.0b013e31819cd612 [DOI] [PubMed] [Google Scholar]
- 45.Heydemann A, Huber JM, Kakkar R, et al. Functional nitric oxide synthase mislocalization in cardiomyopathy. J Molec Cell Cardiol 2004;36:213-23. J Molec Cell Cardiol 2004;36:213-23 https://doi.org/10.1016/j.yjmcc.2003.09.020 10.1016/j.yjmcc.2003.09.020 [DOI] [PubMed] [Google Scholar]
- 46.Fanin M, Pegoraro E, Matsuda-Asada C, et al. Calpain-3 and dysferlin protein screening in limb-girdle muscular dystrophy and myopathy patients. Neurology 2001;56:660-5. https://doi.org/10.1212/wnl.56.5.660 10.1212/wnl.56.5.660 [DOI] [PubMed] [Google Scholar]
- 47.Cacciottolo M, Numitone G, Aurino S, et al. Muscular dystrophy with marked dysferlin deficiency is consistently caused by primary dysferlin gene mutations. Eur J Hum Genet 2011;19:974-80. https://doi.org/10.1038/ejhg.2011.70 10.1038/ejhg.2011.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Angelini C, Fanin M, Brown RH. Phenotypic heterogeneity of dysferlin gene mutation in the same pedigree. Acta Myol 1999;3:61-3. [Google Scholar]
- 49.Cenacchi G, Fanin M, Badiali De Giorgi L, et al. Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J Clin Pathol 2005;58:190-5. https://doi.org/10.1136/jcp.2004.018978 10.1136/jcp.2004.018978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fanin M, Angelini C. Muscle pathology in dysferlin deficiency. Neuropathol Appl Neurobiol 2002;28:461-70. https://doi.org/10.1046/j.1365-2990.2002.00417.x 10.1046/j.1365-2990.2002.00417.x [DOI] [PubMed] [Google Scholar]
- 51.Fanin M, Nascimbeni AC, Angelini C. Muscle atrophy, ubiquitin-proteasome, and autophagic pathways in dysferlinopathies. Muscle Nerve 2014;33:119-26. https://doi.org/10.1002/mus.24167 10.1002/mus.24167 [DOI] [PubMed] [Google Scholar]
- 52.Fanin M., Angelini C. Progress and challenges in diagnosis of dysferlinopathies. Muscle Nerve 2016;54:821-35. https://doi.org/10.1002/mus.25367 10.1002/mus.25367 [DOI] [PubMed] [Google Scholar]
- 53.Palenzuela L, Andreu AL, Gamez J, et al. A novel autosomal dominant limb-girdle muscular dystrophy (LGMD1F) maps to 7q32.1-32.2. Neurology 2003;61:404-6. https://doi.org/10.1212/01.wnl.0000073984.46546.4f 10.1212/01.wnl.0000073984.46546.4f [DOI] [PubMed] [Google Scholar]
- 54.Gamez J, Navarro C, Andreu AL, et al. Autosomal dominant limb-girdle muscular dystrophy: a large kindred with evidence for anticipation. Neurology 2001;56:450-4. https://doi.org/10.1212/wnl.56.4.450 10.1212/wnl.56.4.450 [DOI] [PubMed] [Google Scholar]
- 55.Torella A, Fanin M, Mutarelli M, et al. Next-generation sequencing identifies Transportin 3 as the causative gene for LGMD1F. PLoS One 2013;8:e63536 https://doi.org/10.1371/journal.pone.0063536 10.1371/journal.pone.0063536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Melià MJ, Kubota A, Ortolano S, et al. Limb-girdle muscular dystrophy 1F is caused by a microdeletion in the transportin 3 gene. Brain 2013;136:1508-17. https://doi.org/10.1093/brain/awt074 10.1093/brain/awt074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peterle E, Fanin M, Semplicini C, et al. Clinical phenotype, muscle MRI and muscle pathology of LGMD1F. J Neurol 2013;260:2033-41. https://doi.org/10.1007/s00415-013-6931-1 10.1007/s00415-013-6931-1 [DOI] [PubMed] [Google Scholar]
- 58.Rodríguez-Mora S, De Wit F, García-Perez J, et al. The mutation of Transportin 3 gene that causes limb girdle muscular dystrophy 1F induces protection against HIV-1 infection. PLoS Pathog 2019:15:e1007958 https://doi.org/10.1371/journal.ppat.1007958 10.1371/journal.ppat.1007958 [DOI] [PMC free article] [PubMed] [Google Scholar]