

Abstract
Myasthenia gravis (MG) is an autoimmune disease of the neuromuscular junction which affects all striated muscles, resulting in fluctuating weakness. Approaching MG as a disease with subgroups having different clinical, serological and genetic features is crucial in predicting the progression and planning treatment. Three relatively less frequently seen subtypes of MG are the subject of this review: MG with anti-MuSK antibodies (MuSK MG), non-thymomatous late-onset MG (LOMG), and ocular MG (OMG). In addition to reviewing the literature, mainly from a clinical point of view, our experience in each of the subgroups, based on close to 600 patients seen over a 10 year period, is related. MuSK MG is a severe disease with predominant bulbar involvement. It is more common in women and in early-onset patients. With the use of high dose corticosteroids, azathioprine and more recently rituximab, outcome is favorable, though the patients usually require higher maintenance doses of immunosuppressives. LOMG with onset ≥ 50 years of age is more common in men and ocular onset is common. Frequency of anti-AChR and anti-titin antibodies are high. Although it can be severe in some patients, response to treatment is usually very good. OMG is reported to be more frequent in men in whom the disease has a later onset. Anti-AChR antibodies are present in about half of the patients. Generalization is less likely when symptoms remain confined to ocular muscles for 2 years. Low dose corticosteroids are usually sufficient. Thyroid disease is the most common autoimmune disease accompanying all three subgroups.
Key words: Myasthenia gravis, MuSK MG, Late-onset MG, Ocular MG
Introduction
Myasthenia gravis (MG) is an autoimmune disease of the neuromuscular junction which affects all striated muscles, resulting in fluctuating weakness 1. MG is mostly caused by antibodies against the acetylcholine receptor (AChR) and rarely by antibodies against muscle specific kinase (MuSK). In some of the patients without any detected antibodies (seronegative MG, SN MG), anti-AChR and anti-MuSK antibodies can be found with finer techniques 2. The clinical significance of anti-LRP4 antibodies, many times coexisting with the classical antibodies in the same patient, is unknown.
Approaching MG as a disease with subgroups having different clinical, serological and genetic features 3-5 is crucial in predicting the progression and planning treatment. MG is associated with thymoma in 10-15% of patients. Among those without a thymoma, the largest subgroup consists of young women (age < 50 years) with anti-AChR antibodies. An increasingly important subgroup is MG with later onset (≥ 50 years). A very small subgroup is caused by antibodies against the MuSK antigen. MG usually tends to affect the oculobulbar and extremity muscles in all of these patients. In a small subgroup, it affects only the ocular muscles.
Three relatively less frequently seen subtypes of MG are the subject of this review: MG with anti-MuSK antibodies (MuSK MG), late-onset MG (LOMG), and ocular MG (OMG). Attempt was made to define the subgroups carefully with particular attention given to distinguishing generalized from ocular disease. In addition to reviewing the literature, mainly from a clinical point of view, our experience in each of the subgroups, based on close to 600 patients seen over a 10 year period, is related. To put our data on the subgroups into perspective, it is necessary to describe our cohort first.
Istanbul University MG cohort
Our cohort consisted of 576 patients, derived from our MG Database, who applied for the first time to the Neuromuscular Outpatient Clinic, Neurology Department, Istanbul Medical Faculty, Istanbul University (IU) during a 10 year period between 2001 and 2010. It is an unselected group with consecutive patients, subjected to the same criteria for inclusion in a set period of time.
Some patients with MuSK MG from this cohort were reported in two articles 6,7, they consisted of all MuSK MG patients seen until 2005 6 and until 2009 7. Patients with LOMG in this cohort were also reported 8. A manuscript on ocular MG, again with patients in this cohort, is in preparation.
Evaluating the frequency of the subgroups, thymoma-associated MG (TAMG) was found in 14%. Early-onset (< 50 years) generalized MG was the most common subgroup comprising close to 40% of the cohort. Late-onset (≥ 50 years) generalized MG and ocular MG (OMG) each made up about one fifth of the patients. MuSK MG comprised 7% of the cohort (Fig. 1). Taking both onset age and antibody status into consideration (Fig. 2), it was striking to note that the subgroups of MuSK MG and SN MG were extremely rare in older ages. Also interesting was the fact that thymoma was more common in younger patients in this cohort. Antibody status of ocular MG, about half with anti-AChR antibodies, is not indicated in the figure to avoid confusion. Almost all patients with thymoma had anti-AChR antibodies.
Figure 1.
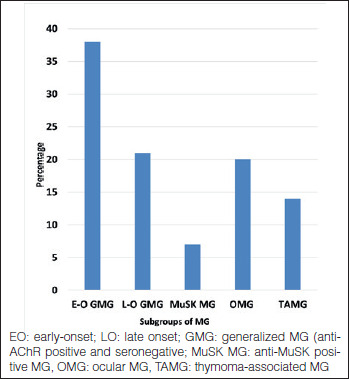
Subgroups of MG (576 patients).
Figure 2.
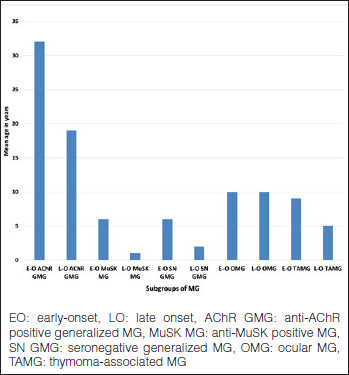
Subgroups of MG according to onset age (576 patients).
Gender differences among the subgroups were noteworthy. Women predominated in early-onset generalized AChR MG and MuSK MG. Women also predominated in seronegative generalized MG, both early-onset and late-onset. Men predominated in late-onset generalized AChR MG. The two genders were comparable in ocular MG and TAMG (Fig. 3).
Figure 3.
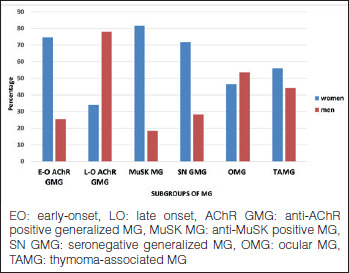
Subgroups of MG. Percentages of gender (576 patients).
Our percentages in general reflected what is reported in the literature, except that thymoma was more common in younger patients in our cohort.
MuSK MG
MG with antibodies to the postsynaptically located MuSK protein is a relatively newly defined disease. It constitutes less than 10% of the MG patients, being more prevalent in the Mediterranean countries as compared to the northern ones in Europe, but more prevalent in the north as compared to the south in China 5. The antibodies are mainly of the IgG4 class as opposed to IgG1 in MG with anti-AChR antibodies (AChR MG) 2. There is a specific association with HLA-DRB1∗14, -DRB1∗16 and -DQB1∗05 HLA 9,10.
Onset age peaks in the late 30’s and it is uncommon in prepubertal and elderly patients 6,11. The disease is reported to be more common in women 11 although female preponderance was found to be comparable to AChR MG and SN MG in one report 6. Clinically, it constitutes the most severe form of MG, paralleled by TAMG 5. Bulbar, neck and respiratory muscles are involved with fast progression of the disease to life-threatening symptoms 11,12. Dropped head syndrome with neck extensor muscle weakness can be the presenting symptom 13,14. Ocular muscles are frequently mildly involved, and conjugate limitation of eye movements which is exceptional in MG can occur in MuSK MG 11. Extremity muscles are usually spared or mildly affected. In those with ocular onset, generalization occurs in a short time. Rare patients generalize after a few years and pure ocular MG is extremely rare. Interestingly, we had one patient with onset at 3 years 15 who remained with purely ocular symptoms for a prolonged period before generalization, similar to another reported patient with MuSK MG 16, and in accordance with the frequent course of prepubertal-onset patients 17.
There are difficulties in diagnosis when the onset is indolent and fluctuations are not evident. At least half of the patients do not respond to anticholinesterases and a few can get worse, fasciculations and cramps are common 14. Decrement is usually not present in the extremity muscles with repetitive nerve stimulation. The muscle with the best yield was found to be the orbicularis oculi 18. Needle EMG may show a myopathic pattern.
Corticosteroids are the most effective drugs, but they are needed in high doses in many patients. Usually a second immunosuppressive is required at an early stage. Plasma exchange is considered to be more beneficial than IVIg in MuSK MG 19. Although the disease is usually severe, many patients eventually do well with appropriate therapy 11,14. We had previously compared all MuSK MG and generalized SN MG patients registered in our MG database with consecutive non-thymomatous 161 AChR MG patients 6. Their outcome with conventional therapy was similar to AChR MG patients; however, they required higher dose of maintenance corticosteroid therapy. Tongue atrophy has been noted in MuSK MG, but the role of refractoriness and the use of long term high dose corticosteroids is questioned 20.
There are MuSK MG patients who are clinically not distinguishable from non-MuSK MG 12. Likewise, some MuSK MG patients can be easily treated. We had reported 7 that fast and good response within 3 months of starting corticosteroids predicted good outcome in MuSK MG patients. These patients, comprising one third of the study population, had pharmacological remission or minimal manifestations on a mean low maintenance dose of 6 mg/day (or as is generally done, 12 mg on alternate days) of prednisolone, usually with additional azathioprine.
Newer drugs such as rituximab have changed the scene in MuSK MG, particularly for refractory patients 21-23. It is considered to be an early therapeutic option in the patients without a satisfactory initial response to conventional immunosuppressives 19. Symptomatic treatment with 3,4-diamynopyridine (3,4-DAP) and albuterol is also being considered 11. Over the years, a better outcome is noted in MuSK MG patients, attributed to early diagnosis and better treatment 11.
It has been difficult to evaluate the effect of thymectomy, because of the confounding effect of steroids, which have usually been started before the surgery. The pathology of the thymus showed involution in most patients 24, further suggesting that the thymus may not play a similar role in MuSK MG as compared to AChR MG. Thymectomy is now not considered to be indicated in MuSK MG. There are exceptional cases with small thymomas 25. In parallel to the other subgroups of MG, thyroid disease was the most frequent autoimmune disease in MuSK MG 11.
One point needs to be emphasized. An important differential diagnosis of MuSK MG is with bulbar amyotrophic lateral sclerosis (ALS), particularly because of fasciculations 26 and myotonic discharges 27 in rare patients with MuSK MG. Absence of definite fluctuations, absence of response to anticholinesterases, and difficulty in finding decrement in classically-examined muscles in repetitive nerve stimulation in some patients with MuSK MG may suggest ALS in the face of bulbar symptoms/head drop without ocular symptoms. Even without fasciculations, the differential diagnosis of MuSK MG and bulbar ALS can be difficult. It is sometimes difficult to distinguish from ALS the speech in MuSK MG which may lack the nasal quality of typical MG. Furthermore, mild fluctuations, mild response to anticholinesterases and decrement can all be seen in ALS. Needle EMG of the extremities is usually normal when the symptoms are confined to bulbar muscles in ALS. Caution is necessary in the diagnosis of some cases with bulbar symptoms.
IU MG cohort: MuSK MG
In our cohort of 576 patients, 38 had MuSK MG. One patient, a man with onset at age 39, had a father with LOMG, who was negative for both anti-AChR and -MuSK antibodies. In the cohort, median age of onset was 34 years (range of 3-66 years). Below age 16, there was only one patient with onset at 3 years 15. Onset age was ≥ 50 in 5 patients. Eighty-two percent were women. Onset with ocular symptoms was present in 15 patients (39%). None had ocular MG throughout the entire period of observation. Onset with bulbar symptoms was seen in 12 patients (32%). Three patients had head drop as an onset symptom. The rest had oculobulbar or extremity onset.
Most of our patients did not respond to anticholinesterases. However, some patients responded to lower doses so that anticholinesterases can still be an option to be used with caution in MuSK MG. Of note is the fact that the initial test dose was very good even in some of the non-responders. About half of the patients had hypersensitivity to anticholinesterases. As reported, we found that repetitive nerve stimulation of the orbicularis oculi muscles yielded the best results, both in the frequency and in the amount of decrement.
Maximum severity and outcome were evaluated in 36 patients excluding the two patients who had been seen only once. Median disease duration was 8 years (range: 1-20 years) and median follow-up was 6 years (range: 1-13 years). Using the Myasthenia Gravis Foundation of America (MGFA) Classification 28, 75% were MGFA Class 3 or above; 31% of these were 4b and 3 had been intubated (MGFA Class 5). Three patients had tongue atrophy; it was notable that two of these patients had been treated late and the response had taken a long time. Four patients had fasciculations, usually related to the beginning of MG or to exacerbations.
Treatment included corticosteroids and azathioprine. Prednisolone at 1 mg/kg/day or 60 mg/day was usually needed and it was continued for a longer period than usual in some patients. A look at the outcome revealed that two patients had died of MG. Twenty patients (56%) had reached MGFA post-intervention status (PIS) 28 of pharmacological remission or minimal manifestations with a mean maintenance dose of about 10 mg/day of prednisolone, but there was none with complete stable remission. Two were unchanged and the rest had improved to some degree.
At the time of this cohort, treatment of MuSK MG was a little different in that we did not use rituximab and had not yet discontinued performing thymectomies. In fact, 23 patients had been thymectomized. No patient in our cohort had a thymoma. In most of these patients, corticosteroids had been started before thymectomy and in a few within 3-4 months after thymectomy so that it was impossible to evaluate the effect of thymectomy properly. Involution of the thymus was the main pathological result in these patients with a few showing mild hyperplasia. Overall, it is not possible to say that thymectomy has any effect in MuSK MG.
Late-onset MG
The prevalence of LOMG has increased in recent years, 29 worldwide 30. It is not clear whether the increase reflects a biological phenomenon31 or has been influenced by increased awareness, availability of the AChR antibody assay 29,32 and longer lifespan 29. LOMG is different from early-onset MG (EOMG) with respect to demographic-clinical characteristics as well as serological properties. Specific HLA associations of LOMG have been discovered 33,34. Comparison between studies on LOMG is somewhat difficult because of different cut-off ages used to distinguish LOMG from EOMG, and the inclusion/exclusion of thymoma. Despite the difficulties, there seems to be consensus on many characteristics of LOMG.
The percentage of men is higher in LOMG 8,17,35-37. Anti-AChR antibodies were present in over 80% 8,17,35-38. A rising frequency with higher decades of the percentage of anti-AChR antibodies 8,37 was reported, with 93% in very-late-onset MG (≥ 65 years) 37. Anti-MuSK antibodies were found to be uncommon after 65-70 years of age 11,37. The implication of these findings is that one has to be extremely cautious when diagnosing MG in double seronegative patients in the elderly and other causes have to be sought diligently.
Anti-titin antibodies were present in one third to over one half of the LOMG patients 8,38-40, being more frequent in LOMG than in EOMG. They increased in higher decades in LOMG 8,40. Anti-titin antibodies were not found to be a poor prognostic factor in LOMG in two studies 8,40.
Ocular onset was more frequent in LOMG 8,17,36,37,41. Myasthenic crisis occurred in 6-11% of the patients 8,17,36,37. Although a higher percentage of patients presented with life-threatening events at onset, they responded better to medications; those over 65 were particularly better than early-onset patients in terms of drug requirements and drug refractoriness 37. At the end of follow-up, over 80% were reported to be improved or better 8,17,35,42. Beneficial effect of thymectomy in LOMG has been reported, but it is usually not advised beyond age 55. Thyroid disease was reported to be the most frequent autoimmune disease accompanying LOMG 8,36.
IU MG cohort: Late-onset MG
In an attempt to understand more about non-thymomatous generalized LOMG and its outcome, we analyzed separately 95 of the LOMG patients with generalized symptoms (ocular MG excluded) who had been followed for ≥ 3 years in the same cohort. Although reported 8, it might be useful to emphasize some of the findings. Men constituted 63% of the patients. Onset was ocular in 62%, bulbar in 23% and in the extremities in 15%. Anti-AChR antibodies were positive in 84% and anti-MuSK antibodies in only 5%. Anti-titin antibodies were present in 61%.
Half of the patients were MGFA Class 3 or above with myasthenic crisis in 6%. Outcome was good with 63% reaching MGFA PIS of complete stable remission, pharmacological remission or minimal manifestations with a mean maintenance dose of about 5 mg/day of prednisolone and a further 24% were improved. Patients in whom azathioprine was added to prednisolone did significantly better than those receiving only prednisolone. Another finding in the study was that many patients with mild disease who received a low maximum dose of prednisolone (≤ 30 mg/day), usually together with azathioprine, had a favorable outcome, implying that low-dose prednisolone with additional azathioprine may be sufficient in mild disease of older people. Thymectomy, done in 12 patients, was not found to be useful although it is not easy to evaluate thymectomy when corticosteroids are used.
Ocular MG
Ocular MG (OMG) refers to patients whose symptoms are confined to ocular muscles (levator palpebrae superioris and extraocular muscles), resulting in ptosis and diplopia; orbicularis oculi muscles are variably weak. Ocular muscles are involved in about 50% of patients at onset of MG. In about half of these patients, generalization occurs to non-ocular muscles, leaving about one fourth of patients with purely ocular symptoms for a prolonged/indefinite period 44-46. Generalization usually occurs within one year, mostly within 2 years and it is uncommon after 2 years 44-46. Generalization is reported to be more likely in anti-AChR positive 47,48 and TAMG 49. Purely ocular MG is more common in Asians, particularly in juvenile-onset patients 50. It is reported to be more frequent in men 51,52. Spontaneous remissions with a mean of 4-5 years are reported to occur, mainly at earlier stages of the disease 46,51.
When a patient presents with ocular symptoms, there are several clinical clues which make the diagnosis of MG more likely. Complete or almost complete unilateral ptosis without pupillary changes, one sided ptosis alternating with ptosis on the other side, definite improvement in the mornings and presence of remissions are very suggestive of the diagnosis of MG. Ptosis can be mild. Eye movement limitation is usually asymmetrical, it can mimick all cranial nerve palsies as well as internuclear ophthalmoplegia.
Diagnosis is easy when anti-AChR antibodies, present in about half of them, are detected. Anti-MuSK antibodies are extremely rare in MG with prolonged pure ocular symptoms. When antibodies are not detected, diagnosis can be difficult in some patients. Response to anticholinesterases may not be present in some patients; on the other hand, other entities such as intracranial mass lesions may show a positive response, usually requiring cranial magnetic resonance imaging for the differential diagnosis 47. Repetitive nerve stimulation is not very useful with purely ocular symptoms. Single fiber EMG is very sensitive, but one has to be careful remembering that it is not specific to MG 53. One other caveat about single fiber EMG is in order: Presence of abnormalities in limb muscles of a patient with OMG does not indicate generalization 54, the diagnosis of generalized MG is clinical. When all tests are negative in a patient with ocular symptoms, a trial of corticosteroids may be necessary.
In the differential diagnosis, mitochondrial myopathy (progressive external ophthalmoplegia, PEO) must be considered. Diplopia is not a feature of PEO; however, ptosis can be asymmetrical and fluctuations can be present, making the differential diagnosis with MG difficult. Oculopharyngeal muscular dystrophy is usually bilateral and without eye movement limitation, at least for a long time. Congenital myasthenic syndromes usually start in infancy and it is then easy to eliminate MG. The distinction with thyroid ophthalmopathy can be difficult. In thyroid ophthalmopathy, ptosis is rare; esotropia (inward deviation of the eye) and hypotropia (downward deviation of the eye) are usually present since muscles causing restriction are medial rectus and inferior rectus. Thus, ptosis and exotropia (outward deviation of the eye) are suggestive of MG in a patient with thyroid ophthalmopathy 55.
Anticholinesterases provide symptomatic treatment for some patients with ptosis, but they are usually not useful for diplopia since the required precise alignment cannot be achieved 50. Corticosteroids have caused debates on whether they are beneficial or not 50. Observational studies 47,56 and one clinical trial 57 with a small number of patients (11 patients) have found them to be effective. Generalization appeared to be less likely in the patients who received immunosuppressives. It is emphasized that low doses are sufficient: About 25 mg/day or twice the dose on alternate days usually results in pharmacological remission. A real concern is the appearance of relapses upon discontinuation of steroids in many patients, necessitating long term administration with small doses. High dose intravenous methylprednisone has also been advocated 58. Azathioprine and mycophenolate mofetil have been found to be beneficial 50 and again lower than the standard doses may be sufficient in OMG. It takes a few months for them to take effect so that they are usually used as additive drugs. However, they can be given as the sole therapeutic agent in selected patients.
Intravenous immunoglobulins do not improve the symptoms 59. Thymectomy, reported to be beneficial in some patients 60, is usually not considered to be indicated in OMG, although some centers might have revised their indications after the advent of videothoracoscopic thymectomy.
Eye patches for diplopia, prisms when eye movement limitation is mild and eyelid crutches for ptosis can be useful. There are rare patients who do not improve and have severe symptoms/signs despite all therapy. In these patients, if the signs are chronic and stable, blepharoplasty can be a good option, taking care not to cause the diplopia to be more disturbing once the ptosis is alleviated.
IU MG cohort: Ocular MG
In our cohort, 101 patients with a median disease duration of 8 years remained with purely ocular symptoms/signs throughout the entire period of observation. Two of the patients had thymomas. In the cohort, almost equal distribution was present regarding gender as well as onset age with very slight preponderance of men and early-onset. In patients with early-onset, women predominated while men predominated in late-onset. About half of the patients were anti-AChR positive.
Low dose prednisolone of 15-30 mg/day was sufficient in many patients. Addition of azathioprine, sometimes also at a low dose, appeared to be beneficial. Azathioprine was used as a single agent successfully in selected patients. We have also found that several courses of intravenous methylprednisone pulse, alone or in addition to oral steroids, was very helpful in difficult cases.
Myasthenia generalized after 2 years in another small group of patients. The majority of the patients were anti-AChR positive. Presence of thymoma was another risk factor for generalization. The two patients with anti-MuSK antibodies also generalized after several years.
Conclusions
In this review, the importance of analyzing the subgroups separately was emphasized because each has its own characteristics and different treatment approaches. Thymectomy, a very important treatment in MG, is not considered to be a widely-accepted therapeutic option in any of these subgroups. Corticosteroids are useful in all of the subgroups; however, while MuSK MG patients need high doses, low doses are usually effective in OMG, and LOMG patients can also respond well to low doses. Additional immunosuppressives are usually needed in order to taper corticosteroids to low maintenance doses and perhaps to be able to discontinue them. With the advent of new therapies, the differences between the subgroups are likely to play a more important role.
Figures and tables
Acknowledgments
All of the patients were seen at the Neuromuscular Unit, Istanbul Medical Faculty, Istanbul University, founded by Coskun Ozdemir, and included, at the time of the data collection, Piraye Serdaroglu-Oflazer, Yesim Gulsen-Parman and Hacer Durmus-Tekce. Many neurology residents chose an aspect of MG as the subject of their theses, led by Feza Deymeer: Aylin Kilic on the development of the MG database and HLA, Ozlem Gungor-Tuncer on MuSK MG, Senay Yıldız-Celik on late-onset MG, Tugba Uyar on ocular MG and Ozlem Canbolat on early-onset MG. Serology was done by Guher Saruhan-Direskeneli and Vuslat Yılmaz. Thymectomies were performed by Alper Toker and Berker Ozkan. The author is grateful to all of them for the unique collaboration over many years.The author also remembers with a lot of fondness and nostalgia all the congresses organized by the late Giovanni Nigro, the late Lucia Comi, and their valuable colleagues.
References
- 1.Drachman DB. Myasthenia gravis. N Engl J Med 1994;330:1797-810. [DOI] [PubMed] [Google Scholar]
- 2.Vincent A, Huda S, Cao M, et al. Serological and experimental studies in different forms of myasthenia gravis. Ann N Y Acad Sci 2018;1413:143-53. https://doi.org/10.1111/nyas.13592 [Erratum in: Ann N Y Acad Sci 2018;1417:130. PMID: 29377162]. 10.1111/nyas.13592 [DOI] [PubMed] [Google Scholar]
- 3.Compston DA, Vincent A, Newsom-Davis J, et al. Clinical, pathological, HLA antigen and immunological evidence for disease heterogeneity in myasthenia gravis. Brain 1980;103:579-601. https://doi.org/10.1093/brain/103.3.579 10.1093/brain/103.3.579 [DOI] [PubMed] [Google Scholar]
- 4.Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol 2015;14:1023-36. https://doi.org/10.1016/S1474-4422(15)00145-3 10.1016/S1474-4422(15)00145-3 [DOI] [PubMed] [Google Scholar]
- 5.Gilhus NE, Tzartos S, Evoli A, et al. Myasthenia gravis. Nat Rev Dis Primers 2019;5:30 https://doi.org/10.1038/s41572-019-0079-y 10.1038/s41572-019-0079-y [DOI] [PubMed] [Google Scholar]
- 6.Deymeer F, Gungor-Tuncer O, Yilmaz V, et al. Clinical comparison of anti-MuSK- vs anti-AChR-positive and seronegative myasthenia gravis. Neurology 2007;68:609-11. https://doi.org/10.1212/01.wnl.0000254620.45529.97 10.1212/01.wnl.0000254620.45529.97 [DOI] [PubMed] [Google Scholar]
- 7.Gungor-Tuncer O, Yilmaz V, Toker A, et al. Prompt response to prednisone predicts benign course in MuSK-MG. Eur Neurol 2017;78:137-42. https://doi.org/10.1159/000479228 10.1159/000479228 [DOI] [PubMed] [Google Scholar]
- 8.Yildiz Celik S, Durmus H, Yilmaz V, et al. Late-onset generalized myasthenia gravis: clinical features, treatment, and outcome. Acta Neurol Belg 2020;120:133-40. https://doi.org/10.1007/s13760-019-01252-x 10.1007/s13760-019-01252-x [DOI] [PubMed] [Google Scholar]
- 9.Niks EH, Kuks JB, Roep BO, et al. Strong association of MuSK antibody-positive myasthenia gravis and HLA-DR14-DQ5. Neurology 2006;66:1772-4. https://doi.org/10.1212/01.wnl.0000218159.79769.5c 10.1212/01.wnl.0000218159.79769.5c [DOI] [PubMed] [Google Scholar]
- 10.Alahgholi-Hajibehzad M, Yilmaz V, Gülsen-Parman Y, et al. Association of HLA-DRB1∗14, -DRB1∗16 and -DQB1∗05 with MuSK-myasthenia gravis in patients from Turkey. Hum Immunol 2013;74:1633-5. https://doi.org/10.1016/j.humimm.2013.08.271 10.1016/j.humimm.2013.08.271 [DOI] [PubMed] [Google Scholar]
- 11.Evoli A, Alboini PE, Damato V, et al. Myasthenia gravis with antibodies to MuSK: an update. Ann N Y Acad Sci 2018;1412:82-9. https://doi.org/10.1111/nyas.13518 10.1111/nyas.13518 [DOI] [PubMed] [Google Scholar]
- 12.Sanders DB, El-Salem K, Massey JM, et al. Clinical aspects of MuSK antibody positive seronegative MG. Neurology 2003;60:1978-80. https://doi.org/10.1212/01.wnl.0000065882.63904.53 10.1212/01.wnl.0000065882.63904.53 [DOI] [PubMed] [Google Scholar]
- 13.Casasnovas C, Povedano M, Jaumà S, et al. Musk-antibody positive myasthenia gravis presenting with isolated neck extensor weakness. Neuromuscul Disord 2007;17:544-6. https://doi.org/10.1016/j.nmd.2007.03.007 10.1016/j.nmd.2007.03.007 [DOI] [PubMed] [Google Scholar]
- 14.Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 2011;44:36-40. https://doi.org/10.1002/mus.22006 10.1002/mus.22006 [DOI] [PubMed] [Google Scholar]
- 15.Gungor-Tuncer O, Orhan EK, Yilmaz V, et al. Prepubertal anti-MuSK positive myasthenia gravis with long remission. Neuromuscul Disord 2014;24:36-9. https://doi.org/10.1016/j.nmd.2013.07.012 10.1016/j.nmd.2013.07.012 [DOI] [PubMed] [Google Scholar]
- 16.Anlar B, Yilmaz V, Saruhan-Direskeneli G. Long remission in muscle-specific kinase antibody-positive juvenile myasthenia. Pediatr Neurol 2009;40:455-6. https://doi.org/10.1016/j.pediatrneurol.2008.11.014 10.1016/j.pediatrneurol.2008.11.014 [DOI] [PubMed] [Google Scholar]
- 17.Deymeer F, Serdaroglu P, Ozdemir C. Juvenile and late-onset myasthenia gravis. Deymeer F, Ed. Neuromuscular diseases: from basic mechanisms to clinical management. Basel: Karger; 2000. [Google Scholar]
- 18.Oh SJ, Hatanaka Y, Hemmi S, et al. Repetitive nerve stimulation of facial muscles in MuSK antibody-positive myasthenia gravis. Muscle Nerve 2006;33:500-4. https://doi.org/10.1002/mus.20498 10.1002/mus.20498 [DOI] [PubMed] [Google Scholar]
- 19.Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology 2016;87:419-25. https://doi.org/10.1212/WNL.0000000000002790 10.1212/WNL.0000000000002790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farrugia ME, Robson MD, Clover L, et al. MRI and clinical studies of facial and bulbar muscle involvement in MuSK antibody-associated myasthenia gravis. Brain 2006;129(Pt 6):1481-92. https://doi.org/10.1093/brain/awl095 10.1093/brain/awl095 [DOI] [PubMed] [Google Scholar]
- 21.Díaz-Manera J, Martínez-Hernández E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012;78:189-93. https://doi.org/10.1212/WNL.0b013e3182407982 10.1212/WNL.0b013e3182407982 [DOI] [PubMed] [Google Scholar]
- 22.Tandan R, Hehir MK, 2nd, Waheed W, et al. Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve 2017;56:185-96. https://doi.org/10.1002/mus.25597 10.1002/mus.25597 [DOI] [PubMed] [Google Scholar]
- 23.Hehir MK, Hobson-Webb LD, Benatar M, et al. Rituximab as treatment for anti-MuSK myasthenia gravis: multicenter blinded prospective review. Neurology 2017;89:1069-77. https://doi.org/10.1212/WNL.0000000000004341 10.1212/WNL.0000000000004341 [DOI] [PubMed] [Google Scholar]
- 24.Leite MI, Ströbel P, Jones M, et al. Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol 2005;57:444-8. https://doi.org/10.1002/ana.20386 10.1002/ana.20386 [DOI] [PubMed] [Google Scholar]
- 25.Saka E, Topcuoglu MA, Akkaya B, et al. Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology 2005. September 13;65(5):782-3; author reply 782-3. doi: 10.1212/wnl.65.5.782 10.1212/wnl.65.5.782 [DOI] [PubMed] [Google Scholar]
- 26.Huijbers MG, Niks EH, Klooster R, et al. Myasthenia gravis with muscle specific kinase antibodies mimicking amyotrophic lateral sclerosis. Neuromuscul Disord 2016;26:350-3. https://doi.org/10.1016/j.nmd.2016.04.004 10.1016/j.nmd.2016.04.004 [DOI] [PubMed] [Google Scholar]
- 27.Magnussen M, Karakis I, Harrison TB. The myotonic plot thickens: electrical myotonia in antimuscle-specific kinase myasthenia gravis. Case Rep Neurol Med 2015;2015:242691 https://doi.org/10.1155/2015/242691 10.1155/2015/242691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaretzki A, 3rd, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology 2000;55:16-23. https://doi.org/10.1212/wnl.55.1.16 10.1212/wnl.55.1.16 [DOI] [PubMed] [Google Scholar]
- 29.Aarli JA. Myasthenia gravis in the elderly: Is it different? Ann N Y Acad Sci 2008;1132:238-43. https://doi.org/10.1196/annals.1405.040 10.1196/annals.1405.040 [DOI] [PubMed] [Google Scholar]
- 30.Murai H, Yamashita N, Watanabe M, et al. Characteristics of myasthenia gravis according to onset-age: Japanese nationwide survey. J Neurol Sci 2011;305:97-102. https://doi.org/10.1016/j.jns.2011.03.004 10.1016/j.jns.2011.03.004 [DOI] [PubMed] [Google Scholar]
- 31.Pakzad Z, Aziz T, Oger J. Increasing incidence of myasthenia gravis among elderly in British Columbia, Canada. Neurology 2011;76:1526-8. https://doi.org/10.1212/WNL.0b013e318217e735 10.1212/WNL.0b013e318217e735 [DOI] [PubMed] [Google Scholar]
- 32.Pedersen EG, Hallas J, Hansen K, et al. Late-onset myasthenia not on the increase: a nationwide register study in Denmark, 1996-2009. Eur J Neurol 2013;20:309-14. https://doi.org/10.1111/j.1468-1331.2012.03850.x 10.1111/j.1468-1331.2012.03850.x [DOI] [PubMed] [Google Scholar]
- 33.Seldin MF, Alkhairy OK, Lee AT, et al. Genome-Wide Association Study of Late-Onset Myasthenia Gravis: confirmation of TNFRSF11A and identification of ZBTB10 and Three Distinct HLA Associations. Mol Med 2016;21:769-81. https://doi.org/10.2119/molmed.2015.00232 10.2119/molmed.2015.00232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saruhan-Direskeneli G, Hughes T, Yilmaz V, et al. Genetic heterogeneity within the HLA region in three distinct clinical subgroups of myasthenia gravis. Clin Immunol 2016;166-167:81-8. https://doi.org/10.1016/j.clim.2016.05.003 10.1016/j.clim.2016.05.003 [DOI] [PubMed] [Google Scholar]
- 35.Evoli A, Batocchi AP, Minisci C, et al. Clinical characteristics and prognosis of myasthenia gravis in older people. J Am Geriatr Soc 2000;48:1442-8. https://doi.org/10.1111/j.1532-5415.2000.tb02635.x 10.1111/j.1532-5415.2000.tb02635.x [DOI] [PubMed] [Google Scholar]
- 36.Zivković SA, Clemens PR, Lacomis D. Characteristics of late-onset myasthenia gravis. J Neurol 2012;259:2167-71. https://doi.org/10.1007/s00415-012-6478-6 10.1007/s00415-012-6478-6 [DOI] [PubMed] [Google Scholar]
- 37.Cortés-Vicente E, Álvarez-Velasco R, Segovia S, et al. Clinical and therapeutic features of myasthenia gravis in adults based on age at onset. Neurology 2020;94:e1171-80. https://doi.org/10.1212/WNL.0000000000008903 10.1212/WNL.0000000000008903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aarli JA, Romi F, Skeie GO, et al. Myasthenia gravis in individuals over 40. Ann N Y Acad Sci 2003;998:424-31. https://doi.org/10.1196/annals.1254.055 10.1196/annals.1254.055 [DOI] [PubMed] [Google Scholar]
- 39.Suzuki S, Utsugisawa K, Nagane Y, et al. Three types of striational antibodies in myasthenia gravis. Autoimmune Dis 2011;2011:740583 https://doi.org/10.4061/2011/740583 10.4061/2011/740583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szczudlik P, Szyluk B, Lipowska M, et al. Antititin antibody in early- and late-onset myasthenia gravis. Acta Neurol Scand 2014;130:229-33. https://doi.org/10.1111/ane.12271 10.1111/ane.12271 [DOI] [PubMed] [Google Scholar]
- 41.Suzuki S, Utsugisawa K, Nagane Y, et al. Clinical and immunological differences between early and late-onset myasthenia gravis in Japan. J Neuroimmunol 2011;230:148-52. https://doi.org/10.1016/j.jneuroim.2010.10.023 10.1016/j.jneuroim.2010.10.023 [DOI] [PubMed] [Google Scholar]
- 42.Slesak G, Melms A, Gerneth F, et al. Late-onset myasthenia gravis. Follow-up of 113 patients diagnosed after age 60. Ann N Y Acad Sci 1998;841:777-80. https://doi.org/10.1111/j.1749-6632.1998.tb11017.x 10.1111/j.1749-6632.1998.tb11017.x [DOI] [PubMed] [Google Scholar]
- 43.Olanow CW, Lane RJ, Roses AD. Thymectomy in late-onset myasthenia gravis. Arch Neurol 1982;39:82-3. https://doi.org/10.1001/archneur.1982.00510140016004 10.1001/archneur.1982.00510140016004 [DOI] [PubMed] [Google Scholar]
- 44.Grob D. Course and management of myasthenia gravis. J Am Med Assoc 1953;153:529-32. https://doi.org/10.1001/jama.1953.02940230001001 10.1001/jama.1953.02940230001001 [DOI] [PubMed] [Google Scholar]
- 45.Oosterhuis HJ. The natural course of myasthenia gravis: a long term follow up study. J Neurol Neurosurg Psychiatry 1989;52:1121-7. https://doi.org/10.1136/jnnp.52.10.1121 10.1136/jnnp.52.10.1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bever CT, Jr, Aquino AV, Penn AS, et al. Prognosis of ocular myasthenia. Ann Neurol 1983;14:516-9. https://doi.org/10.1002/ana.410140504 10.1002/ana.410140504 [DOI] [PubMed] [Google Scholar]
- 47.Sommer N, Sigg B, Melms A, et al. Ocular myasthenia gravis: response to long-term immunosuppressive treatment. J Neurol Neurosurg Psychiatry 1997;62:156-62. https://doi.org/10.1136/jnnp.62.2.156 10.1136/jnnp.62.2.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galassi G, Mazzoli M, Ariatti A, et al. Antibody profile may predict outcome in ocular myasthenia gravis. Acta Neurol Belg 2018;118:435-43. https://doi.org/10.1007/s13760-018-0943-7 10.1007/s13760-018-0943-7 [DOI] [PubMed] [Google Scholar]
- 49.Hong YH, Kwon SB, Kim BJ, et al. Prognosis of ocular myasthenia in Korea: a retrospective multicenter analysis of 202 patients. J Neurol Sci 2008;273:10-4. https://doi.org/10.1016/j.jns.2008.05.023 10.1016/j.jns.2008.05.023 [DOI] [PubMed] [Google Scholar]
- 50.Al-Haidar M, Benatar M, Kaminski HJ. Ocular myasthenia. Neurol Clin 2018;36:241-51. https://doi.org/10.1016/j.ncl.2018.01.003 10.1016/j.ncl.2018.01.003 [DOI] [PubMed] [Google Scholar]
- 51.Grob D, Brunner N, Namba T, et al. Lifetime course of myasthenia gravis. Muscle Nerve 2008;37:141-9. https://doi.org/10.1002/mus.20950 10.1002/mus.20950 [DOI] [PubMed] [Google Scholar]
- 52.Smith SV, Lee AG. Update on ocular myasthenia gravis. Neurol Clin 2017;35:115-23. https://doi.org/10.1016/j.ncl.2016.08.008 10.1016/j.ncl.2016.08.008 [DOI] [PubMed] [Google Scholar]
- 53.Sanders DB, Stålberg EV. AAEM minimonograph #25: single-fiber electromyography. Muscle Nerve 1996;19:1069-83. https://doi.org/10.1002/(SICI)1097-4598(199609)19:9<1069::AID-MUS1>3.0.CO;2-Y [DOI] [PubMed] [Google Scholar]
- 54.Rostedt A, Saders LL, Edards LJ, et al. Predictive value of single-fiber electromyography in the extensor digitorum communis muscle of patients with ocular myasthenia gravis: a retrospective study. J Clin Neuromuscul Dis 2000;2:6-9. https://doi.org/10.1097/00131402-200009000-00003 10.1097/00131402-200009000-00003 [DOI] [PubMed] [Google Scholar]
- 55.Bojikian KD, Francis CE. Thyroid eye disease and myasthenia gravis. Int Ophthalmol Clin 2019;59:113-24. https://doi.org/10.1097/IIO.0000000000000277 10.1097/IIO.0000000000000277 [DOI] [PubMed] [Google Scholar]
- 56.Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol 2003;60:243-8. https://doi.org/10.1001/archneur.60.2.243 10.1001/archneur.60.2.243 [DOI] [PubMed] [Google Scholar]
- 57.Benatar M, Mcdermott MP, Sanders DB, et al. Efficacy of prednisone for the treatment of ocular myasthenia (EPITOME): a randomized, controlled trial. Muscle Nerve 2016;53:363-9. https://doi.org/10.1002/mus.24769 10.1002/mus.24769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ozawa Y, Uzawa A, Kanai T, et al. Efficacy of high-dose intravenous methylprednisolone therapy for ocular myasthenia gravis. J Neurol Sci 2019;402:12-5. https://doi.org/10.1016/j.jns.2019.05.003 10.1016/j.jns.2019.05.003 [DOI] [PubMed] [Google Scholar]
- 59.Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology 2007;68:837-41. https://doi.org/10.1212/01.wnl.0000256698.69121.45 10.1212/01.wnl.0000256698.69121.45 [DOI] [PubMed] [Google Scholar]
- 60.Schumm F, Wiethölter H, Fateh-Moghadam A, et al. Thymectomy in myasthenia with pure ocular symptoms. J Neurol Neurosurg Psychiatry 1985;48:332-7. https://doi.org/10.1136/jnnp.48.4.332 10.1136/jnnp.48.4.332 [DOI] [PMC free article] [PubMed] [Google Scholar]