

Abstract
Becker muscular dystrophy is caused by mutations in the DMD gene that permit significant residual dystrophin protein expression in patient muscle. This is in contrast to DMD gene mutations in Duchenne muscular dystrophy where little or no dystrophin is produced (typically < 3% normal levels). Clinically, Becker muscular dystrophy is extremely variable, from slightly milder than DMD, to asymptomatic hyperCKemia at old age. The factors driving clinical variability in Becker muscular dystrophy have now been studied in some depth, and the findings are likely highly relevant to anticipated clinical findings in exon skipping therapy in DMD. The specific mutations in Becker dystrophy play an important role, and clinical variability is less with high frequency mutations (deletions exons 45-47, 45-48). The percentage of dystrophin content in patient muscle is not well-correlated with clinical findings. Muscle MRI findings (degree of fibrofatty replacement) are very well-correlated with the degree of patient disability, regardless of mutation or muscle dystrophin content. Taken together, data to date suggest that the main determinant driving clinical disability in Becker dystrophy patients is the degree of fibrofatty replacement in muscle. Thus, as with DMD, DMD gene mutations and resulting dystrophin protein abnormalities initiate the disease process, but downstream tissue pathophysiology plays a dominant role in disease progression. Factors influencing the age-dependent rate of fibrofatty replacement of muscles are responsible for much of the clinical variability seen in Becker dystrophy, as well as Duchenne dystrophy. These fibrosis-related factors include genetic modifiers, degree of muscle inflammation, and induction of microRNAs in muscle that bind to dystrophin mRNA and down-regulate dystrophin protein content in patient muscle. Studies to date regarding clinical variability in Becker dystrophy suggest that exon skipping therapy in DMD may show variable efficacy from patient to patient.
Key words: Duchenne muscular dystrophy, Becker muscular dystrophy, dystrophin
Genotype/phenotype correlations in Becker dystrophy (or lack thereof)
Often genotype/phenotype correlations are discussed as ‘good’ or ‘bad’ – there is a strong correlation of specific genotypes with phenotypes, or not strong. The DMD gene is the largest in the human genome, and also complex with multiple transcriptional initiation points (alternative promoters), over 80 exons, all spread out over 2 megabases of the X chromosome (Fig. 1). With such complexity, and the highest spontaneous mutation rate of any gene (1 in 10,000 sperm and eggs), genotype/phenotype correlations might be expected to be more nuanced than “good or bad”. Excellent reviews of genotype/phenotype correlations in the dystrophinopathies have been published for both skeletal muscle and cardiac disease 1-5. Dystrophin isoforms and pathologies in non-muscle tissues have also been well-described, such as retina 6, peripheral nerve 7, vascular smooth muscle 8-9, intestinal smooth muscle 10-11. Assessments of dystrophin mRNA transcript levels in human tissues show relatively high levels in most tissues with smooth muscle layers (visceral and vascular), skeletal muscle, cardiac muscle, and peripheral nerve (Fig. 2). Specific mutations should affect different dystrophin mRNAs, and cell-specific dystrophin function in variable ways. Indeed, the genotype/phenotype correlations of DMD gene mutations with the latter half of the gene showing greater cognitive involvement, and correlation with 3’ mRNAs expression in brain, are quite compelling 12,13.
Figure 1.
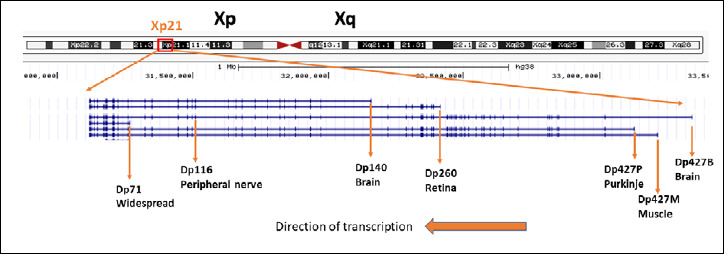
Schematic of the DMD gene. Shown is a genome browser visualization (www.genome.ucsc.edu), with labels added for the 6 different gene promoters, resulting mRNAs, and encoded proteins. Gene mutations in the first half of the gene may affect dystrophin expression driven from the first 3 promoters driving ‘full length’ dystrophin (Dp427B, Dp427M, Dp427P), but leave intact expression of downstream promoters (Dp71, Dp116, Dp140). Mutations located in the second half of the gene may disrupt expression or most or all dystrophin mRNAs and proteins driven from all promoters.
Figure 2.
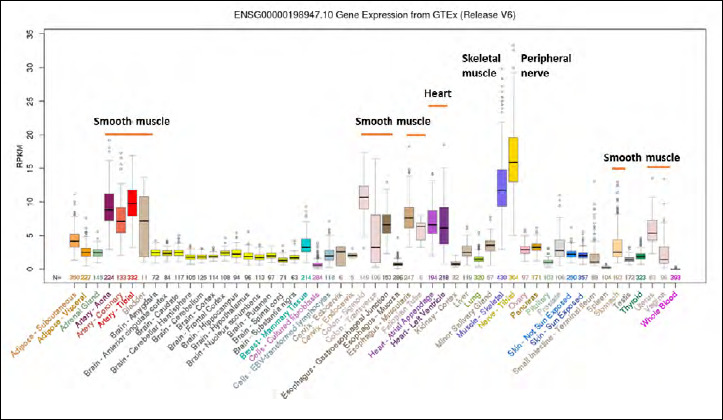
Dystrophin mRNA expression in human tissues. Shown is RNAseq data from multiple human tissues from the genome browser (genome.ucsc.edu). Dystrophin is highly expressed in all types of muscle (skeletal muscle, cardiac muscle, visceral smooth muscle, vascular smooth muscle), peripheral nerve, and some neurons. Dystrophin deficiency leads to pathology in most or all these tissues, but depending on the relative position of the gene promoter relative to the mutation involved.
With DMD and BMD mutations distributed throughout the DMD gene, affecting changes to the dystrophin mRNA and protein in different ways, sometimes isoform-specific, in multiple organ systems (skeletal muscle, cardiac muscle, smooth muscle, peripheral nerves and neurons), things are ‘quite complicated’. Thus, the response to the question of the reliability of genotype/phenotype correlations in the dystrophinopathies is not so much ‘good or bad’, but instead, “Well, it’s complicated”.
The definition of a syndromic disorder is that multiple organ systems are affected. To date, the dystrophinopathies have been defined as a disease restricted to skeletal muscle, and hence called a non-syndromic ‘muscular dystrophy’. It is probably time to re-evaluate this and acknowledge that both DMD and BMD are syndromic disorders, with involvement of multiple organ systems. This is likely an important distinction; the clinical phenotypes of syndromic disorders are acknowledged to be the interactive summation of perturbations of multiple organ systems. It is increasingly likely that the DMD and BMD phenotypes are interactive summation of perturbations of skeletal muscle, vascular smooth muscle, visceral smooth muscle, heart, peripheral nerve and central neurons. A corollary of this logic is that the DMD gene mutations initiate a process, but the downstream events leading to a phenotype are quite complex and variable; typical of syndromic disorders. Or stated in another way, if one views DMD and BMD as non-syndromic muscular dystrophies, then one could view the genotype/phenotype correlations as ‘poor’. On the other hand, if one views DMD and BMD as syndromic disorders, the genotype/phenotype correlations could be considered ‘quite good’.
What are known of drivers of clinical variability?
Becker muscular dystrophy is extremely clinically variable. Certainly, the gene mutation and resulting perturbations of the dystrophin protein are a major component of this variability (e.g. the extent to which dystrophin function is retained). However, it is possible to hold the BMD mutation constant with the most common mutations (deletion exons 45-47, and exons 45-48). Excellent correlative studies of genotype/phenotype, dystrophin content of muscle, MRI findings, and clinical symptoms have now been published in large series of Becker dystrophy patients 14,15. These have shown that Becker patients do become more homogeneous when holding the mutation constant, but there remains extensive clinical variability both between patients with the same mutation, as well as within families with the same mutation. Thus, there are clearly variables downstream of the specific abnormal dystrophin that contribute to clinical variability. These studies also show that MRI findings in skeletal muscle (degree of fibrofatty replacement) is much more predictive of clinical disability (stage of disease) than is dystrophin mutation or dystrophin protein content (% of normal).
The fact that MRI measures of fibrofatty replacement are more predictive of clinical phenotype in Becker dystrophy than genotype (mutation) or biochemistry (dystrophin protein), points to the importance of cellular and tissue events downstream of the biochemical defect. Again, in Becker dystrophy, dystrophin abnormalities initiate a process, but clinical disability results from events downstream (fibrofatty replacement of muscle). The prominent (if not dominant) importance of cellular and tissue pathophysiology far downstream of the initiating dystrophin perturbations is clearly evident in Duchenne muscular dystrophy as well (where the biochemistry – dystrophin null – is held constant). In DMD, different muscle groups show dramatically different MRI findings, and the MRI findings correlate well with histopathology 16-17. DMD is also a quite variable disease given a homogeneous biochemical defect, with marked clinical variability in young boys studied in a highly controlled clinical trial study (4 to < 7 years, steroid naïve) (Fig. 3). Genetic modifiers of DMD (common polymorphisms in other non-DMD genes) have generally been found to involve TGFβ fibrosis and inflammation cascades, consistent with the importance of the downstream progressive histopathology sensitively seen by MRI 18-21. Genetic modifiers affecting disease severity through fibrosis pathways have been successfully replicated in the mouse 22.
Figure 3.
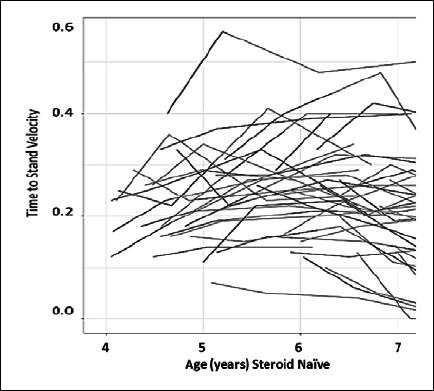
Clinical variability in 48 steroid-naïve Duchenne muscular dystrophy boys, age 4 to < 7 years. Shown is Time to Stand velocity (1/event) in DMD boys enrolled into vamorolone clinical trials. Extensive variability in disease severity is seen, with a 6-fold difference in velocity from the most severe to the mildest patient (from Dang et al., 2020 and Smith et al., 2020, mod.) 38,39.
The importance of pathophysiological processes, specifically early inflammation, later TGFβ, and end-stage fibrofatty replacement as critical drivers of clinical phenotype and disability cannot be disputed. Further, it is now well-established that genetic modifiers of dystrophinopathy in both human and mouse are centered on these downstream pathways. That said, DMD gene mutations and resulting dystrophin production is not irrelevant, as it clearly initiates the process. Critical here is relatively low levels of dystrophin seen with certain out-of-frame ‘leaky’ mutations can mitigate the severity to some degree (~3-4%), although generally not to the point where the patient would be clearly characterized as a mild/moderate Becker patient. For example, an intermediate DMD/BMD phenotype was seen in a patient with an out-of-frame deletion that expressed low levels of dystrophin (~4%), and also had the LTPB4 rare genotype associated with milder disease 23. Also, certain deletions seem generally ‘leaky’ permitting some low-level dystrophin, and many of these patients show later loss of ambulation. For example, exon 44 skippable mutations have about 3-5 year later loss of ambulation 24-27, whereas exon 51 skippable about a 2-year earlier loss of ambulation 26-27. Mutations involving the initial exons of the dystrophin gene (e.g. del exons 3-7) are known to show mRNA translation of dystrophin at alternative AUG initiation codons, and thus result in dystrophin production despite an out-of-frame mutation.
The multiple studies of genetic modifiers of loss of ambulation in DMD have been increasingly robust and relatively corroborative, considering that such genetic studies are challenging in rare diseases (lack of statistical power). Indeed, the ability to identify and replicate genetic modifiers in DMD suggests that the ‘effect size’ of the modifiers is surprisingly large (hence easy to detect). It also speaks to the importance of the TGFβ fibrosis pathways, as the majority of genetic modifiers modulate this pathway. It can be concluded that genetic modifiers lead to differences in the speed of the transition of skeletal muscle from successful regeneration to failure of regeneration (and fibrofatty replacement).
Effect size of genetic modifiers can be quantitated in DMD by the number of years change in mean age at loss of ambulation (Fig. 4). Genetic modifiers to date all show about 1-2 years change in mean age of loss of ambulation (where mean age is LOA at around 11-12 years). One must consider the genotype frequencies, the inheritance model (dominant [SPP1, CD40], or recessive [LTBP4]), and the calculated percentage of DMD boys that have the “at-risk” genotype (Fig. 4). These allele frequencies also vary in different ethnicities and world populations. For example, in China the SPP1 genotype associated with earlier loss of ambulation in Europeans is at a very low allele frequency, and thus does not show significant association with LOA in Chinese DMD boys. However, a different SPP1 promoter at high allele frequency in Chinese DMD boys shows highly significant association with LOA. In effect, one might consider this an independent validation of the importance of SPP1 (osteopontin) in the progression of DMD 21.
Figure 4.
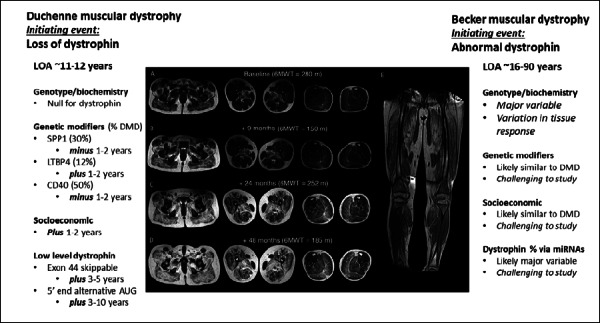
Factors contributing to clinical variability in Duchenne and Becker muscular dystrophies. Shown is a schematic of progressive MRI findings, with gradual fibrofatty replacement over time (from Godi et al., 2016, mod.) 17. The progressive clinical phenotypes of both Duchenne and Becker muscular dystrophies is driven predominantly by the extent of fibrofatty replacement of muscle. The factors driving the fibrofatty replacement are listed for both Duchenne and Becker dystrophy, as discussed in the text. Plus and minus refer to later or earlier age at loss of ambulation.
Data emerging in genetic modifiers of heart involvement DMD and BMD are quite interesting. Dystrophin-deficient cardiac tissue shows distinct pathology and functional deficits compared to skeletal muscle. Heart does not show the repeated bouts of degeneration and regeneration, and fibrotic replacement is slowly progressive and limited initially to basolateral free wall of the left ventricle (likely due to inflammation and death of myocardiocytes where functional load on the heart tissue is highest) 28. Also, dystrophin driven from the brain promoter can compensate for a deleted muscle gene promoter in skeletal muscle but not cardiac muscle 29. One might expect some genetic modifiers to be consistent between dystrophin-deficient heart and skeletal muscle, and some distinct. Consistent with this, genetic modifiers can be found that are specific to heart30, and also shared with skeletal muscle 31.
Pulling variables together into models of DMD and BMD disease progression
For DMD, the initiating event is loss of dystrophin in multiple tissues that likely interact to lead to gradual fibrotic replacement of skeletal muscle, and, later, heart. The initiating event is relatively homogenous, and generally leads to loss of ambulation about 11-12 years of age. Genetic modifiers, low level dystrophin ‘leakiness’ of the mutation, and socioeconomic status32 all contribute to increased severity or decreased severity relative to this ‘average’ (Fig. 4). For each genetic modifier, the inheritance pattern, and the allele frequency can be calculated from the published papers, and define whether the ‘rare allele’ genotype causes earlier loss of ambulation (e.g. minus 1-2 years), or later loss of ambulation (e.g. plus 1-2 years) (Fig. 4). Indeed, all genetic modifiers have been found to change the age of loss of ambulation by 1-2 years, with SPP1 and CD40 rare allele causing a more severe progression (minus 1-2 years) (both dominant inheritance patterns), and LTBP4 rare allele a milder disease progression (plus 1-2 years; recessive inheritance pattern). Likewise, the effect size of low level dystrophin expression due to exon 44 skippable mutations can be quantified (3-5 years later LOA), the effects of alternative AUG use and dystrophin expression in 5’ mutations (plus 3-10 years), and the effects of socioeconomic factors (minus 1-2 years for low socioeconomic status 32 (Fig. 4). This begins to paint a more complete picture of factors influencing clinical severity in DMD, and also points to the multi-variate and complex nature of these factors.
For BMD, the initiating event of abnormal dystrophin is a much greater ‘driver’ of disease variability in onset and progression than it is in DMD where lack of dystrophin is held constant. The great heterogeneity of gene mutations resulting in many different abnormal dystrophin proteins with variable residual function leads to variability in the initiation and progression of disease. Moreover, it could be expected that different dystrophin-expressing tissues and cells may respond differentially to specific abnormal dystrophins. For example, the abnormal dystrophin resulting from a deletion of exons 45-47 may lead to some cellular and physiological abnormalities in skeletal muscle, but different abnormalities in smooth muscle. Thus, the ‘syndromic’ nature of dystrophinopathies may be accentuated in Becker dystrophy due to differential perturbations of different tissues (Fig. 4).
It is highly likely that the same genetic modifiers and effects of socioeconomic status observed in DMD are relevant to BMD as well. That said, it is nearly impossible to study and prove effects of genetic modifiers and socioeconomic status in Becker dystrophy. This is because the major effects of genotype and biochemistry (abnormal dystrophin) on clinical symptoms in Becker dystrophy creates extensive population stratification, leading to precipitous loss of statistical power. To detect the effects of genetic modifiers and socioeconomic status on phenotype in Becker dystrophy would require the study of cohorts of a single Becker mutation (e.g. only those with either of the common BMD mutations; e.g. del 45-47 or del 45-48). One would not want to mix the del 45-47 and del 45-48 patients as they have different dystrophin proteins (and this is too much of a variable). As each of these two genotype groups is only about 15% of all Becker patients, it will be challenging to assemble such cohorts. Until these studies can be done, it is probably safe to assume that genetic and socioeconomic modifiers of DMD are shared in BMD (Fig. 4).
A variable that may drive clinical severity in Becker dystrophy that is not relevant to DMD is effects of microRNAs. The dystrophin mRNA has a very large number (~80) putative microRNA binding sites that have the potential to individually and/or collectively decrease protein translation from the dystrophin mRNA, and thus in turn lead to decreased amounts of dystrophin (in muscle, heart, smooth muscle or nerve) 33-35. It has been well-established that inflammation in muscle leads to induction of inflammation-associated microRNAs, and these in turn bind to the dystrophin mRNA and decrease dystrophin protein content in muscle.
This inflammation/microRNA/dystrophin pathway seems to explain the highly variable dystrophin levels seen in Becker dystrophy patients, even when controlling for the same causative exon 45-47 mutation 33. Further, inflammatory disease of muscle unrelated to dystrophinopathy appears to have the same pathway active, leading to reductions of dystrophin in skeletal muscle secondary to inflammation35. It is intuitively attractive to consider that the microRNA pathway has effects on Becker patient disease severity, assuming that ‘more dystrophin is better’ (Fig. 4). However, there is no evidence for this. Indeed, in contrast, there is little evidence that dystrophin levels between 20-100% normal are in any way correlated with clinical severity in Becker dystrophy 14,36. Muscle biopsy studies include a high degree of ‘sampling error’, where only a small area of a single muscle, in a single stage of disease is studied for dystrophin protein amounts. Thus, a single biopsy is not representative of dystrophin levels in the patient as a whole. The lack of correlations of dystrophin content in Becker patient muscle biopsies with clinical phenotypes does not rule out the potential importance of the microRNA/dystrophin pathway. As with genetic modifiers, carrying out studies to prove the contribution of microRNA-mediated reductions in dystrophin in Becker patients would be important, but studies will be challenged to control for the mutation/protein stratification problem noted above.
Relevance of BMD models of clinical severity to exon skipping therapy in DMD
There are now 3 exon skipping drugs approved by the FDA in the USA, targeted towards exon 51 (Etiplersen, Sarepta), and exon 53 (Golodirsen, Sarepta; Viltepso, NS Pharma). The Sarepta drugs led to a mean ~1% normal dystrophin levels in treated DMD patient muscle (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211970Orig1s000SumR.pdf; https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/206488Orig1s000SumR.pdf).
The NS Pharma drug showed induction of a mean ~6% (https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212154Orig1s000SumR.pdf). The NS Pharma drug has shown preliminary evidence of clinical benefit in 16 boys treated with viltolarsen, with drug-related improvements in multiple clinical motor outcomes 37. All 3 drugs were approved under an “accelerated” pathway dependent on surrogate outcome measures (dystrophin protein by immunoblot).
In the context of the above discussion regarding the variables contributing to clinical variability in both Duchenne and Becker dystrophies, it may be instructive to interpret the status and promise of exon skipping in DMD. First, one can interpret existing exon skipping data in the context of clinically meaningful dystrophin levels. Data to date suggests that dystrophin levels ~3 – 10% of normal levels typically lead to an intermediate phenotype between Duchenne and Becker muscular dystrophies (sometimes called “severe Becker dystrophy”) 14,36. This is consistent with the ~3% dystrophin content of a leaky exon 44 mutation 23. Given this data, the two Sarepta drugs with ~1% dystrophin would might not be expected to show significant clinical benefit. The report of a mean ~6% dystrophin induced from the NS Pharma drug is consistent with preliminary evidence of clinical benefit 37.
Assuming that exon skipping drugs were able to produce 3% or more of Becker-like dystrophin, one expects the extensive variability in patient clinical response given all the variables influencing clinical variability in Becker muscular dystrophy (Fig. 4). Functionalities of specific abnormal dystrophins, levels of dystrophin, microRNA/dystrophin pathways, genetic modifiers, and socioeconomic status are all likely to factor relatively heavily into the clinical response to exon skipping.
Figures and tables
References
- 1.Nigro G, Politano L, Nigro V, et al. Mutation of dystrophin gene and cardiomyopathy. Neuromuscul Disord 1994;4:371-9. https://doi.org/10.1016/0960-8966(94)90073-6 10.1016/0960-8966(94)90073-6 [DOI] [PubMed] [Google Scholar]
- 2.Nigro G, Comi LI, Politano L, et al. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol 1990;26:271-7. https://doi.org/10.1016/0167-5273(90)90082-g 10.1016/0167-5273(90)90082-g [DOI] [PubMed] [Google Scholar]
- 3.Ferlini A, Sewry C, Melis MA, et al. X-linked dilated cardiomyopathy and the dystrophin gene. Neuromuscul Disord 1999;9:339-46. https://doi.org/10.1016/s0960-8966(99)00015-2 10.1016/s0960-8966(99)00015-2 [DOI] [PubMed] [Google Scholar]
- 4.Kamdar F, Garry DJ. Dystrophin-deficient cardiomyopathy. J Am Coll Cardiol 2016;67:2533-46. https://doi.org/10.1016/j.jacc.2016.02.081 10.1016/j.jacc.2016.02.081 [DOI] [PubMed] [Google Scholar]
- 5.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731-40. https://doi.org/10.1016/s1474-4422(03)00585-4 10.1016/s1474-4422(03)00585-4 [DOI] [PubMed] [Google Scholar]
- 6.D’Souza VN, Nguyen TM, Morris GE, et al. A novel dystrophin isoform is required for normal retinal electrophysiology. Hum Mol Genet 1995;4:837-42. https://doi.org/10.1093/hmg/4.5.837 10.1093/hmg/4.5.837 [DOI] [PubMed] [Google Scholar]
- 7.Byers TJ, Lidov HG, Kunkel LM. An alternative dystrophin transcript specific to peripheral nerve. Nat Genet 1993;4:77-81. https://doi.org/10.1038/ng0593-77 10.1038/ng0593-77 [DOI] [PubMed] [Google Scholar]
- 8.Mancinelli R, Tonali P, Romani R, et al. Mechanical properties of smooth muscle portal vein in normal and dystrophin-deficient (mdx) mice. Exp Physiol 1999;84:929-40. PMID: 10502660 [PubMed] [Google Scholar]
- 9.Ito K, Kimura S, Ozasa S, et al. Smooth muscle-specific dystrophin expression improves aberrant vasoregulation in mdx mice. Hum Mol Genet 2006;15:2266-75. https://doi.org/10.1093/hmg/ddl151 10.1093/hmg/ddl151 [DOI] [PubMed] [Google Scholar]
- 10.Barohn RJ, Levine EJ, Olson JO, Mendell JR. Gastric hypomotility in Duchenne’s muscular dystrophy. N Engl J Med 1988;319:15-8. https://doi.org/10.1056/NEJM198807073190103 10.1056/NEJM198807073190103 [DOI] [PubMed] [Google Scholar]
- 11.Singh K, Randhwa G, Salloum FN, et al. Decreased smooth muscle function, peristaltic activity, and gastrointestinal transit in dystrophic (mdx) mice. Neurogastroenterol Motil 2020:e13968. https://doi.org/10.1111/nmo.13968. 10.1111/nmo.13968 Epub ahead of print. PMID: 32789934 [DOI] [PubMed] [Google Scholar]
- 12.Ricotti V, Mandy WP, Scoto M, et al. Neurodevelopmental, emotional, and behavioral problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol 2016;58:77-84. https://doi.org/10.1111/dmcn.12922 10.1111/dmcn.12922 [DOI] [PubMed] [Google Scholar]
- 13.Chaussenot R, Amar M, Fossier P, et al. Dp71-dystrophin deficiency alters prefrontal cortex excitation-inhibition balance and executive functions. Mol Neurobiol 2019;56:2670-84. https://doi.org/10.1007/s12035-018-1259-6 10.1007/s12035-018-1259-6 [DOI] [PubMed] [Google Scholar]
- 14.van den Bergen JC, Wokke BH, Janson AA, et al. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry 2014;85:747-53. https://doi.org/10.1136/jnnp-2013-306350 10.1136/jnnp-2013-306350 [DOI] [PubMed] [Google Scholar]
- 15.Barp A, Bello L, Caumo L, et al. Muscle MRI and functional outcome measures in Becker muscular dystrophy. Sci Rep 2017;7:16060 https://doi.org/10.1038/s41598-017-16170-2 10.1038/s41598-017-16170-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinali M, Arechavala-Gomeza V, Cirak S, et al. Muscle histology vs MRI in Duchenne muscular dystrophy. Neurology 2011;76:346-53. https://doi.org/10.1212/WNL.0b013e318208811f 10.1212/WNL.0b013e318208811f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godi C, Ambrosi A, Nicastro F, et al. Longitudinal MRI quantification of muscle degeneration in Duchenne muscular dystrophy. Ann Clin Transl Neurol 2016;3:607-22. https://doi.org/10.1002/acn3.319 10.1002/acn3.319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bello L, Flanigan KM, Weiss RB, et al. Association study of exon variants in the NF-κB and TGFβ pathways identifies CD40 as a modifier of Duchenne muscular dystrophy. Am J Hum Genet 2016;99:1163-71. https://doi.org/10.1016/j.ajhg.2016.08.023 10.1016/j.ajhg.2016.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss RB, Vieland VJ, Dunn DM, et al. ; United Dystrophinopathy Project. Long-range genomic regulators of THBS1 and LTBP4 modify disease severity in Duchenne muscular dystrophy. Ann Neurol 2018;84:234-5. https://doi.org/10.1002/ana.25283 10.1002/ana.25283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bello L, D’Angelo G, Villa M, et al. Genetic modifiers of respiratory function in Duchenne muscular dystrophy. Ann Clin Transl Neurol 2020;7:786-98. https://doi.org/10.1002/acn3.51046 10.1002/acn3.51046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen M, Wang L, Li Y, et al. Genetic modifiers of Duchenne muscular dystrophy in Chinese patients. Front Neurol 2020;11:721 https://doi.org/10.3389/fneur.2020.00721 10.3389/fneur.2020.00721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mázala DA, Novak JS, Hogarth MW, et al. TGF-β-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight 2020;5:e135703 https://doi.org/10.1172/jci.insight.135703 10.1172/jci.insight.135703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waldrop MA, Gumienny F, El Husayni S, et al. Low-level dystrophin expression attenuating the dystrophinopathy phenotype. Neuromuscul Disord 2018;28:116-21. https://doi.org/10.1016/j.nmd.2017.11.007 10.1016/j.nmd.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van den Bergen JC, Ginjaar HB, Niks EH, et al. Prolonged ambulation in Duchenne patients with a mutation amenable to Exon 44 skipping. J Neuromuscul Dis 2014b;1:91-4. PMID: 27858662 [PubMed] [Google Scholar]
- 25.Bello L, Morgenroth LP, Gordish-Dressman H, et al. DMD genotypes and loss of ambulation in the CINRG Duchenne natural history study. Neurology 2016;87:401-9. https://doi.org/10.1212/WNL.0000000000002891 10.1212/WNL.0000000000002891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ricotti V, Ridout DA, Pane M, et al. The NorthStar ambulatory assessment in Duchenne muscular dystrophy: considerations for the design of clinical trials. J Neurol Neurosurg Psychiatry 2016;87:149-55. https://doi.org/10.1136/jnnp-2014-309405 10.1136/jnnp-2014-309405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang RT, Barthelemy F, Martin AS, et al. DMD genotype correlations from the Duchenne Registry: endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat 2018;39:1193-202. https://doi.org/10.1002/humu.23561 10.1002/humu.23561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marty B, Gilles R, Toussaint M, et al. Comprehensive evaluation of structural and functional myocardial impairments in Becker muscular dystrophy using quantitative cardiac magnetic resonance imaging. Eur Heart J Cardiovasc Imaging 2019;20:906-15. https://doi.org/10.1093/ehjci/jey209 10.1093/ehjci/jey209 [DOI] [PubMed] [Google Scholar]
- 29.Neri M, Valli E, Alfano G, et al. The absence of dystrophin brain isoform expression in healthy human heart ventricles explains the pathogenesis of 5’ X-linked dilated cardiomyopathy. BMC Med Genet 2012;13:20 https://doi.org/10.1186/1471-2350-13-20 10.1186/1471-2350-13-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang X, Shao Y, Araj FG, et al. Heterozygous cystic fibrosis transmembrane regulator gene missense variants are associated with worse cardiac function in patients with Duchenne muscular dystrophy. J Am Heart Assoc 2020;9:e016799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barp A, Bello L, Politano L, et al. Genetic modifiers of Duchenne muscular dystrophy and dilated cardiomyopathy. PLoS One 2015;10:e0141240 https://doi.org/10.1371/journal.pone.0141240 10.1371/journal.pone.0141240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hufton M, Roper H. Variations in Duchenne muscular dystrophy course in a multi-ethnic UK population: potential influence of socio-economic factors. Dev Med Child Neurol 2017;59:837-42. https://doi.org/10.1111/dmcn.13460 10.1111/dmcn.13460 [DOI] [PubMed] [Google Scholar]
- 33.Fiorillo AA, Heier CR, Novak JS, et al. TNF-α-induced microRNAs control dystrophin expression in Becker muscular dystrophy. Cell Rep 2015;12:1678-90. https://doi.org/10.1016/j.celrep.2015.07.066 10.1016/j.celrep.2015.07.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiorillo AA, Tully CB, Damsker JM, et al. Muscle miRNAome shows suppression of chronic inflammatory miRNAs with both prednisone and vamorolone. Physiol Genomics 2018;50:735-45. https://doi.org/10.1152/physiolgenomics.00134.2017 10.1152/physiolgenomics.00134.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinder TB, Heier CR, Tully CB, et al. Muscle Weakness in myositis: MicroRNA-mediated dystrophin reduction in a myositis mouse model and human muscle biopsies. Arthritis Rheumatol 2020;72:1170-83. https://doi.org/10.1002/art.41215 10.1002/art.41215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffman EP, Kunkel LM, Angelini C, et al. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology 1989;39:1011-7. https://doi.org/10.1212/wnl.39.8.1011 10.1212/wnl.39.8.1011 [DOI] [PubMed] [Google Scholar]
- 37.Clemens PR, Rao VK, Connolly AM, et al. Safety, tolerability, and efficacy of viltolarsen in boys with Duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol 2020;77:982-91. https://doi.org/10.1001/jamaneurol.2020.1264 10.1001/jamaneurol.2020.1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dang UJ, Ziemba M, Clemens PR, et al. Serum biomarkers associated with baseline clinical severity in young steroid-naïve Duchenne muscular dystrophy boys. Hum Mol Genet 2020;29:2481-95. https://doi.org/10.1093/hmg/ddaa132 10.1093/hmg/ddaa132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith EC, Conklin LS, Hoffman EP, et al. Efficacy and safety of vamorolone in Duchenne muscular dystrophy: an 18-month interim analysis of a non-randomized open-label extension study. PLoS Med 2020;17:e1003222 https://doi.org/10.1371/journal.pmed.1003222 10.1371/journal.pmed.1003222 [DOI] [PMC free article] [PubMed] [Google Scholar]