

Abstract
Triptolide, a major active component of Triptergium wilfordii Hook. f, is used in the treatment of autoimmune disease. However, triptolide is associated with severe adverse reactions, especially hepatotoxicity, which limits its clinical application. To examine the underlying mechanism of triptolide-induced liver injury, a combination of dose- and time-dependent toxic effects, RNA-seq and metabolomics were employed. Triptolide-induced toxicity occurred in a dose- and time-dependent manners and was characterized by apoptosis and not necroptosis. Transcriptomics profiles of the dose-dependent response to triptolide suggested that PI3K/AKT, MAPK, TNFα and p53 signaling pathways were the vital steps in triptolide-induced hepatocyte apoptosis. Metabolomics further revealed that glycerophospholipid, fatty acid, leukotriene, purine and pyrimidine metabolism were the major metabolic alterations after triptolide exposure. Finally, acylcarnitines were identified as potential biomarkers for the early detection of triptolide-induced liver injury.
Keywords: triptolide, transcriptomics, metabolomics, apoptosis, acylcarnitine
1. Introduction
Triptolide is a diterpene epoxide compound extracted from Triptergium wilfordii Hook. f (TWHF), used as an anti-inflammatory drug for the treatment of rheumatoid arthritis for decades in China (Wang et al., 2016). As a main active ingredient of TWHF, triptolide exhibits multiple pharmacological effects, including immune modulation, anti-tumor and anti-fibrosis (Liu, 2011; Wang et al., 2014; Dai et al., 2019; Chen et al., 2019). Despite its good anti-rheumatoid arthritis effects, post-marketing clinical observations have demonstrated that triptolide can cause severe hepatotoxicity with jaundice, drug-induced hepatitis and abnormal liver function, and it is the main obstacle for its widespread application in patients (Fan et al., 2018; Hou et al., 2018). In addition, there are few available treatments for mitigating idiosyncratic acute poisoning after triptolide administration. A comprehensive understanding the mechanisms of this disease is urgently needed.
The toxic effects of triptolide in liver are manifested histologically as hepatocyte apoptosis, hepatic parenchymal necrosis and bile duct proliferation (Yang et al., 2017; Zhou et al., 2018). Cell death is crucial in maintaining tissue homeostasis by eliminate damaged cells in healthy tissues, but also contributes to pathologies that occur as a result of chemically-induced toxicity (Guicciardi et al.,2013; Bergsbaken et al., 2009). Apoptosis and necroptosis are programmed cell death processes, but are dependent on different characteristics. Apoptosis is mainly triggered by two fundamental pathways: the death receptor pathway and the mitochondrial pathway, and downstream of both pathways is activation of caspase (CASP)-3 (Guicciardi and Gores, 2005; Iorga and Dara, 2019). Necroptosis requires the involvement of RIPK1 and RIPK3, with the later and its substrate MLKL thought to be a more specific regulator of necroptosis than RIPK1 (Khoury et al., 2019). Both apoptosis and necroptosis are widely observed in models of acute liver injury (Iorga and Dara, 2019; Jaeschke et al., 2019). Several studies demonstrated that triptolide could cause liver injury mainly via disrupting normal metabolism and inducing apoptosis or necroptosis (Yao et al., 2008; Li et al., 2014a; Lu et al., 2017; Yuan et al., 2019). However, significant questions remain in understanding the exact molecular mechanisms and metabolic pathways involved in the development and progression of triptolide-induced liver injury.
Multi-omics analysis technologies are increasingly used in identifying potential biomarkers and clarifying the pathogenesis and mechanisms associated with diseases. Compared to each type of omics data, which typically provides a signal list of differences in the pathogenesis of the disease, multi-omics analysis can elucidate potential causative changes that can be further tested by detailed molecular studies, not justly reflecting reactive processes (Hasin et al., 2017). Several studies suggested that triptolide causes liver injury mainly via disrupting normal metabolism or expression of immune response associated genes with metabolomics or transcriptomics (Vliegenthart et al., 2017; Wang et al., 2017). Metabolic alterations after triptolide exposure were recently described by metabolomics (Zhao et al., 2018). Differential expressed proteins and fatty acids both participate in pathogenic pathways including cellular lipolysis and peroxisomal fatty acid β-oxidation with quantitative proteomics and targeted fatty acids analysis (Li et al., 2017a). Others showed that regulation of sphingolipid metabolism was associated with triptolide poisoning in the liver by using integrated targeted sphingolipidomics and transcriptomics (Qu et al., 2015). An integrated analysis of transcriptomics and metabolomics to investigate the dose-dependent response of triptolide-induced liver injury has not been reported.
In the present study, a comprehensive of dose- and time-dependent toxic effects, transcriptomics and metabolomics was performed to study the roles of necroptosis and apoptosis, and to identify the differential expressed genes and metabolites after triptolide exposure. In order to further investigate the potential metabolic alterations and molecular mechanisms, systemic correlation networks of similarities in the two omics data was performed.
2. Materials and methods
2.1. Chemical reagents
Triptolide was purchased from Vicket Biological Technology Inc. (Chengdu, China). Myristoyl-trimethyl D9 carnitine was obtained from Sigma-Aldrich (Burlington, MA, USA). Deceaniylcarnitine, lauroylcarnitine, myristoylcarnitine, palmitoylcarnitine, stearoylcarnitine, acylcarnitine, propionylcarnitine, buylcarnitine, pivaloycarnitine, and 2-methylcarnitine were obtained from Sigma-Aldrich (St. Louis, MO).
2.2. Animal maintenance and treatment
All animal experimental procedures were approved by the National Cancer Institute Animal Care and Use Committee. Six- to eight-week-old male Ripk3−/− and Ripk3+/+ mice on the C57BL/6N background were used in this study, and Ripk3−/− and Ripk3+/+ mice were provided by Vishva Dixit (Genentech, South San Francisco, CA). The Ripk3−/− and Ripk3+/+ mice was confirmed by genotyping with primers, Ripk3 primer-1: 5′-CGCTTTAGAAGCCTTCAGGTTGAC-3′, Ripk3 primer-2: 5′-GCCTGCCCATCAGCAACTC-3′, Ripk3 primer-3: 5′-CCAGAGGCCACTTGT-GTA GCG-3′, which yields a 450 bp electrophoretic band (Ripk3−/− allele) or a 700 bp band (Ripk3+/+ allele). Mice were housed in a pathogen-free animal facility controlled for temperature and light (25°C,12-h light/dark cycle) and fed pelleted NIH-31 chow diet and water ad libitum. All the mice were acclimatized for 1 week in the NCI vivarium before starting the experiments. For triptolide dose-response experiments, wild-type mice were administered with a single oral dosage of 0.4 mg/kg, 0.6 mg/kg or 0.8 mg/kg body weight triptolide or 0.9% saline. Twenty-four h after triptolide treatment, the mice were killed by CO2 asphyxiation. For triptolide time-course experiments, mice were administered a single dosage of 0.8 mg/kg body weight of triptolide or 0.9% saline. Mice were killed at 12, 24, or 48 h. For the Ripk3-null versus their wild-type littermate experiments, Ripk3−/− mice and their littermates on the same C57BL/6N genetic background were orally administered 0.8 mg/kg triptolide. After triptolide exposure for 24 h, the mice were killed. Blood was collected by venipuncture of the caudal vena cava to Serum Separator Tubes (Becton Dickinson and Company, Franklin Lakes, NJ). Liver tissues were excised and collected for histological examination and the rest was quick frozen in liquid nitrogen, and stored at − 80°C until use.
2.3. Serum aminotransferase assays
Serum was obtained by centrifuging for 10 min at 8,000 × g at 4°C. The level of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using commercially available ALT and AST kits (Catachem, Oxford, UK) according to the manufacturer’s instructions.
2.4. Liver histopathology examinations
A part of each fresh liver tissue was kept in 10% formalin solution for 24 h. Then, formalin-fixed liver tissues were embedded in paraffin and sliced into 5 μm thick sections. The sections were stained with the hematoxylin and eosin (H&E). In brief, the paraffin slices were dewaxed in xylene twice for 5 min each, transferred to decreasing gradient of ethanol (100%, 90%, 80% and 70%) for 2 min each, and washed with water for 2 min. Slices were then stained with hematoxylin for 5 min, washed with water for 2 min, transferred into 1% hydrochloric acid ethanol for 30 sec, and washed again for 15 min. Then slices were moved into eosin solution for 2 min, and wipe off the excess dye with water. After washing, the slices were transferred successively into 95% ethanol, 100% ethanol twice, xylene twice for 2 min each. The slices were then dried, sealed with a drop of gum, and then covered with clean glass. Digital images were collected with an ECLIPSE Ci-L microscope (Tokyo, Japan).
2.5. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays
To assess apoptosis in liver tissue sections, TUNEL labeling was carried out using the DeadEnd™ Colorimetric Apoptosis Detection System kit (Promega, USA) according to the manufacturer’s instructions. For tissue preparation section, paraffin-embedded liver slices were washed twice with xylene for 5 min each, immersed in 100% ethanol for 5 min, washed with a decreasing concentration of ethanol (100%, 95%, 85%, 70%,50%) for 3 min each, immersed in 0.85% NaCl for 5 min, and washed with PBS for 5 min. For apoptosis detection section, the slices were transferred into 4% paraformaldehyde in PBS for 15 min, washed twice with PBS for 5 min each, permeabilized with 100 μl of a 20 μg/ml proteinase K solution for 20 min at room temperature, washed with PBS for 5 min, immersed in 4% paraformaldehyde in PBS for 5 min, and washed twice with PBS for 5 min each. After that, 100 μl equilibration buffer was added to the tissue sections on the slices for 10 min at room temperature, and then 100 μl of TdT reaction mix was added to the same sections for 60 min at 37°C in a humidified chamber. The reaction was stopped by immersed slices in 2 × SSC for 15 min. The slices were washed with PBS for 5 min, transferred into 0.3% hydrogen peroxide for 3 min, and washed with PBS for 5 min. 100 μl of streptavidin HRP (diluted 1:500 in PBS) were added to the slices for 30 min at room temperature, and slices were washed three times in PBS for 5 min each. At last, 100 μl of DAB solution were added to the slices 1 min, and slices were washed three times with deionized water for 5 min each. Then, the slices were stained with hematoxylin for 20 sec, washed with running water for 5 min, dehydrated and sealed. Digital images were collected with an ECLIPSE Ci-L microscope (Tokyo, Japan).
2.6. RNA sequences analysis and data processing
Total RNA was extracted from liver tissues of triptolide dose-response experiments with Trizol reagent (Invitrogen, USA) according to the manufacturer’s protocol and purified with a RNeasy micro kit (QIAGEN, German) and RNase-Free DNase Set (QIAGEN, German). RNA concentrations and purities were measured by a NanoDrop spectrophotometer, and RNA integrity assessed by an Agilent bioanalyzer 2100. Samples with a RIN (RNA integrity number) ≥ 7.0 were used in next steps, and two samples (one in control group and another in 0.4 mg/kg triptolide group) were excluded. cDNA libraries from total RNA were prepared using VAHTS Stranded mRNA-seq Library Prep Kit (Vazyme, China) according to manufacturer’s protocol. Libraries were checked for size and purity with Agilent 2200 TapeStation. cDNA libraries were sequenced on an Illumina HiSeq X Ten sequencing machine under standard protocols. All samples had a Q20 (bases of Q ≥20 /all bases of sequencing) of > 96%. Sequence readers were aligned with seqtk and then mapped to the Ensemble Human genome sequence (GRCh38) by Hisat2 (version:2.0.4). Messenger RNA levels was quantified by the value of fragments per kilobase of exon per million mapped reads (FPKM). Differentially-expressed mRNAs were identified as those with q-values < 0.001 and | log2 (fold change) | >1 between control and triptolide-treated groups.
2.7. UPLC-ESI-QTOFMS analysis and data processing
Ultra-performance liquid chromatography equipped with quadrupole time-of-flight mass spectroscopy (UPLC-ESI-QTOFMS, Waters, USA) was applied for metabolomic analysis as previously described (Jiang et al., 2015; Zhao et al., 2018). Liver acylcarnitines were quantified as previously described (Millington D.S., 1900). Briefly, 25 mg liver tissues were added to 100 μl extra-pure water and 400 μl acetonitrile containing 5 nM myristoyl-trimethyl D9 carnitine. The mixtures were homogenized for 30 s and centrifuged at 16,000 g for 15 min at 4°C. Fifty μl of the supernatants were removed to 1.5 ml new tubes, dried under vacuum, re-dissolved in HPLC-grade water containing 1.6% acetonitrile, and assayed using a Waters UPLC-Q/TOF MS system with an electrospray source. An Acquity BEH C18 column (50 × 2.1 mm, 1.7 μm, Waters Crop.) was applied for chromatographic separation. The flow rate was 0.5 mL/min with an aqueous acetonitrile gradient containing 0.1% formic acid over a 10 min run. Mass spectrometry was performed in positive (ESI+) ionization mode. Source and desolvation temperature were set at 120°C and 350°C, respectively. The capillary voltage and cone voltage were set 3000 and 20 V, respectively. Nitrogen was used as both cone gas (50 L/h) and desovation gas (600 L/h). Argon was set as collision gas. A mass range of m/z 50–850 was acquired. The results were calculated according to individual standard curves established as follows: areaanalyte/areainternal.
The operations of date processing have been described in our previous reported work (Zhao et al., 2018). In brief, Masslynx software (Waters Corp.) was used for identifying the original mass spectral data and normalizing the total ion intensity of each chromatogram to acquire a data matrix containing the m/z value, retention time and normalized peak area. SIMCA-P + 14 software (Umetrics, Umea, Sweden) for Partial least squares discriminant analysis (PLS-DA). The potential metabolites were screened based on the variable importance in the projection (VIP) values and Student’s t- test. VIP > 1 and p < 0.05 were considered as statistically significant. Online metabolic databases including Metlin (http://metlin.scripps.edu), HMDB (http://www.hmdb.ca/) and PubChem (http://pubchem.Ncbi.nlm.nih.gov) with exact masses of the metabolites were used to identify the differential metabolites.
2.8. Metabolite and gene set enrichment, pathway and molecular network analysis
The MetaboAnalyst 3.0 was applied to identify the most relevant pathway of the differential metabolites. The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of the differentially expressed genes was carried out using omicshare network platform (https://www.omicshare.com/tools/). The integrated network analysis of above metabolites and genes were constructed using Metscape, a Cytoscape software (v3.1.1) plugin.
2.9. Quantitative real-time PCR analysis (q-PCR)
Total RNA was extracted from frozen liver tissue using TRIzol reagent (Invitrogen, Carlsbad, CA) and reverse-transcribed using qScript™ cDNA SuperMix (Quantabio, Beverly, MA). Real-time PCR was performed with STBR Green PCR master mix (AB Applied Biosystems, Warrington, UK). Primer sequences are listed in Supplemental Table 1. The relative changes of each gene were normalized with their corresponding Gapdh mRNA and expressed as fold change relative to the control group.
2.10. Western blot analysis
Frozen liver tissue was lysed in 1×RIPA buffer containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo-Fisher, Waltham, MA) and PMSF. Protein concentrations were measured with Pierce BCA protein Assay Kit (Pierce, Rockford, IL) and proteins separated on a 4–12% Criterion TGX Precast Gel (Bio-Rad, Hercules, CA) with 135 V of constant voltage for 80 min. Protein was then transferred to polyvinylidene difluoride membranes. Membranes were incubated with 5% BSA for 1 h at room temperature and then incubated with primary antibodies overnight in a shaker at 4 °C. The following antibodies were used: caspase-9 (CASP9, 9508, Cell Signaling Technology), caspase-3 (CASP3,19677–1-AP, Proteintech) and GAPDH (10494–1-AP, Proteintech). After primary antibody incubation, the membranes were washed three times with 1% Tween 20 in TBST (pH 7.5) for 10 min each and incubated with a secondary antibody for 1 h at room temperature. Anti-mouse peroxidase-conjugated second antibody (7076, Cell Signaling Technology) and anti-rabbit peroxidase-conjugated second antibody (7074, Cell Signaling Technology) were used. The signals were detected on a ChemiDoc™ MP System (Bio-Rad, Hercules, CA). The intensities of protein bands in blot imaging were measured on Image J software (NIH, USA). GAPDH was considered as the internal control. The ratio of cleaved and uncleaved forms was used to indicate the relative cleavage level.
2.11. Statistical analysis
Statistical analysis was performed with Prism 7.0 (GraphPad Software, San Diego, CA). All results are expressed as mean ± SEM. Statistical significance was determined by Student’s t-test. P values of less than 0.05 were considered significant.
3. Results
3.1. Dose-dependent manners of triptolide-induced liver injury
The toxic effect of triptolide in the liver was investigated with a series of dosages. Serum ALT and AST levels and histological analysis were used to assess liver toxicity. Administration of triptolide at the low dosage of 0.4 mg/kg did not result in significant differences in ALT and AST levels between the treated and control groups (Figure 1A) while sporadic cellular apoptosis was observed in the livers of this dosage group (Figure 1B), reflected as condensed and deeply eosinophilic cytoplasm (Shown as black arrows). ALT and AST levels were significantly increased after 0.6 mg/kg triptolide treatment (Figure 1A), with loss of hepatic architecture and a small portion of cellular apoptosis and necrosis (Figure 1B). Administration of triptolide at the highest dosage of 0.8 mg/kg resulted in extensive liver damage including disordered arrangement of liver lobule structure and markedly elevated cellular apoptosis. Meanwhile focal necrosis was observed (Figure1B), reflected as loss of hepatocyte cytologic architecture, nuclear pyknosis and karyorrhexis and inflammatory infiltration (Shown as black arrowheads). These results indicated that triptolide exposure could cause liver injury accompanied by hepatocellular apoptosis and necrosis.
Figure 1.

Dose-dependent manners of triptolide-induced liver injury. Mice were treated with 0.9% saline, 0.4 mg/kg, 0.6 mg/kg or 0.8 mg/kg of triptolide, respectively. (A) ALT and AST levels in serum. (B) Micrographs of H&E stained liver sections (200×). Black arrows indicated apoptosis and black arrowheads indicated necrosis. The data were expressed as mean ± SEM (n=8). Statistical analyses were performed with Student’s t tests. **p<0.01.
3.2. Time-dependent manners of triptolide-induced liver injury
In the time course experiments, no difference in ALT and AST levels was found at the 12 h time point (Figure 2A), which was consistent with the histological analysis (Figure 2B). At this time point, the liver structure showed a normal lobular structure with a radiated orderly arrangement of central veins and clear liver sinusoids. At 48 h, the ALT and AST levels reached over 15,000 and 20,000 U/L (Figure 2A), which were coincident with totally destroyed cellular structure (Figure 2B), reflected as a large area of degeneration and disappeared liver sinusoids. Furthermore, liver cell swelling, cytoplasm loss and nuclear pyknosis were also visible. These results indicated that triptolide could cause liver injury in a time-dependent manner, and administration of triptolide at 0.8 mg/kg with 24 h exposure time could cause significant liver damage.
Figure 2.

Time-dependent manners of triptolide-induced liver injury. Mice were treated with 0.8 mg/kg of triptolide and sacrificed at 0, 12, 24, 48 h, respectively. (A) ALT and AST levels in serum. (B) Micrographs of H&E stained liver sections (200×). Black arrows indicated apoptosis and black arrowheads indicated necrosis. The data were expressed as mean ± SEM (n=8). Statistical analyses were performed with Student’s t tests. **p<0.01.
3.2. RIPK3-mediated necroptosis is not involved in triptolide-induced liver injury
After triptolide exposure, damaged cells showed necrotic characteristics, including cell swelling and rupture of the plasma membrane and inflammation, indicating that necrosis may be related to triptolide-induced liver injury. To confirm whether triptolide exposure could induce RIPK3-mediated necroptosis, the expression of Ripk3 and Mlkl mRNAs was measured. After triptolide treatment, the Ripk3 and Mlkl mRNA levels were significantly increased (Figure 3A) indicating that RIPK3-mediated necroptosis may be involved in triptolide-induced liver injury. However, the effects of triptolide in Ripk3−/− and Ripk3+/+ mice did not reveal any significant differences in levels of serum ALT and AST and liver histological analysis (Figure 3B and C). These results indicated that RIPK3-mediated necroptosis was not directly involved in triptolide-induced liver injury, and that the upregulation of Ripk3 and Mlkl mRNA levels may be a consequence of hepatocyte death.
Figure 3.

Role of necrosis regulated by RIPK3 in triptolide-induced hepatotoxicity. (A) Expression of Ripk3 and Mlkl gene in control and mice treated with 0.8 mg/kg of triptolide. (B) Serum ALT and AST levels from control and 0.8 mg/kg triptolide-treated mice. (C) Micrographs of H&E stained liver sections (200×). Black arrows indicated apoptosis and black arrowheads indicated necrosis. The data were expressed as mean ± SEM (n=8). Statistical analyses were performed with Student’s t tests. *p <0.05, **p<0.01.
3.3. Role of apoptosis in triptolide-induced liver injury
Since the structural and morphological features of apoptosis were seen in liver sections stained with H&E after triptolide treatment, the potential role of apoptosis was investigated. TNFα, an apoptosis inductor, was significantly increased at the mRNA (Figure 4A) and protein (Figure 4B) levels after 0.8 mg/kg triptolide treatment. Apoptotic cells undergoing DNA damage stained brown were also increased in numbers in the TUNEL assay (Figure 4C). In addition, the relative cleavage levels (See Materials and methods) of CASP3 proteins (p17 and p19), the executor of apoptosis, was significantly induced upon triptolide exposure (Figure 4D and E). The relative cleavage levels of CASP9 proteins (p36), the upstream protein of CASP3 was also markedly up-regulated in triptolide-treated mice (Figure 4D and E). These results suggested that apoptosis was involved in triptolide-induced liver injury.
Figure 4.

Role of apoptosis in triptolide-induced hepatotoxicity. (A) QPCR measurement of the expression of Tnfα gene. (B) Plasma cytokine level of TNFα. (C) Micrographs of TUNEL assay in liver sections (200×). Black arrows indicated positive cells in TUNEL assay. (D) Western blotting analysis of expression of CASP9, cleaved CASP9, CASP3, cleaved CASP3 and GAPDH in the liver. (E) The ratio of cleaved form to full from of CASP9 and CASP3. The data were expressed as mean ± SEM (n=5–8). Statistical analyses were performed with Student’s t tests. **p<0.01.
3.4. Transcriptomic alterations induced by triptolide treatment
To elucidate the pathogenesis of triptolide-induced liver injury, gene expression profiles were developed from the control and three dosages of triptolide groups by RNA-seq analysis. PCA was used to explore dose-response relationships revealing a clear separation between the control and three dosages of triptolide groups (Figure 5A). Compared to the control group, the number of differentially expressed genes (DEGs) increased from 1234 to 5839 and 7312 with triptolide exposure dosages raised from 0.4 to 0.6 and 0.8 mg/kg (Figure 5B), respectively. Increased mRNAs ranged from 59% to 69% of the total DEGs in each dosage group (versus control group). This indicated a slight trend towards upregulation against down-regulation after triptolide treatment. A Venn diagram was performed to represent the DEGs profiles in different dosage groups revealing 767 DEGs detected in different dosage groups (Figure 5C, Supplemental Table 2), suggesting that these mRNAs might change in response to triptolide in a dose-dependent manner, and the relevant pathway might play important roles in triptolide-induced liver injury.
Figure 5.

Transcriptomic analysis of liver tissues in control and mice treated with 0.4 mg/kg, 0.6mg/kg and 0.8 mg/kg of triptolide. (A) Scores scatter plot of liver transcriptome in control mice and mice treated with three dosages of triptolide as determined by PCA. (B) The number of regulated genes after triptolide exposure; the red color represents up-regulated gene and the green color represents down-regulated gene. (C) Venn diagram of regulated genes in dose-related modules.
3.5. Biological pathways induced by triptolide treatment
Then omicshare network platform (https://www.omicshare.com/tools/) was constructed to explore the potential biological significance of DEGs. The functional categories of DEGs based on GO analysis were shown in Supplemental Figure 1. The top 20 KEGG pathways in each dosage group (versus control group) are represented in Figure 6. As expected, metabolic pathways were in the top of 20 KEGG pathways in different dosage groups. Compared to the control group, we also found the second top pathway affected by 0.4 mg/kg triptolide exposure in mice were systemic lupus erythematosus (Figure 6A), which indicated that this dosage of triptolide might also show therapeutic actions. Fifteen pathways were found both existing in 0.6 mg/kg and 0.8 mg/kg triptolide groups (versus control group), as shown in Figure 6B and C. Since several pathways were involved in apoptosis signaling, including PI3K/AKT, MAPK, TNFα and p53 signaling pathways, the KEGG pathway for apoptosis was revealed (Figure 7). The expression of Akt, p53 and Bcl2 were validated by q-PCR (Supplemental Figure 2). The results showed that mRNA levels of Akt and p53 were significantly increased by two-fold, and Bcl2 slightly increased (p=0.057) in the 0.8 mg/kg triptolide group, compared with the control group. This suggested that various targets in apoptosis signaling were tightly associated with triptolide-induced liver injury.
Figure 6.

KEGG pathway enrichment of regulated genes in dose-related modules. (A) Top 20 pathway enrichment in 0.4 mg/kg of triptolide versus control. (B) Top 20 pathway enrichment in 0.6 mg/kg of triptolide versus control. (C) Top 20 pathway enrichment in 0.8 mg/kg of triptolide versus control.
Figure 7.

KEGG pathway analysis of apoptosis signaling based on DEGs of 0.8 mg/kg of triptolide versus control group. The red rectangle nodes represent up-regulated genes and the green rectangle nodes represent down-regulated genes.
3.6. Alterations in metabolism induced by triptolide treatment
To further clarify changed metabolic pathways of triptolide-induced liver injury, UPLC-ESI-QTOFMS analysis coupled with multivariate data analysis was used to profile liver metabolites. PLS-DA plots were applied to analyze data sets revealing that liver metabolite profiles were separated among the control and three dosage groups in positive mode (Figure 8A) and negative mode (Figure 8B), respectively. Orthogonal partial least-square discrimination analysis (OPLS-DA) was further applied to identify differential metabolites between each dosage group and control group. There were 44, 54 and 70 metabolites differently regulated in the three dosage groups (versus control group), respectively. Thirty-four differential metabolites were found in different dosage groups (Table 1), which may indicate the most important metabolic pathways in triptolide-induced liver injury.
Figure 8.

Metabolomic analysis of liver tissue in control and mice treated with 0.4 mg/kg, 0.6mg/kg and 0.8 mg/kg of triptolide. (A) Scores scatter plot of liver metabolites in control mice and mice treated with three dosages of triptolide as determined by PLS-DA in positive mode. (B) Scores scatter plot of liver metabolites in control mice and mice treated with three dosages of triptolide as determined by PLS-DA in negative mode. (C) Quantitation of acetylcarnitines in the liver. The data were expressed as mean ± SEM (n=8). Statistical analyses were performed with Student’s t tests. *p <0.05, **p<0.01.
Table 1.
List of 34 differential metabolites identified in different dosage groups by metabolomics
| NO. | Metabolites | Ion | m/z | Retention time (min) | Triptolide (0.4 mg/kg) vs control | Triptolide (0.6 mg/kg) vs control | Triptolide (0.8 mg/kg) vs control | Metabolic pathway |
|---|---|---|---|---|---|---|---|---|
| 1 | Choline | M+H | 104.108 | 4.36 | ↓ | ↓ | ↓ | Glycerophospholipid metabolism |
| 2 | Uracil | M-H | 111.0203 | 0.99 | ↑ | ↑ | ↑ | Pyrimidine metabolism |
| 3 | Pyroglutamic acid | M-H | 128.0352 | 4.67 | ↑ | ↑ | ↑ | Glutathione metabolism |
| 4 | Malic acid | M-H | 132.0299 | 4.77 | ↑ | ↑ | ↑ | Citric acid cycle |
| 5 | Creatine | M+H | 132.0778 | 3.84 | ↓ | ↓ | ↓ | Glycine, serine and threonine metabolism |
| 6 | Indan-1-ol | M+H | 135.0824 | 0.37 | ↑ | ↑ | ↑ | Arachidonic acid metabolism |
| 7 | Adenine | M+H | 136.0638 | 4.38 | ↑ | ↑ | ↑ | Purine metabolism |
| 8 | 1-Methylnicotinamide | M+H | 137.0719 | 2.58 | ↓ | ↓ | ↓ | Nicotinate and nicotinamide metabolism |
| 9 | L-Glutamate | M-H | 146.0459 | 4.68 | ↑ | ↑ | ↑ | Glutathione metabolism |
| L-Glutamate | M+H | 148.0614 | 4.68 | ↑ | ↑ | ↑ | Glutathione metabolism | |
| 10 | 5-Aminopentanamide | M-H | 151.0612 | 1.77 | ↑ | ↑ | ↑ | Lysine degradation |
| 11 | Guanine | M+H | 152.058 | 2.38 | ↑ | ↑ | ↑ | Purine metabolism |
| 12 | L-Carnitine | M+H | 162.1132 | 3.8 | ↑ | ↑ | ↓ | β-oxidation of fatty acid metabolism |
| 13 | Citrulline | M+H | 176.1047 | 4.06 | ↑ | ↑ | ↑ | Arginine biosynthesis |
| 14 | Phosphocholine | M+H | 184.0751 | 4.35 | ↓ | ↓ | ↓ | Glycerophospholipid metabolism |
| 15 | N-Acetyl-D-glucosamine | M+H | 204.0871 | 5.12 | ↑ | ↑ | ↑ | Amino sugar and nucleotide sugar metabolism |
| 16 | Propionylcarnitine | M+H | 218.1392 | 2.59 | ↑ | ↑ | ↑ | b-oxidation of fatty acid metabolism |
| 17 | Uridine | M-H | 243.0619 | 0.99 | ↑ | ↑ | ↑ | Pyrimidine metabolism |
| 18 | Malonylcarnitine | M+H | 248.113 | 4.62 | ↑ | ↑ | ↑ | b-oxidation of fatty acid metabolism |
| 19 | Palmitic acid | M-H | 255.2325 | 0.41 | ↑ | ↑ | ↑ | Fatty acid metabolism |
| 20 | Glycerophosphocholine | M+H | 258.11 | 4.35 | ↓ | ↓ | ↓ | Glycerophospholipid metabolism |
| 21 | Nicotinate D-ribonucleoside | M+H | 273.1215 | 2.31 | ↑ | ↑ | ↑ | Nicotinate and nicotinamide metabolism |
| 22 | Saccharopine | M+H | 277.1402 | 5.35 | ↑ | ↑ | ↑ | Lysine degradation |
| 23 | 3-Methylglutarylcarnitine | M+H | 290.1595 | 4.66 | ↑ | ↑ | ↑ | b-oxidation of fatty acid metabolism |
| 24 | 5’-Methylthioadenosine | M+H | 298.098 | 0.54 | ↓ | ↓ | ↓ | Cysteine and methionine metabolism |
| 25 | Uridine 5’-monophosphate | M-H | 323.0278 | 5.26 | ↑ | ↓ | ↓ | Pyrimidine metabolism |
| 26 | Inosinic acid | M-H | 347.0392 | 5.38 | ↓ | ↓ | ↓ | Purine metabolism |
| 27 | S-Adenosylhomocysteine | M-H | 383.114 | 4.35 | ↑ | ↑ | ↑ | Cysteine and methionine metabolism |
| S-Adenosylhomocysteine | M+H | 385.1296 | 4.35 | ↑ | ↑ | ↑ | Cysteine and methionine metabolism | |
| 28 | Uridine diphosphate | M+H | 405.0083 | 5.3 | ↓ | ↓ | ↓ | Pyrimidine metabolism |
| 29 | Cytidine diphosphate choline | M+H | 489.1128 | 5.3 | ↑ | ↑ | ↑ | Glycerophospholipid metabolism |
| 30 | LysoPC (18:2) | M+H | 520.34 | 0.45 | ↑ | ↑ | ↑ | Glycerophospholipid metabolism |
| 31 | Uridine diphosphate glucose | M-H | 565.0461 | 5.28 | ↓ | ↓ | ↓ | Carbohydrate metabolism |
| 32 | Dephospho-CoA | M+H | 688.1567 | 4.19 | ↓ | ↓ | ↓ | Pantothenate and CoA biosynthesis |
| 33 | Glycogen | M+H | 689.2074 | 5.79 | ↓ | ↓ | ↓ | Starch and sucrose metabolism |
| 34 | Inositol-P-ceramide | M+H | 971.7257 | 0.34 | ↓ | ↓ | ↓ | sphingolipid metabolism |
shows up-regulated metabolites compared with control group
shows down-regulated metabolite compared with control group
Among significantly changed metabolites, three acylcarnitines (propionylcarnitine, malonylcarnitine and 3-methylglutarylcarnitine) were found to be increased after the three dosage of triptolide treatment. To determine whether levels of acylcarnitines in the liver were affected by triptolide treatment, levels of acylcarnitines were measured by using specific ion monitoring with authentic standards on an ACQUITY UPLC system equipped with a XEVO triple quadrupole tandem mass spectrometer. The levels of five long-chain acylcarnitines (deceaniylcarnitine, lauroylcarnitine, myristoylcarnitine, palmitoylcarnitine, and stearoylcarnitine) and five short-chain acylcarnitines (acylcarnitine, propionylcarnitine, buylcarnitine, pivaloycarnitine, and 2-methylcarnitine) were significantly increased at the highest dosage of 0.8 mg/kg triptolide exposure (Figure 8C). An increase of proprotylcarnitine, 2-methylcarnitine, and decanoylcarnitine was even detected after treatment with the lowest dosage of 0.4 mg/kg triptolide. These observations showed that acetylcarnitines were markedly increased by triptolide treatment in the liver.
In order to determine the most important metabolic pathways, MetaboAnalyst was applied to analyze the 34 mutual metabolites changed in different dosage groups (Figure 9 and Table 2). Six of the metabolic pathways with p <0.1 were considered as the most relevant pathways, including glycerophospholipid metabolism, pyrimidine metabolism, arginine biosynthesis, nicotinate and nicotinamide metabolism, pantothenate and CoA biosynthesis and glutathione metabolism, which were involved in lipid metabolism and amino acid metabolism.
Figure 9.

Summary of pathways analysis of differential metabolites represented in different dosage groups with Metaboanalyst. The color and size of circles were depended on P value and pathway impact value analyzed by MetaboAnalyst, respectively. The bigger and the closer to the top right corner of the plot represented the more important metabolic pathways.1: Glycerophospholipid metabolism 2: Pyrimidine metabolism 3: Arginine biosynthesis 4: Nicotinate and nicotinamide metabolism 5: Pantothenate and CoA biosynthesis 6: Glutathione metabolism 7: D-Glutamine and D-glutamate metabolism 8: Cysteine and methionine metabolism 9: purine metabolism 10: Amino sugar and nucleotide sugar metabolism 11: Arginine and proline metabolism.
Table 2.
Analyzed metabolic pathways with p <0.1 of 34 differential metabolites in metabolomics
| Total | Expected | Hits | Raw p | −log p | Holm adjust | FDR | Impact | |
|---|---|---|---|---|---|---|---|---|
| Glycerophospholipid metabolism | 36 | 0.69323 | 4 | 0.004269 | 5.4565 | 0.35856 | 0.24049 | 0.10258 |
| Pyrimidine metabolism | 39 | 0.751 | 4 | 0.005726 | 5.1628 | 0.47525 | 0.24049 | 0.21627 |
| Arginine biosynthesis | 14 | 0.26959 | 2 | 0.028239 | 3.5671 | 1 | 0.67617 | 0.34518 |
| Nicotinate and nicotinamide metabolism | 15 | 0.28884 | 2 | 0.032198 | 3.4358 | 1 | 0.67617 | 0.13816 |
| Pantothenate and CoA biosynthesis | 19 | 0.36587 | 2 | 0.050009 | 2.9955 | 1 | 0.84015 | 0.2 |
| Glutathione metabolism | 28 | 0.53918 | 2 | 0.099441 | 2.3082 | 1 | 1 | 0.02675 |
3.7. Network analysis of transcriptomics and metabolomics data
To explore the systemic mechanism of triptolide-induced liver injury, a latent relationship network of mutual differential metabolites was built and DEGs detected in different groups with Metscape. Those metabolites and DEGs were mainly clustered in metabolic pathways of glycerophospholipid metabolism, fatty acid biosynthesis, leukotriene metabolism, purine metabolism and pyrimidine metabolism (Figure 10). The integrated analysis of transcriptomics and metabolomics provided potential implications for understanding the systemic mechanism of triptolide-induced liver injury.
Figure 10.
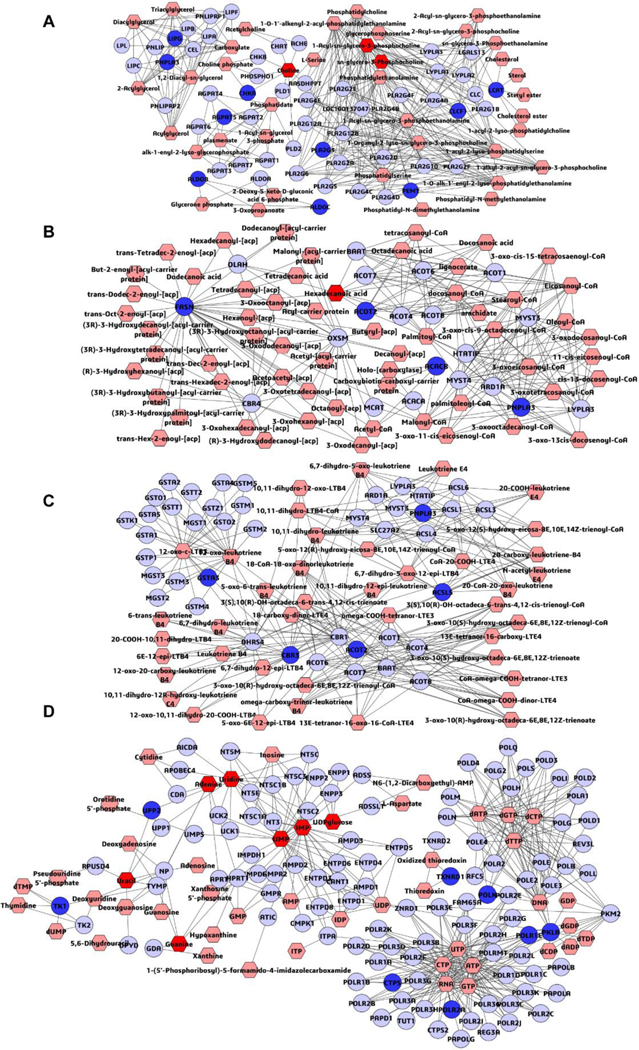
Metabolomic correlation networks of the differential metabolites (hexagons) and genes (rounds) represented in different dosage groups. Input metabolites were shown in red and input genes were shown in blue. The metabolic networks of glycerophospholipid metabolism (A), fatty acid biosynthesis (B), leukotriene metabolism (C), purine metabolism and pyrimidine metabolism (D).
4. Discussion
The triptolide-containing Chinese traditional herb medicine has been widely used in clinic for the treatment of autoimmune disorders (Hou et al., 2019), but triptolide-induced liver injury has drawn increasing attention in recent years. The therapeutic dosage of TWHF tablets was about 4–6 tablets/d (equivalent to 0.4–0.6 mg/d triptolide) (Qu et al., 2015). Previous reports showed that the plasma concentration of triptolide in humans was around 0.15–0.4 μM (Yao et al., 2006). In acute toxicity studies, the LD50 value of triptolide by gavage was reported to be 0.788 mg/kg in Kunming mice (Ding et al., 2004). Based on those reports and our previous research (Zhao et al., 2018), in order to metabolically describe triptolide-induced liver injury, three dosages (0.4 mg/kg, 0.6 mg/kg and 0.8 mg/kg) of triptolide were used to induce dose-dependent toxic effects, including slight, general and severe liver toxicity. The mice on C57/BL/6N background did not die at the high dose of 0.8 mg/kg after 24 h triptolide treatment. This dose in mice was equivalent to 0.08 mg/kg in humans (4 mg in a 50-kg person), within 10-fold of its clinal use. Results from serum aminotransferase assays and liver histology were accord with the increasing dosages. In the dose- and time-course experiments, cell death occurred in a dose- and time-dependent manner. The potential roles of necroptosis and apoptosis, two programmed cell death signaling pathways, were investigated in triptolide-induced liver injury in vivo. Transcriptomics and metabolomics were performed to gain clues to the mechanism of liver damage. These studies demonstrated insights into the potential mechanism of triptolide-induced liver injury and may help provide new strategies for the treatment of acute liver toxicity.
Necroptosis, also known as programmed necrosis, is a kind of programmed cell death and morphologically resembles necrosis (Vandenabeele, 2010). Receptor-interacting serine-threonine kinases (RIPK) 3 has a crucial role in necroptosis with its signaling downstream of mixed lineage kinase domain-like protein (MLKL). The role of necroptosis in models of acute liver injury was analyzed in several studies, but the results were inconsistent (Li et al., 2014b; Deutsch et al., 2015). In the present study, after triptolide exposure, damaged cells showed necrotic characteristics, including nuclear pyknosis and karyorrhexis and inflammatory infiltration and the expression of Ripk3 and Mlkl mRNA were also signification increased in triptolide exposure group, which indicated the potential of necroptosis. Ripk3−/− and Ripk3+/+ mice were treated with triptolide to evaluate its effect on necroptosis. Unexpectedly, serum aminotransferase assays and liver histological examinations showed no significant differences between Ripk3−/− and Ripk3+/+ mice. Previous study also pointed out the similar issue in drug-induced liver injury, which using Ripk3−/− mice in the acetaminophen-induced liver injury model did not reveal any significant difference between Ripk3−/− and Ripk3+/+ mice (Dara et al., 2015). And the increased expression of Ripk3 and Mlkl may be a consequence of triptolide-induced cell death. RIPK3-mediated necroptosis may be not the mediator of triptolide-induced liver injury.
Apoptosis is characterized by cellular shrinkage, chromatin condensation and CASP3 activation (Schwabe and Luedde, 2018). Studies have shown that apoptosis is associated with liver damage (Guicciardi and Gores, 2005). Triptolide-induced liver injury studies previously revealed that triptolide exposure could significantly increase Tnfα mRNA or TNFα protein levels (Vliegenthart et al., 2017; Wang et al., 2018). TNFα, a classical apoptosis inducer, is produced extensively during sepsis-induced liver injury, acetaminophen-induced liver injury, and hepatic ischemia reperfusion injury (Du et al., 2016). Treatment of cultured human liver-derived L-02 cells with triptolide revealed that triptolide could induce apoptosis by loss of mitochondrial membrane potential and release of cytochrome c associated with activation of CASP9 and CASP3 (Yao et al., 2008). In addition, chronic triptolide exposure induced liver apoptosis through mitochondrial injury and activation of CASP3 (Zhou et al., 2018). In the present study, consistent with the previous work, Tnfα mRNA in the liver and TNFα protein in serum were significantly elevated in the triptolide treatment group. TUNEL assay results showed that apoptotic cells occurred after 0.8 mg/kg triptolide exposure and the active CASP9 (p36) and CASP3 (p19 and 17) protein levels were also significantly increased. These results indicated that triptolide exposure could induced apoptosis through a TNFα-dependent pathway in the liver, which were consistent with the omics studies.
Based on transcriptomics data, except for the TNFα-dependent pathway, PI3K/AKT, MAPK and p53 were also suggested to be partly responsible for apoptosis. PI3K/AKT signaling pathway is an essential pathway in regulating the cell cycle, apoptosis and DNA binding. PI3K (phosphatidyl-3-phosphate kinase), an intracellular phosphatidylinositol kinase, could affect its key downstream effector AKT achieving its anti-apoptosis effect. Previous reports have shown that the activation of PI3K/AKT signaling was involved in various diseases, and activated AKT could stimulate the expression of Bax and CASP9 expression or activate NF-κB pathway to promote resistance of cancer cells to apoptosis (Yang et al., 2019). However, the potential role of PI3K/AKT pathway in drug-induced liver injury was still unclear. In this study, the expression of Akt was validated by q-PCR (Supplemental Figure 2), which showed about two-fold increase in the 0.8 mg/kg triptolide group, compared with the control group. This result was accordance with a recent report showing that D-galactosamine/lipopolysaccharide could induce acute liver damage through activation of PI3K/AKT pathway with increased expression levels of PI3K, AKT, and p-AKT, and PI3K agonist group could promoted cell apoptosis (Li et al., 2017b). Besides, compared with the control group, we found 164 DEGs in PI3K/AKT pathway were significantly changed in the 0.8 mg/kg triptolide group as shown in heat map (Supplemental Figure 3) and 12 DEGs (Gys2, Klk1b4, Itga5, Eif4e, Creb3l4, Egfr, Mdm2, Myc, Cdkn1a, Pik3r3, Hsp90aa1 and Igf1) in PI3K/AKT pathway were detected in different dosage groups, indicating its potential role in triptolide-induced liver. And this result provides a clue for us to make further exploring. MAPK (mitogen-activated protein kinase) has been reported to be associated with drug-induced liver injury, which plays a vital role in the mediating cellular inflammatory response and apoptosis (Omar et al., 2016). The MAPK family consists of three major proteins, ERK, JNK and p38 which are highly linked to apoptosis and cell death (Nakagawa and Maeda, 2012). Triptolide was reported to induce cell cycle arrest at the G1/S phase and cell apoptosis by MAPK/ERK and p53 signaling (Yuan et al., 2019). PI3K/AKT and MAPK signaling to modulate the Bcl2 proteins which play a key role in regulating intrinsic apoptosis (Wang et al., 2013; Xie et al., 2016). Additionally, p53 in known to promote apoptosis by transcriptional upregulation of Noxa, Puma, Bid and Bax (Baskin-Bey and Gores, 2005). BCL2 is an anti-apoptosis protein, while NOXA, PUMA, BID and BAX possess pro-apoptotic properties. The imbalance of these proteins leads to the releasing cytochrome c from mitochondria to cytosol, which further activates CASP8, CASP9 and ultimately CASP3, triggering apoptotic cell death. In this study, 5 DEGs (Mapk8ip3, Gadd45b, Map2ka6, Rps6ka3 and Zak) in MAPK signaling and 2 mRNAs (Gtse1 and Zmat3) in p53 signaling were detected in different dosage groups. The pro-apoptotic gene p53 was increased by two-fold, while Bcl2 slightly increased but no significance by q-PCR validation (Supplemental Figure 2). Additionally, in the 0.8 mg/kg triptolide group, the pro-apoptotic genes such as Bim, Bid, Bax, and Noxa were significantly increased, suggesting that apoptosis occurred. The increased expression of pro-apoptosis genes could further induce cell apoptosis by activating the CASP9 and CASP3. These results were also confirmed by western blot results which showed that CASP9 (p36) and CASP3 (p19 and 17) were induced in the 0.8 mg/kg triptolide group.
To further define the changed metabolic pathways and molecular pathways predicted by transcriptomics, metabolomics was used for the identification of endogenous biomarkers involved in triptolide-induced toxicity and finding metabolic clues to uncover potential mechanisms. An untargeted metabolomics approach was employed to determine the global liver metabolic profiles. Among the significantly changed metabolites, what drew our attention was that several acylcarnitines were significantly changed in the liver from triptolide-treated mice. Acylcarnitines, conjugated derivatives of fatty acids that are transported into mitochondria to undergo fatty acid β-oxidation, were recently reported to be a biomarker of liver injury and mitochondrial dysfunctions in several models of drug-induced liver injury (Chen et al., 2009; McGill et al., 2014; Ramachandran et al., 2018). If the β-oxidation is affected, acylcarnitines would be accumulated within the mitochondria and then be released when the mitochondria suffered the membrane permeability transition (MPT) (McGill et al., 2014). Inducting opening of the MPT led to the release of pro-apoptotic factors such as cytochrome c, resulting in the activation of CASP9 and 3 and cell apoptosis. Serum acylcarnitines levels were significantly increased during acetaminophen toxicity in mice (Chen et al., 2009) and in children (Bhattacharyya et al., 2014). Recently, triptolide exposure disrupted endogenous peroxisome proliferator-activated receptor α (PPARα) causing the accumulation of acylcarnitines in serum (Hu et al., 2019). In the current study, triptolide significantly increased ten acylcarnitines metabolites in the liver. This result was at least partly suggested that elevating of acylcarnitines after triptolide exposure in the serum reflect their increased levels in the liver. And our finding further confirmed that acylcarnitines would be a potential biomarker for the early detection of triptolide-induced liver injury.
Integrated omics data by profiling metabolomic correlation networks of the metabolites and genes, identified five metabolic pathways, including glycerophospholipid metabolism, fatty acid biosynthesis, leukotriene metabolism, purine metabolism and pyrimidine metabolism, which deserve further attention. This result provided a systemic observation into the dysregulated pathway and clarified the molecular mechanism of triptolide-induced.
5. Conclusion
In the current study, a dose- and time-dependent toxic effects of triptolide were investigated. With the integrated application of the transcriptomics and metabolomics, we provide a deep insight into the systemic mechanism of triptolide-induced liver injury. The results obtained demonstrate that triptolide exhibits apoptosis by regulating PI3K/AKT signaling pathway, MAPK signaling pathway, TNFα signaling pathway and p53 signaling pathway. The metabolomic pathways, including glycerophospholipid metabolism, fatty acid biosynthesis, leukotriene metabolism, purine metabolism and pyrimidine metabolism, play vital roles in triptolide-induced liver injury. Moreover, acylcarnitines were identified as potential biomarkers for the early detection of triptolide-induced liver injury. The current work provides a new reference for studies on drug-induced liver injury and guidance for the development of new treatments in triptolide poisoning in the future.
Supplementary Material
Highlights:
An integrated application of transcriptomics and metabolomics methods was conducted to reveal mechanism of triptolide-induced liver toxicity
Triptolide-induced toxicity occurred in a dose- and time-dependent manners
Apoptosis might be responsible for triptolide-induced liver toxicity and not necroptosis
PI3K/AKT, MAPK, TNFα and p53 signaling pathways might be the vital steps in triptolide-induced hepatocyte apoptosis
Acylcarnitines could be considered as potential biomarkers
Acknowledgments
Funding
This work was supported by the National Cancer Institute Intramural Research Program, Hebei Science and Technology Department in China (17392501D), the National Science Foundation of China (81973469), and the Doctoral Scientific Research Start-up Foundation from Henan University of Chinese Medicine (RSBSJJ2018–13).
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- Bim
Bcl2-like 11
- Bax
Bcl2-associated X protein
- CCK-8
cell counting kit-8
- DMARD
disease modifying antirheumatic drug
- ESI
electrospray ionization; MS, mass spectroscopy
- FPKM
fragments per kilobase of exon per million mapped reads
- H&E
hematoxylin and eosin
- MAPK
mitogen-activated protein kinase
- MLKL
mixed lineage kinase domain-like protein
- MPT
membrane permeability transition
- OPLS-DA
Orthogonal partial least-square discrimination analysis
- PI3K
phosphatidylinositol-3 kinases
- PLS-DA
partial least squares discriminant analysis
- RIN
RNA integrity
- RIPK3
receptor-interacting serine-threonine kinases 3
- Tnfα
tumor necrosis factor alpha
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- TWHF
Triptergium wilfordii Hook. f
- UPLC
ultra-performance liquid chromatography
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Conflict of Interests
The authors declared that there was no conflict of interest with any commercial or financial relationships.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baskin-Bey ES, Gores GJ, 2005. Death by association: BH3 domain-only proteins and liver injury. Am J Physiol Gastrointest Liver Physiol 289(6), G987–990. doi: 10.1152/ajpgi.00371.2005. [DOI] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT, 2009. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7(2), 99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Yan K, Pence L, Simpson PM, Gill P, Letzig LG, et al. , 2014. Targeted liquid chromatography-mass spectrometry analysis of serum acylcarnitines in acetaminophen toxicity in children. Biomark Med 8(2), 147–159. doi: 10.2217/bmm.13.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Krausz KW, Shah YM, Idle JR, Gonzalez FJ, 2009. Serum metabolomics reveals irreversible inhibition of fatty acid beta-oxidation through the suppression of PPARalpha activation as a contributing mechanism of acetaminophen-induced hepatotoxicity. Chem Res Toxicol 22(4), 699–707. doi: 10.1021/tx800464q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Wang JM, Wang D, Wu R, Hou HW, 2019. Triptolide inhibits migration and proliferation of fibroblasts from ileocolonic anastomosis of patients with crohn’s disease via regulating the miR-16–1/HSP70 pathway. Mol Med Rep 19(6), 4841–4851. doi: 10.3892/mmr.2019.10117. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dai J, Sun Y, Chen D, Zhang Y, Yan L, Li X, et al. , 2019. Negative regulation of PI3K/AKT/mTOR Axis regulates fibroblast proliferation, apoptosis and autophagy play a vital role in triptolide-induced epidural fibrosis reduction. Eur J Pharmacol 864, 172724. doi: 10.1016/j.ejphar.2019.172724. [DOI] [PubMed] [Google Scholar]
- Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, et al. , 2015. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 62(6), 1847–1857. doi: 10.1002/hep.27939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, et al. , 2015. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis 6(5), e1759. doi: 10.1038/cddis.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Wu J, Tong J, Yuan X, Chen J, Shi G, 2004. Acute toxicity of triptolide and its mechanism (Article in Chinese). Zhong Yao Cai 27(2), 115–118. [Google Scholar]
- Du K, Ramachandran A, Jaeschke H, 2016. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol 10, 148–156. doi: 10.1016/j.redox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan D, Guo Q, Shen J, Zheng K, Lu C, Zhang G, et al. , 2018. The effect of triptolide in rheumatoid arthritis: from basic research towards clinical translation. Int J Mol Sci 19(2). doi: 10.3390/ijms19020376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guicciardi ME, Gores GJ, 2005. Apoptosis: a mechanism of acute and chronic liver injury. Gut 54(7), 1024–1033. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guicciardi ME, Malhi H, Mott JL, Gores GJ, 2013. Apoptosis and necrosis in the liver. Compr Physiol 3(2), 977–1010. doi: 10.1002/cphy.c120020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin Y, Seldin M, Lusis A, 2017. Multi-omics approaches to disease. Genome Biol 18(1), 83. doi: 10.1186/s13059-017-1215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W, Liu B, Xu H, 2019. Triptolide: Medicinal chemistry, chemical biology and clinical progress. Eur J Med Chem 176, 378–392. doi: 10.1016/j.ejmech.2019.05.032. [DOI] [PubMed] [Google Scholar]
- Hou Z, Chen L, Fang P, Cai H, Tang H, Peng Y, et al. , 2018. Mechanisms of triptolide-induced hepatotoxicity and protective effect of combined use of isoliquiritigenin: possible roles of nrf2 and hepatic transporters. Front Pharmacol 9, 226. doi: 10.3389/fphar.2018.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu DD, Zhao Q, Cheng Y, Xiao XR, Huang JF, Qu Y, et al. , 2019. The protective roles of PPARalpha activation in triptolide-induced liver injury. Toxicol Sci. doi: 10.1093/toxsci/kfz146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorga A, Dara L, 2019. Cell death in drug-induced liver injury. Adv Pharmacol 85, 31–74. doi: 10.1016/bs.apha.2019.01.006. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Ramachandran A, Chao X, Ding WX, 2019. Emerging and established modes of cell death during acetaminophen-induced liver injury. Arch Toxicol 93(12), 3491–3502. doi: 10.1007/s00204-019-02597-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, et al. , 2015. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun 6, 10166. doi: 10.1038/ncomms10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury MK, Gupta K, Franco SR, Liu B, 2019. Necroptosis in the pathophysiology of disease. Am J Pathol. doi: 10.1016/j.ajpath.2019.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Shen F, Guan C, Wang W, Sun X, Fu X, et al. , 2014a. Activation of Nrf2 protects against triptolide-induced hepatotoxicity. PLoS One 9(7), e100685. doi: 10.1371/journal.pone.0100685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, et al. , 2014b. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 5, e1278. doi: 10.1038/cddis.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hu T, Tie C, Qu L, Zheng H, Zhang J, 2017a. Quantitative proteomics and targeted fatty acids analysis reveal the damage of triptolide in liver and kidney. Proteomics 17(22). doi: 10.1002/pmic.201700001. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu L, Luo N, Wang YQ, Gao HM, 2017b. Inhibition of PI3K/AKt/mTOR signaling pathway protects against D-galactosamine/lipopolysaccharide-induced acute liver failure by chaperone-mediated autophagy in rats. Biomed Pharmacother 92, 544–553. doi: 10.1016/j.biopha.2017.05.037. [DOI] [PubMed] [Google Scholar]
- Liu Q, 2011. Triptolide and its expanding multiple pharmacological functions. Int Immunopharmacol 11(3), 377–383. doi: 10.1016/j.intimp.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Lu Y, Xie T, Zhang Y, Zhou F, Ruan J, Zhu W, et al. , 2017. Triptolide Induces hepatotoxicity via inhibition of CYP450s in Rat liver microsomes. BMC Complement Altern Med 17(1), 15. doi: 10.1186/s12906-016-1504-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Li F, Sharpe MR, Williams CD, Curry SC, Ma X, et al. , 2014. Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after acetaminophen overdose in mice and humans. Arch Toxicol 88(2), 391–401. doi: 10.1007/s00204-013-1118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millington DS, Kodo N, Norwood DL, Roe CR, 1900. Tandem mass spectrometry: A new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J. lnher. Metab. Dis 13, 321–324. doi: 10.1007/bf01799385. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Maeda S, 2012. Molecular mechanisms of liver injury and hepatocarcinogenesis: focusing on the role of stress-activated MAPK. Patholog Res Int 2012, 172894. doi: 10.1155/2012/172894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar HA, Mohamed WR, Arab HH, Arafa SA, 2016. Tangeretin alleviates cisplatin-induced acute hepatic injury in rats: targeting MAPKs and apoptosis. PLoS One 11(3), e0151649. doi: 10.1371/journal.pone.0151649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Qu F, Jia Z, Wang C, Wu C, Zhang J, 2015. Integrated targeted sphingolipidomics and transcriptomics reveal abnormal sphingolipid metabolism as a novel mechanism of the hepatotoxicity and nephrotoxicity of triptolide. J Ethnopharmacol 170, 28–38. doi: 10.1016/j.jep.2015.05.010. [DOI] [PubMed] [Google Scholar]
- Ramachandran A, Duan L, Akakpo JY, Jaeschke H, 2018. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: current understanding and future perspectives. J Clin Transl Res 4(1). doi: 10.18053/jctres.04.201801.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe RF, Luedde T, 2018. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 15(12), 738–752. doi: 10.1038/s41575-018-0065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G,2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11(10):700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Vliegenthart ADB, Wei C, Buckley C, Berends C, de Potter CMJ, Schneemann S, et al. , 2017. Characterization of triptolide-induced hepatotoxicity by imaging and transcriptomics in a novel zebrafish model. Toxicol Sci 159(2), 380–391. doi: 10.1093/toxsci/kfx144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Jiang Q, Feng XH, Zhang HD, Ge L, Luo CG, et al. , 2016. Tripterygium wilfordii Hook F versus conventional synthetic disease-modifying anti-rheumatic drugs as monotherapy for rheumatoid arthritis: a systematic review and network meta-analysis. BMC Complement Altern Med 16, 215. doi: 10.1186/s12906-016-1194-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Xu D, Li L, Xing X, Liu L, Ismail Abdelmotalab M, et al. , 2018. Possible role of hepatic macrophage recruitment and activation in triptolide-induced hepatotoxicity. Toxicol Lett 299, 32–39. doi: 10.1016/j.toxlet.2018.08.017. [DOI] [PubMed] [Google Scholar]
- Wang X, Jiang Z, Xing M, Fu J, Su Y, Sun L, Zhang L, 2014. Interleukin-17 mediates triptolide-induced liver injury in Mice. Food Chem Toxicol, 71, 33–41. doi: 10.1016/j.fct.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang ZZ, Wu Y, Ke JJ, He XH, Wang YL, 2013. Quercetin postconditioning attenuates myocardial ischemia/reperfusion injury in rats through the PI3K/Akt pathway. Braz J Med Biol Res 46(10), 861–867. doi: 10.1590/1414-431X20133036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu JQ, Xu JD, Zhu H, Kong M, Zhang GH, et al. , 2017. UPLC/ESI-QTOF-MS-based metabolomics survey on the toxicity of triptolide and detoxication of licorice. Chinese Journal of Natural Medicines 15(6), 474–480. doi: 10.1016/s1875-5364(17)30071-7. [DOI] [PubMed] [Google Scholar]
- Xie CQ, Zhou P, Zuo J, Li X, Chen Y, Chen JW, 2016. Triptolide exerts pro-apoptotic and cell cycle arrest activity on drug-resistant human lung cancer A549/Taxol cells via modulation of MAPK and PI3K/Akt signaling pathways. Oncol Lett 12(5), 3586–3590. doi: 10.3892/ol.2016.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Sun L, Wang L, Hassan HM, Wang X, Hylemon PB, et al. , 2017. Activation of Sirt1/FXR signaling pathway attenuates triptolide-induced hepatotoxicity in rats. Front Pharmacol 8, 260. doi: 10.3389/fphar.2017.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhao J, Song X, Li L, Li F, Shang J, et al. , 2019. Amygdalin reduces lipopolysaccharide-induced chronic liver injury in rats by down-regulating PI3K/AKT, JAK2/STAT3 and NF-κB signalling pathways. Artif Cells Nanomed Biotechnol 47(1), 2688–2697. doi: 10.1080/21691401.2019.1634084. [DOI] [PubMed] [Google Scholar]
- Yao J, Jiang Z, Duan W, Huang J, Zhang L, Hu L, et al. , 2008. Involvement of mitochondrial pathway in triptolide-induced cytotoxicity in human normal liver L-02 cells. Biol Pharm Bull 31(4), 592–597. [DOI] [PubMed] [Google Scholar]
- Yao J, Zhang L, Zhao X, Hu L, Jiang Z, 2006. Simultaneous determination of triptolide, wilforlide A and triptonide in human plasma by high-performance liquid chromatography-electrospray ionization mass spectrometry. Biol Pharm Bull 29, 1483–1486. [DOI] [PubMed] [Google Scholar]
- Yuan Z, Zhang H, Hasnat M, Ding J, Chen X, Liang P, et al. , 2019. A new perspective of triptolide-associated hepatotoxicity: Liver hypersensitivity upon LPS stimulation. Toxicology 414, 45–56. doi: 10.1016/j.tox.2019.01.005. [DOI] [PubMed] [Google Scholar]
- Zhao J, Xie C, Mu X, Krausz KW, Patel DP, Shi X, et al. , 2018. Metabolic alterations in triptolide-induced acute hepatotoxicity. Biomed Chromatogr 32(10), e4299. doi: 10.1002/bmc.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Zhou C, Feng Z, Liu Z, Zhu H, Zhou X, 2018. Triptolide-induced hepatotoxicity can be alleviated when combined with Panax notoginseng saponins and Catapol. J Ethnopharmacol 214, 232–239. doi: 10.1016/j.jep.2017.12.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.