

Abstract
Cisplatin is a potent drug used in about 40% of cancer treatment but also leads to severe deafness in 60-80% of the cases. Although the mechanism is known to be related to the accumulation of reactive oxygen species (ROS), no drug or FDA approved treatment is currently available to prevent cisplatin ototoxicity. With this study, we show for the first time that honokiol (HNK), a pleiotropic poly-phenol prevents cisplatin-induced hearing loss. HNK also improves the wellbeing of the mice during the treatment, determined by the increase in the number of surviving animals. In a transgenic tumor mouse model, HNK does not hinder cisplatin’s antitumor effect. The mechanism is related to the activation of sirtuin 3, a deacetylase in mitochondria essential for ROS detoxification. We expect a paradigm shift in cisplatin chemotherapy based on the current study and future clinical trials, where honokiol is applied to reduce side effects including hearing loss.
Keywords: Chemotherapy, hearing protection, cochlea, reactive oxygen species, tumor-bearing mice
Introduction
Cisplatin is a potent antitumor drug used in ~40% of cancer chemotherapy regimens [1,2]. According to the data from the National Cancer Institute (NCI), cisplatin is prescribed for an estimated 10-20% of all cancer patients, along with other similar platinum-based drugs. Unfortunately, these drugs have significant sequelae that limit their usage and dosage [3-5], including nausea, vomiting, liver damage, kidney failure, hearing loss, tinnitus, and vertigo, etc. In the USA, the prevalence of hearing loss during chemotherapy with cisplatin is 60-80% [4,6], that is, about 100-300 thousand new cases annually. The mechanism of cisplatin ototoxicity is closely related to the accumulation of reactive oxygen species (ROS) [3-5]. Outer hair cells (OHCs) are most vulnerable to this cumulated oxidative damage, which are eventually lost through apoptosis [4,7]. Consequently, the majority of the proposed otoprotective agents are exogenous antioxidants that work as free radical scavengers [8-10]. Some of them, including sodium thiosulfate [11], N-acetylcysteine [12], and amifostine [13,14] have been tested in clinical practice (for a review, see [3]). Others, including neurotrophins [15], hormones [12], molecules involved in endogenous metabolism [16], and modulators of cell signaling pathways [17] have also been proposed. Since the antioxidants potentially can also protect tumor cells and interfere with the therapeutic effects of cisplatin [8,10], local drug delivery through trans-tympanic injection is proposed [18-20]. However, no drug has been approved by the FDA for systemic application in clinic yet.
Honokiol (HNK) is a multi-functional polyphenol extracted from an Asian herbal medicine (magnolia bark) with anti-angiogenic and antitumor effects [21,22]. It is synergistic with cisplatin for tumor suppression as shown in (pre-) clinical studies [21,23,24]. HNK also protects various tissues and organs in vivo against oxidative stress, including the brain [25-27], heart [28-30], kidney [31], and liver [32]. The molecular mechanism underlying the activity of HNK is through the direct activation of a protein from the silent information regulator family, sirtuin 3 (SIRT3) [29]. SIRT3 is the primary NAD+-dependent deacetylase [33] in mitochondria involved in multiple intracellular metabolic processes [34]. SIRT3-mediated protein deacetylation via the activation of Manganese Superoxide Dismutase (MnSOD) is essential for ROS reduction and detoxification [35]. Having both tumor suppressive and normal tissue protective effects, HNK is an ideal candidate for hearing protection during cisplatin chemotherapy. For the first time, we show in this study that HNK protects against cisplatin ototoxicity both in vitro and in vivo. Cisplatin treatment in tumor-bearing mice was not compromised by HNK.
Materials and methods
Cell culture, treatment, and cell counting
A primary cochlear cell line, the House Ear Institute-Organ of Corti 1 (HEI-OC1) cells (from Jing Zheng’s lab) [36] and 3 cancel cell lines: colon cancer (HCT116), cervical cancer (HeLa) and prostate cancer (C4-2B), were used in the study. HEI-OC1, HCT116, and HeLa Cells (from David Gius’ lab) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, Carlsbad, CA) with 10% Fetus Bovine Serum (FBS), while C4-2B cells were cultured in DMEM/F-12 with 5% FBS. For cell counting, the cells were trypsinized (0.25%, 1 min), triturated by pipetting up and down, resuspended, and re-seeded 1 day before in 24-well plates with a confluence of 2 × 105/well, counted with a hematocytometer (Fisher Scientific, Pittsburgh, PA). The cells were then treated with cisplatin (0, 50, 100 µM) and HNK (0, 5, 10, 25 µM) mixtures (both from Sigma Aldrich, Port Washington, WI, or Tocris Bioscience, Minneapolis, MN) dissolved in DMSO, each with 3 replicates on one plate. After another 24 or 48 hours, the cells in each well were digested, triturated, and resuspended again in DMEM, in a precise total volume of 500 µl. Precisely 9 µl of culture cell suspension was taken and mixed with an equal amount of trypan blue, and 9 μl of the mixture was transferred to the hemocytometer and the numbers of cells were counted in 5 out of the 9 squares. Cell sampling was repeated to a total of 10 squares for each well. The total amount of cells in each well was calculated with the following equation: Total cells per well = Total cells in 10 squares (/μl) * 500 (μl) * 2 (dilution by trypan blue). In the case of high cell confluence, a 1:1 to 1:4 dilution in DMEM was applied before the mixing with trypan blue. In the case of low cell confluence, the volume of the resuspension was decreased to 200-250 µl. The equation for cell number calculation was adjusted accordingly. The entire procedure was repeated for 3 times.
Western blot analysis
The general procedure of western blotting was described in detail in previous publications [29]. Briefly, HEI-OC1 cells were seeded in 100-150 mm dishes with a density of 8-20 × 105 per dish and were cultured for 24 hours. Cisplatin (0, 50, 100 µM) and HNK (0, 5, 10 µM) were then added (separately or in combination) and the cells cultured for another 24 hours. Cells were then collected and homogenized on ice in lysis buffer containing 50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100 and supplemented with protease inhibitor cocktails (Thermo Fisher Scientific Inc., Rockford, IL). After incubating for 30 minutes, the lysates were collected by centrifuging at 10,000 rpm, 4°C for 10 min. The amount of total protein was determined using a BCA protein assay kit (Pierce, Rockford, IL). An equal amount of total protein extracts was mixed with 5 × sample loading buffer and boiled for 5 min. Protein (30-50 mg) was resolved on SDS/PAGE (12% gels) and transferred on to PVDF membranes. Membranes were blocked in 5% (w/v) non-fat dried skimmed milk powder or 5% (v/v) horse serum in TBST (50 mM Tris/HCl, 150 mM NaCl, pH 7.4, and 0.1% Tween 20) for 30 min at room temperature. The membranes were then incubated overnight at 4°C with primary antibodies at the following dilutions: anti-cleaved poly ADP ribose polymerase (PARP), 1:1000; anti-SIRT3, 1:1000; anti-SIRT2, 1:1000; anti-actin/Tubulin, 1:8000. The membranes were then washed 4 times with TBST and incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. After washing for 40 min with TBST, the membranes were developed using an ECL substrate kit. The procedure was repeated at least 3 times for all the treatment groups. ImageJ software (http://rsbweb.nih.gov/ij/) was used to quantify the Western blot band intensity.
Animal groups and drug administration
Adult C57BL/6 mice (The Jackson Laboratory, Bar Harbor, Maine) of both sexes (80 in total) between 6-8 weeks of age were used for the in vivo studies. All procedures were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Northwestern University. Animals were randomly divided into groups with different doses of cisplatin (0, 15, 20 mg/kg) and honokiol (0, 10, 20 mg/kg), and no sexual difference except weight was observed in the study. Cisplatin was dissolved in saline and HNK (both from Sigma Aldrich, Port Washington, WI, or Tocris Bioscience, Minneapolis, MN) in corn oil, and both were administered through intraperitoneal (i.p.) injection on 2 consecutive days. HNK (or coin oil for controls) was delivered 1 hour prior to cisplatin (or saline). Animals were hydrated with Ringer’s Lactated Solution 25 µl/g once daily for 1 week. The overall wellbeing of the animals was monitored through weighing and observing their activity on daily basis throughout the study so that animals in critical condition, e.g., weight loss greater than 25%, would be removed from the study and euthanized. Data collection was partially single-blinded, meaning that the treatment was done without the awareness of the personnel who collected the ABR data. Specifically, this include 3 animals each in cisplatin 15 mg/kg, cisplatin 20 mg/kg, cisplatin 15 + HNK 20 mg/kg, and cisplatin 20 + HNK 20 mg/kg groups.
For cisplatin chemotherapy regimen, female MMTV-PyMT carrier mice (Jackson Laboratory) were used and their tumor growth was monitored every 2 days since palpable. The size of each tumor was measured with a caliper, and the volume calculated using the following formula [37]: (length × width2)/2. When the total volume of all tumors reaches 500 mm3, a chemotherapy regimen was performed consisting of 3 cycles. In each cycle, 4 doses of HNK and/or cisplatin were administered across 4 days, followed by a 10-day recovery interval (Figure 6A). The entire chemotherapy regimen lasted for 42 days [38]. A total of 13 animals were randomly assigned into 4 groups: Cis-only group (n=4) was given cisplatin at a daily dose of 4 mg/kg. HNK-only group (n=2) was given HNK at a daily dose of 10 mg/kg. Cis + HNK group (n=4) was given both cisplatin (4 mg/kg/day) and HNK (10 mg/kg/day), where HNK is delivered 1 hour prior to cisplatin. Control group (n=3) was given the same amounts of vehicles only. Cisplatin was dissolved in saline, and HNK in corn oil, both with a concentration of 2 mg/ml. They were both be given through i.p. injection. Animals were also hydrated daily with saline for the first 7 days of each cycle with a dose of 25 µl/g. Animals were weighed and their health monitored daily for stress and pain. During the experiment, animals with a weight loss over 25%, a total tumor size over 1500 mm2, a tumor size reaching the IACUC predetermined limit of 20 mm along one axis, or a tumor burden visibly affecting the host, were euthanized for humane reasons.
Figure 6.
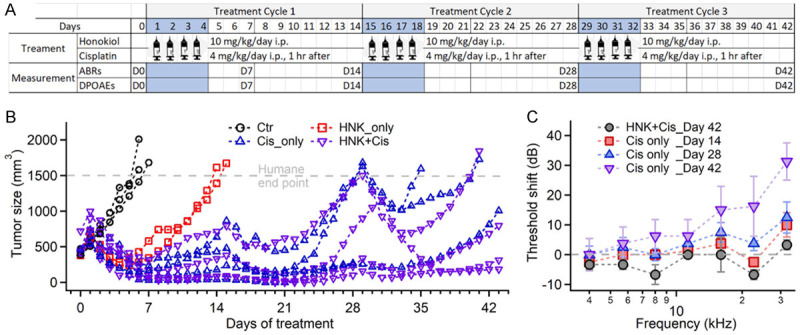
A. Chemotherapy regimen for Tumor-bearing mice. B. Tumor growth during different treatment plans. Each curve represents one animal. Once the tumor size reached 1500 mm3, the animal is euthanized (with 2 exceptions in the plot). C. ABR threshold shift of HNK + cisplatin and cisplatin-only groups, normalized by the data recorded on Day 0. Data represent mean ± SEM.
Auditory brainstem response (ABR) measurement
The cochlear function was evaluated through ABR measurements at day 0 (baseline before the treatment), days 3, 7, and 14 (after treatment), respectively. The detailed method for ABR measurements has been described in our previous publications [39]. Briefly, animals were anesthetized with an intraperitoneal (i.p.) injection of a mixture of ketamine (100 mg/kg) and xylazine (2 mg/kg), diluted 1:20 in RLS. Maintenance doses of ketamine were given when the paw withdrawal reflex was positive. The level of anesthesia was tested every 15 minutes. The body temperature of the animals was maintained at 37°C using a water-filled heating pad and monitored every 15 minutes along with other vital signs including O2 saturation, respiratory and pulse rates. Acoustic stimuli were delivered with a Beyer DT770-Pro headphone, which has been calibrated with a Brüel and Kjær 1/8-inch microphone. Acoustic clicks (50 µs) and tone pips (5 ms, including a rise/fall time of 1 ms, frequency range: 4-32 kHz, 2 steps/Octave) were delivered via the speculum of the speaker, which was placed directly in the ear canal of the animals (quasi free field). The highest sound level for all the acoustic stimuli was ≤107 dB SPL (sound pressure level re 20 µPa), which was attenuated in 5 dB steps, and each stimulus was repeated 256 times or until the signal/noise ratio ≥4. To record the ABRs to the different acoustic stimuli, three hypodermic needles were placed at the bulla (low), vertex (high), and the body (common ground). The electrodes were connected to an ISO-80 differential amplifier, set to 80 dB amplification. The high- and low-pass filter of the amplifier (slop: -12 dB/octave) was set to 300 Hz and 3 kHz, respectively. The traces were further filtered and amplified by a frequency devices filter (Hewlett Packard, corner frequencies, 0.3 and 3 kHz; slope: -48 dB/octave, gain 20 dB). ABR threshold was defined as the minimal sound level required for an ABR response with a root mean square (RMS) 1.5 times above the noise floor. The stimulation signal generation and data acquisition were controlled by custom-written software in Testpoint. The sampling rate was set at 250 kHz. During data acquisition, the stimuli and the corresponding responses were visually monitored with an oscilloscope. A confounding factor introducing noise into the measurements is the electrocardiogram (ECG), which is usually at least three times larger in amplitude (~25 µV) than the ABR. The program automatically rejected recordings that contained the ECG by rejecting traces with peak-to-peak amplitudes larger than 15 µV.
Cardiac perfusion and dissection of the cochlea
At the conclusion of the physiological study, the animals were euthanized, cardiac perfused with 4% paraformaldehyde (PFA), and the cochleae were harvested and followed by an in-cochlea PFA injection for better fixation. The left cochlea was decalcified using 10% EDTA in 1X PBS (diluted from 10X PBS, Invitrogen) for 2-3 days. Full-length cochlear surface preparation was then dissected out in 1X PBS and cut into 5 pieces at specific locations for immune-staining, following the instructions from the video published online by the Massachusetts Eye and Ear Infirmary (https://vimeo.com/144531710). The coil of the cochlea was cut into 5 segments and the tectorial membrane was removed during this process. The average length of each segment (S1-5, from apex to base) was (in mm): 1.41±0.15 (n=6), 1.49±0.16 (n=16), 1.36±0.15 (n=14), 0.86±0.21 (n=6), and 0.65±0.19 (n=6), respectively, determined later by the length of the inner hair cells (IHC) row in confocal microscopy images. The full length of the cochlea, determined by 5 full-length measurements, was 5.89±0.09 mm.
Immunofluorescence histochemistry (IFHC)
IFHC was performed afterwards following standard protocols described in previous publications. Briefly, for OHC staining, cochlear sections were put in small vials, added in 50 µl blocking solution (1% goat serum + 1% Triton X-100 in 1X PBS), and incubated for 1 hour at room temperature. Primary antibody (rabbit anti-prestin, 1:1000 dilution in blocking solution) was then added and incubated overnight at 4°C. After washing 3 times with 1X PBS, a mix of the secondary antibody (goat anti-rabbit Alexa-488, 1:500 dilution in blocking solution), phalloidin-Alexa 546 (for actin, 1:250), and Hoechst (for nuclei, 1:1000) was added and incubated away from light for 1 hour at room temperature. The sections were then washed for another 3 times with 1X PBS and mounted on slides. For synapse and SIRT3 expression, cochlear sections were first soaked in 30% sucrose on a shaker for 20 minutes, and then transferred to dry ice for 10-15 minutes or until the sucrose was completely frozen. The samples were then allowed to thaw at room temperature and washed 3 times with 1X PBS, rinsing for 20 minutes in between on a shaker. After blocking with 5% serum + 1% Triton, the primary antibody mix, including rabbit anti-Myosin VIIa (Proteus Biosciences, 1:200), mouse (IgG1) anti-CtBP2 (BD transduction Labs, 1:200), or mouse (IgG1) anti-SIRT3 (Novus Biologicals, 1:200), and mouse (IgG2a) anti-GluR2 (Millipore, 1:500), was added and incubated overnight at 37°C. After rinsing for 3 times in PBS (10 minutes each), the secondary antibody mix, including duck anti-rabbit (IgGH-L) AF647 (Jackson Immuno Research, 1:200), goat anti-mouse (IgG1) AF568 (Thermo Fisher, 1:200), goat anti-mouse (IgG2a) AF488 (Thermo Fisher, 1:200), and Hoechst (1:1000), was added and incubated away from light for 2 hours at 37°C. The sections were then washed for another 3 times with 1X PBS and mounted on slides. To make a flat surface preparation for confocal microscopy, S3 was sometimes cut in half right before mounting (Figure S3).
Confocal microscopy
Confocal images were taken in the Center for Advanced Microscopy/Nikon Imaging Center at Northwestern University, using a Nikon A1 Laser Scanning or Nikon W1 Dual CAM Spinning Disk imaging setup. Appropriate excitation and emission settings were used and fixed for all the panels in the same images presented in the paper [40]. Confocal images were processed using imageJ (NIH Image) and Imaris (Oxford Instruments, Abingdon, Oxfordshire, England) afterwards. OHC loss was counted on S2 and S3.
Data analysis
The results were uniformly formatted and processed using MatLab (The Mathworks, Inc., Natik, MA). Averages and standard errors of the mean (SEM) were calculated and presented for all the acquired data. Each replicate was derived from an individual animal. ABR thresholds for different animal groups and days post-treatment were all normalized with those acquired before the treatment (Day 0). Since animals tend to completely lose response to certain frequencies after cisplatin treatment, those with a completely lost response were assigned to be 10 dB over the maximal output of the speaker at that frequency for statistical analysis. One-way ANOVA (two tails) was performed in Matlab for all the ABR threshold data. If the ANOVA indicated differences among the means, the Tukey’s Honestly-Significant Difference post-hoc test was followed. The finalized data were plotted using IGOR Pro (Wavemetrics, Lake Oswego, OR). The tests are part of a statistical package provided by IGOR Pro. Statistical decisions were made for a probability of 0.05. During the data analysis, 5 animals from different groups were excluded because of existent hearing loss determined during the baseline hearing test on Day 0.
Results
HNK prevents cisplatin ototoxicity both in vitro and in vivo
HNK protects cultured primary cochlear cells
The protective effect of HNK was first tested on a primary cochlear cell line, the House Ear Institute-Organ of Corti 1 (HEI-OC1) cells. They are derived from a long-term culture of immortomouse cochleae [36]. Although undifferentiated, HEI-OC1 cells express both hair cell and supporting cell markers [41] and therefore are widely used for ototoxic and oto-protective drug screening [36,41]. In our study, HEI-OC1 cells were treated with cisplatin and HNK. As shown in Figure 1A, HNK treatment did not affect cell survival rates as no significant difference was observed between the treated and non-treated cells for up to 48 hours (one-way ANOVA, two tails, P=0.09). In contrast, cisplatin treatment caused a dose-dependent cell loss (Figure 1B) as cell viability decreased to 56% (50 µM) or 32% (100 µM) 24 hours after treatment (P<0.01 for both doses compared to the untreated cells, one-way ANOVA followed by Tukey’s Honestly-Significant Difference post-hoc test, same in the following). With a cisplatin dose of 50 µM, HNK application (5-25 µM) improved cell survival rate from 56% to over 80% at 24 hours, and even reached 100% (increased by 78%) at 10 µM (Figure 1C, the black curve, P<0.01 for all the doses). At 48 hours, the cell survival rate also improved from 36% to 55% (increased by 53%) with 5-10 µM of HNK (the red curve, P<0.01 for all the doses). Please note here that all the data of 24 hours are normalized to the untreated cells at 24 hours, and all the data of 48 hours are normalized to the untreated cells at 48 hours. The number of cells at 48 hours increased by about 60% compared to that of 24 hours, estimated from 2 trials of studies.
Figure 1.
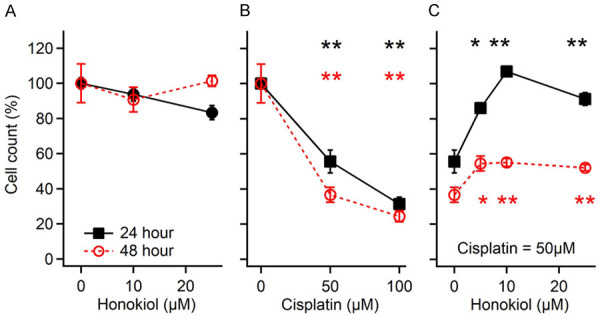
Survival of cultured House Ear Institute-Organ of Corti 1 (HEI-OC1) cells after honokiol (A), cisplatin (B), and their co-treatment (C). *: P<0.05; **: P<0.01, compared to the first dot of each plot. Data represent mean ± SEM.
HNK prevents cisplatin-induced hearing threshold elevation
C57BL/6J mice were used in the in vivo study to evaluate the ototoxicity of cisplatin and the otoprotective effect of HNK. Animals were treated with 2 doses of cisplatin (with a total amount of 15 or 20 mg/kg) and/or HNK (10 or 20 mg/kg) on 2 consecutive days. Auditory Brainstem Response (ABR) thresholds started to elevate at high frequencies (32 kHz, 15 dB) on Day 3 after cisplatin treatment (20 mg/kg) (Figure 2A). A severe threshold elevation developed in most animals by Day 7 at frequencies lower than 8 kHz and higher than 22 kHz (14-38 dB). The difference was statistically significant as determined by one-way ANOVA (two tails, F(1,256)=4.21, P=0.04) followed by Tukey’s Honestly-Significant Difference post-hoc test (same in the following). Most animals were removed from the study after Day 8 because of deteriorated health and were euthanized for humane reasons. In fact, data on Day 14 are missing in Figure 2A because only 1 out of 19 animals remained in the study, with a 25-dB threshold elevation at 32 kHz. However, when the animals were pre-treated with HNK one hour prior to cisplatin, the ABR threshold elevation was significantly reduced (Figure 2B). A 15 dB threshold elevation was observed at 32 kHz but not at any other frequency on Day 14 (P<0.01 for 32 kHz, P>0.35 for all other frequencies). Furthermore, 8 out of 12 animals remained in the study. A lower dose of cisplatin (15 mg/kg) was also tested as shown in Figure S1A and S1B. A 21 dB threshold elevation was observed on Day 14 at 32 kHz but not at any other frequencies (Figure S1A, P<0.01 for 32 kHz). This was also prevented by HNK 20 mg/kg pretreatment (Figure S1B). The dose-dependent threshold shift is plotted in Figure 2C and 2D, normalized by the data from Day 0. Significant changes between the cisplatin 20 mg/kg group and the HNK 20 + cisplatin 20 mg/kg group were observed at all frequencies (P<0.01). The baseline hearing of all the animal groups and the changes at the end of the study are plotted in Figure S1C and S1D. No significant difference in the hearing baseline was observed among the different groups (Figure S1C, P=0.08). The averaged ABR threshold shifts of the different groups at the end of the study are also plotted together for direct comparison (Figure S1D).
Figure 2.
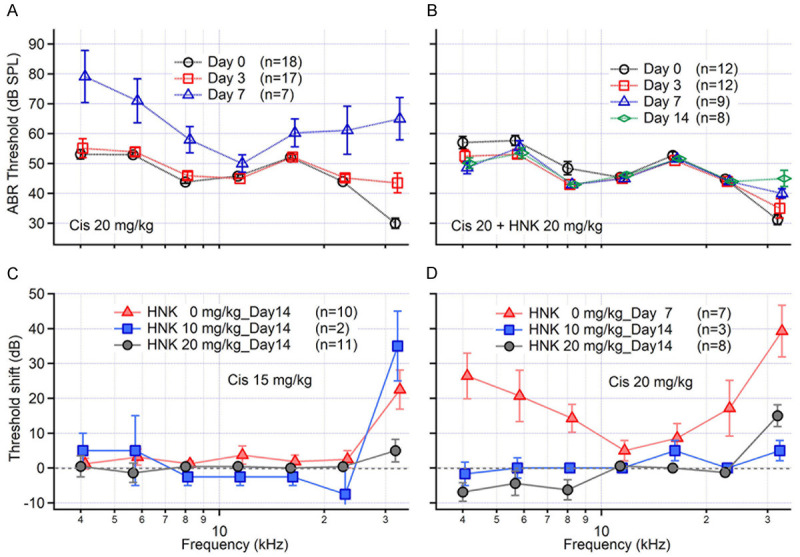
Threshold changes of the auditory brainstem response (ABR) after HNK and/or cisplatin treatment. A. Cisplatin (Cis, 20 mg/kg) treatment induced a severe ABR threshold elevation at most frequencies on day 7. Day 14 data is missing because most (18 out of 19) animals were moved out of the study. B. Pre-treatment with honokiol (HNK, 20 mg/kg) significantly reduced cisplatin-induced ABR threshold elevation. C and D. Dose-response curves of Cis 15 and 20 mg/kg with different HNK doses. Cisplatin 15 mg/kg induced an ABR threshold elevation only at 32 kHz, which was also prevented by HNK pretreatment. Data represent mean ± SEM.
HNK prevents the decrease of otoacustic emissions and OHC loss
Cisplatin-induced hearing loss is marked by OHC loss. Distortion product otoacustic emission (DPOAE) provides a measurement of cochlear nonlinearity, which is associated with OHC function. In a separate small group of animals, DPOAEs were measured before and after cisplatin and HNK treatment (Figure S2) as an estimation of OHC function. Cisplatin (15 mg/kg) reduced the DPOAE magnitude (2f1-f2) on Day 3 (an example is shown in Figure S2A). The DPOAE magnitude decrease was mostly at frequencies above 16 kHz (30-50 dB, Figure S2B), although the ABR threshold only showed an approximately 5 dB elevation on average in this frequency range (Figure S1A). This DPOAE magnitude decrease is prevented by HNK (10 and 20 mg/kg) pretreatment in a dose-dependent manner (Figure S2C-E). With the pretreatment of HNK 10 mg/kg, the DPOAE amplitude decrease was 5-12 dB at frequencies over 27 kHz (Figure 2D). Complete protection was achieved with the pretreatment of 20 mg/kg HNK (Figure S2E).
The protection of OHC by HNK against cisplatin ototoxicity was also verified with immunostaining performed on the harvested cochleae after completion of the physiological studies. Immunostaining of the full length of the cochlea showed OHC loss from the apex to the base when treated with cisplatin. These changes were reduced by the application of HNK (Figure S3). Artifacts caused by fixation, dissection, and immunostaining were more severe at the base and apex. Therefore, we counted the OHC loss at the regions involving segments 2 and 3 (S2 and S3) using 6 animals each, as shown in Figure 3. Corresponding frequency ranges of S2 and S3 are 9.5-19.1 kHz and 19.1-36.5 kHz, respectively. They were determined using the published frequency map based on the measurements from CBA-J mice [42]. Examples of S3 from differently treated animals are shown in Figure 3. Only a few OHCs were missing in both the control (Figure 3A) and HNK treated (Figure 3B) samples as indicated by the arrows. On the other hand, samples from animals treated with cisplatin only (15 mg/kg) revealed numerous OHC loss scattered along the entire sample (Figure 3C). Samples from animals treated with both cisplatin (15 mg/kg) and HNK (20 mg/kg) lost fewer OHCs (Figure 3D). The average OHC loss of S2 and S3 in cisplatin 15 mg/kg group was 14.5±4.9 and 22.5±6.4, respectively (Figure 3E). In cisplatin 15 mg/kg + HNK 20 mg/kg group, it was 7.7±2.1 and 9.7±4.0, respectively (Figure 3F). Statistical analysis shows that the difference is significant for both segments (P=0.01 for both S2 and S3, student t-test, two tails). Corresponding immunostaining and confocal imaging for samples from the cisplatin 20 mg/kg group is not available, since most of the animals were removed from the study for humane reasons on Day 8.
Figure 3.
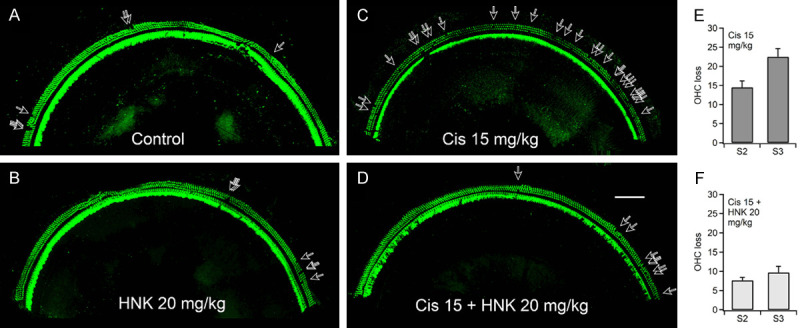
(A-D) Confocal images of the middle turns (Segment 3, frequency range: 19.1-36.5 kHz) immuno-stained with Myosin 7A, showing hair cell loss after HNK and/or cisplatin treatment. Severe OHC was induced by treatment of cisplatin 15 mg/kg (C), which is largely reduced by pre-treatment of HNK 20 mg/kg (D). Scale bar: 100 µm. (E and F) Histograms showing the average OHC loss in the two cochlear segments, S2 (9.5-19.1 kHz) and S3, of cisplatin 15 mg/kg and cisplatin 15 + HNK 20 mg/kg groups. Data represent mean ± SEM.
HNK protects the overall health of the animals
As described above, cisplatin treatment at 20 mg/kg correlated with weight loss greater than 25% and required the animals to be removed from the study and euthanized for humane reasons. As shown in Figure 4A, animals started to be removed on Day 3 (the red circles with solid line), and only one animal (5%) reached Day 14. When pretreated with HNK 20 mg/kg, however, the survival of the animals was improved dramatically. On Day 6, one animal was removed and 67% of the animals survived until the end of the study (the red dots with dashed line). A lower cisplatin dose of 15 mg/kg also resulted in 25% of animals being lost on Day 5, while the other 75% survived until the end (the blue hollow square with solid line). With HNK treatment, the animal survival rate increased to 92% (the blue solid square with dashed line). The health-protective effect of HNK against cisplatin treatment also showed in the weight change of the animals. Weight loss occurred immediately after cisplatin application (Figure 4B). With a dose of 20 mg/kg, the weight loss increased until reaching a maximum on Day 7 (the red dots with dashed line). Animals treated with HNK regained weight up to 90% of the baseline at the end of the study (the red dots with dashed line). The weight loss of HNK treated groups was slightly smaller than that for the cisplatin-only groups (-2.3±1.0% on average for the first 7 days), while statistical analysis still showed a significance (Figure 4B, P=0.04, paired t-test, two tails).
Figure 4.
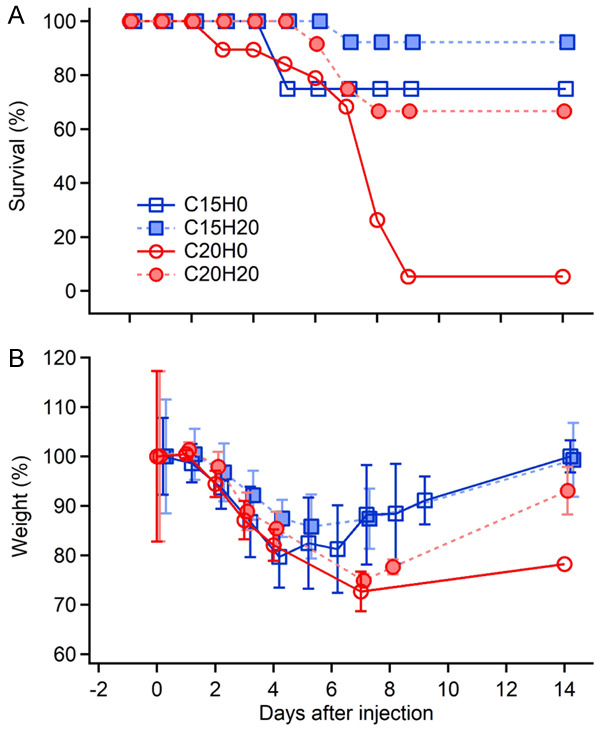
Animal survival (A) and weight loss (B) after the treatment with cisplatin alone and cisplatin + HNK. Cisplatin 20 mg/kg alone (C20H0, the red circle with solid lines) induced a severe animal loss (only 5% survived after day 8) and weight loss. Animal survival rate is largely improved by pretreatment of 20 mg/kg HNK (C20H20, the red dots with dashed lines, 66.7% survived till the end of the study). Cisplatin 15 mg/kg also causes a 25% animal loss (the blue hollow squares with solid lines), which decreased to 5% with 20 mg/kg HNK pretreatment (the blue squares with dashed lines). Data represent mean ± SEM.
HNK does NOT protect tumor cells against cisplatin toxicity
Effects on tumor cell growth in cell cultures
To test whether HNK protects tumor cells during cisplatin treatment, three tumor cell lines are selected, including C2-4B (prostate cancer), HCT116 (colon cancer), and HeLa (cervical cancer) cells. The results are shown in Figure 5. The three tumor cell lines responded differently to cisplatin and/or HNK treatment. Prostate cancer cells (green triangles) were the most sensitive to cisplatin among the three, which were almost completely killed by 50 µM cisplatin treatment (Figure 5A, survival rate 2.2±1.7%, P<0.01, one-way ANOVA followed by paired t-test, two-tails, same in the following). Interestingly, they were not sensitive to HNK, for none of the HNK doses induced a significant cell loss (Figure 5B). When co-treated with cisplatin (50 mg/kg) and HNK, no protection from HNK was observed. Cell survival at HNK 5, 10, and 25 µM were 2.6±1.8%, 2.6±1.6%, and 1.6±1.4%, respectively. Cervical cancer (blue squares) and colon cancer (red dots) cell survival rates decreased to ~80% at 50 µM cisplatin, though not significantly, and dropped down to 0.8±0.2% and 3.2±2.1% at 100 µM cisplatin, respectively (Figure 5A). When treated with HNK, cervical cancer cells were only sensitive at a high dose (25 µM), while colon cancer cells showed significant changes at both doses (10 and 25 µM) (Figure 5B). When co-treated with cisplatin 50 µM and HNK, further decrease of cell survival was observed on both cell lines (Figure 5C), which reached 3.3±3.8% and 0.8±1.3% at 25 µM HNK, respectively. The results indicated that HNK had no protective effects on these tumor cell lines. Instead, a synergistic effect with cisplatin was observed on cervical cancer and colon cancer cell lines.
Figure 5.
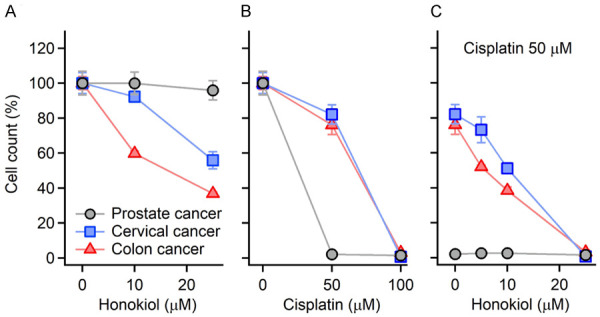
The sensitivity of three tumor cell lines to cisplatin and/or HNK. The three tumor cell lines are C2-4B (prostate cancer, black circles), HeLa (cervical cancer, blue squares), and HCT116 (colon cancer, red triangles) cells. Different sensitivities to cisplatin (A), HNK (B), and their combinations (C) are observed. Note that a synergistic effect of cisplatin 50 µM and HNK 25 µM is observed. Data represent mean ± SEM.
Effects on tumor growth in tumor-bearing mice during chemotherapy
A transgenic mouse model expressing mouse mammary tumor virus (MMTV) polyomavirus middle T agent (PyMT) oncogene was used for testing the effects of HNK and cisplatin treatment on tumor growth in vivo [43]. As stated by Jackson Laboratories, female MMTV-PyMT transgenic mice develop palpable mammary tumors with a mean latency of 53 days. In our study, the female carriers developed mammary tumors to the criteria of intervention (total size of ≥500 mm3) at the age of 62.0±10.8 days (N=13). The animals were then treated with 3 cycles of cisplatin and HNK [17,38]. In each cycle, 4 doses of HNK (10 mg/kg/day) and/or cisplatin (4 mg/kg/day) were administered over 4 days, followed by a 10-day recovery interval (Figure 6A). The tumor growth of all the animals is plotted in Figure 6B. In the control group (N=3), tumors grew fast and reached a total size of ≥1500 mm3 in 6 days. The animals were then removed from the study for humane reasons (the black curves). When normalized with the tumor size on Day 1, before the treatment, the tumor size increased 3.1-fold (from 578±27 mm3 to 1795±302 mm3) on Day 6. In HNK-only group (N=2), the tumor size decreased 2.5-fold (from 650±70 mm3 to 264±26 mm3) after 4 doses of HNK treatment. The tumor growth restarted quickly and reached the size limit on Day 14.5 (red traces with square markers). In cisplatin-only group (N=4), the tumor size decreased 4.5-fold (from 640±97 mm3 to 111±110 mm3) on Day 7, and increased afterwards, to 382±311 mm3 on Day 15, before the start of the next cycle (blue traces with triangle-up markers). The same trend of tumor growth was found in the 2nd and 3rd cycles, although the tumor-suppressive effect became less efficient. By the end of the 2nd cycle (Day 29, right before the treatment of the 3rd cycle), the tumor size was 1188±806 mm3. Two out of three animals reached the size limit before the end of the 3rd cycle. In the HNK + cisplatin group (N=4), the changes in tumor growth were similar to the cisplatin-only group. The size of the tumors decreased 4.8-fold (from 789±204 mm3 to 162±110 mm3) on Day 7 and gradually increased afterwards to 335±334 mm3 on Day 15 (purple traces with triangle-down markers). Two out of four animals reached the humane end point before the end of the 3rd cycle. The difference between the cisplatin-only and the HNK + cisplatin groups was significant (paired t-test, P<0.01, t-value =2.01, df =43). More detailed analysis shows that the difference was mainly on the 2nd and 3rd cycle, where the tumor size of the HNK + cisplatin group was apparently smaller than that in the cisplatin-only group (P=0.09 for the 1st cycle and P<0.01 for the 2nd and 3rd cycle). The results clearly demonstrated that both cisplatin and HNK can inhibit mammary tumor growth with different efficiencies. More importantly, the tumor-suppressive effect of cisplatin was not affected in the 1st cycle and improved in the 2nd and 3rd cycles with HNK co-treatment. Examples of tumor size measurements are shown in Figure S4.
Effects on ABR threshold in tumor-bearing mice during chemotherapy
Hearing impairment was also induced in these tumor-bearing mice during cisplatin chemotherapy. As shown in Figure 6C, cisplatin treatment induced a progressive hearing loss over time (Days 14, 28, and 42, the colored curves) at frequencies 22.6 and 32 kHz. Threshold shifts in the cisplatin-only group at these two frequencies on Day 42 were 16.3±20.2 and 31.3±12.5, respectively. The difference are statistically significant as determined by one-way ANOVA followed by Tukey’s Honestly-Significant Difference post-hoc test (P=0.05, F(1,47)=4.07 for 22 kHz, and P<0.01, F(1,47)=10.52 for 32 kHz, respectively). Elevated thresholds were also observed at almost all other frequencies, although the differences were not significant. On the other hand, in the HNK + cisplatin group, this threshold shift was largely suppressed. No significant threshold shift was detected at these two frequencies (P=0.22 and 0.06, respectively) or any other frequencies (Figure S5 and the black curve in Figure 6C). Note that the data on Day 42 include ABR recordings taken on the last day, which is before day 42, when the animals were removed from the study.
SIRT3 expression increases in OHCs and HEI-OC1 cells treated with HNK and cisplatin
SIRT3 is expressed in the cochlea and activated by HNK
To verify the expression of SIRT3, C57BL/6 mice were given 2 doses of HNK (20 mg/kg) on 2 consecutive days and were euthanized 1 hour after the 2nd injection. The cochleae were harvested and immunostaining was performed with primary antibodies for SIRT3 and Myosin VIIA. As shown in Figure 7, animals in the control group (WT_Ctr, the 1st row in Figure 7) showed a baseline SIRT3 signal, which was higher in OHCs than in IHCs, supporting cells, and pillar cells, as indicated in the panel showing SIRT3 staining. After treatment with HNK, the expression level of SIRT3 increased as shown in the 2nd row of Figure 7 (WT_HNK). Most of the OHCs showed bright SIRT3 staining, as did some IHCs. Colocalization with Myosin VIIa staining in the merged view indicated that the increase of SIRT3 expression was mainly shown in OHCs and IHCs. To verify the results, cochlear samples from SIRT3 knock-out mice (SIRT3-/-) with C57BL/6 background were used as a negative control. A weak background was also observed in cochlear cells (the 3rd row, S3KO_Ctr), which was not increased by HNK application (the 4th row, S3KO_HNK).
Figure 7.
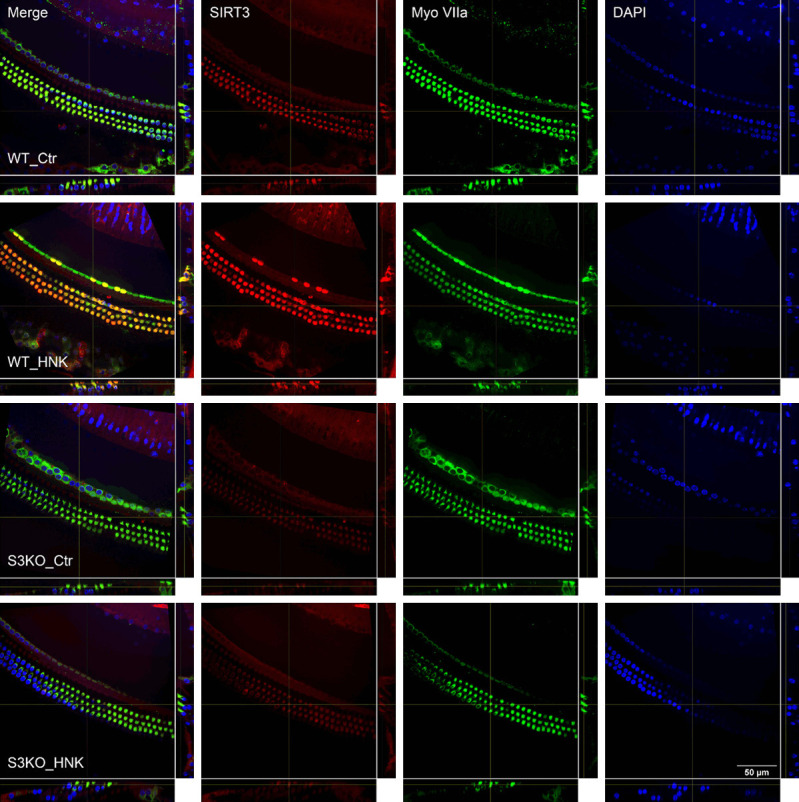
Confocal imaging showing SIRT3 expression in the cochlear segment 2 (frequency range 9.5-19.1 kHz) 2 days after different treatments. WT_Ctr: wild type mice without treatment; WT_HNK: wild type mice treated with 20 mg/kg HNK; S3KO_Ctr: SIRT3 knockout mice without treatment; S3KO_HNK: SIRT3 knockout mice treated with 20 mg/kg HNK. Scale bar: 50 µm.
SIRT3 is activated by HNK and cisplatin in HEI-OC1 cells
To gain a relative quantification of SIRT3 activation by HNK, western blot was performed on the cultured HEI-OC1 cells treated with cisplatin and HNK, as shown in Figure 8. SIRT3 expression levels were relatively low in non-treated HEI-OC1 cells (C0H0, which means cisplatin 0 and HNK 0 µM, and so on), and increased 1.6±0.3 fold with HNK treatment (C0H10). Interestingly, SIRT3 expression also increased during cisplatin treatment (2.8±1.1 and 2.7±1.1 fold for C50H0 and C100H0, respectively). No further increase was observed in the co-application of HNK and cisplatin (2.6±1.1 for C50H10). As a control, the expression level of SIRT2, another SIRT family member expressed in the nucleus and cytoplasm, increased slightly with HNK treatment (1.3±0.4 for C0H10), but not during cisplatin or cisplatin + HNK treatment. It is known that cisplatin treatment leads to apoptosis because of DNA damage and growth arrest due to its influence on cell cycle check points [44]. To assess the involvement of the apoptotic pathway in cisplatin treatment, cleaved Poly (ADP-ribose) polymerase (PARP, a marker for late apoptosis and DNA damage) expression level was also measured and was shown to have significantly increased during cisplatin treatment (3.3±2.6 for C50H0), and the co-treatment with HNK (2.2±2.1 for C50H10).
Figure 8.
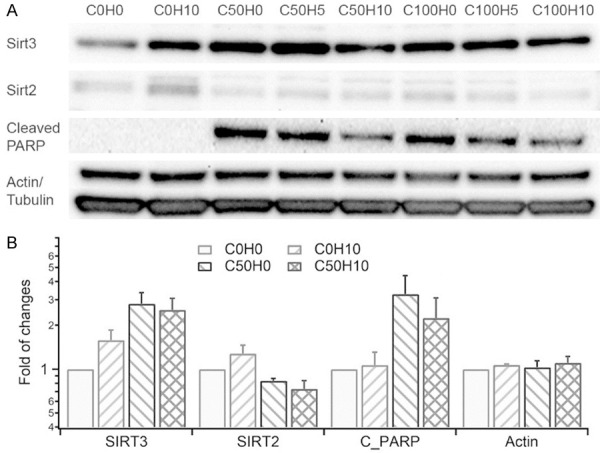
(A) Western blot and (B) Histograms showing expression levels of SIRT3, SIRT2, and cleaved-PARP 24 hours after cisplatin and HNK treatments in HEI-OC1 cells. The fold change was normalized to the expression level of each protein in the non-treated cells (C0H0). Data represent mean ± SEM.
SIRT3-/- with C57BL/6 background show severe early-onset hearing loss and abnormal morphology of synapses
To examine whether SIRT3 is involved in the mechanism of HNK hearing protection, we performed the studies on SIRT3-/- mice. Unfortunately, SIRT3-/- mice suffer from a severe early-onset hearing loss at most frequencies, as shown in Figure 9A. At the age of 8 weeks (the black dots with the solid line), all the animals lost response to the highest (32 kHz) and the lowest (4 kHz) frequencies measured. Significant changes between the wild type and SIRT3-/- mice were found at all frequencies measured except 16 kHz (P=0.09 for 16 kHz, and P<0.001 for all other frequencies, one-way ANOVA followed by Tukey’s honestly significant difference post hoc test). The average ABR threshold shift across all frequencies measured was 21.3±17.6 dB. To determine when this hearing loss developed and how it progressed, the hearing of these mice at the ages of 6 and 12 weeks were also measured. Hearing loss has already developed at 6 weeks (the red triangles, average ABR threshold shift: 17.7±15.2 dB) and became even more apparent at 12 weeks (the blue squares, average ABR threshold shift: 29.3±12.1 dB). The cochleae were harvested for morphological examinations after the physiological studies. In Figure 9, IFHC with Myo7A shows no significant hair cell loss in SIRT3-/- animals compared to the wild type mice (Figure 9B and 9C). IFHC with the postsynaptic marker GluR2 shows ribbon synapses of the wild type mice evenly distributed at the basal part of the IHCs (Figure 9B), while the synapses of the SIRT3-/- mice are more clustered at the bottom of the IHCs (Figure 9C).
Figure 9.
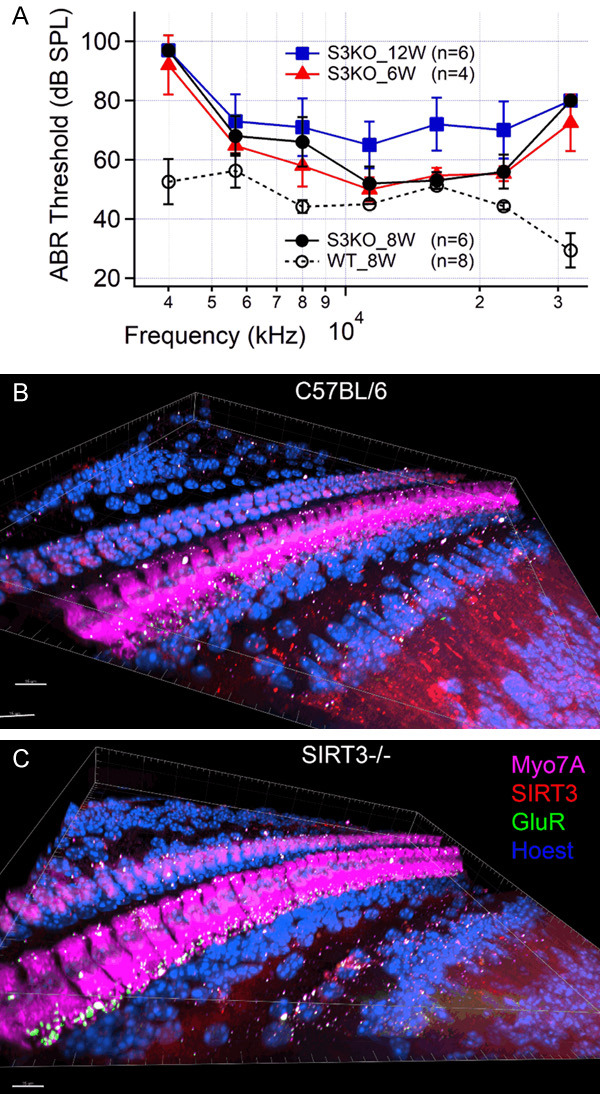
A. Changes of ABR threshold of SIRT3 knockout mice (S3KO) over time (from 6 to 12 weeks) and the comparison with that of the wild type (WT) animals (8 weeks). Note that an early-onset hearing loss is observed in S3KO mice as early as 6 weeks. B and C. Confocal images showing the hair cells, SIRT3 expression, and synapses of the wild type and S3KO mice at ~32 kHz region. Note that SIRT3 knockout mice have no significant hair cell loss. Instead, abnormal aggregation of the synapses of S3KO mice is observed, indicating possible synaptopathy in these mice. Scale bar: 15 µm. Data represent mean ± SEM.
Discussion
Our experiments have shown for the first time that HNK, a multi-functional polyphenol, prevents cisplatin-induced hearing loss and synergizes with cisplatin in tumor suppression. Animals treated with HNK are in better overall conditions as judged by the survival rate of the animals during cisplatin treatment. We have also shown that the function of HNK is associated with the activation of SIRT3 in the cochlea. Together, the results suggest that HNK is a promising agent for hearing protection in clinical cisplatin chemotherapy.
Benefit of HNK as a candidate for hearing protection in cisplatin chemotherapy
As a widely used drug in solid tumor chemotherapy, cisplatin is a potent and first-line medicine [1,2]. However, cisplatin exposure can result in various adverse effects including nausea, vomiting, kidney failure, and hearing and balance-related issues such as significant hearing loss, tinnitus, and vertigo [3-5]. The incidence of hearing loss in chemotherapy involving cisplatin can be as high as 60-80%, or even higher for children [4,6]. A recent study shows that cisplatin accumulates and resides in the cochlea indefinitely following chemotherapy, suggesting that cisplatin may be a continuous long-period risk factor for cochlear function [45]. With the improvement of prognosis after cancer treatment, the demand for hearing protection in chemotherapy becomes more prominent and urgent. According to a recent review in clinical practice and research, there are 3 prerequisites for any treatment for hearing protection in cisplatin chemotherapy [3,46]. First, it should not interfere with the antitumor effect of cisplatin. Second, it should not be toxic to the tissue or have other adverse effects. Third, it should cross the blood-cochlea barrier and be accessible inside the cochlea. Until now, none of the suggested candidates for hearing protection has satisfied all three criteria or has been approved for clinical application by the FDA.
Cisplatin exerts its cytotoxic effects mainly in two ways. It causes DNA inter-strand and intra-strand crosslinking. Platinum-DNA adduct formation damages the DNA, blocking replication, and transcription and accounting for 5-10% of cisplatin’s cytotoxic effects [47,48]. Cisplatin is especially toxic to fast proliferating cells such as tumor cells. Toxic effects are also produced as cisplatin binds to other proteins, especially thiol group-containing bio-active molecules such as tripeptide glutathione (GSH). About 75-85% of cisplatin is found in this form, which inhibits ROS detoxification, eventually leading to apoptosis, even in normal cells [44,47,48]. ROS is normally generated in the mitochondria at physiological levels as a signaling molecule [49], while excessive ROS formation under oxidative stress is cytotoxic and a universal mechanism leading to apoptosis [50,51]. It is also a causative factor for hair cell loss in a variety of hearing impairments (e.g., noise-induced (NIHL) [52], drug-induced (DIHL) [4,7], and age-related hearing loss (ARHL) [53,54]; for reviews, see [50,55]). The majority of proposed otoprotective agents have been exogenous antioxidants that work as free radical scavengers to detoxify ROS [8-10]. Clinical trials have been conducted for statins, sodium thiosulfate, amifostine [13,14], etc. However, none of these candidates fulfills the three prerequisites. The major concern is the interference with cisplatin’s antitumor action, either by deactivating cisplatin or protecting the tumor cells [8,10]. Other problems include toxicity [56] and the blood-cochlear barrier [18]. These deficiencies essentially hindered the possibility of systemic application of these candidates. Nevertheless, local delivery through trans-tympanic injection has been tested for some candidates [18-20]. Promising results were observed in some animal studies or clinical trials (e.g., sodium thiosulfate [18]), but not in others (e.g., dexamethasone [19,20]). In addition, other adverse effects of cisplatin, such as neurotoxicity and nephrotoxicity, still persist.
The antitumor effect of HNK and its synergism with cisplatin has been intensively studied [21,23,24]. It has also been demonstrated in vivo that HNK protects the brain [25-27], heart [28-30], kidney [31], and liver [32] against oxidative stress. In our study, we showed for the first time that systemic administration of HNK also protects against cisplatin-induced hearing loss in both wild type and tumor-bearing mice. Furthermore, we also observed synergistic effects between HNK and cisplatin in tumor suppression as previously reported. Therefore, interference with the therapeutic effects of cisplatin is not likely a concern during chemotherapy for mammary tumors. According to the literature, HNK also has high systemic bioavailability with no known cytotoxicity [57]. It has been shown that HNK is permeable to the blood-brain-barrier and blood-cerebrospinal fluid barrier [58,59], a strong indication of its permeability to the blood-cochlear barrier.
HNK is approved by the European Food Safety Authority (EFSA) as a food supplement. In the USA it is exempted from an Investigational New Drug (IND) application to the Food and Drug Administration (FDA) for a clinical test. In fact, clinical studies on its effects on asthma and anti-angiogenesis have been performed in Japan [21], and a few other clinical tests of honokiol (and/or magnolia bark extraction) are also ongoing in the US. Test on other animal tumor models and application for an Internal Review Board (IRB) approval is ongoing at the Institute of the current study.
In our view, hearing protection is the most valuable effect of HNK, preventing OHC loss. Considering ROS accumulation and mitochondrial dysfunction both contribute to OHC death in various types of hearing impairments (e.g., DIHL [60], NIHL [52,61], and ARHL [53,62]), studies on the otoprotective effect of HNK can also provide insights into hearing protection in general.
Role of SIRT3 in cancer treatment and hearing protection
It has been shown that HNK activates SIRT3 [29], the primary NAD+-dependent deacetylase in mitochondria [33], which is involved in multiple intracellular metabolic processes [34,63]. SIRT3-mediated protein deacetylation via the activation of Manganese Superoxide Dismutase (MnSOD) [35,63] and isocitrate dehydrogenase 2 (IDH2) [33] are essential for ROS reduction and detoxification, as well as for improving mitochondrial function. In normal tissue, this is the underlying mechanism protecting cells from oxidative damage [64]. In tumor cells, on the other hand, SIRT3 expression is usually low and the SIRT3-MnSOD-ROS axis is dysregulated in tumor cells, which promotes metabolic reprogramming and malignancy [65,66]. SIRT3 activation can reverse these processes, thereby inhibiting tumor cell proliferation [66]. One possible mechanism of the role of SIRT3 in tumor suppression is through the inhibition of the nuclear hypoxia-inducible factor 1α (HIF-1α) [67]. HIF-1α is a marker gene for tumor cells which is usually overexpressed [67]. It is critical for ROS detoxification, metabolism, survival, and proliferation. It also activates genes for glycolysis and angiogenesis [66,67]. In other words, SIRT3 plays an important role in both normal tissue protection and tumor suppression and is therefore a promising target in cancer research.
Several studies have suggested the involvement of SIRT3 in hearing protection, including NIHL [68], DIHL [69], and ARHL [53,54]. Evidence includes increased IDH2 expression level [70], decreased ROS level [54], and increased SIRT3 expression in cultured cells [69], which are mostly indirect. Drugs and treatments used in these studies for SIRT3 activation and hearing protection might have limitations. Specifically, the dose of nicotinamide riboside required for hearing protection in noise is 2000 mg/kg/day [68], which is too high for clinical application. Adjudin, which activates SIRT3 and protects against gentamycin ototoxicity [69], is a male contraceptive. Caloric restriction to activate SIRT3 for hearing protection against noise [53,54] is apparently not suitable for cisplatin treatment. Our study provides direct evidence of the expression of SIRT3 in the cochlea and its activation by HNK (Figure 7). It is also effective (10 mg/kg/day) in activating SIRT3 and suitable for systemic application for hearing protection.
SIRT3 can be an intrinsic protective mechanism against cisplatin cytotoxicity since it is also activated by cisplatin in HEI-OC1 cell culture study (Figure 8). However, HNK application did not induce further increase in SIRT3 activation in these cells, which is worth further investigation as it might disfavor the dominant role of SIRT3 activation in the hearing protective effect of HNK. Unfortunately, we do not have strong evidence on this argument yet. What we would like to point out is that, cisplatin was in a much higher dose than honokiol in HEI-OC1 cell culture, which might contribute to the higher activation of SIRT3 and account for failing to show further increase with HNK co-treatment. Furthermore, in the in vivo studies, HNK was given 1 hour before the application of cisplatin for pre-activation of SIRT3, which might be critical for HNK to exert its hearing protective function. In addition, SIRT3 activation was shown in other tissues to be the mechanism underlying the protective effect of HNK against cisplatin cytotoxicity [29], while lack of SIRT3 aggravates cisplatin nephrotoxicity [71]. Therefore, it is reasonable to assume the important role of SIRT3 activation in the hearing protective effects as shown in our studies. Nevertheless, further studies correlate the dose of cisplatin, SIRT3 expression level, and severity of tissue damage are ongoing and will help to clarify this open question.
We also noticed that discrepancies existed in different studies regarding the hearing of the SIRT3-/- mice which may affect our understanding on the role of SIRT3 in hearing protection. In one study, knock out of SIRT3 on a WldS mouse model overexpressing NAD+ biosynthetic enzyme abolished its resistance to the noise trauma induced by 2 hours one-octave band noise exposure at 90 dB SPL. These mice are with a C57BL/6 background. The authors concluded that SIRT3 mediated the hearing protection in NIHL [68]. In a different study, SIRT3-/- mice with an FVB background and their wild type littermates showed a similar elevation in threshold and subsequent recovery after a 30-minutes one-octave band (8-16 kHz) noise exposure at 105 dB SPL. These results indicate that endogenous SIRT3 had an insignificant role in hearing recovery after mild noise trauma [40]. Our results, however, show that SIRT3-/- mice with a C57BL/6 background developed a severe hearing loss as early as 6 weeks (Figure 9A). Personal communications with the authors of the other two studies reasoned the difference to be potentially the difference in the gene background.
Other potential mechanisms involve in HNK hearing protection
As discussed above, the results from our experiments support the view that elevated SIRT3 levels are necessary to prevent cisplatin-induced hearing loss. However, some of our findings and reports in the literature are equivocal and suggest that HNK has additional effects beyond the activation of SIRT3. HNK can suppress the expression of caspase-3 and caspase-9 and upregulate phosphorylated-Akt and -Erk 1/2 [72]. HNK may even theoretically work as a ROS scavenger directly by attacking peroxide with its phenolic hydroxyl group [21]. More studies are needed to clarify the potential pathways activated by HNK. SIRT3-/- mice have early onset of hearing loss but do not show corresponding hair cell loss, which would explain the threshold elevations in young mice. A possible explanation is the apparent synaptopathy in SIRT3-/- mice (Figure 8). Improved general health in HNK and cisplatin treatment groups also suggests that HNK also protects other vital organs, such as the liver [32] and kidney [31]. Whether and how much the improved physical status of the animals contributes to preserving hearing function should also be investigated in future studies. Disturbance on the spiral ganglion neurons and the potential protection of HNK to the auditory neurons is also worth further investigation.
Acknowledgements
Imaging work was performed at the Northwestern University Center for Advanced Microscopy generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. SIRT3-/- mice were provided by Dr. Gius’ lab. We thank Dr. Mary Ann Cheatham for the permission and instructions on DPOAE measurements. We also thank Dr. Lyubov Czech, Dr. Frederic Depreux from Dr. Donna Whitlon’s lab, and Dr. Satoe Homma from Dr. Kazuaki Homma’s lab for the assistance and suggestions in IFHC and other aspects of the study. This work is supported by the Hearing Health Foundation through a 2017 Emerging Research Grants Award to XT. DG is supported by 2R01CA152601-A1, 1R01CA152799-01A1, 1R01CA168292-01A1, 1R01CA214025-01, the Avon Breast Cancer Foundation, the Lynn Sage Cancer Research Foundation, the Zell Family Foundation, and the Chicago Biomedical Consortium, as well the Searle Funds at The Chicago Community Trust.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Galanski M, Jakupec MA, Keppler BK. Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr Med Chem. 2005;12:2075–2094. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- 2.Johnstone TC, Park GY, Lippard SJ. Understanding and improving platinum anticancer drugs--phenanthriplatin. Anticancer Res. 2014;34:471–476. [PMC free article] [PubMed] [Google Scholar]
- 3.Rybak LP, Mukherjea D, Jajoo S, Ramkumar V. Cisplatin ototoxicity and protection: clinical and experimental studies. Tohoku J Exp Med. 2009;219:177–186. doi: 10.1620/tjem.219.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schacht J, Talaska AE, Rybak LP. Cisplatin and aminoglycoside antibiotics: hearing loss and its prevention. Anat Rec (Hoboken) 2012;295:1837–1850. doi: 10.1002/ar.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rybak LP. Mechanisms of cisplatin ototoxicity and progress in otoprotection. Curr Opin Otolaryngol Head Neck Surg. 2007;15:364–369. doi: 10.1097/MOO.0b013e3282eee452. [DOI] [PubMed] [Google Scholar]
- 6.Benedetti Panici P, Greggi S, Scambia G, Baiocchi G, Lomonaco M, Conti G, Mancuso S. Efficacy and toxicity of very high-dose cisplatin in advanced ovarian carcinoma: 4-year survival analysis and neurological follow-up. Int J Gynecol Cancer. 1993;3:44–53. doi: 10.1046/j.1525-1438.1993.03010044.x. [DOI] [PubMed] [Google Scholar]
- 7.Huth ME, Ricci AJ, Cheng AG. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. Int J Otolaryngol. 2011;2011:937861. doi: 10.1155/2011/937861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howell SB, Taetle R. Effect of sodium thiosulfate on cis-dichlorodiammineplatinum(II) toxicity and antitumor activity in L1210 leukemia. Cancer Treat Rep. 1980;64:611–616. [PubMed] [Google Scholar]
- 9.Rybak LP, Kelly T. Ototoxicity: bioprotective mechanisms. Curr Opin Otolaryngol Head Neck Surg. 2003;11:328–333. doi: 10.1097/00020840-200310000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Dickey DT, Wu YJ, Muldoon LL, Neuwelt EA. Protection against cisplatin-induced toxicities by N-acetylcysteine and sodium thiosulfate as assessed at the molecular, cellular, and in vivo levels. J Pharmacol Exp Ther. 2005;314:1052–1058. doi: 10.1124/jpet.105.087601. [DOI] [PubMed] [Google Scholar]
- 11.Rolland V, Meyer F, Guitton MJ, Bussières R, Philippon D, Bairati I, Leclerc M, Côté M. A randomized controlled trial to test the efficacy of trans-tympanic injections of a sodium thiosulfate gel to prevent cisplatin-induced ototoxicity in patients with head and neck cancer. J Otolaryngol Head Neck Surg. 2019;48:4. doi: 10.1186/s40463-019-0327-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarafraz Z, Ahmadi A, Daneshi A. Transtympanic injections of n-acetylcysteine and dexamethasone for prevention of cisplatin-induced ototoxicity: double blind randomized clinical trial. Int Tinnitus J. 2018;22:40–45. doi: 10.5935/0946-5448.20180007. [DOI] [PubMed] [Google Scholar]
- 13.Marina N, Chang KW, Malogolowkin M, London WB, Frazier AL, Womer RB, Rescorla F, Billmire DF, Davis MM, Perlman EJ, Giller R, Lauer SJ, Olson TA Children’s Oncology Group. Amifostine does not protect against the ototoxicity of high-dose cisplatin combined with etoposide and bleomycin in pediatric germ-cell tumors: a Children’s Oncology Group study. Cancer. 2005;104:841–847. doi: 10.1002/cncr.21218. [DOI] [PubMed] [Google Scholar]
- 14.Fouladi M, Chintagumpala M, Ashley D, Kellie S, Gururangan S, Hassall T, Gronewold L, Stewart CF, Wallace D, Broniscer A, Hale GA, Kasow KA, Merchant TE, Morris B, Krasin M, Kun LE, Boyett JM, Gajjar A. Amifostine protects against cisplatin-induced ototoxicity in children with average-risk medulloblastoma. J. Clin. Oncol. 2008;26:3749–3755. doi: 10.1200/JCO.2007.14.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowers WJ, Chen X, Guo H, Frisina DR, Federoff HJ, Frisina RD. Neurotrophin-3 transduction attenuates cisplatin spiral ganglion neuron ototoxicity in the cochlea. Mol Ther. 2002;6:12–18. doi: 10.1006/mthe.2002.0627. [DOI] [PubMed] [Google Scholar]
- 16.Scasso F, Sprio AE, Canobbio L, Scanarotti C, Manini G, Berta GN, Bassi AM. Dietary supplementation of coenzyme Q10 plus multivitamins to hamper the ROS mediated cisplatin ototoxicity in humans: a pilot study. Heliyon. 2017;3:e00251. doi: 10.1016/j.heliyon.2017.e00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teitz T, Fang J, Goktug AN, Bonga JD, Diao S, Hazlitt RA, Iconaru L, Morfouace M, Currier D, Zhou Y, Umans RA, Taylor MR, Cheng C, Min J, Freeman B, Peng J, Roussel MF, Kriwacki R, Guy RK, Chen T, Zuo J. CDK2 inhibitors as candidate therapeutics for cisplatin- and noise-induced hearing loss. J Exp Med. 2018;215:1187–1203. doi: 10.1084/jem.20172246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Lloyd Faulconbridge RV, Fetoni A, Guitton MJ, Pujol R, Puel JL. Local application of sodium thiosulfate prevents cisplatin-induced hearing loss in the guinea pig. Neuropharmacology. 2003;45:380–393. doi: 10.1016/s0028-3908(03)00194-1. [DOI] [PubMed] [Google Scholar]
- 19.Hill GW, Morest DK, Parham K. Cisplatin-induced ototoxicity: effect of intratympanic dexamethasone injections. Otol Neurotol. 2008;29:1005–1011. doi: 10.1097/MAO.0b013e31818599d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calli C, Pinar E, Oncel S, Alper Bagriyanik H, Umut Sakarya E. Recovery of hearing in Cisplatin-induced ototoxicity in the Guinea pig with intratympanic dexamethasone. Indian J Otolaryngol Head Neck Surg. 2012;64:46–50. doi: 10.1007/s12070-011-0160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fried LE, Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal. 2009;11:1139–1148. doi: 10.1089/ars.2009.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujita M, Itokawa H, Sashida Y. Studies on the components of Magnolia obovata Thunb. 3. Occurrence of magnolol and honokiol in M. obovata and other allied plants. Yakugaku Zasshi. 1973;93:429–434. doi: 10.1248/yakushi1947.93.4_429. [DOI] [PubMed] [Google Scholar]
- 23.Cheng N, Xia T, Han Y, He QJ, Zhao R, Ma JR. Synergistic antitumor effects of liposomal honokiol combined with cisplatin in colon cancer models. Oncol Lett. 2011;2:957–962. doi: 10.3892/ol.2011.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Chen L, He X, Fan L, Yang G, Chen X, Lin X, Du L, Li Z, Ye H, Mao Y, Zhao X, Wei Y. Enhancement of therapeutic effectiveness by combining liposomal honokiol with cisplatin in ovarian carcinoma. Int J Gynecol Cancer. 2008;18:652–659. doi: 10.1111/j.1525-1438.2007.01070.x. [DOI] [PubMed] [Google Scholar]
- 25.Maruyama Y, Kuribara H, Morita M, Yuzurihara M, Weintraub ST. Identification of magnolol and honokiol as anxiolytic agents in extracts of saiboku-to, an oriental herbal medicine. J Nat Prod. 1998;61:135–138. doi: 10.1021/np9702446. [DOI] [PubMed] [Google Scholar]
- 26.Liou KT, Shen YC, Chen CF, Tsao CM, Tsai SK. Honokiol protects rat brain from focal cerebral ischemia-reperfusion injury by inhibiting neutrophil infiltration and reactive oxygen species production. Brain Res. 2003;992:159–166. doi: 10.1016/j.brainres.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 27.Lin YR, Chen HH, Ko CH, Chan MH. Differential inhibitory effects of honokiol and magnolol on excitatory amino acid-evoked cation signals and NMDA-induced seizures. Neuropharmacology. 2005;49:542–550. doi: 10.1016/j.neuropharm.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 28.Lo YC, Teng CM, Chen CF, Chen CC, Hong CY. Magnolol and honokiol isolated from Magnolia officinalis protect rat heart mitochondria against lipid peroxidation. Biochem Pharmacol. 1994;47:549–553. doi: 10.1016/0006-2952(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 29.Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP, Gius D, Gupta MP. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. 2015;6:6656. doi: 10.1038/ncomms7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsai SK, Huang SS, Hong CY. Myocardial protective effect of honokiol: an active component in Magnolia officinalis. Planta Med. 1996;62:503–506. doi: 10.1055/s-2006-957957. [DOI] [PubMed] [Google Scholar]
- 31.Chiang CK, Sheu ML, Lin YW, Wu CT, Yang CC, Chen MW, Hung KY, Wu KD, Liu SH. Honokiol ameliorates renal fibrosis by inhibiting extracellular matrix and pro-inflammatory factors in vivo and in vitro. Br J Pharmacol. 2011;163:586–597. doi: 10.1111/j.1476-5381.2011.01242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiu JH, Ho CT, Wei YH, Lui WY, Hong CY. In vitro and in vivo protective effect of honokiol on rat liver from peroxidative injury. Life Sci. 1997;61:1961–1971. doi: 10.1016/s0024-3205(97)00836-9. [DOI] [PubMed] [Google Scholar]
- 33.Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem. 2012;287:14078–14086. doi: 10.1074/jbc.M112.355206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giralt A, Villarroya F. SIRT3, a pivotal actor in mitochondrial functions: metabolism, cell death and aging. Biochem J. 2012;444:1–10. doi: 10.1042/BJ20120030. [DOI] [PubMed] [Google Scholar]
- 35.Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalinec GM, Webster P, Lim DJ, Kalinec F. A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol Neurootol. 2003;8:177–189. doi: 10.1159/000071059. [DOI] [PubMed] [Google Scholar]
- 37.Park J, Morley TS, Scherer PE. Inhibition of endotrophin, a cleavage product of collagen VI, confers cisplatin sensitivity to tumours. EMBO Mol Med. 2013;5:935–948. doi: 10.1002/emmm.201202006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roy S, Ryals MM, Van den Bruele AB, Fitzgerald TS, Cunningham LL. Sound preconditioning therapy inhibits ototoxic hearing loss in mice. J Clin Invest. 2013;123:4945–4949. doi: 10.1172/JCI71353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan X, Jahan I, Xu Y, Stock S, Kwan CC, Soriano C, Xiao X, Garcia-Anoveros J, Fritzsch B, Richter CP. Auditory neural activity in congenitally deaf mice induced by infrared neural stimulation. Sci Rep. 2018;8:388. doi: 10.1038/s41598-017-18814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel S, Shah L, Dang N, Tan X, Almudevar A, White PM. SIRT3 promotes auditory function in young adult FVB/nJ mice but is dispensable for hearing recovery after noise exposure. PLoS One. 2020;15:e0235491. doi: 10.1371/journal.pone.0235491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walters BJ, Diao S, Zheng F, Walters BJ, Layman WS, Zuo J. Pseudo-immortalization of postnatal cochlear progenitor cells yields a scalable cell line capable of transcriptionally regulating mature hair cell genes. Sci Rep. 2015;5:17792. doi: 10.1038/srep17792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller M, von Hunerbein K, Hoidis S, Smolders JW. A physiological place-frequency map of the cochlea in the CBA/J mouse. Hear Res. 2005;202:63–73. doi: 10.1016/j.heares.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Breglio AM, Rusheen AE, Shide ED, Fernandez KA, Spielbauer KK, McLachlin KM, Hall MD, Amable L, Cunningham LL. Cisplatin is retained in the cochlea indefinitely following chemotherapy. Nat Commun. 2017;8:1654. doi: 10.1038/s41467-017-01837-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hazlitt RA, Min J, Zuo J. Progress in the development of preventative drugs for cisplatin-induced hearing loss. J Med Chem. 2018;61:5512–5524. doi: 10.1021/acs.jmedchem.7b01653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mehmood RK. Review of Cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments. Oncol Rev. 2014;8:256. doi: 10.4081/oncol.2014.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomez-Ruiz S, Maksimovic-Ivanic D, Mijatovic S, Kaluderovic GN. On the discovery, biological effects, and use of Cisplatin and metallocenes in anticancer chemotherapy. Bioinorg Chem Appl. 2012;2012:140284. doi: 10.1155/2012/140284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamogashira T, Fujimoto C, Yamasoba T. Reactive oxygen species, apoptosis, and mitochondrial dysfunction in hearing loss. Biomed Res Int. 2015;2015:617207. doi: 10.1155/2015/617207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong AC, Ryan AF. Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front Aging Neurosci. 2015;7:58. doi: 10.3389/fnagi.2015.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohlemiller KK, Wright JS, Dugan LL. Early elevation of cochlear reactive oxygen species following noise exposure. Audiol Neurootol. 1999;4:229–236. doi: 10.1159/000013846. [DOI] [PubMed] [Google Scholar]
- 53.Someya S, Prolla TA. Mitochondrial oxidative damage and apoptosis in age-related hearing loss. Mech Ageing Dev. 2010;131:480–486. doi: 10.1016/j.mad.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng AG, Cunningham LL, Rubel EW. Mechanisms of hair cell death and protection. Curr Opin Otolaryngol Head Neck Surg. 2005;13:343–348. doi: 10.1097/01.moo.0000186799.45377.63. [DOI] [PubMed] [Google Scholar]
- 56.Church MW, Blakley BW, Burgio DL, Gupta AK. WR-2721 (Amifostine) ameliorates cisplatin-induced hearing loss but causes neurotoxicity in hamsters: dose-dependent effects. J Assoc Res Otolaryngol. 2004;5:227–237. doi: 10.1007/s10162-004-4011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arora S, Singh S, Piazza GA, Contreras CM, Panyam J, Singh AP. Honokiol: a novel natural agent for cancer prevention and therapy. Curr Mol Med. 2012;12:1244–1252. doi: 10.2174/156652412803833508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin JW, Chen JT, Hong CY, Lin YL, Wang KT, Yao CJ, Lai GM, Chen RM. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro Oncol. 2012;14:302–314. doi: 10.1093/neuonc/nor217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang X, Duan X, Yang G, Zhang X, Deng L, Zheng H, Deng C, Wen J, Wang N, Peng C, Zhao X, Wei Y, Chen L. Honokiol crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L intracerebral gliosarcoma model and human U251 xenograft glioma model. PLoS One. 2011;6:e18490. doi: 10.1371/journal.pone.0018490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alam SA, Ikeda K, Oshima T, Suzuki M, Kawase T, Kikuchi T, Takasaka T. Cisplatin-induced apoptotic cell death in Mongolian gerbil cochlea. Hear Res. 2000;141:28–38. doi: 10.1016/s0378-5955(99)00211-7. [DOI] [PubMed] [Google Scholar]
- 61.Hu BH, Henderson D, Yang WP. The impact of mitochondrial energetic dysfunction on apoptosis in outer hair cells of the cochlea following exposure to intense noise. Hear Res. 2008;236:11–21. doi: 10.1016/j.heares.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Someya S, Xu J, Kondo K, Ding D, Salvi RJ, Yamasoba T, Rabinovitch PS, Weindruch R, Leeuwenburgh C, Tanokura M, Prolla TA. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc Natl Acad Sci U S A. 2009;106:19432–19437. doi: 10.1073/pnas.0908786106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV Jr, Weissman S, Verdin E, Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hart PC, Mao M, de Abreu AL, Ansenberger-Fricano K, Ekoue DN, Ganini D, Kajdacsy-Balla A, Diamond AM, Minshall RD, Consolaro ME, Santos JH, Bonini MG. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun. 2015;6:6053. doi: 10.1038/ncomms7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y, Huang J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014;5:e1047. doi: 10.1038/cddis.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schumacker PT. SIRT3 controls cancer metabolic reprogramming by regulating ROS and HIF. Cancer Cell. 2011;19:299–300. doi: 10.1016/j.ccr.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown KD, Maqsood S, Huang JY, Pan Y, Harkcom W, Li W, Sauve A, Verdin E, Jaffrey SR. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014;20:1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quan Y, Xia L, Shao J, Yin S, Cheng CY, Xia W, Gao WQ. Adjudin protects rodent cochlear hair cells against gentamicin ototoxicity via the SIRT3-ROS pathway. Sci Rep. 2015;5:8181. doi: 10.1038/srep08181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.White K, Kim MJ, Han C, Park HJ, Ding D, Boyd K, Walker L, Linser P, Meneses Z, Slade C, Hirst J, Santostefano K, Terada N, Miyakawa T, Tanokura M, Salvi R, Someya S. Loss of IDH2 accelerates age-related hearing loss in male mice. Sci Rep. 2018;8:5039. doi: 10.1038/s41598-018-23436-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim D, Park W, Lee S, Kim W, Park SK, Kang KP. Absence of Sirt3 aggravates cisplatin nephrotoxicity via enhanced renal tubular apoptosis and inflammation. Mol Med Rep. 2018;18:3665–3672. doi: 10.3892/mmr.2018.9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu F, Yao H, Zheng F, Tang S, Lin X, Li L, Zhou J, Li H. Protective effects of honokiol against oxidative stress-induced apoptotic signaling in mouse podocytes treated with H2O2. Exp Ther Med. 2018;16:1278–1284. doi: 10.3892/etm.2018.6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.