

Abstract
Increased expression levels of constitutively active androgen receptor splice variants (AR-Vs) cause alterations in AR signaling, resulting in drug resistance and failed hormone therapy among patients with advanced prostate cancers. Several available compounds targeting the androgen axis and AR signaling have not demonstrated efficacy in preventing prostate cancer recurrence. Here, we investigated whether a new agent, 6-[6-ethoxy-5-ispropoxy-3,4-dihydroisoquinolin-2[1H)-yl]-N-[6-methylpyridin-2-yl]nicotinamide (EIQPN), has the potential for treating advanced prostate cancer. EIQPN interacted with the AR-activation fragment-1 (AF-1) domain and blocked its androgen-independent activity, robustly decreased the protein levels of AR and variants in prostate cancer cells by inducing AR protein degradation, and inhibited the androgen-independent proliferation of various AR-positive prostate cancer cells. In xenograft mouse models, EIQPN blocked the tumor growth of androgen-independent prostate cancer cells. Overall, these findings indicate that EIQPN could serve as a novel therapeutic agent for advanced recurrent prostate cancers.
Keywords: Prostate cancer, androgen receptor, splice variant, androgen-independent activity, protein degradation
Introduction
Prostate cancer is a predominant lethal malignancy among males [1] and the second most common cause of male cancer death in the USA [2]. Androgen-deprivation therapy (ADT) through either medical or surgical castration has been the standard treatment for prostate cancer. However, cancer usually recurs after a couple of years, and progresses to advanced diseases such as castration-resistant prostate cancer (CRPC) [3-5].
Androgen receptor (AR) and its signaling play important roles in the cell proliferation, metastasis, and progression of prostate cancers [6,7]. The AR comprises functional NH2-terminal transactivation (NTD), DNA-binding (DBD), and ligand-binding (LBD) domains. The AR-NTD contains most of the phosphorylated amino acid residues that regulate AR protein stabilization, cellular localization, and transcriptional activity [8-10]. A major activating domain of AR (activation fragment-1, AF-1), which is located within the NTD, has ligand-independent activity, whereas activation fragment-2 (AF-2) located within the LBD is associated with ligand-dependent activity [8,9,11]. The AF-1 in AR-NTD is essential for AR transcriptional activity, both androgen-dependent and -independent activity [12]. Androgen receptor signaling is activated via the NH2/COOH terminal (N/C) interaction, subcellular localization, and recruitment to androgen-response elements (AREs) in target gene promoters [6,13].
Changes in AR signaling lead to development of advanced CRPC. Several proposed mechanisms underlying CRPC are AR amplification and mutations that lead to increased AR activity, altered levels of AR coregulator expression, ligand-independent AR activation by outlaw pathways, and altered steroidogenesis [14,15]. Another recently proposed mechanism of progression to CRPC after failed androgen deprivation might involve an association between some recurrent cancers and the increased expression levels of constitutively active AR splice variants (AR-Vs) that lack the LBD [16]. Among the various AR-Vs that have been identified in prostate cancer cell lines and tumors, AR-V7 (AR3) with androgen-independent transactivation is the most prevalent and studied variant [16-18].
Since androgens play critical roles in prostate cancers, compounds targeting the androgen axis, including casodex (bicalutamide, BIC), abiraterone (CYP17 inhibitor) and enzalutamide (MDV-3100), have been developed to treat prostate cancers [19-22]. However, even the next generation AR signaling inhibitors such as enzalutamide could not prevent the progression of prostate cancer to CRPC [23]. Among the proposed mechanisms responsible for this resistance, the expression of AR-Vs that have lost the LBD and are therefore constitutively active has received considerable focus [24,25]. A few compounds such as EPI-001 and UT-155 that target the AR-NTD have recently been developed as candidates to treat advanced prostate cancers [26,27]. Currently, however, no drugs that target the AR-NTD and therefore become effective against these AR-Vs are clinically available.
In previous structure-activity relationship studies to develop AR antagonists [28,29], we found that 6-[6-ethoxy-5-ispropoxy-3,4-dihydroisoquinolin-2[1H)-yl]-N-[6-methylpyridin-2-yl]nicotinamide (EIQPN) did not bind to AR-LBD unlike conventional AR antagonists. Therefore, in the present study, we investigated the effects of EIQPN on AR function and expression, as well as on prostate cancer cell proliferation and tumor growth in a CWR22rv xenograft mouse model, aiming to develop an agent for treating advanced prostate cancer. We found that EIQPN inhibited AR-AF1 activity and markedly decreased protein levels of AR and AR-Vs in prostate cancer cells. Furthermore, EIQPN inhibited the androgen-independent proliferation of AR-positive prostate cancer cells in vitro and in vivo. These findings suggest that EIQPN has potential for treating recurrent prostate cancers by blocking and downregulating the signaling pathways in CRPC/androgen-independent prostate cancer (AIPC).
Materials and methods
Reagents
We purchased the following reagents from the described suppliers: radiolabeled dihydrotestosterone ([3H]5α-DHT; PerkinElmer Life and Analytical Sciences Inc., Waltham, MA, USA), forskolin (FSK) and chloroquine diphosphate salt (CQ; Sigma-Aldrich Corp., St. Louis, MO, USA), carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG-132; A.G. Scientific Inc., San Diego, CA, USA), human IL-6 (R&D Systems Inc., Minneapolis, MN, USA), 5α-dihydrotestosterone (DHT; Sigma-Aldrich), enzalutamide and bicalutamide (MDV-3100 and BIC; Sequoia Research Products Ltd., Newbury, England, UK). 6-(6-ethoxy-5-isopropoxy-3,4-dihydro-1H-isoquinolin-2-yl)-N-(6-methyl-pyridin-2-yl)-nicotinamide (EIQPN) was synthesized and provided by Medigene Co. (Daejeon, Republic of Korea).
Plasmids
The plasmids pARE2-TATA-Luc, GAL4.AR-LBD658-919, VP16.AR1-660, 5xGAL4-luc3, pcR3.1 SRC-1, pcDNA3.AR, and pEGFP-AR have been previously described [28,30]. For androgen-independent activity studies, the LBDs were removed from pcDNA3.AR and pEGFP-AR to generate pcDNA3.AR-NTD and GFP-AR-NTD, respectively. We constructed pB3.AR-AF1 to overexpress and purify AR-AF1 protein for in vitro protein-substrate interaction assay. The AR-AF1 domain (amino acids 110~485) was amplified from a human cDNA library and inserted into pB3 at a SmaI site to generate pB3.AR-AF1. Table 1 lists the primers for cloning of AR-AF1.
Table 1.
Primers
| Cloning | AR-AF1 LIC Forward: GGCGGTGGTGGCGGCATGGATGAGGAACAGCAACCT |
| AR-AF1 LIC Reverse: TCTTCTCCTTTGCGCCCCTACCGAGTGTAGCCGTAGGG | |
| ChIP | ARE-2 Forward: ACAATCTCCTGAGTGCTGGTGT |
| ARE-2 Reverse: GCAGAGGAGACATGCCCAG | |
| ARE-3 Forward: TGAGAAACCTGAGATTAGGA | |
| ARE-3 Reverse: GTTCCTCCAGAGTAGGTCTGTTTTC | |
| β-actin Forward: TCCTCCTCTTCCTCAATCTCG | |
| β-actin Reverse: AAGGCAACTTTCGGAACGG | |
| RT-PCR | Gapdh Forward: ATCACCATCTTCCAGGAGCGAG |
| Gapdh Reverse: GAGATGATGACCCTTTTGGCTCC | |
| PSA Forward: GGCCAGGTATTTCAGGTCAG | |
| PSA Reverse: TCGTGGCTGGAGTCATCAC | |
| TMPRss2 Forward: CGCCAGAGCATTGTG | |
| TMPRss2 Reverse: GCGGCTGTCACGATCC |
Cell lines
The LNCaP (CRL-1740) and HEK 293T (CRL-11268) cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). The C4-2 and CWR22rv cell lines were kindly provided by Dr. C. Jung (Chonnam National University Medical School), and PPC1 and DU145 cells were a kind gift from Dr. S. Back (Seoul National University). LNCaP, C4-2, and CWR22rv cells were maintained in RMPI-1640 media (HyClone Laboratories Inc., South Logan, UT, USA) with 5% Gibco fetal bovine serum (FBS; Thermo Fisher Scientific Inc., Waltham, MA, USA). The PPC1 and HEK 293T cells were cultured in Dulbecco’s Modified Eagle’s Media (DMEM; HyClone) with 10% FBS. Media contained 5% charcoal-stripped serum (cFBS) for starvation and steroid studies. All cell lines were maintained in media containing 100 units/mL penicillin and 100 mg/mL streptomycin (P/S; Thermo Fisher) under a 5% CO2 atmosphere at 37°C.
Cell transfection and reporter assays
Cells were transfected with the expression constructs, luciferase reporter, and pRSV-LacZ (Takara Bio USA Inc., Mountain View, CA, USA) using Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA), starved, and then stimulated with androgen, growth factors, or AR-antagonists for 24 h. Cells were lysed in luciferase lysis buffer (0.2 M Tris-Cl (pH 8.0), 0.2% Triton X100, and 1% NP-40) at 25°C for 15 min. Luciferase activity was analyzed using Beetle Luciferin (Promega Inc., Madison WI, USA) and a Centro XS3 LB960 Luminometer (Berthold Technologies U.S.A. LLC., Oakridge, TN, USA). Results were normalized to β-galactosidase activity measured at a wavelength of 405 nm using a Versa Max microplate reader (Molecular Devices LLC., San Diego, CA, USA).
Subcellular localization
HEK 293T cells were transiently transfected with GFP-AR-FL or GFP-AR-NTD, starved for 12 h, then incubated with 10 µM EIQPN, MDV-3100, or BIC with or without 10 nM DHT for 2 h. The cells were fixed with 3.7% paraformaldehyde in PBS, stained with TOPRO-3. Subcellular localization of ARs was analyzed using a Nikon A1 laser-scanning Leica TCS SPE confocal microscope equipped with an ACS APO × 63/1.30 numerical aperture immersion objective. Images were analyzed using the ImageJ software (http://imagej.nih.gov/ij, ImageJ 1.46r, Wayne Rasband, National Institutes of Health, USA).
Chromatin immunoprecipitation (ChIP)
To examine the effect of EIQPN on a recruitment of AR to AREs, we conducted ChIP assays as described with modifications [31]. After starvation for 48 h, LNCaP cells were incubated with vehicle or 10 µM EIQPN and 10 nM DHT for 4 h, crosslinked with 1% formaldehyde (Sigma-Aldrich) for 10 min, sonicated to shear DNA, and then processed for ChIP assays. Table 1 lists the primers used for ChIP.
Competitive radioligand binding assays
To determine whether EIQPN interacts with the LBD of AR, we performed a competitive radioligand binding assay as described [32]. HEK 293T cells were transiently transfected with pcDNA3.AR, starved for 12 h, and then incubated with 5 nM [3H]5α-DHT (~0.667 µCi) and various concentrations of competing unlabeled ligands (DHT and EIQPN) for 2 h. The cells were washed and harvested in lysis buffer (2% SDS, 10% glycerol, and 10 mM Tris, pH 6.8), then radioactivity was measured using QuantaSmart™ -1.31 (Serial #: 428238, PerkinElmer Life and Analytical Sciences Inc.).
AR-AF1 protein purification
We induced AR-AF1 protein with 0.2 mM IPTG for 4 h in BL21 E. coli. Cell pellets were sonicated in 50 mM Tris-Cl pH 7.5 containing protein inhibitors, and the supernatant was collected. The AR-AF1 protein was purified via immobilized metal affinity (IMAC) and anion exchange (Q-Sepharose) chromatography. Fractions containing AR-AF1 protein were identified by western blotting, and protein concentrations were determined by measuring λ at 280 nm. The purified AR-AF1 proteins were then used for in vitro protein-substrate interaction.
In vitro protein-substrate interaction
To determine whether EIQPN interacts with AR-AF1 protein, the interaction reactions were performed using fluorescence spectroscopy as described with modifications [33,34]. The intrinsic fluorescence was measured at excitation (λEx) and emission (λEm) wavelengths of 278 and 300-400 nm, respectively, using a Shimadzu RF-5301 spectrofluorimeter (Shimadzu Corporation, Kyoto, Japan). EIQPN (5 µM) was added to the AR-AF1 binding reaction. Conformational changes in the protein were evaluated as changes in quantum yield and/or peak shifts compared with those of native AR-AF1. Changes in the intrinsic fluorescence spectrum of AR-AF1 denatured with 8 M urea was also monitored.
3D computational modeling
We analyzed 3D interactions between AR-AF1 and EIQPN and EPI-001. A putative 3D structure of the docking region within the TAU-5 domain of AF-1 was determined using the SWISS-MODEL online server (https://swissmodel.expasy.org/). Both EIQPN and EPI-001 were directly drawn and converted to 3D via the 1-Click docking online server by mcule (https://mcule.com/apps/1-click-docking/); then, each 3D-modeled docking region generated by SWISS-MODEL was docked with a specific docking center. More negative scores suggest a higher affinity. We analyzed interaction scores of the compounds at the same amino acid residue.
RNA isolation and RT-PCR
Total RNA was isolated from cells using the TRI Reagent® (Molecular Research Center Inc., Cincinnati, OH, USA), and reverse transcribed using Oligo d(T)15 (ELPiS, Daejeon, Korea) and M-MLV Reverse transcriptase kits (Promega Inc.) according to the manufacturer’s instructions. Levels of mRNA were analyzed by RT-PCR using Taq polymerase. Table 1 lists the primers for RT-PCR.
Western blotting
Cells were sonicated in 25 mM Tris-Cl (pH 8.0) containing protease inhibitors. The protein levels were analyzed by western blotting. Proteins were separated by SDS-PAGE, and transblotted onto PortranTM Premium 0.2 µM nitrocellulose membranes (GE Healthcare, Chicago, IL, USA). Nonspecific binding was blocked, then the membranes were incubated with primary antibodies (Table 2) overnight at 4°C, washed, incubated with HRP-conjugated secondary antibodies for 1 h, and then visualized on X-ray films using the ECLTM Western Blotting Analysis System (GE Healthcare).
Table 2.
Antibodies and sources
| Antibodies | Source |
|---|---|
| Anti-AR (PG-21) | Millipore |
| Anti-AR (C-19), Anti-PSA (C-19), Anti-HA (F-7), Anti-GAPDH, Rabbit-anti-goat | Santa Cruz |
| Anti-cleaved caspase 3 | Cell Signaling |
| Goat anti-rabbit, Goat anti-mouse | Thermo Fisher |
Cell proliferation assays
The LNCaP, C4-2 and CWR22rv cells were seeded onto 96-well plates and starved for 12 h, then incubated with 10 µM of EIQPN, MDV-3100, or BIC with or without 1 nM DHT for 5 days. The PPC1 and DU145 cells were incubated with 10 µM EIQPN, MDV-3100, or BIC for 5 days. For non-androgen-stimulated cell growth, LNCaP cells were starved for 12 h, and incubated with 10 µM EIQPN, MDV-3100, or BIC, with or without IL-6 (50 ng/mL) for 5 days. Cell growth (%) and numbers were determined using MTS and trypan-blue staining, respectively.
Xenograft animal model
We generated CWR22rv xenograft mouse models to examine the in vivo effect of EIQPN on androgen-independent prostate tumorigenesis. Healthy 4-week-old male NOD.CB17-PrkdcSCID/J mice (mean weight [± SD], 20 g; KRIBB, Daejeon, Korea) were gently injected with 100 µL of anesthetic (Zoletil 50:Rompun:saline buffer [20:10:270]) and warmed using a veterinary system. CWR22rv cells (4 × 106/site [intact] or 6 × 106/site [castrated] mixed 1:1 with Matrigel) were subcutaneously implanted into the shoulders of the mice. After 1 week, the animals were injected i.p. with 50 µL of vehicle or 25 mg/kg EIQPN in DMSO:PEG300 (2:8) three times per week for 7 weeks. The intact (n = 9 per group) and castrated (n = 7 per group) mice were euthanized with CO2 for tumor dissection. All animals were maintained inside a cleaned bench in an animal room with a 12/12 h light/dark cycle and controlled temperature at 25°C. The cages, water, food, and bedding were sterilized and replaced weekly. The mice were injected with ultra-fine II short needles (U-100 INSULIN 30 gauge 5/16”, 8 mm) to minimize pain and lesions. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Chonnam National University (permit number: 2012-44).
Statistical analysis
All graphs were created using GraphPad Prims (GraphPad software Inc., Version 5.0, La Jolla, CA, USA). Data are presented as means ± SEM of more than three independent experiments. Statistical significance was calculated using one-way ANOVA with Tukey post hoc tests and two-tailed t-tests. Values with P < 0.05 were considered statistically significant.
Results
Novel EIQPN inhibits androgen-independent AR transactivation in prostate cancer cells
While developing new antagonists of AR, we identified the new compound EIQPN (Figure 1A) that inhibited androgen-independent AR activity in addition to androgen-dependent activity. In hypothesis-testing mechanistic studies, EIQPN inhibited the transactivation of endogenous AR activated by the androgen DHT and the non-androgen forskolin (FSK) and IL-6 in LNCaP cells (Figure 1B-D). Moreover, EIQPN blocked endogenous AR transactivation in AIPC cell line CWR22rv that expresses full-length AR (AR-FL) and AR-Vs (Figure 1E).
Figure 1.

EIQPN inhibits androgen-independent AR transactivation as well as androgen-dependent. (A) Chemical structure of (6-[6-ethoxy-5-ispropoxy-3,4-dihydroisoquinolin-2[1H)-yl]-N-[6-methylpyridin-2-yl]nicotinamide (EIQPN). (B-E) EIQPN inhibits endogenous AR transactivation in prostate cancer cells. LNCaP (B-D) and CWR22rv (E) cells were transiently transfected with pARE2-TATA-luc reporter and incubated with 10 µM EIQPN and with or without 10 nM DHT (B), 50 ng/mL IL-6 (C), or 50 µM FSK (D). (F, G) EIQPN inhibits exogenous AR transactivation in PPC1 cells. Cells were transiently transfected with full-length AR (AR-FL) (F) or AR N-terminal domain (AR-NTD) (G) expression construct along with pARE2-TATA-luc, and incubated with 10 µM EIQPN, MDV-3100, or BIC, and with and without 10 nM DHT. Luciferase activity was normalized to β-galactosidase activity. Data are shown as means ± SEM of at least three independent experiments. *, P < 0.01; **, P < 0.001; one-way ANOVA with Tukey post hoc tests. ns, not significant. (H) EIQPN dose-dependently inhibits AR-NTD transactivation. HEK 293T cells overexpressing AR-NTD were incubated with various doses of EIQPN or BIC. Inhibitory concentration (IC50) was obtained by nonlinear regression analysis.
We further analyzed the effects of EIQPN on androgen-independent AR transactivation by comparing its effects on AR-NTD that lacks the LBD, with those on AR-FL in PPC1 cells. We found that EIQPN inhibited the transactivation of exogenous AR-FL to an extent comparable to that of the same concentration of the established AR antagonists MDV-3100 and BIC in an androgen-dependent manner (Figure 1F). Notably, EIQPN blocked the androgen-independent transactivation of AR-NTD with an IC50 of 0.7865 µM, whereas MDV-3100 and BIC did not (Figure 1G, 1H). These results suggest that unlike conventional AR antagonists, EIQPN targets AR-NTD and inhibits androgen-independent AR transactivation as well as androgen-dependent transactivation.
Activation of AR at various stages is blocked by EIQPN
To elucidate the mechanism through which EIQPN inhibits AR transactivation, we investigated the effects of EIQPN on the AR activation steps, comprising N/C interaction, subcellular localization, and AR recruitment to AREs. In N/C interaction assays using AR NH2-terminal (VP16.AR1-660) and COOH-terminal (GAL4.AR658-919) constructs, EIQPN blocked the N/C interaction of AR under induction by DHT or FSK more than MDV-3100 and BIC (Figure 2A, 2B).
Figure 2.

EIQPN blocks AR activation at various stages. (A, B) EIQPN inhibits the N/C interaction of AR. PPC1 cells co-transfected with AR N-terminal (VP16/AR1-660) and C-terminal (GAL4/AR-LBD658-919) domain constructs along with 5xGAL4-luc3 reporter, were incubated with 10 µM EIQPN, MDV-3100, or BIC, and 10 nM DHT (A) or 50 µM FSK (B). (C, D) EIQPN blocks AR nuclear translocation. HEK 293T cells overexpressing GFP-AR-FL (C) and GFP-AR-NTD (D) were incubated with 10 µM EIQPN, MDV-3100, or BIC, and with or without 10 nM DHT for 2 h. Subcellular localization of ARs was detected as green fluorescent protein (GFP) signals. Nuclei were stained blue with TOPRO-3. Images were acquired using a confocal microscope. Scale bars, 25 µm. (E) EIQPN prevents AR recruitment to ARE. Recruitment of AR protein to ARE-2 and ARE-3 within PSA promoter was determined by ChIP assays using anti-AR (C-19) antibody. LNCaP cells were incubated with 10 µM EIQPN and 10 nM DHT for 2 h. Changes in AR enrichment at ARE was examined by PCR. The loading control was β-actin. (F, G) EIQPN inhibits coactivator recruitment to AR. PPC1 cells co-transfected with AR expression construct and SRC-1 or empty vector (EV) along with pARE2-TATA-luc were incubated with 10 µM EIQPN, MDV-3100, or BIC, and with or without 10 nM DHT (F) or 50 µM FSK (G). Data represent means ± SEM of at least three independent experiments. *, P < 0.01, and **, P < 0.001; one-way ANOVA with Tukey post hoc tests.
We then investigated whether EIQPN androgen-dependently and -independently alters the subcellular localization of AR by overexpressing GFP-AR-FL and GFP-AR-NTD in HEK 293T cells. We found that AR-FL, which is located in the cytoplasm in the absence of DHT, was completely translocated to the nucleus by stimulation with DHT. However, EIQPN blocked the DHT-induced nuclear translocation of AR-FL, promoting AR protein aggregation in the cytoplasm (Figure 2C). The localization of AR-NTD, which was distributed in the cytoplasm and nucleus under starvation, was barely affected by EIQPN, whereas the antiandrogens, MDV-3100 and BIC, induced the nuclear import of AR-NTD (Figure 2D).
We also assessed whether EIQPN alters the recruitment of AR to AREs. The DHT-induced recruitment of AR to the ARE-3, which is located within the distal major enhancer ~4 kb upstream of the transcription start site of the AR target PSA gene in LNCaP cells, was prevented by EIQPN (Figure 2E). This major enhancer’s activity is more sensitive to androgen stimulation than that of the proximal enhancer containing ARE-2 [6,13]. In addition, overexpressed SRC-1 enhanced AR transactivation under starvation, and DHT or FSK induction, whereas EIQPN blocked this AR co-activation, suggesting the inhibition of coactivator recruitment to AR (Figure 2F, 2G). Notably, coactivator recruitment was inhibited more by EIQPN than by MDV-3100 and BIC (Figure 2F, 2G). These results suggest that EIQPN blocks AR activation at various stages.
EIQPN interacts with the AF-1 domain of AR, but not with the LBD
We aimed to elucidate the molecular mechanism through which EIQPN inhibited AR activity. We first determined whether EIQPN interacts with the LBD and/or NTD domain of AR using competitive radioligand binding assays and protein-substrate interaction assays in vitro based on fluorescence spectroscopy. In competitive radioligand binding assays, 0.1 to 100 µM EIQPN did not bind to the AR-LBD, as binding signals were undetectable (< 15%). In contrast, DHT, a high affinity AR ligand, bound to the AR-LBD with a K d of 0.001 µM (Figure 3A). This finding appeared to contradict the inhibition of androgen-dependent AR activity by EIQPN (Figure 1B, 1F), because binding or interaction with the LBD is important for androgen-dependent AR transactivation. Therefore, we speculated that EIQPN binds to or interacts with the NTD, preferably the AF-1 domain of AR, to inhibit the transactivation of AR-FL and AR-NTD.
Figure 3.

EIQPN interacts with AR-AF1 domain, but not with LBD. (A) EIQPN does not bind AR-LBD. HEK 293T cells overexpressing AR-FL were incubated with 5 nM [3H]5α-DHT and various concentrations of unlabeled ligand (DHT or EIQPN). K d values were obtained by nonlinear regression analysis. (B) Schema of cloned AR-AF1 and docking region within TAU-5 domain. AR-AF1 domain (amino acids 110-485) was cloned for protein-substrate interaction assays in vitro. Docking region (H385-G410) contains 26 amino acids of TAU-5 domain. (C) Conformational change in AR-AF1 protein caused by interaction with EIQPN. Changes in intrinsic fluorescence spectrum of native or urea-denatured AR-AF1 protein caused by EIQPN were monitored using fluorescence spectroscopy. (D-F) Computational modeling of EIQPN interaction with AF-1 domain. Molecular PDB model of docking region of AR-AF1 was obtained using SWISS-MODEL. Novel interaction between EIQPN and docking region was determined by 1-Click docking. Summary of docking interaction scores (D). Images show 3D structure of H385-G410 TAU-5 docking region (orange) and EIQPN interacting in a K389/E391/P393/W400 pocket (E). EPI-001 was the positive control (F).
Interactions between protein and substrates often cause protein conformational changes that can be measured by fluorescence spectroscopy [26,27,35]. We investigated whether EIQPN interacts with AR-AF1. We prepared AR-AF1 domain proteins (amino acids 110~485) containing TAU-1 and TAU-5 domains but not the glutamine-rich region (Figure 3B), and assayed protein-substrate interactions in vitro (Figure 3C). The steady-state intrinsic fluorescence spectrum for native AF-1 protein was characterized by an emission maximum (peak) at 333 nm due to tryptophan residues and a tyrosine shoulder at ~309 nm. EIQPN caused quantum yield decrease and tryptophan peak shift from 333 nm to 343-345 nm (Figure 3C). These results indicate that the tryptophan residues became losing energy and its signal was quenched due to the interaction with EIQPN. Meanwhile, the steady-state fluorescence spectrum of AF-1 protein denatured with urea showed two distinct peaks with decreased intensity. Unlike native AF-1 protein, adding EIQPN to denatured AF-1 protein did not cause a peak shift. These results suggest that the binding of EIQPN to the AR-AF1 domain requires some AF-1 secondary structures.
We also computationally modeled an interaction site within the TAU-5 domain of AR-AF1 (docking region) (Figure 3B), based on the results of the present protein-substrate, and previous EPI-001-TAU-5 interaction analyses [36]. EPI-001 binds to the TAU5 domain of AR-AF1 and inhibits AR-NTD activity [27]. The modeling results showed a lower affinity score for EIQPN (-6.0) than EPI-001 (-5.6) at the docking region (H385-G410 TAU-5) (Figure 3D-F), suggesting that EIQPN has higher interaction affinity than EPI-001. The modeled image shows the 3D structure of the TAU-5 domain (orange), and EIQPN and EPI-001 interacting with the TAU-5 domain at a K389/E391/P393/W400 pocket, which might result in a reduction of quantum yield and a red-shift of tryptophan peak (Figure 3C, 3E, 3F). These results suggest that EIQPN interacts with the AR-AF1 domain and induces conformational changes that eventually result in the loss of AR transactivation in both androgen-independent and -dependent manners.
EIQPN decreases AR protein levels
We found that EIQPN robustly decreased the protein levels of endogenous ARs in various AR-positive prostate cancer cells. In androgen-dependent (LNCaP) and -independent (CWR22rv) prostate cancer cells, 10 µM EIQPN reduced the protein levels of AR and AR-Vs > 90% (Figure 4A, 4B). The same concentration of the control compounds, MDV-3100 and BIC, showed a little effect on AR protein levels in LNCaP cells but significantly enhanced those in CWR22rv cells as previously reported in several other cell lines [17,37,38]. In addition, EIQPN reduced the protein levels of both endogenous and exogenous AR-FL and AR-NTD overexpressed in LNCaP, and DU145 but not in PPC1 and HEK 293T cells (Figure 4C-F).
Figure 4.
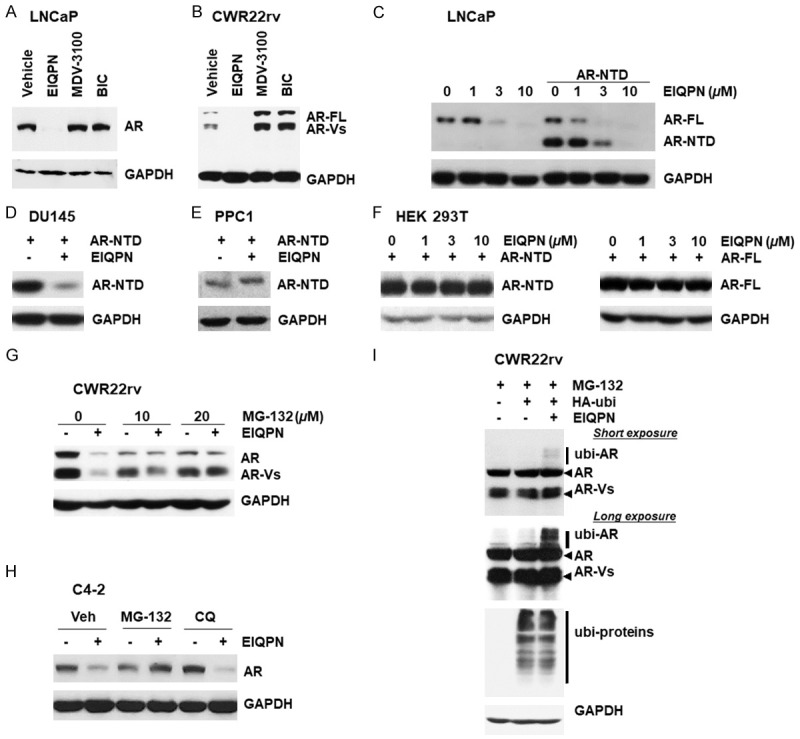
EIQPN reduces the AR protein levels. (A, B) Protein levels of AR and AR-Vs are decreased by EIQPN. LNCaP (A) and CWR22rv (B) cells were incubated with 10 µM EIQPN, MDV-3100, or BIC. Amounts of AR proteins were analyzed by western blotting, with GAPDH as loading control. (C) Western blots show decreased protein levels of endogenous AR and exogenous AR-NTD in LNCaP cells incubated with various doses of EIQPN. (D) Western blots show decreased protein levels of exogenous AR-NTD in DU145 cells incubated with 10 µM EIQPN. (E, F) Western blots show no effects of 10 µM EIQPN on protein levels of exogenous AR-NTD in PPC1 cells (E) and of different doses of EIQPN on exogenous AR-NTD (left) and AR-FL (right) in HEK 293T cells (F). (G, H) Ubiquitin-proteasome inhibitor MG-132 fully recovered AR protein levels decreased by EIQPN. CWR22rv (G) and C4-2 (H) cells cultured with or without 10 µM EIQPN for 16 h were incubated with MG-132 or chloroquine (CQ) for 8 h. (I) EIQPN significantly enhances ubiquitinated AR (ubi-AR) levels. CWR22rv cells were transfected with HA-tagged E3 ubiquitin ligase Mdm2 (HA-ubi) expression construct and incubated with 10 µM EIQPN for 2 h in the presence of 10 µM MG-132.
We then explored the mechanism through which EIQPN decreased AR protein levels. The ubiquitin-proteasome pathway mainly regulates AR protein degradation in prostate cancers, although the lysosomal pathway is also involved in AR protein degradation [8]. Blocking the ubiquitin-proteasome pathway with MG-132, but not the lysosomal pathway with chloroquine (CQ), fully recovered AR protein levels that were decreased by EIQPN in CWR22rv and C4-2 cells (Figure 4G, 4H). These results suggest that protein degradation of AR in cells incubated with EIQPN occurs mainly through the ubiquitin-proteasome pathway.
We investigated EIQPN-induced changes in ubiquitinated AR (ubi-AR) levels in CWR22rv cells transiently transfected with an HA-tagged E3 ubiquitin ligase Mdm2 expression construct to understand more about AR degradation promoted by EIQPN. Levels of ubi-AR were significantly enhanced in cells incubated with, than without 10 µM EIQPN for 2 h (Figure 4I), confirming that EIQPN mediates AR protein degradation via the ubiquitin-proteasome pathway.
Proliferation of prostate cancer cells is inhibited by EIQPN
Because EIQPN negatively affected AR signaling in prostate cancer cells, we explored the effects of EIQPN on the survival and proliferation of various prostate cancer cell lines. The proliferation of LNCaP cells under androgen-independent and -dependent stimulation with IL6 and DHT, respectively, was more effectively inhibited by EIQPN than by MDV-3100 and BIC (Figure 5A, 5B). Furthermore, EIQPN also inhibited the androgen-independent proliferation of castration-resistant (C4-2) and androgen-independent, advanced prostate cancer cells (CWR22rv), with IC50 values of 1.100 and 1.514 µM, respectively (Figure 5C-F). In contrast, MDV-3100 exerted some, whereas BIC exerted no significant effects on the androgen-independent proliferation of both cell lines. Meanwhile, EIQPN, like MDV-3100 and BIC, did not affect the proliferation of AR-negative DU145 prostate cancer cells (Figure 5G). Taken together, these results suggest that EIQPN inhibits the androgen-independent and-dependent proliferation of prostate cancer cells.
Figure 5.
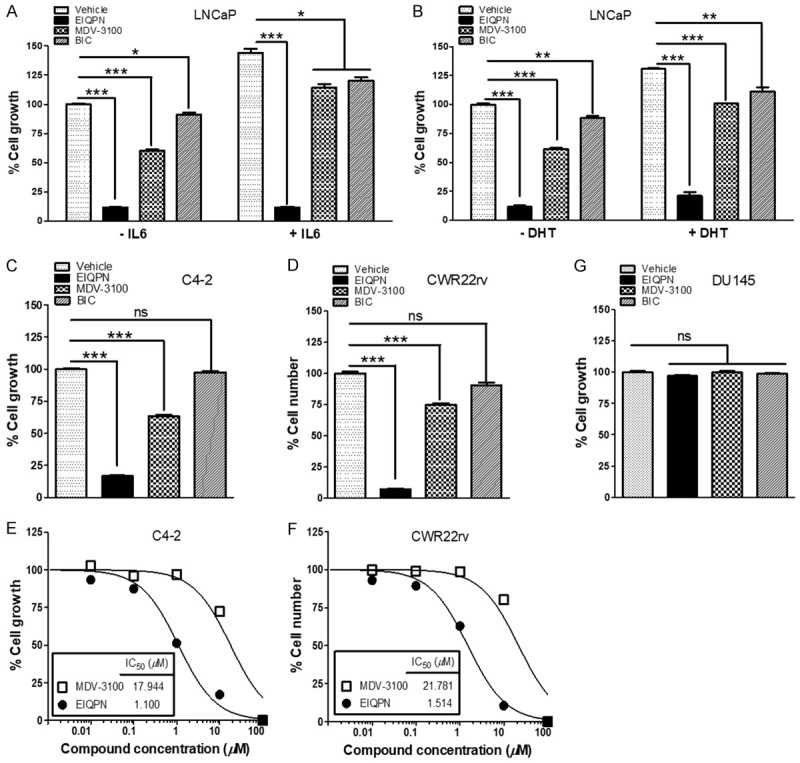
EIQPN inhibits androgen-dependent and -independent growth of AR-positive prostate cancer cells. (A-F) Proliferation of AR-positive prostate cancer cells is inhibited by EIQPN. LNCaP (A, B), C4-2 (C) and CWR22rv (D) cells were incubated for 5 days with 10 µM EIQPN, MDV-3100, or BIC, and with or without 50 ng/mL IL-6 (A) or 1 nM DHT (B). C4-2 (E) and CWR22rv (F) cells were incubated with different doses of EIQPN or MDV-3100. Half maximal inhibitory concentrations (IC50) were determined by nonlinear regression analyses. Cell growth was assessed by MTS assays (A-C and E) or counting cells stained with trypan blue (D and F). (G) Proliferation of AR-negative prostate cancer cells is not affected by EIQPN. DU145 cells were incubated with 10 µM EIQPN, MDV-3100, or BIC, then growth was assessed using MTS assays. Data represent means ± SEM of at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed t-test analysis. ns, not significant.
Formation and growth of CWR22rv xenograft tumors in vivo are inhibited by EIQPN
The effects of EIQPN on prostate tumorigenesis in vivo were examined using CWR22rv xenograft mouse models. Compared with control mice, EIQPN (~10 mg/kg/day, i.p.) caused an approximate 3-week delay in tumor formation and inhibited tumor growth by ~65% and ~90% in intact and castrated mice, respectively (Figure 6A-D). The absence of significant changes in the body and testis weight of intact and castrated mouse models suggest minimal toxicity (Figure 6A, 6B).
Figure 6.

CWR22rv xenograft tumor growth in vivo is inhibited by EIQPN. (A-D) Tumor growth in intact (A, C) and castrated (B, D) mouse xenograft models is prevented by EIQPN. CWR22rv cells were injected into shoulders of intact and castrated 4-week-old, male NOD.CB17-PrkdcSCID/J mice. After 1 week, mice were injected i.p. with 25 mg/kg EIQPN in DMSO:PEG300 (2:8) or vehicle three times per week for 7 weeks. Two days after the final injection, tumor (left), testis (middle) and body (right) weight of intact (A) and castrated (B) mice were assessed. Intact mice, n = 9; castrated mice, n = 7. (C, D) Representative tumors dissected from intact (C) and castrated (D) mice. Scale bars, 1 cm. (E, F) Levels of cleaved caspase 3 (cleaved C3) and AR are significantly increased and decreased, respectively, by EIQPN in tumors dissected from intact (E) and castrated (F) mice. Protein band signals were scanned by Image Studio Lite, and visualized by GraphPad Prism 5.0. (G, H) EIQPN downregulated expression of AR target genes, PSA and TMPRss2, in tumors from intact (G) and castrated (H) mice. Levels of mRNA were analyzed by RT-PCR. Data represent means ± SEM of at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed t-test analysis. ns, not significant.
Molecular analysis of tumors dissected from intact and castrated mice showed that EIQPN significantly increased and reduced protein levels of the apoptosis marker, cleaved caspase 3, and AR, respectively (Figure 6E, 6F). Furthermore, EIQPN also tended to downregulate expression of the AR target genes, PSA and TMPRss2, especially in tumors from castrated mice (Figure 6G, 6H). These results were consistent with the negative effects of EIQPN on AR signaling in prostate cancer cells, which resulted in the inhibition of cell survival and proliferation. These results suggest that EIQPN inhibits the formation and growth of advanced, androgen-independent prostate tumors.
Discussion
Failed castration and antiandrogen therapy are associated with the increased expression of constitutively active AR-Vs without the LBD [16,24,25]; hence, AR-NTD is an alternative target for the development of compounds to treat advanced prostate cancers. EPI-001 interacts with the TAU-5 domain in the AF-1 region of the AR-NTD [36]. It blocks the androgen-independent function of TAU-5 and prevents 433WHTLF437/AF2 interaction, which attenuates androgen-dependent AR transactivation [26,27]. The present study showed that EIQPN interacted with the AR-AF1 domain in the same manner as EPI-001 and caused conformational changes. Such interaction likely prevented AR N/C interaction and/or the recruitment of AR and coactivator SRC-1 to AREs, resulting in the loss of androgen-dependent and -independent AR activity. Additionally, the interaction between EIQPN and the AF-1 domain blocked the nuclear translocation of both androgen-induced AR-FL and androgen-independent AR-NTD.
However, unlike EPI-001, EIQPN interaction with the AF-1 domain not only blocked AR activity, but also induced protein degradation and reduced AR protein levels in most prostate cancer cell lines. The ubiquitin-proteasome pathway is probably the main mechanism through which EIQPN promoted AR protein degradation. Androgen receptors are similarly degraded by EIQPN, galeterone and IRC117539, that is, via the ubiquitin-proteasome pathway [39]. However, > 10 µM galeterone is needed to induce AR degradation in androgen-dependent LNCaP and LAPC4 cells, and AR T878A mutation is necessary for proteasome-dependent degradation [40]. Meanwhile, IRC117539 exerted antiandrogenic effects on the prostate by inducing castration-like histopathological changes. Nevertheless, IRC117539 (25 mg/kg/day for 50 days) reduced LNCaP-derived tumor volumes only by 42% in intact Balb/c nude mice, whereas EIQPN (3 × 25 mg/kg/week (~10 mg/kg/day) for 49 days reduced CWR22rv xenograft tumor weight by ~65% and ~90% in intact and castrated mice, respectively.
The selective and potent AR antagonist UT-155 also behaves like EIQPN. It targets the AR-NTD, promote AR protein degradation potentially through the ubiquitin-proteasome pathway and inhibiting CWR22rv cell growth with an IC50 of ~1 µM [41]. However, UT-155 (100 mg/kg/day s.c.) for 20 days and EIQPN (~10 mg/kg/day i.p.) for 49 days reduced ~25% [41] and ~90%, respectively, of CWR22rv xenograft tumor weight in castrated mice. These findings indicate that EIQPN is comparable with other agents that inhibit AR function and expression as well as prostate cancer cell proliferation.
In conclusion, EIQPN inhibited AR-AF1 activity and reduced AR protein levels, thereby inhibiting prostate cancer cell survival and proliferation. A functional study found that EIQPN delayed and inhibited advanced androgen-independent prostate tumorigenesis in intact and castrated xenograft mouse models in vivo. These findings suggest EIQPN could serve as a novel agent to treat and prevent the recurrence of advanced prostate cancers.
Acknowledgements
We thank Dr. C. Jung for the kind gifts of C4-2 and CWR22rv cell lines. We are also grateful to Drs. J. J. Palvino and E. M. Wilson for providing pARE2-TATA-Luc and GAL4.AR-LBD658-919/VP16.AR1-660/5xGAL4-luc3, respectively. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2014R1A2A1A11051396 and NRF-2020R1A2C1006705).
Disclosure of conflict of interest
None.
Abbreviations
- A (ALA)
alanine
- ADT
androgen-deprivation therapy
- AF-1
activation fragment-1
- AF-2
activation fragment-2
- AR
androgen receptor
- ARE
androgen-response element
- AR-V
androgen receptor variant
- CRPC
castration-resistant prostate cancer
- DBD
DNA-binding domain
- E (GLN)
glutamate
- EIQPN
6-[6-ethoxy-5-ispropoxy-3,4-dihydroisoquinolin-2[1H)-yl]-N-[6-methylpyridin-2-yl]nicotinamide)
- G (GLY)
glycine
- H (HIS)
histidine
- K (LYS)
lysine
- L (LEU)
leucine
- LDB
ligand-binding domain
- NTD
NH2-terminal transactivation domain
- P (PRO)
proline
- PSA
prostate-specific antigen
- TAU
ligand-dependent transcriptional activation unit
- TMPRss2
transmembrane serine protease 2
- W (TRP)
tryptophan
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol. 2015;67:470–479. doi: 10.1016/j.eururo.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohler JL. Castration-recurrent prostate cancer is not androgen-independent. Adv Exp Med Biol. 2008;617:223–234. doi: 10.1007/978-0-387-69080-3_21. [DOI] [PubMed] [Google Scholar]
- 5.Sharifi N. Mechanisms of androgen receptor activation in castration-resistant prostate cancer. Endocrinology. 2013;154:4010–4017. doi: 10.1210/en.2013-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai C, Heemers H, Sharifi N. Androgen signaling in prostate cancer. Cold Spring Harb Perspect Med. 2017;7:a030452. doi: 10.1101/cshperspect.a030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan MH, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. 2015;36:3–23. doi: 10.1038/aps.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Steen T, Tindall DJ, Huang H. Posttranslational modification of the androgen receptor in prostate cancer. Int J Mol Sci. 2013;14:14833–14859. doi: 10.3390/ijms140714833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anbalagan M, Huderson B, Murphy L, Rowan BG. Post-translational modifications of nuclear receptors and human disease. Nucl Recept Signal. 2012;10:e001. doi: 10.1621/nrs.10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dar JA, Masoodi KZ, Eisermann K, Isharwal S, Ai J, Pascal LE, Nelson JB, Wang Z. The N-terminal domain of the androgen receptor drives its nuclear localization in castration-resistant prostate cancer cells. J Steroid Biochem Mol Biol. 2014;143:473–480. doi: 10.1016/j.jsbmb.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bevan CL, Hoare S, Claessens F, Heery DM, Parker MG. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol Cell Biol. 1999;19:8383–8392. doi: 10.1128/mcb.19.12.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995;270:7341–7346. doi: 10.1074/jbc.270.13.7341. [DOI] [PubMed] [Google Scholar]
- 13.Dai Y, Ngo D, Forman LW, Qin DC, Jacob J, Faller DV. Sirtuin 1 is required for antagonist-induced transcriptional repression of androgen-responsive genes by the androgen receptor. Mol Endocrinol. 2007;21:1807–1821. doi: 10.1210/me.2006-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 15.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12:1665–1671. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 16.Sprenger CC, Plymate SR. The link between androgen receptor splice variants and castration-resistant prostate cancer. Horm Cancer. 2014;5:207–217. doi: 10.1007/s12672-014-0177-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2014;33:3140–3150. doi: 10.1038/onc.2013.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sette C. Alternative splicing programs in prostate cancer. Int J Cell Biol. 2013;2013:458727. doi: 10.1155/2013/458727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ang JE, Olmos D, de Bono JS. CYP17 blockade by abiraterone: further evidence for frequent continued hormone-dependence in castration-resistant prostate cancer. Br J Cancer. 2009;100:671. doi: 10.1038/sj.bjc.6604904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, Larson S, Fleisher M, Sawyers CL. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed A, Ali S, Sarkar FH. Advances in androgen receptor targeted therapy for prostate cancer. J Cell Physiol. 2014;229:271–276. doi: 10.1002/jcp.24456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vander Ark A, Cao J, Li X. Mechanisms and approaches for overcoming enzalutamide resistance in prostate cancer. Front Oncol. 2018;8:180. doi: 10.3389/fonc.2018.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wadosky KM, Koochekpour S. Androgen receptor splice variants and prostate cancer: from bench to bedside. Oncotarget. 2017;8:18550–18576. doi: 10.18632/oncotarget.14537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang G, Sadar MD. Amino-terminus domain of the androgen receptor as a molecular target to prevent the hormonal progression of prostate cancer. J Cell Biochem. 2006;98:36–53. doi: 10.1002/jcb.20802. [DOI] [PubMed] [Google Scholar]
- 26.Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, McEwan IJ, Plymate S, Andersen RJ, Sadar MD. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948–2960. doi: 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Banuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 28.Song CH, Yang SH, Park E, Cho SH, Gong EY, Khadka DB, Cho WJ, Lee K. Structure-based virtual screening and identification of a novel androgen receptor antagonist. J Biol Chem. 2012;287:30769–30780. doi: 10.1074/jbc.M112.379107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang SH, Song CH, Van HTM, Park E, Khadka DB, Gong EY, Lee K, Cho WJ. SAR based design of nicotinamides as a novel class of androgen receptor antagonists for prostate cancer. J Med Chem. 2013;56:3414–3418. doi: 10.1021/jm3014103. [DOI] [PubMed] [Google Scholar]
- 30.Hong CY, Park JH, Ahn RS, Im SY, Choi HS, Soh J, Mellon SH, Lee K. Molecular mechanism of suppression of testicular steroidogenesis by proinflammatory cytokine tumor necrosis factor alpha. Mol Cell Biol. 2004;24:2593–2604. doi: 10.1128/MCB.24.7.2593-2604.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyd KE, Farnham PJ. Coexamination of site-specific transcription factor binding and promoter activity in living cells. Mol Cell Biol. 1999;19:8393–8399. doi: 10.1128/mcb.19.12.8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Bellis A, Quigley CA, Cariello NF, el-Awady MK, Sar M, Lane MV, Wilson EM, French FS. Single base mutations in the human androgen receptor gene causing complete androgen insensitivity: rapid detection by a modified denaturing gradient gel electrophoresis technique. Mol Endocrinol. 1992;6:1909–1920. doi: 10.1210/mend.6.11.1480178. [DOI] [PubMed] [Google Scholar]
- 33.Hoang HN, Tran TT, Jung CH. The activation of glycerol dehydrogenase from escherichia coli by ppGpp. Bull Korean Chem Soc. 2020;41:133–138. [Google Scholar]
- 34.Reid J, Kelly SM, Watt K, Price NC, McEwan IJ. Conformational analysis of the androgen receptor amino-terminal domain involved in transactivation. Influence of structure-stabilizing solutes and protein-protein interactions. J Biol Chem. 2002;277:20079–20086. doi: 10.1074/jbc.M201003200. [DOI] [PubMed] [Google Scholar]
- 35.Lavery DN, McEwan IJ. Structural characterization of the native NH2-terminal transactivation domain of the human androgen receptor: a collapsed disordered conformation underlies structural plasticity and protein-induced folding. Biochemistry. 2008;47:3360–3369. doi: 10.1021/bi702221e. [DOI] [PubMed] [Google Scholar]
- 36.De Mol E, Fenwick RB, Phang CTW, Buzón V, Szulc E, de la Fuente A, Escobedo A, García J, Bertoncini CW, Estébanez-Perpiñá E, McEwan IJ, Riera A, Salvatella X. EPI-001, a compound active against castration-resistant prostate cancer, targets transactivation unit 5 of the androgen receptor. ACS Chem Biol. 2016;11:2499–2505. doi: 10.1021/acschembio.6b00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kregel S, Chen JL, Tom W, Krishnan V, Kach J, Brechka H, Fessenden TB, Isikbay M, Paner GP, Szmulewitz RZ, Vander Griend DJ. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget. 2016;7:26259–74. doi: 10.18632/oncotarget.8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuruma H, Matsumoto H, Shiota M, Bishop J, Lamoureux F, Thomas C, Briere D, Los G, Gleave M, Fanjul A, Zoubeidi A. A novel antiandrogen, compound 30, suppresses castration-resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo. Mol Cancer Ther. 2013;12:567–76. doi: 10.1158/1535-7163.MCT-12-0798. [DOI] [PubMed] [Google Scholar]
- 39.Auvin S, Öztürk H, Abaci YT, Mautino G, Meyer-Losic F, Jollivet F, Bashir T, de Thé H, Sahin U. A molecule inducing androgen receptor degradation and selectively targeting prostate cancer cells. Life Sci Alliance. 2019;2:e201800213. doi: 10.26508/lsa.201800213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alyamani M, Li Z, Berk M, Li J, Tang J, Upadhyay S, Auchus RJ, Sharifi N. Steroidogenic metabolism of galeterone reveals a diversity of biochemical activities. Cell Chem Biol. 2017;24:825–832. e826. doi: 10.1016/j.chembiol.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ponnusamy S, Coss CC, Thiyagarajan T, Watts K, Hwang DJ, He Y, Selth LA, McEwan IJ, Duke CB, Pagadala J, Singh G, Wake RW, Ledbetter C, Tilley WD, Moldoveanu T, Dalton JT, Miller DD, Narayanan R. Novel selective agents for the degradation of androgen receptor variants to treat castration-resistant prostate cancer. Cancer Res. 2017;77:6282–6298. doi: 10.1158/0008-5472.CAN-17-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]