

Abstract
EGFR tyrosine kinase inhibitors (TKIs) are the first-line drugs for NSCLC. But, the acquired resistance limited their efficacy, so that the patients deteriorate eventually. Therefore, it is necessary to clarify the mechanism of the acquired resistance and overcome it for effective NSCLC therapy. In this experimental study, a stable gefitinib resistant lung adenocarcinoma cell line (PC9/GR) infected with shRNA-c-kit-homo-1386 were established; c-kit siRNA and c-kit inhibitors were used to block c-kit signaling; the acquired resistance of PC9/GR cells and the effects of c-kit siRNA and c-kit inhibitors on the growth and invasion of PC9/GR cells were investigated with CCK-8 assay, colony formation and cell invasion assays in vitro; the tumor growth inhibition effects of c-kit inhibitors on PC9/GR cell generated tumors were tested in vivo; the mechanisms involved in the acquired resistance reverse, growth and invasion inhibition effects of c-kit siRNA and c-kit inhibitors on PC9/GR cells were evaluated with qRT-PCR, Western blot and immunohistochemistry staining. The proliferation, colony formation, and invasion of PC9/GR cells were decreased by c-kit siRNA and inhibitors in vitro significantly; c-kit inhibitors suppressed the tumor growth of PC9/GR cell generated tumors in vivo. In the stable shRNA-c-kit transfected PC9/GR cells, the protein expressions of c-kit signaling and stemness phenotype related proteins, including ALDH1A1, Oct4, Sox2 and ABCG2 were decreased, and EMT phenotype related protein expressions including vimentin, N-cadherin, and Slug, were downregulated and with upregulation of E-cadherin; c-kit inhibitors reduced stemness phenotype related protein expressions, downregulated EMT phenotype related protein expressions including vimentin, N-cadherin, and Slug, with upregulation of E-cadherin, and the stemness related protein expressions of c-kit, ALDH1A1, ABCG2 and EMT-related proteins of vimentin and slug were decreased in the imatinib treated tumor tissues. The findings of this study indicated that c-kit signaling mediated the acquired gefitinib resistance, cell growth, invasion, stemness and EMT phenotype of PC9/GR cells. Targeting c-kit signaling with c-kit siRNA and small molecule c-kit inhibitors might overcome the acquired gefitinib resistance, and inhibit PC9/GR cell growth in vitro and in vivo.
Keywords: c-kit, NSCLC, gefitinib resistance, growth inhibition, stemness, EMT
Introduction
Lung cancer is one of the common malignant tumors and has become the main cause of human death from cancers [1,2]. It is classified as non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), according to the pathological characteristics. NSCLC accounts for more than 85% of all lung cancer cases [3]. NSCLC patients carrying an epidermal growth factor receptor (EGFR) mutation often initially have response to EGFR tyrosine kinase inhibitor (EGFR-TKI) treatment; the therapeutic outcomes of patients with NSCLC were improved greatly, compared to conventional chemotherapy. But unfortunately, the patients treated with EGFR-TKIs may acquire drug resistance due to many factors [3,4]. Gefitinib, one of epidermal-growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) has been used as the front-line treatment for non-small cell lung cancer (NSCLC) therapy, it is effective for NSCLC patients with EGFR sensitive mutations who can tolerate the side effects of chemotherapy [5-7]. Most patients are sensitive when they received primary EGFR-TKI treatment, and tumor regression was obvious, but after 8-10 months of progression-free survival, the patients got resistant to EGFR-TKI drugs inevitably [8,9]. This kind of drug resistance also occurred in the third-generation TKI treatment, which led to tumor reoccurrence. Therefore, it is very important to clarify the mechanism of EGFR-TKI resistance and explore the treatment strategy for EGFR-TKI resistant NSCLC patients [7].
Previous investigations showed that the mechanism of EGFR-TKI resistance might be associated with target gene mutation, bypass signaling pathway activation, histological transformation [10], acquisition of stem-like properties and elevated expression cancer stem cell markers [11,12], epithelial-mesenchymal transition (EMT) [13-15] and PI3K/Akt/mTOR signaling pathway activation [16]. Although these mechanisms seem to be responsible for acquired resistance, all mechanisms have not been revealed [11].
Among the potential mechanisms of EGFR-TKI resistance [9-16], cancer stem cell properties seem to play a substantial role [17]. Cancer stem cells in tumor microenvironment possess stemness phenotype, having potential for multiple differentiation, selfrenewal, and epithelial-mesenchymal transition, which are responsible for tumor initiation, drug resistance, cancer relapse and metastasis [12-15,17,18]. Thus, targeting the cancer stem cells in NSCLC and attenuating their stemness properties and epithelial-mesenchymal transition may overcome the drug resistance, and prevent from tumor growth and metastasis [17-20].
The c-kit gene is a proto-oncogene; its product is a transmembrane glycoprotein, which belongs to the receptor tyrosine kinase family [21]. Previous studies showed that c-kit gene expression products and their ligands are important regulators for the growth and development of various human tissue cells, and are closely related to the occurrence of various solid tumors and leukemia [22,23]. c-kit expression is correlated with poor prognosis, faster tumor growth, and early lymph node metastasis in NSCLC [24]. Recent investigations indicated that the proportion of lung cancer stem cells in EGFR/TKI-resistant cell lines increased significantly; SCF/c-kit expression was upregulated in NSCLC CSCs, which is related to the stemness phenotype, survival of lung CSCs, drug resistance, recurrence and metastasis of NSCLC, blocking SCF-c-kit signaling could improve the antitumor efficacy of chemotherapy of NSCLC [25,26]. Furthermore, the tumor xenografts of human skin squamous cell carcinoma (A431) developed drug resistance after 25-week consecutive gefitinib therapy, resulting from c-kit overexpression [27]; and c-kit was overexpressed in afatinib resistant H1975 lung cancer cell clones [28]; It was suggested that c-kit might be involved in EGFR-TKIs resistance in NSCLC therapeutics.
In light of the research results mentioned above, it could be speculated that c-kit plays an important role in gefitinib resistance for NSCLC therapy. But, its exact effect and mechanism of activity is unknown. To confirm this speculation, this experimental study was performed with the parent non-small cell lung cancer cell line PC9 (with EGFR 19 exon mutation) and its gefitinib resistant pattern cell line, PC9/GR to test whether targeting c-kit could inhibit gefitinib resistant NSCLC cell growth in vitro and in vivo and to explore its potential mechanism of action.
Materials and methods
Cell lines and cell culture
The NSCLC cell line PC9 was were purchased from Shanghai Cell Bank, Chinese Academy of Sciences (Shanghai, China), cultured in 1640 medium supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2. Gefitinib-resistant lung cancer PC9/GR cell line was generated from the parental PC9 cells and was kindly presented by Professor Luo Feng (Lung Cancer Center, Laboratory of Lung Cancer, West China Hospital of Sichuan University). Briefly, PC9 cells were cultured in the medium supplemented with 2 μM gefitinib for six months to acquire resistance to gefitinib, and the drug resistance was measured through CCK-8 assay [29].
Reagents and antibodies
Gefitinib (ZD1839), cisplatin (S1166), docetaxel (S1148), imatinib (S2475) and anlotinib (S8726) were purchased from Selleck Chemicals (Houston, TX, USA). Reagents were prepared and stored according to the manufacturer’s protocols. The following primary antibodies were used: Rabbit mAb GAPDH (2118), c-kit (3074), SCF (2093), P-c-Kit (Tyr703, 3073), P- Erk1/2 (4370), ALDH1A1 (36671), Oct4 (2890), Sox2 (3579), vimentin (5741), E-cadherin (3195), N-cadherin (13116), Slug (9585), and ABCG2 (42078) from Cell Signaling Technology (Danvers, MA). The secondary HRP-conjugated goat anti-rabbit IgG (#CW0103S) was purchased from Beijing ComWin Biotech Co., Ltd (Beijing, China).
Cell viability assay
The cell viability was determined by Cell Counting Kit-8 (CCK-8, Dojindo, Japan), according to the manufacturer’s protocols. 3000 PC9 or PC9/GR cells were seeded into the 96-well plates, After 24 h of incubation; the cells were exposed to various concentrations of test agents as indicated for 72 h. Then, the absorbance was measured at 450 nm was measured by a Microplate Reader (SpectraMax 190, Molecular Device, USA). Cell viability was calculated as the percentage of absorbance, comparing treated cells with untreated cells, and three independent experiments were repeated.
Colony formation assay
PC9 or PC9/GR or modified PC9/GR cells were inoculated into six-well plates at 300 cells per well, respectively, incubated for 12-14 days, and terminated in the presence of macroscopic clones. The cells were fixed in 4% paraformaldehyde for 15 minutes, stained with 0.1% crystal violet for 10-20 min, the cell aggregates with ≥50 cells were scored as a colony, and the data was analyzed. Three independent experiments were performed.
Cell invasion assay
The cells were harvested and re-suspended in serum-free medium as single cell solutions. The filters of 24-transwell with 8 μm pores (Corning, Costar, USA), were pre-coated with matrigel (BD Biosciences, NJ, USA). 200 μl cell suspension containing 20,000 cells was loaded into the upper chambers; 500 µl medium with 10% FBS was added into the lower chamber as a chemoattractant. After 24 h incubation at 37°C, the cells on the upside of the filters were removed using a cotton swab, and the cells that penetrated through the filter were fixed with 4% paraformaldehyde for 10 min and stained with 0.1% crystal violet for 10 min. Images were taken under an optical phase contrast microscope. The penetrated cells in 6 non-overlapping random fields per well were counted. Three independent experiments were repeated.
Cell transfection
Small interfering RNA (siRNA) targeting c-kit including siRNA-c-kit-homo-1386, siRNA-c-kit-homo-365, siRNA-c-kit-homo-1684, and negative control siRNA (si-NC) were obtained from GenePharma (Shanghai, China). PC9/GR cells were seeded at a density of 1×105 cells per well in a 6-well microplate and incubated for 24 h. Then siRNA-c-kit or siRNA-NC (30 nM) was transfected into PC9/GR cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. After 48 h of siRNA transfection, transfection efficiency was evaluated using qRT-PCR and Western blot. The experiment was performed in triplicate.
Vector construction and transfection
The lentiviral vector of c-kit interference was constructed by inserting a shRNA-c-kit-homo-1386 CCCAGAGCCCACAATAGAT and shRNA-c-kit-NC TTCTCCGAACGTGTCACGT fragment into a lentiviral shuttle vector (LV3/GFP). c-kit knockdown was achieved using specific shRNA-c-kit-homo-186 targeting c-kit. The packing and purification of the lentiviral vectors were performed by the GenePharma Company (Shanghai, China). The stable cells infected with the lentiviral vectors were screened with 50 µg/ml puromycin for 2 weeks. The transfection efficiency was measured using Western blot. The stale shRNA-c-kit-homo-1386 transfected PC9/ER cells were used for the further experiments.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from PC9/GR and PC9/GR transfected with c-kit siRNA cells by TRIzol reagent (Life Technologies, Shanghai, China) according to manufacturer’s instruction. cDNA was synthesized using the PrimeScript RT kit (GeneCopoeia, Inc., USA) from 1 μg of RNA. The relative quantification of transcripts data calculation were performed using the ΔΔCt method based on SYBR Green Master Mix (GeneCopoeia, Inc., USA) and LightCycler 480 device (Roche Diagnostics). Expression of target genes in the transcript was normalized using GAPDH expression levels. No nonspecific products were detected. The primer sequences are as follows: c-kit-1386: forward, 5’-CCCACAGCCCACAAUAGAUTT-3’ and reverse, 5’-AUCUAUUGUGGGCUCUGGGTT-3’; c-kit-365: forward, 5’-GCAAAUACACGUGCACCAATTT-3’ and reverse, 5’-UUGGUGCACGUGUAUUUGCTT-3’; c-kit-1684: forward, 5’-GCUGGCAUGAUGUGCAUUATT-3’ and reverse, 5’-UAAUGCACAUCAUGCCAGCTT-3’.
Western blot
Proteins were extracted with RIPA buffer and protein protease inhibitor (Beyotime Institute of Biotechnology, Nanjing, China) from cells. Protein concentration was determined using BCA protein assay kit (Beyotime Institute of Biotechnology, Nanjing, China). The different proteins were separated by 10-15% SDS-PAGE and transferred onto PVDF membranes. The membranes carrying proteins were blocked with 5% milk medium for 1 h, and then incubated with specific primary antibodies overnight at 4°C, and antibody recognition was detected with HRP-labeled goat anti-rabbit secondary antibody for 2 hours. The protein bands were visualized with chemiluminescence system (Millipore, Billerica, MA). The densitometry of the western blot bands were quantified by Image J software, and normalized to GAPDH.
PC9/GR cell growth inhibition test in vivo
24 five-week-old male nude mice (BALB/C-nu/nu) were inoculated with 1×106 PC9/GR cells. One week later, the tumor bearing mice were divided into 4 groups randomly and each group containing 6 mice, three test group mice were administrated with 75 mg/kg of gefitinib, 80 mg/kg of imatinib and 10 mg/kg of anlotinib i.p., 3 times weekly for 4 weeks, respectively; the control mice were injected with the same volume of normal saline; In the experimental process, animals were weighted, and tumor size were measured every two days, tumor volumes were calculated using a standard formula (length × width2 ×0.5) [30]. At the end of experiment, the mice were sacrificed by carbon dioxide asphyxiation. The tumor masses were dissected and weighed. The tumor inhibitory rates were calculated using the formula: tumor inhibitory rate (%) = (mean tumor weight of the control group-mean tumor weight of the treated group)/mean tumor weight of the control group ×100%. All animals were maintained under standard conditions according to the guidelines of the Institutional Animal Care and Use Committee of Sichuan University; the animal experiments were performed with an approved protocol by this committee (Permit Number: 2020188A).
Immunohistochemistry staining
The tumor tissues of the control and imatinib treated groups were fixed in 4% paraformaldehyde for 24 h, embedded with paraffin and sliced into 4 μm sections. The sections were dewaxed and hydrated. The tumor tissues were incubated with primary antibodies, including, rabbit mAb c-kit, ALDH1A1, vimentin, Slug, and ABCG2 overnight at 4°C, followed by incubation with abiotinylated goat anti-rabbit secondary antibody and diaminobenzidine substrate. And then, the slides were counterstained with hematoxylin. Staining results were observed under microscope, 3 images were taken randomly, and the average optical density (AOD) was measured with Image-pro Plus 6.0.
Statistical analysis
All data were presented as mean ± SED, and analyzed with SPSS19.0, Image-pro Plus 6.0 and GraphPad Prism 8.0. Statistical significance was determined with two-tailed student’s t test or 1-way analysis of variance (ANOVA). P values ≤ 0.05 (*) and ≤ 0.01 (**) were considered statistically significant for all tests.
Results
The biological characterization of gefitinib resistant NSCLC cells (PC9/GR) is different from that of the parent PC9 cells
The parent PC9 cells maintained epithelial morphology (Figure 1A), while PC9/GR cells exhibited mesenchymal morphology (Figure 1B); PC9/G cells were more resistant to gefitinib than its parental PC9 cells, the IC50 was increased significantly; the drug resistance index is 40 (Figure 1C; P<0.01); PC9/GR cells were also less sensitive to cisplatin and docetaxel (Figure 1D, 1E; P<0.01) and generated more colonies (Figure 1F; P<0.01) compared to the parent PC9 cells; the expressions of stemness related proteins, including ALDH1A1, Oct4, Sox2, and drug resistance protein, ABCG2 were upregulated significantly (Figure 1G, 1H; P<0.01); the invasiveness of PC9/GR cells was increased significantly (Figure 1I-K; P<0.01), with upregulation of EMT related proteins, including vimentin, N-cadherin, and Slug, and downregulation of E-cadherin (Figure 1L, 1M; P<0.01) compared to the parent PC9 cells.
Figure 1.
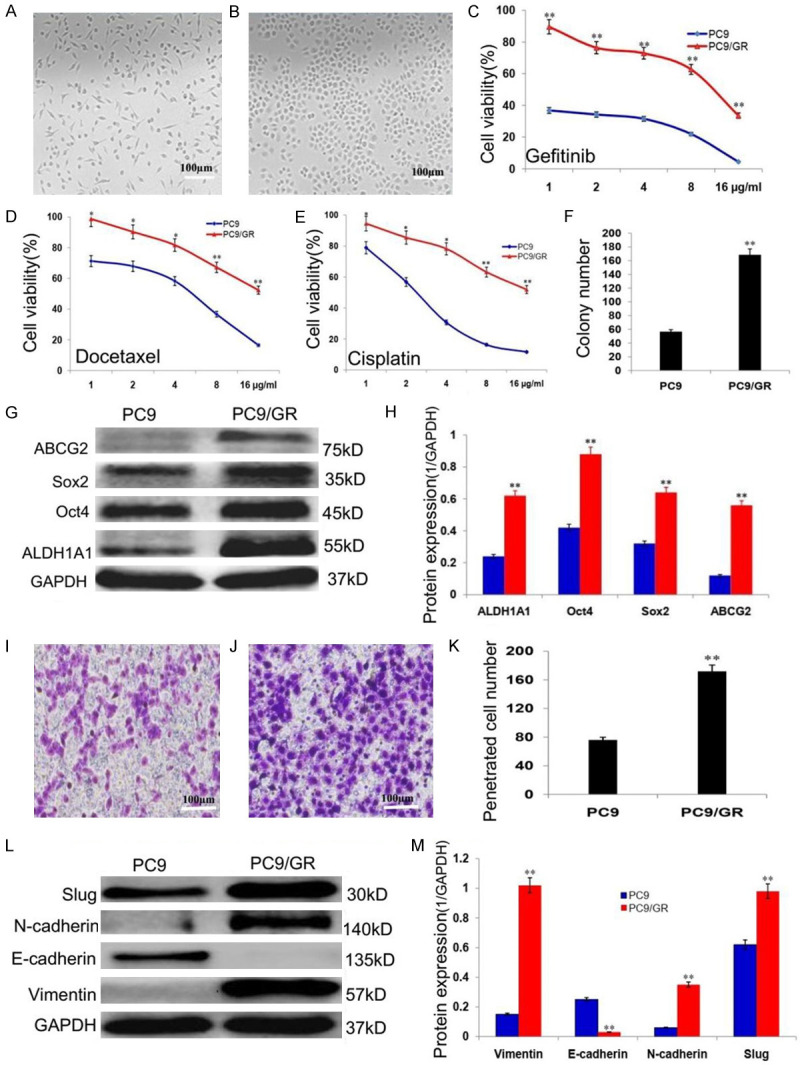
The biological characterization of the gefitinib resistant lung adenocarcinoma cells (PC9/GR). A. The epithelial morphology of lung adenocarcinoma cells (PC9). B. The mesenchymal morphology of the gefitinib resistant lung adenocarcinoma cells (PC9/GR). C. The sensitivities of PC9 and PC9/GR cells to gefitinib. D. The sensitivities of PC9 and PC9/GR cells to cisplatin. E. The sensitivities of PC9 and PC9/GR cells to docetaxel. F. The histogram shows the PC9 and PC9/GR cells generated colony numbers. G. Western blot analysis of the stemness related protein expressions of ALDH1A1, Oct4, Sox2 and ABCG2 in PC9 and PC9/GR cells. H. The histogram shows the significant increase of stemness protein expressions in PC9/GR cells, compared to the parent PC9 cells. I. The PC9 cells penetrated transwell membrane. J. The PC9/GR cells penetrated transwell membrane. K. The histogram shows the penetrated cell number of PC9 and PC9/GR cells. L. Western blot analysis of the EMT related protein expressions of vimentin, N-cadherin, Slug and E-cadherin in PC9 and PC9/GR cells. M. Histogram shows the significant increase of vimentin, N-cadherin, Slug protein expressions and decrease of E-cadherin in PC9/GR cells, compared to the parent PC9 cells. *P<0.05, **P<0.01.
c-kit signaling activation in PC9/GR cells and c-kit RNA interference
c-kit, P-c-kit, SCF and P-Erk1/2 protein expressions were increased in PC9/GR cells significantly (Figure 2A-C; P<0.01), compared to the parent PC9 cells, which indicated that c-kit signaling pathway was activated in PC9/GR cells; siRNA-c-kit-homo-1386, -365, and -1684 exhibited down-regulations of c-kit mRNA and protein expression (Figure 2D, 2E) to varying degrees, while siRNA-c-kit-homo-1386 was the most effective one on down-regulations of c-kit mRNA and protein expressions (Figure 2D, 2E; P<0.01); therefore, C9/GR cells were transfected with siRNA-c-kit-homo-1386 (shc-kit), the stable shc-kit transfected cells (PC9/GR-shc-kit) were screened. c-kit, P-c-kit, SCF and P-Erk1/2 protein expressions were decreased in the stable shc-kit transfected PC9/GR cells significantly, compared to the control PC9/GR cells (Figure 2F-H; P<0.01).
Figure 2.

c-kit signaling activation in PC9/GR cells and c-kit RNA interference. A. Western blot analysis and the histogram show the c-kit protein expression was increased in PC9/GR cells, compared to the parent PC9 cells. B. Western blot analysis of c-kit signaling related protein expressions of P-c-kit, SCF and P-Erk1/2 in PC9 and PC9/GR cells. C. The histogram shows the significant increase of P-c-kit, SCF and P-Erk1/2 protein expressions in PC9/GR cells, compared to the parent PC9 cells. D. The histogram shows the relative c-kit mRNA expressions after interference with siRNA-c-kit-homo-1386, -365, and -1684 in PC9/GR cells. E. Western blot analysis and the histogram show the relative c-kit protein expressions after interference with siRNA-c-kit-homo-1386, -365, and -1684 in PC9/GR cells. F. Western blot analysis and the histogram show the c-kit protein expression was decreased in PC9/GR-shc-kit cells. G. Western blot analysis of c-kit signaling related protein expressions of P-c-kit, SCF and P-Erk1/2 in PC9/GR and PC9/GR-shc-kit cells. H. The histogram shows the significant increase of P-c-kit, SCF and P-Erk1/2 in PC9/GR-shc-kit cells cells, compared to the control PC9/GR cells. *P<0.05, **P<0.01.
The effects of c-kit RNA interference on the growth and invasion of PC9/GR cells
The resistance to gefitinib and colony formation efficiency of the stable shRNA-c-kit transfected PC9/GR cells (PC9/GR-shc-kit) were decreased significantly (Figure 3A, 3B; P<0.01); and the expressions stemness-related proteins, ALDH1A1, Oct4, Sox2 and/or drug resistance protein, ABCG2 were decreased significantly (Figure 3C, 3D; P<0.01) compared to the control PC9/GR cells; the invasiveness of PC9/GR-shc-kit cells was reduced significantly (Figure 3E-G; P<0.01), with down-regulation of EMT related proteins, including vimentin, N-cadherin, and Slug, and upregulation of E-cadherin (Figure 3H, 3I; P<0.01), compared to the control PC9/GR cells.
Figure 3.
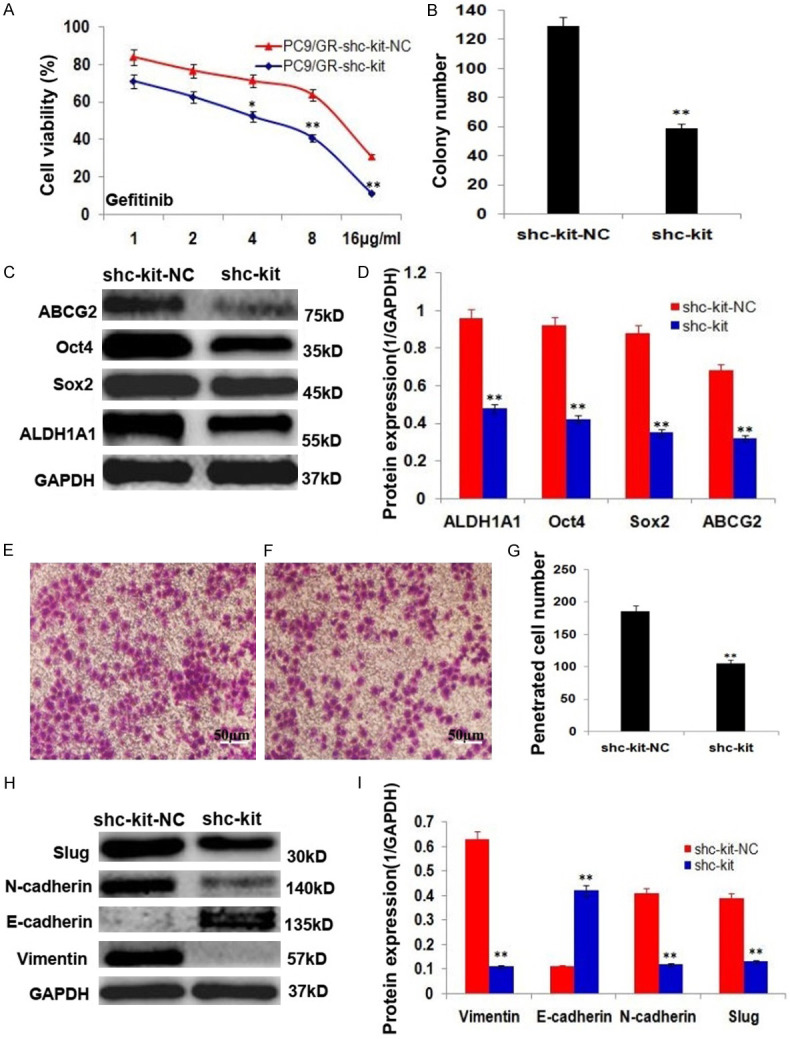
c-kit RNA interference inhibited the growth and invasion of PC9/GR cells and its mechanisms of action. A. PC9/GR-shc-kit cells were more sensitive to gefitinib than PC9/GR cells. B. The histogram shows the control PC9/GR and PC9/GR-shc-kit cells generated colony numbers. C. Western blot analysis of the stemness related protein expressions of ALDH1A1, Oct4, Sox2 and ABCG2 in the control PC9/GR and PC9/GR-shc-kit cells. D. The histogram shows the significant decrease of stemness protein expressions in PC9/GR-shc-kit cells, compared to the control PC9/GR cells. E. The PC9/GR cells penetrated transwell membrane. F. The PC9/GR-shc-kit cells penetrated transwell membrane. G. The histogram shows the penetrated cell number of the control PC9/GR and PC9/GR-shc-kit cells. H. Western blot analysis of the EMT related protein expressions of vimentin, N-cadherin, Slug and E-cadherin in the control PC9/GR and PC9/GR-shc-kit cells. I. The histogram shows the significant decrease of vimentin, N-cadherin, Slug protein expressions and increase of E-cadherin in PC9/GR-shc-kit cells, compared to the control PC9/GR cells. *P<0.05, **P<0.01.
The effects of c-kit inhibitors (imatinib and anlotinib) on the growth and invasion of PC9/GR cells
The parent PC9 is most sensitive to c-kit inhibitors (imatinib and anlotinib) among the parent PC9, PC9/GR and PC9/GR-shc-kit cells, PC9/GR-shc-kit cells were more sensitive than PC9/GR cells, exhibited lower cell growth and colony formation efficiency (Figure 4A-D; P<0.01), and the expressions stemness-related proteins, including ALDH1A1, Oct4, Sox2 and/or drug resistance protein, ABCG2 were reduced significantly (Figure 4E, 4F; P<0.01) compared to the control PC9/GR cells; imatinib and anlotinib suppressed the invasion of PC9/GR cells (Figure 5A-D; P<0.01), with decreasing expression of EMT related proteins, including vimentin, N-cadherin, and Slug, and increasing E-cadherin protein expression (Figure 5E, 5F; P<0.01).
Figure 4.

c-kit inhibitors (imatinib and anlotinib) inhibited the growth of PC9/GR cells and their mechanisms of action. A. Imatinib inhibited the growth of the parent PC9, the control PC9/GR and PC9/GR-shc-kit cells in vitro (*, PC9/GRvs. PC9/GR; #, PC9/GR-shc-kit vs. PC9/GR). B. The histogram shows the imatinib treated PC9/GR cells generated colony numbers. C. Anlotinib inhibited the growth of the parent PC9, the control PC9/GR and PC9/GR-shc-kit cells in vitro (*, PC9/GRvs. PC9/GR; #, PC9/GR-shc-kit vs. PC9/GR). D. The histogram shows the anlotinib treated PC9/GR cells generated colony numbers. E. Western blot analysis of the stemness related protein expressions of ALDH1A1, Oct4, Sox2 and ABCG2 in imatinib and anlotinib treated PC9/GR cells. F. The histogram shows the significant decrease of stemness protein expressions in imatinib and anlotinib treated PC9/GR cells, compared to the control PC9/GR cells.
Figure 5.
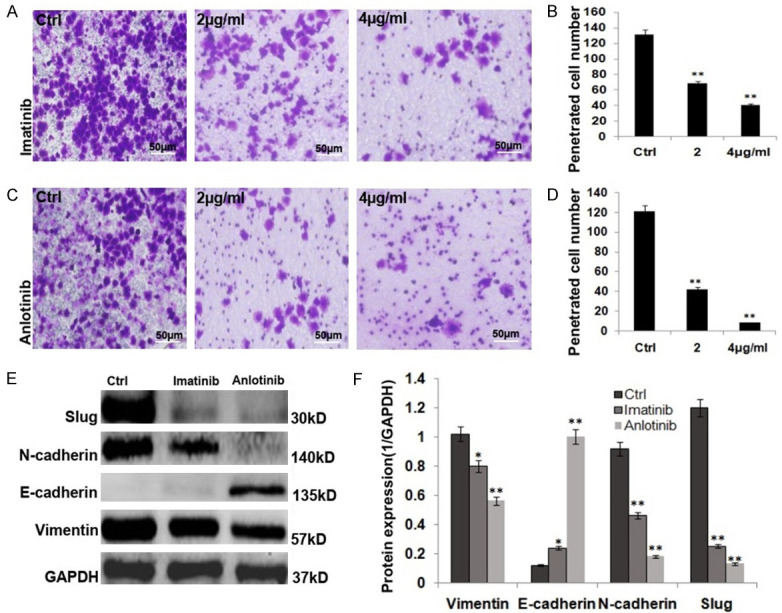
c-kit inhibitors (imatinib and anlotinib) inhibited the invasion of PC9/GR cells and their mechanisms of action. A. Imatinib inhibited the invasion of PC9/GR cells. B. The histogram shows the penetrated cell number of the imatinib treated PC9/GR cells. C. Anlotinib inhibited the invasion of PC9/GR cells. D. The histogram shows the penetrated cell number of the anlotinib treated PC9/GR cells. E. Western blot analysis of the EMT related protein expressions of vimentin, N-cadherin, Slug and E-cadherin in the imatinib and anlotinib treated PC9/GR. F. The histogram shows the significant decrease of vimentin, N-cadherin, Slug protein expressions and increase of E-cadherin in the imatinib and anlotinib treated PC9/GR cells.
c-kit inhibitors (imatinib and anlotinib) inhibited PC9/GR cell generated tumor growth in vivo
In vivo experiment, gefitinib, anlotinib and imatinib inhibited the growth of PC9/GR cell generated tumors (Figure 6). The mean tumor weight of control, gefitinib, imatinib, and anlotinib treated groups were 1.04±0.48, 0.77±0.32, 0.14±0.02, 0.41±0.11 g (Figure 6C, 6D), respectively; the tumor inhibitory rate of gefitinib, anlotinib and imatinib was 26.0% (P>0.05), 86.5% (P<0.01), 60.6% (P<0.01), respectively. No significant difference of mean body weight increase between the control and treated animals was observed (Figure 6A).
Figure 6.

c-kit inhibitors (imatinib and anlotinib) inhibited PC9/GR cell generated tumor growth. A. The mean body weight of each group mice. B. Tumor growth of each group mice. C. Tumor masses of each group mice. D. The histogram shows the mean tumor weight of each group. *P<0.05, **P<0.01.
Immunohistochemistry staining showed that the stemness related protein expressions of c-kit, ALDH1A1, ABCG2 and EMT-related proteins of vimentin and slug were lower in the tumor tissues of the imatinib treated group, compared with those in the tumor tissues of control group (Figure 7).
Figure 7.
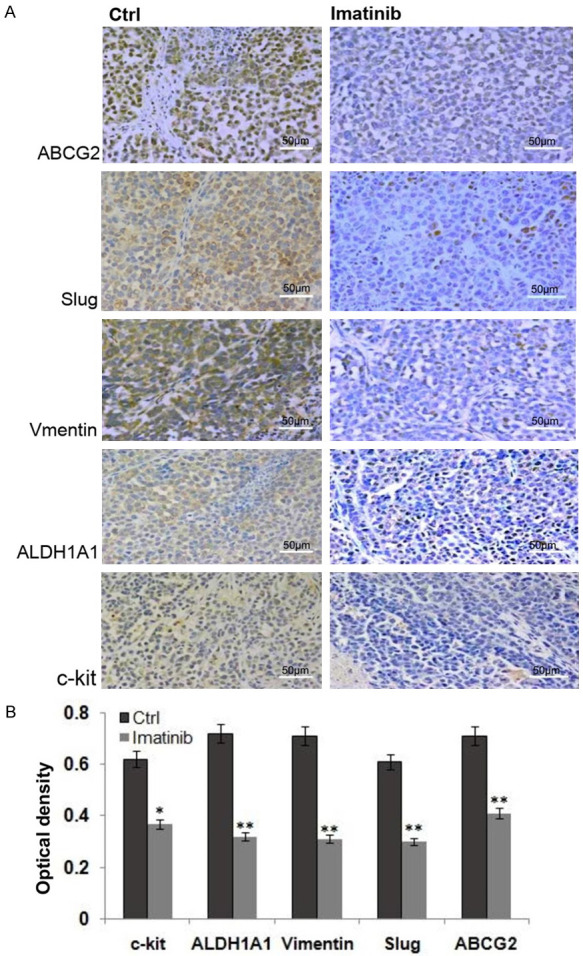
Immunohistochemistry staining the tumor tissues of the control and the imatinib treated group. A. Immunohistochemistry staining c-kit, ALDH1A1, ABCG2 and EMT-related proteins of vimentin and slug in the tumor tissues of the control and the imatinib treated group. B. The histogram shows that the stemness related protein expressions of c-kit, ALDH1A1, ABCG2 and EMT-related proteins of vimentin and slug in the tumor tissues of the imatinib treated group were lower than those in the tumor tissues of the control group.
Discussion
Lung cancer is the commonest and deadliest malignant tumor worldwide, non-small cell lung cancer (NSCLC) account for a large proportion of all lung cancer cases, has become the leading cause of human death from cancers [1-3].
The discovery of epidermal growth factor receptor (EGFR) as a tumorignic driver and the subsequent development of EGFR tyrosine kinase inhibitor (EGFR-TKI) therapy represent a revolutionary change in treatment for advanced NSCLC [31]. Gefitinib is the front-line drug for NSCLC therapeutics, but their response is limited by the acquired resistance [7,8,31]. Even though, the new generations of EGFR-tyrosine kinase inhibitors (EGFR-TKI) were used for the treatment of NSCLC patients harboring EGFR mutations, TKI resistance often occurs as a result of additional EGFR mutations [32] and many other factors [3,4]. The acquired resistance significantly limited the efficacy of EGFR-TKIs, the current chemotherapeutic strategies for NSCLCs and survival of NSCLC patients. Therefore, it is necessary to overcome this resistance for effective NSCLC therapy. However, the mechanisms of the acquired resistance to EGFR-TKIs are still unclear [11,31].
In the present experimental study, gefitinib resistant lung adenocarcinoma cells (PC9/GR) were resistant to gefitinib, cisplatin and docetaxel with higher proliferative potential and drug resistance protein (ABCG2) expression, compared to the parent PC9 cells; PC9/GR cells displayed phenotypes of stemness and epithelial-mesenchymal transition (EMT) (Figure 1A). These features suggested that temness and EMT might be responsible for the acquired resistance of PC9/GR cells [33-37]. Moreover, c-kit protein expression was upregulated (Figure 2A), and c-kit signaling was activated in PC9/GR cells (Figure 2B, 2C). It is suggested that c-kit signaling might be involved in the acquired resistance of PC9/GR cells.
The c-kit gene is a key proto-oncogene; its expression is associated with the prognosis, tumor growth, and metastasis in NSCLC [24]. Recent investigations indicated that the lung cancer stem cell proportion in EGFR/TKI-resistant cell lines has increased significantly; c-kit expression was upregulated in NSCLC CSCs [25,26], gefitinib treated tumor xenografts of human skin squamous cell carcinoma (A431) [27], afatinib resistant H1975 lung cancer cell clones [28], and gefitinib treated head and neck squamous cell carcinoma cells [38]. These findings indicated that c-kit might be involved in the stemness phenotype, survival of lung CSCs, drug resistance, recurrence and metastasis of NSCLC, targeting c-kit signaling could improve the antitumor efficacy of chemotherapy of NSCLC [25,26].
To confirm whether targeting c-kit signaling could inhibit PC9/GR cell growth in vitro and in vivo, PC9/GR cells were treated with c-kit siRNA and c-kit inhibitors, imatinib and anlotinib.
In this experimental study, c-kit mRNA and protein and c-kit signaling related protein expressions were decreased in the stable shRNA-c-kit transfected PC9/GR cells (Figure 2D-H), shRNA-c-kit reduced the acquired resistance to gefitinib, cell proliferation, colony formation, invasion of PC9/GR cells significantly; the stemness and EMT phenotypes of the stable shRNA-c-kit transfected PC9/GR cells were abated, compared to the control PC9/GR cells (Figure 3). These findings suggested that c-kit siRNA could reverse the acquired resistance to gefitinib of PC9/GR cells and suppress their growth in vitro through attenuating their stemness and EMT phenotypes.
c-kit inhibitors, imatinib and anlotinib also exhibited higher growth and colony formation inhibition effects on the stable shRNA-c-kit transfected PC9/GR cells than the control PC9/GR cells (Figure 4A-D); the stemness and EMT phenotypes of PC9/GR cells were also attenuated by imatinib and anlotinib treatment (Figures 4E, 4F, 5); both imatinib and anlotinib treatment inhibited PC9/GR cell generated tumor growth in vivo (Figure 6); with downregulation of protein expressions of c-kit, ALDH1A1, ABCG2 and EMT-related proteins of vimentin and slug in the imatinib treated tumor tissues (Figure 7).
Imatinib and anlotinib are c-kit inhibitors [39-42]. Imatinib has chemoprevention effect on experimental lung carcinogenesis by activating the intrinsic or mitochondrial pathway of apoptosis [43], primary cultures of CD117 highly expressed NSCLC tumors were sensitive to imatinib [44]; anlotinib has efficacy and safety for advanced lung cancer therapy [42], it can overcome the acquired resistance to gefitinib in NSCLC without EGFR T790M mutation (HCC827/GR cells) via FGFR1 signaling, and suppress the growth of HCC827 GR cells in vitro and in vivo [44]. But, it is unknown whether imatinib and anlotinib could reverse the acquired gefitinib resistance of PC9/GR cells with T790M mutation and inhibit their growth in vitro and in vivo [38-45].
Herein, c-kit inhibitors, imatinib and anlotinib inhibited the acquired resistance to gefitinib, cell proliferation, colony formation, invasion of PC9/GR cells significantly in vitro, and reduced the stemness and EMT phenotypes of PC9/GR cells (Figures 4, 5); furthermore, imatinib and anlotinib exhibited significant higher growth inhibition effects on PC9/GR cell generated tumors, with reduced expressions of stemness related proteins of c-kit, ALDH1A1, ABCG2 and EMT-related proteins of vimentin and slug in the imatinib treated tumor tissues. These results showed that imatinib and anlotinib could overcome the acquired resistance to gefitinib of PC9/GR cells and inhibit their growth in vitro and in vivo through attenuating their stemness and EMT phenotypes.
According to the findings in this experimental investigation, it could be believed that c-kit was overexpressed and c-kit signaling was activated in gefitinib resistant PC9/GR cells, which endow PC9/GR cells with the acquired resistance to EGFR-TKIs and/or other chemotherapeutic drugs, stemness and EMT phenotypes. Therefore, c-kit signaling, stemness and EMT phenotypes might be involved in the acquired resistance to EGFR-TKIs in NSCLC therapeutics.
In summary, c-kit signaling mediated the acquired gefitinib resistance, cell growth, invasion, stemness and EMT phenotype of gefitinib resistant PC9/GR cells. c-kit signaling played pivotal role in the acquired resistance to EGFR-TKIs. Targeting c-kit signaling with c-kit siRNA and small molecule c-kit inhibitors to reverse the acquired drug resistance might be a novel strategy for EGFR-TKI resistant NSCLC therapy (Figure 8), which needs to be investigated further.
Figure 8.
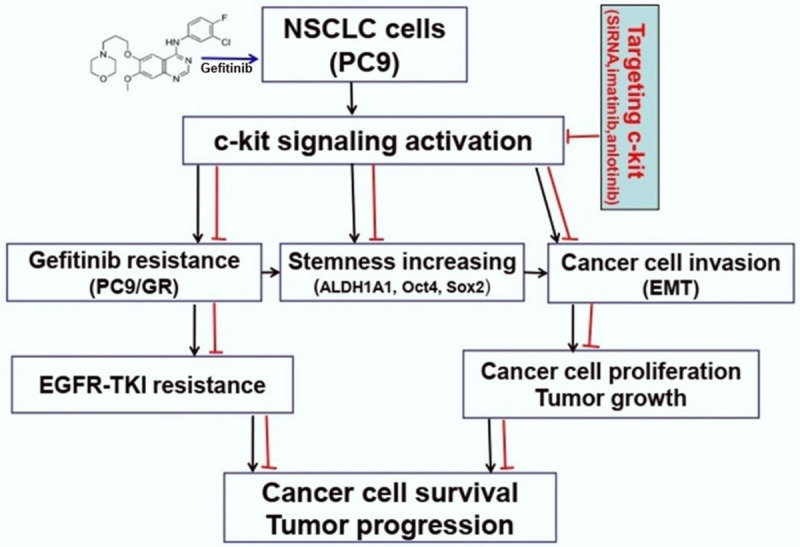
A schematic overview of targeting c-kit with c-kit siRNA and c-kit inhibitors inhibits gefitinib resistant lung adenocarcinoma cell growth in vitro and in vivo and the potential mechanisms of their activity involved.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 81802512).
Disclosure of conflict of interest
None.
Abbreviations
- NSCLC
Non-small cell lung cancer
- EGFR
Epidermal growth factor receptor
- EMT
Epithelial-mesenchymal transition
- siRNA
Small interfering RNA
- FBS
Fetal bovine serum
- CCK-8
Cell counting kit-8
- qRT-PCR
Quantitative real time polymerase chain reaction
- SDS
Sodium dodecyl sulfate
- PAGE
Polyacrylamide gel electrophoresis
- PVDF
Polyvinylidene fluoride
- HRP
Horseradish peroxidase
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- IC50
Half maximal inhibitory concentration
- CSC
Cancer stem cells
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 3.Wang F, Meng F, Wong SCC, Cho WCS, Yang S, Chan LWC. Combination therapy of gefitinib and miR-30a-5p may overcome acquired drug resistance through regulating the PI3K/AKT pathway in non-small cell lung cancer. Ther Adv Respir Dis. 2020;14:1753466620915156. doi: 10.1177/1753466620915156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergonzini C, Leonetti A, Tiseo M, Giovannetti E, Peters GJ. Is there a role for dacomitinib, a second-generation irreversible inhibitor of the epidermal-growth factor receptor tyrosine kinase, in advanced non-small cell lung cancer? Expert Opin Pharmacother. 2020;15:1–11. doi: 10.1080/14656566.2020.1746269. [DOI] [PubMed] [Google Scholar]
- 5.Riely GJ, Politi KA, Miller VA, Pao W. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res. 2006;12:7232–7241. doi: 10.1158/1078-0432.CCR-06-0658. [DOI] [PubMed] [Google Scholar]
- 6.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 7.Kim JH, Ko ES, Kim D, Park SH, Kim EJ, Rho J, Seo H, Kim MJ, Yang WM, Ha IJ, Park MJ, Lee JY. Cancer cell-specific anticancer effects of Coptis chinensis on gefitinib-resistant lung cancer cells are mediated through the suppression of Mcl-1 and Bcl-2. Int J Oncol. 2020;56:1540–1550. doi: 10.3892/ijo.2020.5025. [DOI] [PubMed] [Google Scholar]
- 8.Deng QF, Fang QY, Ji XX, Zhou SW. Cyclooxygenase-2 mediates gefitinib resistance in non-small cell lung cancer through the EGFR/PI3K/AKT axis. J Cancer. 2020;11:3667–3674. doi: 10.7150/jca.42850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanović A, Alfaro V. Gefitinib: current status in the treatment of non-small cell lung cancer. Drugs Today (Barc) 2004;40:809–827. doi: 10.1358/dot.2004.40.10.863742. [DOI] [PubMed] [Google Scholar]
- 10.Gao J, Li HR, Jin C, Jiang JH, Ding JY. Strategies to overcome acquired resistance to EGFR TKI in the treatment of non-small cell lung cancer. Clin Transl Oncol. 2019;21:1287–1301. doi: 10.1007/s12094-019-02075-1. [DOI] [PubMed] [Google Scholar]
- 11.Shien K, Toyooka S, Yamamoto H, Soh J, Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, Tsukuda K, Takigawa N, Kiura K, Gazdar AF, Lam WL, Miyoshi S. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2013;73:3051–3061. doi: 10.1158/0008-5472.CAN-12-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Codony-Servat J, Codony-Servat C, Cardona AF, Giménez-Capitán A, Drozdowskyj A, Berenguer J, Bracht JWP, Ito M, Karachaliou N, Rosell R. Cancer stem cell biomarkers in EGFR-mutation-positive non-small-cell lung cancer. Clin Lung Cancer. 2019;20:167–177. doi: 10.1016/j.cllc.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 13.Weng CH, Chen LY, Lin YC, Shih JY, Lin YC, Tseng RY, Chiu AC, Yeh YH, Liu C, Lin YT, Fang JM, Chen CC. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene. 2019;38:455–468. doi: 10.1038/s41388-018-0454-2. [DOI] [PubMed] [Google Scholar]
- 14.Raoof S, Mulford IJ, Frisco-Cabanos H, Nangia V, Timonina D, Labrot E, Hafeez N, Bilton SJ, Drier Y, Ji F, Greenberg M, Williams A, Kattermann K, Damon L, Sovath S, Rakiec DP, Korn JM, Ruddy DA, Benes CH, Hammerman PS, Piotrowska Z, Sequist LV, Niederst MJ, Barretina J, Engelman JA, Hata AN. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene. 2019;38:6399–6413. doi: 10.1038/s41388-019-0887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poh ME, Liam CK, Rajadurai P, Chai CS. Epithelial-to-mesenchymal transition (EMT) causing acquired resistance to afatinib in a patient with epidermal growth factor receptor (EGFR)-mutant lung adenocarcinoma. J Thorac Dis. 2018;10:E560–E563. doi: 10.21037/jtd.2018.06.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Qu Z, Yao H, Sun L, Harata-Lee Y, Cui J, Aung TN, Liu X, You R, Wang W, Hai L, Adelson DL, Lin L. An effective drug sensitizing agent increases gefitinib treatment by down regulating PI3K/Akt/mTOR pathway and up regulating autophagy in non-small cell lung cancer. Biomed Pharmacother. 2019;118:109169. doi: 10.1016/j.biopha.2019.109169. [DOI] [PubMed] [Google Scholar]
- 17.Del Re M, Arrigoni E, Restante G, Passaro A, Rofi E, Crucitta S, De Marinis F, Di Paolo A, Danesi R. Concise review: resistance to tyrosine kinase inhibitors in non-small cell lung cancer: the role of cancer stem cells. Stem Cells. 2018;36:633–640. doi: 10.1002/stem.2787. [DOI] [PubMed] [Google Scholar]
- 18.Bora-Singhal N, Mohankumar D, Saha B, Colin CM, Lee JY, Martin MW, Zheng X, Coppola D, Chellappan S. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci Rep. 2020;10:4722. doi: 10.1038/s41598-020-61295-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao W, Wang L, Huang H, Li X, Wang P, Mi K, Cheng J, Liu H, Gu C, Huang L, Huang J. All-trans retinoic acid reduces cancer stem cell-like cell-mediated resistance to gefitinib in NSCLC adenocarcinoma cells. BMC Cancer. 2020;20:315. doi: 10.1186/s12885-020-06818-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terzuoli E, Costanza F, Ciccone V, Ziche M, Morbidelli L, Donnini S. mPGES-1 as a new target to overcome acquired resistance to gefitinib in non-small cell lung cancer cell lines. Prostaglandins Other Lipid Mediat. 2019;143:106344. doi: 10.1016/j.prostaglandins.2019.106344. [DOI] [PubMed] [Google Scholar]
- 21.André C, Martin E, Cornu F, Hu WX, Wang XP, Galibert F. Genomic organization of the human c-Kit gene: evolution of the receptor tyrosine kinase subclass III. Oncogene. 1992;7:685–691. [PubMed] [Google Scholar]
- 22.Lennartsson J, Ronnstrand L. Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev. 2012;92:1619–1649. doi: 10.1152/physrev.00046.2011. [DOI] [PubMed] [Google Scholar]
- 23.Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435–443. doi: 10.7150/ijbs.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorelik E, Lokshin A, Levina V. Lung cancer stem cells as a target for therapy. Anticancer Agents Med Chem. 2010;10:164–171. doi: 10.2174/187152010790909308. [DOI] [PubMed] [Google Scholar]
- 25.Guo YN, Liang L, Ren S, Wu M, Shi DC, Mo WJ, Chen G. CD117 expression is correlated with poor survival of patients and progression of lung Carcinoma: a meta-analysis with a panel of 2645 patients. Pol J Pathol. 2019;70:63–78. doi: 10.5114/pjp.2019.87098. [DOI] [PubMed] [Google Scholar]
- 26.Levina V, Marrangoni A, Wang T, Parikh S, Su Y, Herberman R, Lokshin A, Gorelik E. Elimination of human lung cancer stem cells through targeting of the stem cell factor-c-kit-autocrine signaling loop. Cancer Res. 2010;70:338–346. doi: 10.1158/0008-5472.CAN-09-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Yang X, Zhao B, Cai Z. Acquired resistance to EGFR tyrosine kinase inhibitor in A431 squamous cell carcinoma xenografts is mediated by c-Kit pathway transduction. Tumour Biol. 2015;36:2993–2999. doi: 10.1007/s13277-014-2932-7. [DOI] [PubMed] [Google Scholar]
- 28.Booth L, Roberts JL, Tavallai M, Webb T, Leon D, Chen J, McGuire WP, Poklepovic A, Dent P. The afatinib resistance of in vivo generated H1975 lung cancer cell clones is mediated by SRC/ERBB3/c-KIT/c-MET compensatory survival signaling. Oncotarget. 2016;7:19620–19630. doi: 10.18632/oncotarget.7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu W, Wang JP, Meng QZ, Zhu F, Hao XF. MiR-142-5p reverses the resistance to gefitinib through targeting HOXD8 in lung cancer cells. Eur Rev Med Pharmacol Sci. 2020;24:4306–4313. doi: 10.26355/eurrev_202004_21011. [DOI] [PubMed] [Google Scholar]
- 30.Tsai CH, Lin FM, Yang YC, Lee MT, Cha TL, Wu GJ, Hsieh SC, Hsiao PW. Herbal extract of Wedelia chinensis attenuates androgen receptor activity and orthotopic growth of prostate cancer in nude mice. Clin Cancer Res. 2009;15:5435–5444. doi: 10.1158/1078-0432.CCR-09-0298. [DOI] [PubMed] [Google Scholar]
- 31.Chen JA, Riess JW. Advances in targeting acquired resistance mechanisms to epidermal growth factor receptor tyrosine kinase inhibitors. J Thorac Dis. 2020;12:2859–2876. doi: 10.21037/jtd.2019.08.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang YC, Pan KF, Lee WJ, Chang JH, Tan P, Gu CC, Chang WM, Yang SF, Hsiao M, Hua KT, Chien MH. Circulating proteoglycan endocan mediates EGFR-driven progression of non-small cell lung cancer. Cancer Res. 2020;80:3292–3304. doi: 10.1158/0008-5472.CAN-20-0005. [DOI] [PubMed] [Google Scholar]
- 33.Chandimali N, Huynh DL, Zhang JJ, Lee JC, Yu DY, Jeong DK, Kwon T. MicroRNA-122 negatively associates with peroxiredoxin-II expression in human gefitinib-resistant lung cancer stem cells. Cancer Gene Ther. 2019;26:292–304. doi: 10.1038/s41417-018-0050-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang F, Wang J, Wang X, Wei N, Liu H, Zhang X. CD146-mediated acquisition of stemness phenotype enhances tumour invasion and metastasis after EGFR-TKI resistance in lung cancer. Clin Respir J. 2019;13:23–33. doi: 10.1111/crj.12976. [DOI] [PubMed] [Google Scholar]
- 35.Weng CH, Chen LY, Lin YC, Shih JY, Lin YC, Tseng RY, Chiu AC, Yeh YH, Liu C, Lin YT, Fang JM, Chen CC. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene. 2019;38:455–468. doi: 10.1038/s41388-018-0454-2. [DOI] [PubMed] [Google Scholar]
- 36.Liu L, Zhu H, Liao Y, Wu W, Liu L, Liu L, Wu Y, Sun F, Lin HW. Inhibition of Wnt/β-catenin pathway reverses multi-drug resistance and EMT in Oct4 +/Nanog + NSCLC cells. Biomed Pharmacother. 2020;127:110225. doi: 10.1016/j.biopha.2020.110225. [DOI] [PubMed] [Google Scholar]
- 37.Si J, Ma Y, Bi JW, Xiong Y, Lv C, Li S, Wu N, Yang Y. Shisa3 brakes resistance to EGFR-TKIs in lung adenocarcinoma by suppressing cancer stem cell properties. J Exp Clin Cancer Res. 2019;38:481. doi: 10.1186/s13046-019-1486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kramer B, Kneissle M, Birk R, Rotter N, Aderhold C. Tyrosine kinase inhibition in HPV-related squamous cell carcinoma reveals beneficial expression of cKIT and Src. Anticancer Res. 2018;38:2723–2731. doi: 10.21873/anticanres.12514. [DOI] [PubMed] [Google Scholar]
- 39.Pham DDM, Guhan S, Tsao H. KIT and melanoma: biological insights and clinical implications. Yonsei Med J. 2020;61:562–571. doi: 10.3349/ymj.2020.61.7.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim JL, Lee DH, Jeong S, Kim BR, Na YJ, Park SH, Jo MJ, Jeong YA, Oh SC. Imatinib-induced apoptosis of gastric cancer cells is mediated by endoplasmic reticulum stress. Oncol Rep. 2019;41:1616–1626. doi: 10.3892/or.2018.6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen G, Zheng F, Ren D, Du F, Dong Q, Wang Z, Zhao F, Ahmad R, Zhao J. Anlotinib: a novel multi-targeting tyrosine kinase inhibitor in clinical development. J Hematol Oncol. 2018;11:120. doi: 10.1186/s13045-018-0664-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shao L, Wang W, Song Z, Zhang Y. The efficacy and safety of anlotinib treatment for advanced lung cancer. Onco Targets Ther. 2019;12:6549–6554. doi: 10.2147/OTT.S205674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puri S, Kaur G, Piplani H, Sanyal SN, Vaish V. Imatinib modulates pro-inflammatory microenvironment with angiostatic effects in experimental lung carcinogenesis. Inflammopharmacology. 2020;28:231–252. doi: 10.1007/s10787-019-00656-8. [DOI] [PubMed] [Google Scholar]
- 44.Donnenberg AD, Zimmerlin L, Landreneau RJ, Luketich JD, Donnenberg VS. KIT (CD117) expression in a subset of non-small cell lung carcinoma (NSCLC) patients. PLoS One. 2012;7:e52885. doi: 10.1371/journal.pone.0052885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lian Z, Du W, Zhang Y, Fu Y, Liu T, Wang A, Cai T, Zhu J, Zeng Y, Liu Z, Huang JA. Anlotinib can overcome acquired resistance to EGFR-TKIs via FGFR1 signaling in non-small cell lung cancer without harboring EGFR T790M mutation. Thorac Cancer. 2020;11:1934–1943. doi: 10.1111/1759-7714.13485. [DOI] [PMC free article] [PubMed] [Google Scholar]