

Abstract
Classification of multicategory survival-outcome is important for precision oncology. Machine learning (ML) algorithms have been used to accurately classify multi-category survival-outcome of some cancer-types, but not yet that of lung adenocarcinoma. Therefore, we compared the performances of 3 ML models (random forests, support vector machine [SVM], multilayer perceptron) and multinomial logistic regression (Mlogit) models for classifying 4-category survival-outcome of lung adenocarcinoma using the TCGA. Mlogit model overall performed similar to SVM and multilayer perceptron models (micro-average area under curve=0.82), while random forests model was inferior. Surprisingly, transcriptomic data alone and clinico-transcriptomic data appeared sufficient to accurately classify the 4-category survival-outcome in these patients, but no models using clinical data alone performed well. Notably, NDUFS5, P2RY2, PRPF18, CCL24, ZNF813, MYL6, FLJ41941, POU5F1B, and SUV420H1 were the top-ranked genes that were associated with alive without disease and inversely linked to other outcomes. Similarly, BDKRB2, TERC, DNAJA3, MRPL15, SLC16A13, CRHBP and ACSBG2 were associated with alive with progression and GAL3ST3, AD2, RAB41, HDC, and PLEKHG1 associated with dead with disease, respectively, while also inversely linked other outcomes. These cross-linked genes may be used for risk-stratification and future treatment development.
Keywords: Lung adenocarcinoma, cause-specific mortality, survival, machine learning, multilabel classification, transcriptomic
Introduction
Lung cancer is the most common cause of cancer deaths among men or women in the U.S.A., accounting for 135,720 deaths in 2020 [1], although its trend in age-standardized, sex- and race-adjusted morality was downward in the past 5 years [2]. Thanks to better treatments, an increasing number of lung cancer patients died of non-cancer causes in the past decade [3], but the overall survival of lung cancer remains dismal. Besides development of additional targeted therapies, a better risk stratification and subsequent treatment decision-making are urgently needed to reduce the deaths of lung cancer. It is probably equally important to deescalate the treatment intensity to reduce non-cancer deaths in other lung cancer patients.
Many clinical and genomic features have been identified for the prognostication of lung adenocarcinoma [4-8]. The advances in machine learning (ML) algorithm also helped develop several gene-signatures for survival prediction in lung adenocarcinoma patients [9-15]. However, most of these works have been focused on binary survival outcomes, either disease-free survival or overall survival. To fully realize the potential power of ML, we may use ML to stratify the risks of cancer death, non-cancer death and being alive among lung cancer patients. This is statistically a multilabel classification problem. One of the common approaches to classify the lung cancer patients is using clinical or transcriptomic data [5,10,11,16-21]. Indeed, we showed that random forests (RF) model outperformed the conventional multinomial logistic regression (Mlogit) model in classifying 5-category outcomes of lung cancer patients, using clinical data of a large population-based dataset [22]. However, it is still unclear whether other ML models and the transcriptomic data alone are sufficient for classifying multicategory outcomes of lung cancer. Therefore, we compared the performances of 3 ML and Mlogit models in classifying 4-category outcomes of lung adenocarcinoma, using transcriptomic data lone, clinical data alone and combined clinic and transcriptomic data.
Material and methods
We extracted the lung adenocarcinoma cases from the cancer genome atlas (TCGA, legacy Version) from the cBioPortal website (Figure 1) [23]. No exclusion-criteria were used. The outcome was the 4-category survival-outcome based on the vital status and disease-free status, including alive with no progression, alive with disease, dead with no known disease, and dead with disease. We first dichotomized the RNAseq data (here referred as transcriptomic data) using the normalized Z scores based on their expression in all patients. We then identified and included only the genes that were statistically associated with the 4-category outcome. After identification and removal of the correlated genes and clinical features, we merged the two datasets into one single dataset (referred as clinico-transcriptomic data). The clinical, transcriptomic, and clinico-transcriptomic data were then subject to the tuning of ML models and conventional Mlogit models, respectively.
Figure 1.
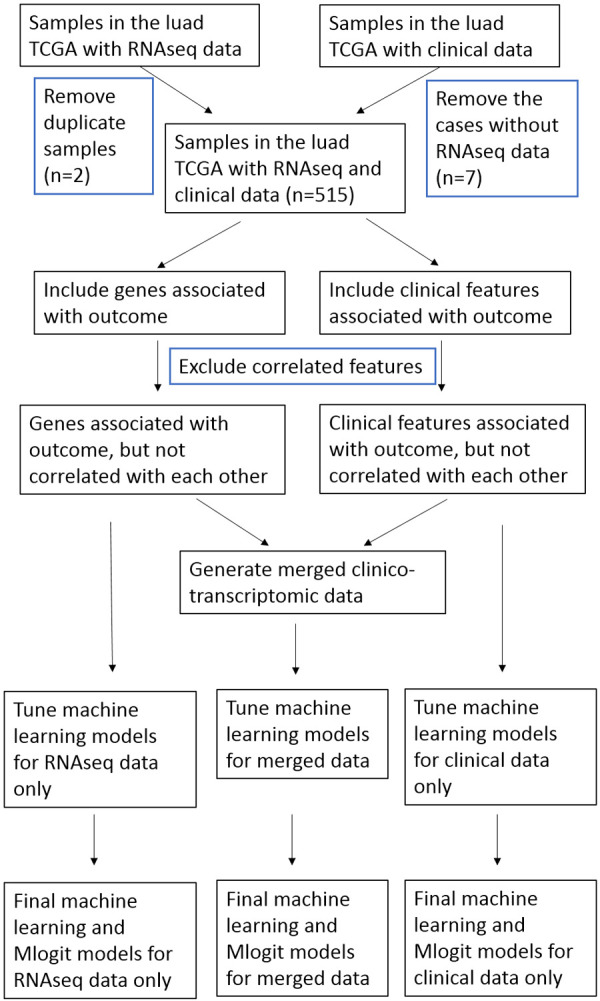
Study flow. We extracted the lung adenocarcinoma cases in the cancer genome atlas (TCGA), and classified the patient survival into 4 categories, including alive with no progression, alive with disease, dead with no known disease and dead with disease. We used random forests, support vector machine, and multilayer perceptron to classify the 4-category outcomes. The 5-fold cross-validation approach was used during the tuning and modelling of the machine learning algorithms.
The informed consent could not be and was not obtained for the TCGA patients due to de-identified nature of the dataset. Because we used de-identified, publicly available cases, this study was deemed exempt from review by an institutional review board. Moreover, a set of policies were developed by National Cancer Institute and National Human Genome Research Institute to protect the privacy of participants donating specimens to TCGA, including the TCGA’s informed consent policy, data access policy and information about Health Insurance Portability and Accountability Act Privacy Rule compliance (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga/history/policies/tcga-human-subjects-data-policies.pdf). Thus, the TCGA data collection was supervised by respective funding agencies and their ethical review committees.
We used the RF, support vector machine (SVM) with linear or radial basis function (RBF) kernel, and multilayer perceptron (MLP) models of the sklearn library [24]. The linear support vector classifier (SVC) was used as a reference, and differed from linear SVM in their kernels (liblinear vs libsvm in SVM) [24]. Specifically, we tuned the number of estimators, and the number of splits for RF, the C and gamma values for SVM, and the C and number of hidden layers for MLP models. The performance metrics for all models were precision, accuracy, recall and F1. The cross_validation library was used to conduct 5-fold cross validation in the ML model-tuning process. Otherwise, the sample split library was used to spilt samples in a proportion of 4:1 (i.e., 80% of the samples for training, and 20% of the samples for testing) as described before [24-26]. The parameters that produced the best accuracy were chosen as the final ML model, while the default settings were also used to avoid over-tuning or missing the proper parameter range. The true positive rate and the true negative rate/sensitivity were calculated using the OneVsRestClassifier library (sklearn). The receiver operator characteristics curve and area under the curve were calculated using the ROC library. The micro-average and macro-average metrics were computed as defined before [24].
The ML and Mlogit processes were conducted using python version 3.6.9. The cut off of P value of 0.05 was used to select the clinical or transcriptomic features that were correlated with the 4-category outcome. The Rho of 0.9 was used to select the transcriptomic or clinical features that were correlated with each other. The first identified feature of the two correlated features would be removed.
We used the Erichr app to conduct gene set enrichment analyses (GSEA) to identify the pathways and related diseases [27], that were associated with the ranked up-regulated or down-regulated transcriptomic features. We interrogated the ranked gene list for their enrichment in 6 domains, including BioPlanet (2019), Kyoto Encyclopedia of Genes and Genomes (KEGG) Human (2019), UK Biobank GWAS v1, gene ontologies (GO) Molecular Function, GO Cellular Component, and GO Biological Process. A P value less than 0.05 was considered statistically significant.
Results
Among the 522 patients in the TCGA lung adenocarcinoma dataset, 7 patients did not have any transcriptomic data. Among the 517 samples of RNA sequencing, 2 of them had duplicates and the duplicates were removed (Figure 1). Therefore, 515 cases/samples were included in the study (Table 1). Among the 17 clinical features, 16 were found uniquely correlated with the 4-category outcome as shown by the correlation study. In the 20,113 genes that were subject to the RNA sequencing, 2,887 genes were found significantly associated with the 4-category outcome and included in the analysis. The correlation study showed that 2,631 genes were uniquely associated with the outcome and used in the study after removing the first correlated genes.
Table 1.
Baseline characteristics of the included patients with lung adenocarcinoma in the TCGA
| Alive no progression, % | Alive with disease, % | Dead with disease, % | Dead with no known disease, % | All, % | |
|---|---|---|---|---|---|
| n | 252 | 76 | 107 | 80 | 515 |
| Sex | |||||
| Female | 55.56 | 51.32 | 57.94 | 45.00 | 53.79 |
| Male | 44.44 | 48.68 | 42.06 | 55.00 | 46.21 |
| Race | |||||
| Black | 11.11 | 9.21 | 11.21 | 6.25 | 10.10 |
| Other | 13.49 | 19.74 | 8.41 | 21.25 | 14.56 |
| White | 75.40 | 71.05 | 80.37 | 72.50 | 75.34 |
| Age (65+ yr) | |||||
| No | 47.22 | 34.21 | 42.06 | 37.50 | 42.72 |
| Yes | 52.78 | 65.79 | 57.94 | 62.50 | 57.28 |
| pT category | |||||
| T1 | 42.46 | 25.00 | 22.43 | 23.75 | 32.82 |
| T2 | 48.81 | 61.84 | 61.68 | 55.00 | 54.37 |
| T3 | 5.95 | 13.16 | 12.15 | 11.25 | 9.13 |
| T4 | 2.78 | 0.00 | 3.74 | 10.00 | 3.69 |
| pN category | |||||
| N0 | 73.41 | 75.00 | 48.60 | 46.25 | 64.27 |
| N1 | 13.10 | 13.16 | 32.71 | 22.50 | 18.64 |
| N2 | 10.32 | 9.21 | 18.69 | 26.25 | 14.37 |
| N3 | 0.40 | 1.32 | 0.00 | 0.00 | 0.39 |
| NX | 2.78 | 1.32 | 0.00 | 5.00 | 2.33 |
| pM category | |||||
| M0 | 65.48 | 63.16 | 73.83 | 67.50 | 67.18 |
| M1 | 2.78 | 3.95 | 4.67 | 12.50 | 4.85 |
| MX | 31.75 | 32.89 | 21.50 | 20.00 | 27.96 |
| Kras gene analysis | |||||
| No | 50.79 | 31.58 | 55.14 | 47.50 | 48.35 |
| Yes | 11.90 | 13.16 | 13.08 | 10.00 | 12.04 |
| Not Available | 37.30 | 55.26 | 31.78 | 42.50 | 39.61 |
| Kras mutation presence | |||||
| No | 7.14 | 11.84 | 7.48 | 5.00 | 7.57 |
| Yes | 5.16 | 1.32 | 4.67 | 5.00 | 4.47 |
| Not Available | 87.70 | 86.84 | 87.85 | 90.00 | 87.96 |
| Alk translocation presence | |||||
| No | 42.86 | 22.37 | 50.47 | 40.00 | 40.97 |
| Yes | 5.95 | 6.58 | 9.35 | 5.00 | 6.60 |
| Not Available | 51.19 | 71.05 | 40.19 | 55.00 | 52.43 |
| ECOG score | |||||
| 0 | 19.05 | 18.42 | 23.36 | 7.50 | 18.06 |
| 1 | 16.67 | 21.05 | 19.63 | 26.25 | 19.42 |
| 2 | 3.97 | 5.26 | 2.80 | 5.00 | 4.08 |
| 3 | 0.40 | 0.00 | 0.93 | 1.25 | 0.58 |
| Not Available | 59.92 | 55.26 | 53.27 | 60.00 | 57.86 |
| Radiation treatment, adjuvant | |||||
| No | 31.35 | 26.32 | 21.50 | 25.00 | 27.57 |
| Yes | 1.19 | 1.32 | 6.54 | 2.50 | 2.52 |
| Not Available | 67.46 | 72.37 | 71.96 | 72.50 | 69.90 |
| Targeted molecular therapy | |||||
| None given | 26.59 | 17.11 | 21.50 | 17.50 | 22.72 |
| Given | 5.95 | 10.53 | 5.61 | 10.00 | 7.18 |
| Not Available | 67.46 | 72.37 | 72.90 | 72.50 | 70.10 |
| Surgery | |||||
| No | 30.16 | 25.00 | 17.76 | 18.75 | 25.05 |
| Yes | 69.84 | 75.00 | 82.24 | 81.25 | 74.95 |
| History of other cancer | |||||
| No | 82.94 | 80.26 | 83.18 | 81.25 | 82.33 |
| Yes | 17.06 | 19.74 | 16.82 | 18.75 | 17.67 |
| Smoking history | |||||
| No | 31.35 | 28.95 | 33.64 | 35.00 | 32.04 |
| Yes | 68.65 | 71.05 | 66.36 | 65.00 | 67.96 |
TCGA, the cancer genome atlas; ECOG, Eastern Cooperative Oncology Group.
We tuned the 3 ML models using the transcriptomic, clinical and clinico-transcriptomic data, respectively (Figure 2). During the tuning of RF and RBF-SVM models, the transcriptomic data alone and clinic-transcriptomic data produced similar accuracy heatmaps, while the MLP and linear SVM models had different accuracy metrics for the three different datasets. The best accuracy appeared to be present in the models using transcriptomic data, reaching to 0.528 in RF model, 0.608 in MPL model, 0.581 in RBF SVM model and 0.583 in linear SVM model.
Figure 2.

Tuning of the machine learning algorithms to classify the 4-category survival outcome. We tuned the random forests (RF), support vector machine (SVM) with linear or radial basis function kernel, and multilayer perceptron (MLP) to classify the 4-category outcomes using 5-fold cross-validation (Heatmap graphs: A-C, RF models; D-F, MLP models; G-I, radial basis function SVM; and J-L, linear SVM models). The left column was the data produced using transcriptomic features alone, middle column using clinical features alone, and the right column using clinico-transcriptomic features (merged dichotomized clinical and transcriptomic features).
We computed performance metrics of the three ML and Mlogic models based on the 5-fold cross validation, including accuracy, precision, recall/sensitivity, and F1 (Table 2). We also included the linear SVC model, whose kernel was different from the linear SVM model. For transcriptomic data, Mlogit had the best accuracy and linear SVC had the best recall. For clinical data, linear SVM and RBF SVM both had the best accuracy, while MLP had the best recall. For clinico-transcriptomic data, linear SVC and linear SVM had the best accuracy, and linear SVC had the best recall.
Table 2.
Performance of machine learning and multinomial logistic regression models in classifying 4-category survival of TCGA lung adenocarcinoma patients using transcriptomic, clinical or clinico-transcriptomic data
| Model | Accuracy | Precision | Recall/sensitivity | F1 |
|---|---|---|---|---|
| Transcriptomic data | ||||
| Mlogit | 0.592±0.041 | 0.586±0.067 | 0.487±0.037 | 0.508±0.043 |
| Random Forest | 0.489±0.005 | 0.122±0.001 | 0.250±0.000 | 0.164±0.001 |
| MLP | 0.573±0.055 | 0.578±0.075 | 0.454±0.045 | 0.473±0.049 |
| Linear SVC | 0.577±0.043 | 0.544±0.056 | 0.490±0.052 | 0.502±0.055 |
| Linear SVM | 0.588±0.045 | 0.604±0.088 | 0.465±0.046 | 0.487±0.055 |
| RBF SVM | 0.581±0.041 | 0.614±0.096 | 0.448±0.046 | 0.470±0.059 |
| Clinical data | ||||
| Mlogit | 0.433±0.051 | 0.210±0.065 | 0.250±0.042 | 0.212±0.046 |
| Random Forest | 0.497±0.007 | 0.273±0.123 | 0.259±0.009 | 0.183±0.017 |
| MLP | 0.476±0.011 | 0.215±0.037 | 0.263±0.017 | 0.206±0.023 |
| Linear SVC | 0.441±0.033 | 0.174±0.030 | 0.241±0.023 | 0.191±0.022 |
| Linear SVM | 0.489±0.005 | 0.122±0.001 | 0.250±0.000 | 0.164±0.001 |
| RBF SVM | 0.489±0.005 | 0.122±0.001 | 0.250±0.000 | 0.164±0.001 |
| Clinico-transcriptomic data | ||||
| Mlogit | 0.573±0.049 | 0.582±0.084 | 0.476±0.046 | 0.496±0.046 |
| Random Forest | 0.489±0.005 | 0.122±0.001 | 0.250±0.000 | 0.164±0.001 |
| MLP | 0.546±0.071 | 0.599±0.104 | 0.443±0.098 | 0.445±0.096 |
| Linear SVC | 0.583±0.040 | 0.580±0.070 | 0.490±0.047 | 0.508±0.045 |
| Linear SVM | 0.583±0.040 | 0.608±0.094 | 0.460±0.046 | 0.475±0.053 |
| RBF SVM | 0.489±0.005 | 0.122±0.001 | 0.250±0.000 | 0.164±0.001 |
Note: Data presented as mean ± standard deviation from 5 cross-validation study. TCGA, the cancer genome atlas; Mlogit, multinomial logistic regression; MLP, multiple layer perceptron; SVC, support vector classifier; SVM, support vector machine; RBF, Radial basis function.
To more reliably assess the performance of these models, we used one over the rest classifier to generate receiver operator characteristics curve, and computed the AUC (Figure 3, next page). For transcriptomic and clinico-transcriptomic data, the Mlogit, linear SVM and MLP models all achieved a micro-average AUC of 0.82, while the RF model only reached the AUC of 0.75. For clinical data set, the RF model reached a micro-average AUC of 0.69, which was slightly higher than those of Mlogit and MLP models, but still lower than most of the AUC produced using transcriptomic or clinic-transcriptomic data.
Figure 3.

Receiver operator characteristics curves and the areas under the curve of the final models. Largely based on the tuning data, we chose the final models of multinomial logistic regression (Mlogit), linear support vector machine (SVM), multilayer perceptron (MLP) and random forests (RF) models to classify the 4-category outcomes. The operator characteristics curves and the areas under the curve were produced using 5-fold cross-validation and the OneVsRestClassifier function. (A-C, Mlogit model; D-F, linear SVM models; G-I, MLP models; and J-L, RF models). The left column was the data produced using transcriptomic features alone, middle column using clinical features alone, and the right column using clinico-transcriptomic features (merged dichotomized clinical and transcriptomic features). The micro-average was the calculated metrics globally by counting the total true positives, false negatives and false positives. The macro-average was the calculated metrics for each label, and find their unweighted mean. This does not take label imbalance into account.
There were 1340 genes (positively) linked to alive without progression, 1304 genes (positively) linked to alive with disease, 135 genes (positively) linked to dead with no known disease, and 1298 genes (positively) linked to dead with disease according to the Mlogit model (Table 3). To identify the genes that were important for classifying the 4-category outcome, we compared the top-ranked genes by their positive and inverse associations with 4-category outcome. Nine of the top 25 ranked genes in the alive without disease group were also in the list of the bottom-25 ranked genes in other outcome-groups, including NDUFS5, P2RY2, PRPF18, CCL24, ZNF813, MYL6, FLJ41941, POU5F1B, and SUV420H1 in ranking order. Similarly, BDKRB2, LOC100133738 (TERC), DNAJA3, MRPL15, SLC16A13, CRHBP and ACSBG2 in the alive with progression group were bottom-25 ranked genes in the other groups, so were GAL3ST3, AD2, RAB41, HDC, and PLEKHG1 in the dead with disease group. Interestingly, only FBXO15, IPMK, and PCDHB8 of the top-25 ranked genes in the death with no known disease were cross-linked to the bottom-25 ranked genes in other groups. The GSEA revealed the pathways, disease/clinical presentations and GO that were enriched in the 4 outcome-groups (Figure 4), as well as the related genes (Supplementary Figure 1A and 1B).
Table 3.
The genes of top- and bottom-ranked coefficients for their associations with the 4-category outcomes in the multinomial logistic regression model

|
Figure 4.

Gene set enrichment analyses on the top ranked genes based on the coefficient of the multinomial logistic regression model. We conducted the gene-set enrichment analyses for the top-ranked genes based on their associations with respective outcome/groups (coefficient as the metric), using the web-based Enrichr algorithm and p-value based ranking. The length of the bar indicates the degree of the gene-enrichment. The top two rows show the results of pathway analyses using Kyoto Encyclopedia of Genes and Genomes (KEGG) and BioPlanet (2019) algorithms, the third row shows the result of disease-related analysis using algorithm of the UK Biobank genome-wide association study (GWAS) v1, and the bottom 3 rows show the result of analyses using the gene ontologies (GO) algorithms. The far-left column was the alive with no progression group, middle-left column the alive with disease, the middle-right column the dead with no known disease, and the far-right column the dead with disease group, respectively. The lighter shade indicates P<0.01 for gene-enrichment, darker shade indicates P<0.05, and gray shade indicates P≥0.05.
Discussion
We here compared the performances of the 3 ML and Mlogit models for classifying the 4-category survival outcomes of lung adenocarcinoma patients using the TCGA data. We found that Mlogit model overall performed similar to the 3 ML models (micro-average AUC=0.82). Surprisingly, transcriptomic data alone appeared to be sufficient to successfully classify the 4-category outcome in lung adenocarcinoma patients, and more useful than clinical data alone for the classification. We also identified a set of genes that were associated with one of the 4-category outcomes, and inversely linked to other outcomes.
The major strength of this study is the first-time classification of the multicategory outcome in lung adenocarcinoma using ML models. The past studies have used ML and other models to classify the causes of death in lung adenocarcinoma patients, but only for the binary survival-outcomes [5,10,11,16-21]. The in-depth outcome-classification will help select the patients who might need only lower doses of chemotherapy or radiotherapy, and increase treatment dosages or use other treatment modalities in the patients who died of cancer. Indeed, three genes were linked to death with disease and inversely linked to alive without disease, while 5 genes were linked to the alive without disease, and inversely linked to death with disease. These genes in our view could help classify cancer-recurrence risks for more effective prevention of cancer deaths and de-escalation of treatment intensity.
Moreover, in contrary to the common understanding that ML helps multilabel prediction, we for the first time showed that ML models were not all superior to Mlogit model in this sample set of 515 lung cancer cases and 2631 differential expressed features. This finding is consistent with some of the previous ML studies [28,29], but in contrast with others on cancer-outcomes [22,30,31]. We believe that the differences might be attributable to the sample size of ~500, although we thought that the large number of features may benefit from ML algorithms, but it did not. Therefore, we recommend to compare the performances of ML models with Mlogit or other conventional statistical models. However, the potential overfitting of Mlogit regression model must also be noted and carefully evaluated in an external validation set. On the other hand, linear SVM and MLP models seemed very useful in classifying multilabel data in this study, but the RF model did not perform well likely due to the small sample size and feature composition.
Furthermore, transcriptomic data alone appear sufficient for classifying 4-category outcome in this study, and more useful than the clinical data alone. To our surprise, we were not able to identify synergistic effects of combining the clinical and transcriptomic data, and the models using only clinical data performed poorly. It is noteworthy that clinical data could be used to reach a reasonably good prediction accuracy using large population-based datasets [25,30] and others [32-34]. The difference may be attributable to the availability of more targeted therapies and better understanding of molecular aspects of lung adenocarcinoma than prostate cancer [35]. Indeed, the guidelines for non-small cell carcinoma of the lung, including lung adenocarcinoma, recently expanded the already-long list of targeted- and immunotherapies [35].
In addition, few of the previous ML studies on transcriptomic data of lung adenocarcinoma used cross-validation approach [26], while some used small-size external validation cohorts [9-11,19]. We here used k-fold (k=5 in our case) cross-validation to increase rigor of our study. Briefly, in the k-fold cross-validation, one randomly and evenly splits the samples into k-portions, and after shuffling conducts k rounds of modelling using the k-1 portions as the training set and the last one portion as the test set. It has been used to effectively measure the performance and validate findings of large datasets [22,25,36,37]. The use of k-fold cross-validation and subsequent evaluation of performance metrics were methodologically rigorous for the randomized repeats. Thus, this approach as used in this study is particularly useful when external validation set is unavailable or not feasible.
Finally, we identified several genes and biological processes that were associated with or useful for classifying the 4-category outcomes of lung adenocarcinoma. For example, P2RY2 is a gene important for lung fibrosis [38], and regulating the proliferation of lung carcinoma cells [39], but its roles in lung cancer development or progression are otherwise unknown. Additional studies on these cross-linked genes are needed, and might reveal novel targets of lung adenocarcinoma therapy. Another example is the genes that are linked to some clinical, socioeconomic and behavioral characteristics in the UK Biobank GWAS study such as smoking, and self-reported eye/eyelid problem, which have been shown important for all-cause mortality or other deaths [40,41].
This study has several limitations. First, we could not validate our findings in another large-size transcriptomic data set with the 4-category outcome, which to our knowledge is yet available. Our internal 5-fold cross-validation approach may in part address this limitation. The cross-link to the large sample size study such as UK Biobank also confirmed some of our findings. Nonetheless, additional validation studies are needed. Second, the sample size of 500 is large for transcriptomic studies, but might be too small for optimal performance of some ML models. We would like to confirm our findings using another large transcriptomic dataset with the 4-category outcome. However, large size clinico-transcriptomic studies of multicategory outcomes are expensive to carry out and largely unavailable. Thus, additional transcriptomic studies are recommended to have a large sample-size and detailed clinical-outcomes such as specific causes of death. Finally, treatment data were included in the TCGA but might be limited or misinterpreted by the data collectors. Despite our slight concern in this regard, the TCGA data have been widely used for external validation, primary study or in silico (secondary) analysis [40,42-45]. Nonetheless, caution should be used when applying our findings to patient care.
In summary, we here show that transcriptomic features alone could be used to accurately classify 4-category survival-outcome in the patients with lung adenocarcinoma using Mlogit regression or ML models such as linear SVM and MLP. These findings may help better classify these patients for choosing the right treatment options. Our findings also reveal several genes and pathways that are important for the different, specific survival-outcome in these patients, and their potential biological significance. Additional studies are warranted to confirm and understand our findings.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Hu X, Lin Y, Qin G, Zhang L. Underlying causes of death with changing mortality among adults in the United States, 2013-2017. doi: 10.14218/ERHM.2020.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaorsky NG, Churilla TM, Egleston BL, Fisher SG, Ridge JA, Horwitz EM, Meyer JE. Causes of death among cancer patients. Ann Oncol. 2017;28:400–407. doi: 10.1093/annonc/mdw604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shang G, Jin Y, Zheng Q, Shen X, Yang M, Li Y, Zhang L. Histology and oncogenic driver alterations of lung adenocarcinoma in Chinese. Am J Cancer Res. 2019;9:1212–1223. [PMC free article] [PubMed] [Google Scholar]
- 5.Yu J, Hu Y, Xu Y, Wang J, Kuang J, Zhang W, Shao J, Guo D, Wang Y. LUADpp: an effective prediction model on prognosis of lung adenocarcinomas based on somatic mutational features. BMC Cancer. 2019;19:263. doi: 10.1186/s12885-019-5433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez-Rueda H, Martinez-Ledesma E, Martinez-Torteya A, Palacios-Corona R, Trevino V. Integration and comparison of different genomic data for outcome prediction in cancer. BioData Min. 2015;8:32. doi: 10.1186/s13040-015-0065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D, Shin H, Song YS, Kim JH. Synergistic effect of different levels of genomic data for cancer clinical outcome prediction. J Biomed Inform. 2012;45:1191–1198. doi: 10.1016/j.jbi.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Fernandez FG, Force SD, Pickens A, Kilgo PD, Luu T, Miller DL. Impact of laterality on early and late survival after pneumonectomy. Ann Thorac Surg. 2011;92:244–249. doi: 10.1016/j.athoracsur.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 9.Xue L, Bi G, Zhan C, Zhang Y, Yuan Y, Fan H. Development and validation of a 12-gene immune relevant prognostic signature for lung adenocarcinoma through machine learning strategies. Front Oncol. 2020;10:835. doi: 10.3389/fonc.2020.00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma B, Geng Y, Meng F, Yan G, Song F. Identification of a sixteen-gene prognostic biomarker for lung adenocarcinoma using a machine learning method. J Cancer. 2020;11:1288–1298. doi: 10.7150/jca.34585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JS, Chun SH, Park S, Lee S, Kim SE, Hong JH, Kang K, Ko YH, Ahn YH. Identification of novel microRNA prognostic markers using cascaded Wx, a neural network-based framework, in lung adenocarcinoma patients. Cancers (Basel) 2020;12:1890. doi: 10.3390/cancers12071890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao X, Shi R, Zhao T, Wang Y. Immune landscape and a novel immunotherapy-related gene signature associated with clinical outcome in early-stage lung adenocarcinoma. J Mol Med (Berl) 2020;98:805–818. doi: 10.1007/s00109-020-01908-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Shi K, Huang W, Weng W, Zhang Z, Guo Y, Deng T, Xiang Y, Ni X, Chen B, Zhou M. The DNA methylation profile of non-coding RNAs improves prognosis prediction for pancreatic adenocarcinoma. Cancer Cell Int. 2019;19:107. doi: 10.1186/s12935-019-0828-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Zhang Q, Gao Z, Xin S, Zhao Y, Zhang K, Shi R, Bao X. A novel 4-gene signature for overall survival prediction in lung adenocarcinoma patients with lymph node metastasis. Cancer Cell Int. 2019;19:100. doi: 10.1186/s12935-019-0822-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho HJ, Lee S, Ji YG, Lee DH. Association of specific gene mutations derived from machine learning with survival in lung adenocarcinoma. PLoS One. 2018;13:e0207204. doi: 10.1371/journal.pone.0207204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu D, Zhang J, Xu H, Zhang Y, Chen W, Gao R, Dehmer M. Multi-scale supervised clustering-based feature selection for tumor classification and identification of biomarkers and targets on genomic data. BMC Genomics. 2020;21:650. doi: 10.1186/s12864-020-07038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rana HK, Akhtar MR, Islam MB, Ahmed MB, Lio P, Huq F, Quinn JMW, Moni MA. Machine learning and bioinformatics models to identify pathways that mediate influences of welding fumes on cancer progression. Sci Rep. 2020;10:2795. doi: 10.1038/s41598-020-57916-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kweon S, Lee JH, Lee Y, Park YR. Personal health information inference using machine learning on RNA expression data from patients with cancer: algorithm validation study. J Med Internet Res. 2020;22:e18387. doi: 10.2196/18387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Fu J, Wang Z, Lv Z, Fan Z, Lei T. Screening key lncRNAs for human lung adenocarcinoma based on machine learning and weighted gene co-expression network analysis. Cancer Biomark. 2019;25:313–324. doi: 10.3233/CBM-190225. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Deng H, Xin S, Zhang K, Shi R, Bao X. Prognostic and predictive value of three DNA methylation signatures in lung adenocarcinoma. Front Genet. 2019;10:349. doi: 10.3389/fgene.2019.00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.R K, R GR. Accuracy enhanced lung cancer prognosis for improving patient survivability using proposed gaussian classifier system. J Med Syst. 2019;43:201. doi: 10.1007/s10916-019-1297-2. [DOI] [PubMed] [Google Scholar]
- 22.Deng F, Zhou H, Lin Y, Heim J, Shen L, Li Y, Zhang L. Predict multicategory causes of death in lung cancer patients using clinicopathologic factors. medRxiv. 2020 doi: 10.1016/j.compbiomed.2020.104161. 2020.2009.2025.20201095. [DOI] [PubMed] [Google Scholar]
- 23.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V. Scikit-learn: machine learning in Python. J Machine Learning Res. 2011;12:2825–2830. [Google Scholar]
- 25.Deng F, Huang J, Yuan X, Cheng C, Zhang L. Performance and efficiency of machine learning algorithms for analyzing rectangular biomedical data. bioRxiv. 2020 doi: 10.1038/s41374-020-00525-x. 2020.2009.2013.295592. [DOI] [PubMed] [Google Scholar]
- 26.Herrmann M, Probst P, Hornung R, Jurinovic V, Boulesteix AL. Large-scale benchmark study of survival prediction methods using multi-omics data. Brief Bioinform. 2020 doi: 10.1093/bib/bbaa167. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–7. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Ploeg T, Austin PC, Steyerberg EW. Modern modelling techniques are data hungry: a simulation study for predicting dichotomous endpoints. BMC Med Res Methodol. 2014;14:137. doi: 10.1186/1471-2288-14-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S, Park T, Kon M. Cancer survival classification using integrated data sets and intermediate information. Artif Intell Med. 2014;62:23–31. doi: 10.1016/j.artmed.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Deng F, Zeng F, Shanahan AJ, Li WV, Zhang L. Predicting long-term multicategory cause of death in patients with prostate cancer: random forest versus multinomial model. Am J Cancer Res. 2020;10:1344–1355. [PMC free article] [PubMed] [Google Scholar]
- 31.Hanson HA, Martin C, O’Neil B, Leiser CL, Mayer EN, Smith KR, Lowrance WT. The relative importance of race compared to health care and social factors in predicting prostate cancer mortality: a random forest approach. J Urol. 2019;202:1209–1216. doi: 10.1097/JU.0000000000000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartholomai JA, Frieboes HB. Lung cancer survival prediction via machine learning regression, classification, and statistical techniques. Proc IEEE Int Symp Signal Proc Inf Tech. 2018;2018:632–637. doi: 10.1109/ISSPIT.2018.8642753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lynch CM, van Berkel VH, Frieboes HB. Application of unsupervised analysis techniques to lung cancer patient data. PLoS One. 2017;12:e0184370. doi: 10.1371/journal.pone.0184370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynch CM, Abdollahi B, Fuqua JD, de Carlo AR, Bartholomai JA, Balgemann RN, van Berkel VH, Frieboes HB. Prediction of lung cancer patient survival via supervised machine learning classification techniques. Int J Med Inform. 2017;108:1–8. doi: 10.1016/j.ijmedinf.2017.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ettinger DS, Wood DE, Aggarwal C, Aisner DL, Akerley W, Bauman JR, Bharat A, Bruno DS, Chang JY, Chirieac LR, D’Amico TA, Dilling TJ, Dobelbower M, Gettinger S, Govindan R, Gubens MA, Hennon M, Horn L, Lackner RP, Lanuti M, Leal TA, Lin J, Loo BW Jr, Martins RG, Otterson GA, Patel SP, Reckamp KL, Riely GJ, Schild SE, Shapiro TA, Stevenson J, Swanson SJ, Tauer KW, Yang SC, Gregory K OCN. Hughes M. NCCN guidelines insights: non-small cell lung cancer, version 1.2020. J Natl Compr Canc Netw. 2019;17:1464–1472. doi: 10.6004/jnccn.2019.0059. [DOI] [PubMed] [Google Scholar]
- 36.Hussain L, Ahmed A, Saeed S, Rathore S, Awan IA, Shah SA, Majid A, Idris A, Awan AA. Prostate cancer detection using machine learning techniques by employing combination of features extracting strategies. Cancer Biomark. 2018;21:393–413. doi: 10.3233/CBM-170643. [DOI] [PubMed] [Google Scholar]
- 37.Agranoff D, Fernandez-Reyes D, Papadopoulos MC, Rojas SA, Herbster M, Loosemore A, Tarelli E, Sheldon J, Schwenk A, Pollok R, Rayner CF, Krishna S. Identification of diagnostic markers for tuberculosis by proteomic fingerprinting of serum. Lancet. 2006;368:1012–1021. doi: 10.1016/S0140-6736(06)69342-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muller T, Fay S, Vieira RP, Karmouty-Quintana H, Cicko S, Ayata K, Zissel G, Goldmann T, Lungarella G, Ferrari D, Di Virgilio F, Robaye B, Boeynaems JM, Blackburn MR, Idzko M. The purinergic receptor subtype P2Y2 mediates chemotaxis of neutrophils and fibroblasts in fibrotic lung disease. Oncotarget. 2017;8:35962–35972. doi: 10.18632/oncotarget.16414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schafer R, Sedehizade F, Welte T, Reiser G. ATP- and UTP-activated P2Y receptors differently regulate proliferation of human lung epithelial tumor cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L376–85. doi: 10.1152/ajplung.00447.2002. [DOI] [PubMed] [Google Scholar]
- 40.Li D, Yang W, Zhang Y, Yang JY, Guan R, Xu D, Yang MQ. Genomic analyses based on pulmonary adenocarcinoma in situ reveal early lung cancer signature. BMC Med Genomics. 2018;11:106. doi: 10.1186/s12920-018-0413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomez SL, Chang ET, Shema SJ, Fish K, Sison JD, Reynolds P, Clément-Duchêne C, Wrensch MR, Wiencke JL, Wakelee HA. Survival following non-small cell lung cancer among Asian/Pacific Islander, Latina, and Non-Hispanic white women who have never smoked. Cancer Epidemiol Biomarkers Prev. 2011;20:545–554. doi: 10.1158/1055-9965.EPI-10-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sherafatian M, Arjmand F. Decision tree-based classifiers for lung cancer diagnosis and subtyping using TCGA miRNA expression data. Oncol Lett. 2019;18:2125–2131. doi: 10.3892/ol.2019.10462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Ge D, Gu J, Xu F, Zhu Q, Lu C. A large cohort study identifying a novel prognosis prediction model for lung adenocarcinoma through machine learning strategies. BMC Cancer. 2019;19:886. doi: 10.1186/s12885-019-6101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao X, Luo H, Krawczyk M, Wei W, Wang W, Wang J, Flagg K, Hou J, Zhang H, Yi S, Jafari M, Lin D, Chung C, Caughey BA, Li G, Dhar D, Shi W, Zheng L, Hou R, Zhu J, Zhao L, Fu X, Zhang E, Zhang C, Zhu JK, Karin M, Xu RH, Zhang K. DNA methylation markers for diagnosis and prognosis of common cancers. Proc Natl Acad Sci U S A. 2017;114:7414–7419. doi: 10.1073/pnas.1703577114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kourou K, Exarchos TP, Exarchos KP, Karamouzis MV, Fotiadis DI. Machine learning applications in cancer prognosis and prediction. Comput Struct Biotechnol J. 2015;13:8–17. doi: 10.1016/j.csbj.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.