

Abstract
Activating mutations of the KRAS gene are one of the major genomic alterations associated with tumorigenesis of non-small cell lung cancer (NSCLC). Thus far, treatment of KRAS-mutant NSCLC remains an unmet medical need. We determined the in vivo treatment responses of 13 KRAS mutant and 14 KRAS wild type NSCLC patient-derived xenografts (PDXs) to agents that target known NSCLC vulnerabilities: the MEK inhibitor trametinib, the MDM2 inhibitor KRT-232, and the BCL-XL/BCL-2 inhibitor navitoclax. The results showed that the tumor regression rate after single agent therapy with KRT-232, trametinib and navitoclax was 11%, 10% and 0%, respectively. Combination therapies of trametinib plus KRT-232 and trametinib plus navitoclax led to improved partial response rates over single-agent activity in a subset of PDX models. Tumor regression was observed in 23% and 50% of PDXs after treatment with trametinib plus KRT-232 and trametinib plus navitoclax, respectively. The disease control rates in KRAS-mutant PDXs tested were 90%-100% after treatment with trametinib plus KRT-232 or plus navitoclax. A correlation analysis of treatment responses and genomic and proteomic biomarkers revealed that sensitivity to KRT-232 was significantly associated with TP53 wild-type or STK11 mutant genotypes (P<0.05). The levels of several proteins, including GSK3b, Nrf2, LKB1/pS334, and SMYD3, were significantly associated with sensitivity to trametinib plus navitoclax. Thus, the combination of trametinib plus KRT-232 or navitoclax resulted in improved efficacy compared with the agents alone in a subgroup of NSCLC PDX model with KRAS mutations. Expanded clinical trials of these targeted drug combinations in NSCLC are warranted.
Keywords: NSCLC, target therapy, combination therapy, MEK, MDM2, Bcl2
Introduction
The mitogen-activated protein kinase (MAPK) pathway is one of the most commonly activated oncogenic pathways in a vast array of cancers, including non-small cell lung cancer (NSCLC). Constitutive activation of the MAPK pathway resulting from upstream activating mutations of oncogenes such as KRAS, BRAF, and EGFR is one of the major drivers of uncontrolled cell proliferation, cell de-differentiation, and tumorigenesis. The MAPK kinases (MEK1 and MEK2) function as gatekeepers in the MAPK pathway because they are the only known mediators that convey activating signals from RAF to ERK. Consequently, MEK inhibitors have been intensively investigated, both preclinically and clinically, for the treatment of NSCLC [1]. The combination of the MEK inhibitor trametinib and the BRAF inhibitor dabrafenib was approved by the United States Food and Drug Administration for the treatment of BRAF V600E mutant NSCLC in 2017 [2,3]. Trametinib combined with chemoradiation therapy, erlotinib, navitoclax, lapatinib, and the immune checkpoint inhibitor pembrolizumab are also undergoing clinical trials for the treatment of NSCLC, with or without KRAS mutations [1]. The combination of the MEK inhibitor selumetinib and chemotherapy has been investigated in clinical trials for the treatment of KRAS mutant or KRAS wild-type (wt)/unknown NSCLC [4-6]. A randomized phase III trial showed that selumetinib plus docetaxel resulted in a higher objective response rate than did placebo and docetaxel in KRAS mutant NSCLC but did not improve the median progression-free survival or median overall survival [4]. The MEK inhibitors cobimetinib and binimetinib, in combination with BRAF, EGFR, or immune checkpoint blockade inhibitors or chemotherapy, are also being studied in various clinical trials for the treatment of NSCLC, with and without KRAS mutations [1,7].
We previously reported that treatment of NSCLC cells with selumetinib inhibited cell proliferation and induced apoptosis by increasing the expression of the pro-apoptotic BCL2 family proteins Bim, PUMA, and NOXA through enhanced nuclear translocation of FOXO3a [8]. Because the interaction between pro-apoptotic and anti-apoptotic BCL2 family proteins strictly controls the intrinsic apoptotic process, the overall ratio of pro-apoptotic and anti-apoptotic BCL2 family proteins in cancer cells is a key factor in the apoptotic response to targeted therapy. We hypothesized that pharmaceutical interventions that further change the balance of pro-apoptotic and anti-apoptotic BCL2 family proteins inside cancer cells will enhance the anti-cancer activity of MEK inhibitors. To test this hypothesis and to identify predictive biomarkers for personalized therapy, we tested whether combinations of MEK inhibitors with the MDM2 inhibitor KRT-232 (AMG 232) or BCL-XL/BCL2 inhibitor, navitoclax, lead to enhanced therapeutic effects in NSCLC PDX models. KRT-232 is a potent MDM2 inhibitor [9] that enhances TP53 activity by blocking the interaction between MDM2 and TP53 and inducing TP53-mediated pro-apoptotic protein expression [10]. A phase I study of KRT-232 in patients with advanced TP53 wt solid tumors or multiple myeloma revealed that it has acceptable safety and dose-proportional pharmacokinetics and has induced stable disease in some patients [11]. The combination of KRT-232 and trametinib was evaluated in a phase Ib study in patients with relapsed or refractory acute myeloid leukemia [12], and the results suggested that the pharmacokinetics of KRT-232 and trametinib were not affected by co-administration [12]. Navitoclax is a potent inhibitor of BCL-XL and BCL-BCL2 [13] and is currently under clinical investigation for the treatment of hematologic malignancies and solid tumors, including in combination therapy with trametinib for the treatment of solid tumors with KRAS or NRAS mutations [1].
Patient-derived xenografts (PDXs) are increasingly being used in preclinical anti-cancer drug development because of their ability to recapitulate the histological and molecular biological features of human primary tumors and predict clinical treatment responses [14,15]. Over the past several years, we have generated about 200 NSCLC PDX models from surgically resected specimens, pleural fluid drainage samples, and biopsy samples. Histological and molecular characterizations on some of these models showed that they recapitulate the features of human primary tumors in their histology, genomic alterations, and tumor microenvironment [16-18]. Here we report the in vivo anti-cancer activity of trametinib, KRT-232, and navitoclax, in combination and as single agents, and biomarkers associated with treatment responses. The combination of trametinib and KRT-232 or navitoclax resulted in improved efficacy in a subgroup of NSCLC PDX models with KRAS mutations, supporting the feasibility of these combinations for the treatment of KRAS mutant NSCLC.
Materials and methods
Therapeutic agents
KRT-232 was provided by Amgen, Inc., and Kartos Therapeutics, Inc. Navitoclax was provided by Abbvie, Inc., both obtained through the Division of Cancer Treatment and Diagnosis of the National Cancer Institute. Trametinib was obtained from Selleck Chemicals.
NSCLC PDXs
The generation and passage of NSCLC PDXs, verification of PDX provenance by DNA fingerprint analysis, genomic characterization by next generation sequencing, and histological characterization of PDXs were performed as we previously described [16,17]. Next generation sequencing was performed at MD Anderson Moon Shot platform Cancer Genomics Laboratory. All clinical samples and data were collected with informed patient consent under a research protocol approved by the Institutional Review Board at The University of Texas MD Anderson Cancer Center (Houston, Texas). All animal studies were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals (National Institutes of Health Publication 85-23) and the institutional guidelines of MD Anderson.
Reverse-phase protein array (RPPA) assay
A small piece of tissue from each PDX model was lysed in RPPA lysis buffer and subjected to a RPPA assay at the Functional Proteomics RPPA Core facility at MD Anderson, as previously reported [19,20]. In brief, serially diluted lysates were arrayed on nitrocellulose-coated slides. Each slide was probed with a validated primary antibody and then with a biotin-conjugated secondary antibody. After performing a colorimetric reaction of horseradish peroxidase and its substrate 3,3’-diaminobenzidine, we scanned the slides and quantified the signals on the basis of spot intensity. We used 196 antibodies specific for human proteins and protein phosphorylation sites to determine baseline protein levels in this study.
Animal experiments
In vivo drug testing studies was performed with randomizing animals in treatment groups and recording results in al blinded manner. Each PDX was subcutaneously inoculated into the dorsal flanks of female nude mice or non-obese diabetic/severe combined immunodeficiency (NOD-SCID) mice with null mutations of the gene encoding interleukin-2 receptor g (NSG). Subcutaneous tumors were measured with calipers, and tumor volume was calculated according to the formula V=ab2/2, where a is the largest diameter and b is the smallest. Mice were added to treatment groups (n=3-5/group) randomly when tumors reached 200 mm3 in size (about 7-9 mm in diameter). The following treatment regimens were used for each testing agent: (1) 45 mg/kg of KRT-232, oral gavage once a day for 5 days/week for 3 weeks; (2) 0.1 mg/kg of trametinib, oral gavage once a day for 4 weeks; and (3) 100 mg/kg of navitoclax, oral gavage once a day for 3 days/week for 3 weeks. Treatment with solvent was used as the control. For combination therapies, animals were treated with the same doses and regimens as for the single-agent therapy except that the 2 drugs were administered in combination. Tumor growth and body weight were monitored every 2 to 3 days. Tumor volume changes were calculated with an R software program, with tumor volume at the beginning of treatment set to a baseline of 0. The experiment was ended and the mice were euthanized when the tumors reached 15~18 mm in diameter. A total of 28 PDX models were tested for 6 treatment groups and one solvent control in this study. However, for each treatment group, the numbers of PDX models tested varied from 8 to 22 models. Some models were tested for certain treatment groups depending on numbers of animals available to be enrolled to the study.
Statistical analysis
For tumor volume data, the time points were transferred from the exact date to the relative days from the treatment start point (day 0 or t0) for each mouse. For individual mice, the tumor volume change for each time point was calculated as a relative level of tumor growth change from the baseline: δt = (Vt - V0)/V0 × 100, where Vt is the tumor volume at time t and V0 is the tumor volume at baseline. For animals for which there was no tumor volume measurement at time t (t 0<t<t 1), we imputed the missing tumor volume data by linear interpolation: δt=δ0+β(t-t 0), where β = (δt1 - δt0)/(t 1 - t 0). The adjusted area under the curve (aAUC) was defined as the mean percentage change over time, which was computed as the area under the tumor growth curve from the baseline up to time t, divided by t. We determined the aAUC values at 21 days and the last observation time point for each individual mouse, and used the aAUC at 21 days for comparison among groups because the treatments with both KRT-232 and navitoclax ended by day 21.
For drug response data, at a given time (t=21 days), each animal was classified into 1 of 3 groups by comparing tumor volume changes, as calculated by aAUC0-21 day or at day 21 after treatment initiation compared with the baseline: partial response ([PR], tumor regression ≥ 30% or δt≤-30%), stable disease ([SD], tumor growth was significantly suppressed when compared with animals treated with solvent control (P<0.05) but δt>-30%), or progressive disease [PD], tumor volume was not significantly different from control group (P>0.05). For the PDX model, 1-way ANOVA or 2-sample t-tests were performed to test for the difference in tumor volume change (δt) and aAUC at a given time point between the treatment and control groups, if applicable. For association studies of mutation and RPPA data, we combined PR and SD into a sensitive group and compared it with the resistant group PD using Fisher’s exact tests for mutation data and 2-sample t-tests for RPPA data. A multiplicity adjustment was performed by controlling the false discovery rate, as described previously [21]. Fisher exact test was used to compare the difference in treatment responses among different models. All statistical analyses were performed using R software version 3.5.2. A P value <0.05 was regarded as significant.
Results
Combination of KRT-232 and trametinib resulted in greater anti-cancer activity than did single agents in NSCLC PDXs
We determined the in vivo activity of the combination of KRT-232 and trametinib in 22 NSCLC PDX models, including 11 KRAS mutant and 11 KRAS wild type NSCLC PDXs. Figure 1 shows examples of tumor volume changes in mice treated with solvent (control), single agents, and combination therapy over time. We observed 2 patterns of tumor growth: 1) combination therapy resulted in significantly more anti-cancer activity than did single-agent therapy, as shown in PDXs TC211, TC494, TC255, and TC383; and 2) combination therapy resulted in either similar tumor volume changes as did single-agent therapy in PDX TC453 or having no activity as did solvent or single-agent treatment in PDX TC680.
Figure 1.
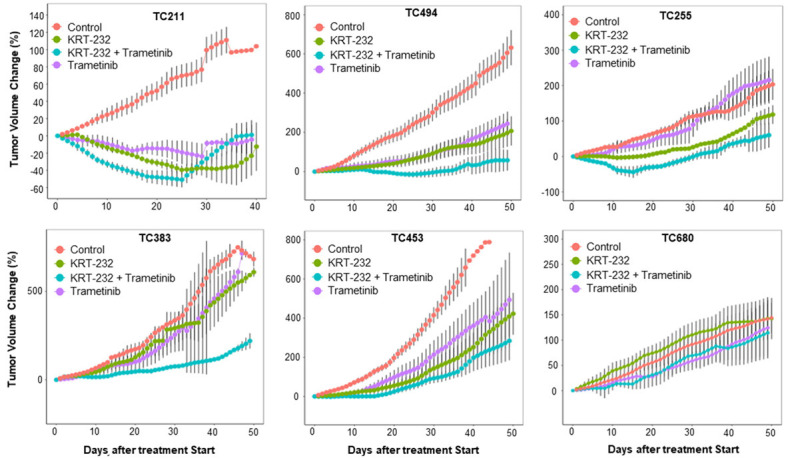
Effect of trametinib and KRT-232 combination therapy in NSCLC PDXs. NSCLC PDXs were treated with trametinib, KRT-232, or both, as described in the Materials and Methods. Mice treated with vehicles were used as controls. Tumor volume changes after treatment were determined by an R software program. The Y axis shows the mean ± standard error (n=3-5/group) of tumor volume changes for each PDX, with the starting tumor volume (about 200 mm3) and treatment starting day set as 0. The tumor volume changes for 6 PDXs are shown as examples. The combination therapy had more activity than did single-agent therapy in PDX TC211, TC494, TC255, and TC383, but not in TC453 (TP53 wt/KRAS mutated) and TC680 (TP53 wt/KRAS wt).
To categorize treatment responses, we determined tumor volume changes at the end of treatment (day 21) and the aAUC from the start of treatment (day 0) to the end of treatment (AUC0-21 day) to measure duration of response (Figure 2). We used the criteria to categorize treatment responses into three groups PR, SD, and PD, as described in Materials and Methods. On the basis of these criteria, 5 (23%), 10 (46%), and 7 (32%) of 22 PDXs treated with KRT-232 plus trametinib had a PR, SD, and PD, respectively (Table 1). In the 11 KRAS mutant PDXs, the PR, SD, and PD rates were 36% (4), 55% (6), and 9% (1), respectively (Table 2).
Figure 2.
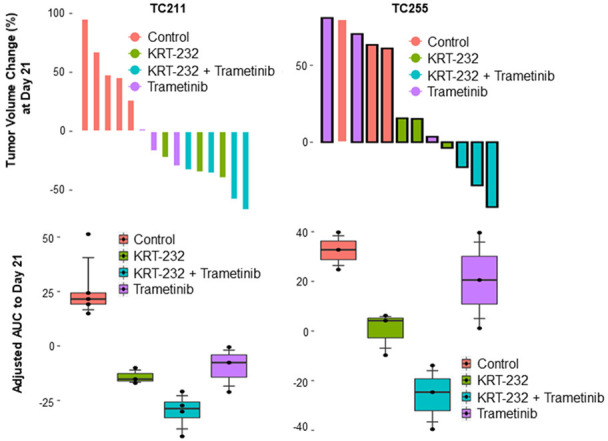
Treatment responses are presented as waterfall graphs and aAUC graphs. Tumor volume changes for each individual mouse at day 21 after treatment started are presented as a waterfall graph in the top panel for PDX TC211 and TC255. The box plots (bottom panel) shows the aAUC, with overall tumor volume changes from day 0 to day 21. The values represent the median (line inside box) and the third and first quartile (box) ± 1.5 x the interquartile range from the top and bottom of the box (error bar). The aAUC in the combination group is significantly different from the control and single-agent groups (P<0.05).
Table 1.
Summary of treatment responses in lung PDX models
| Treatment | PR (%) | SD (%) | Resistance (%) | Total |
|---|---|---|---|---|
| KRT-232 + Trametinib | 5 (23) | 10 (46) | 7 (32) | 22 |
| KRT-232 + Navitoclax | 1 (9) | 9 (82) | 1 (9) | 11 |
| Trametinib + Navitoclax | 5 (50) | 3 (30) | 2 (20) | 10 |
| KRT-232 | 2 (11) | 5 (28) | 11 (61) | 18 |
| Navitoclax | 0 (0) | 4 (50) | 4 (50) | 8 |
| Trametinib | 2 (10) | 10 (48) | 9 (43) | 21 |
Table 2.
Summary of treatment responses in KRAS mutant lung PDX models
| Treatment | PR (%) | SD (%) | Resistance (%) | Total |
|---|---|---|---|---|
| KRT-232 + Trametinib | 4 (36) | 6 (55) | 1 (9) | 11 |
| KRT-232 + Navitoclax | 1 (20) | 4 (80) | 0 (0) | 5 |
| Trametinib + Navitoclax | 5 (71) | 2 (29) | 0 (0) | 7 |
| KRT-232 | 1 (13) | 4 (50) | 3 (38) | 8 |
| Navitoclax | 0 (0) | 2 (50) | 2 (50) | 4 |
| Trametinib | 1 (10) | 7 (70) | 2 (20) | 10 |
PR: Tumor Regression ≥ 30%. SD: Tumor growth inhibition (tumor regression <30%; Tumor growth significantly suppressed vs controls).
Combination of navitoclax and trametinib resulted in greater anti-cancer activity than did single agents in KRAS mutant NSCLC PDXs
We determined the efficacy of combined treatment with navitoclax and trametinib in 10 NSCLC PDXs, including 7 KRAS mutant PDXs. Figure 3 shows the responses to combination therapy versus single-agent therapy in 3 selected KRAS mutant PDXs. Combination therapy resulted in significantly greater in vivo activity, as determined by the aAUC0-21 day, in all the 3 PDX models shown in Figure 3 (P<0.05). Of 10 PDX models treated with navitoclax plus trametinib 5 (50%), 3 (30%), and 2 (20%) NSCLC PDXs experienced a PR, SD, and PD, respectively (Table 1). All 7 KRAS mutant PDXs also responded to this combination treatment (PR, 5 of 7 [71%], or SD, 2 of 7 [29%]) (Table 2).
Figure 3.
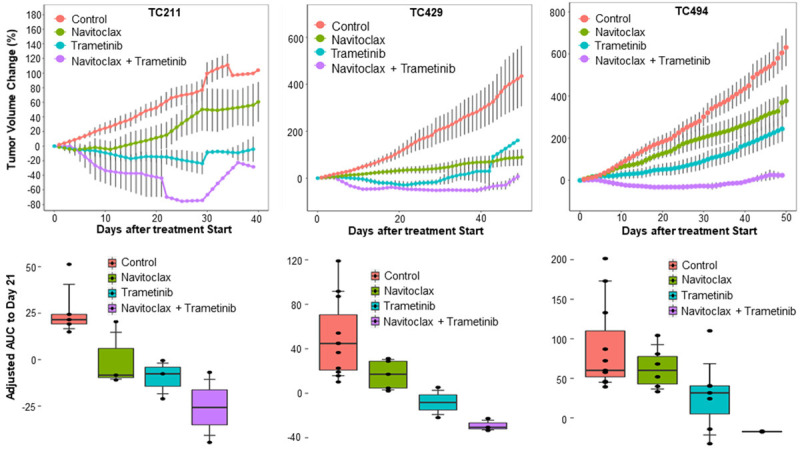
Combined effect of trametinib and navitoclax in NSCLC PDXs. The tumor volume changes and aAUC for 3 KRAS mutant PDXs are presented after treatment with trametinib, navitoclax, or both. The tumor volume changes and aAUC were determined as described in Figures 1 and 2. All 3 PDXs had an enhanced response to the combination therapy compared with single-agent therapies.
KRT-232 and navitoclax combined did not result in greater anti-cancer activity than did single agents in NSCLC PDXs
We tested 11 NSCLC PDX models to determine the in vivo anti-cancer activity of KRT-232 plus navitoclax combination therapy, including 4 TP53 mutant and 5 KRAS mutant PDXs. All KRAS mutant models tested were TP53 wild type. The combination led to 1 PR, 9 SD, and 1 PD. We did not observe any significant improvement in anti-cancer activity with combination therapy compared with single-agent activity (Figure 4). In three KRAS mutant PDXs that were tested for all six treatment groups, combination therapies of trametinib plus KRT-232 and/or trametinib plus navitoclax had enhanced anticancer activity when compared with all other groups (Figure 5).
Figure 4.
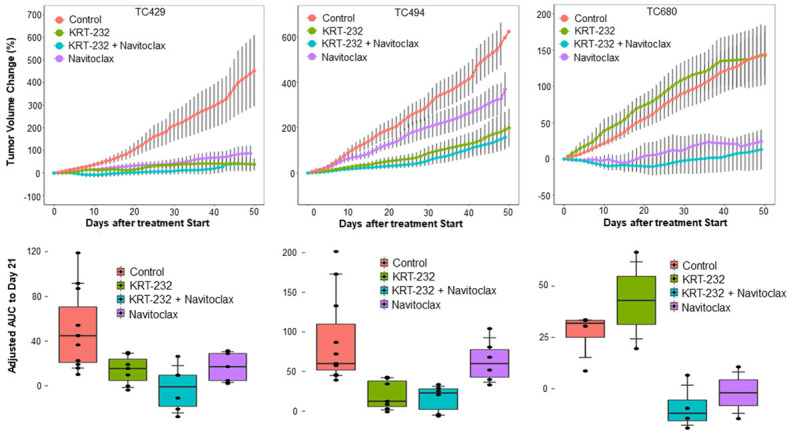
Combination effect of KRT-232 and navitoclax in NSCLC PDXs. The tumor volume changes and aAUC for 3 KRAS mutant PDXs are presented after treatment with KRT-232, navitoclax, or both. The tumor volume changes and aAUC were determined as described in Figures 1 and 2. The treatment responses to combination therapy were not significantly different to those to single-agent therapies in all 3 PDXs.
Figure 5.
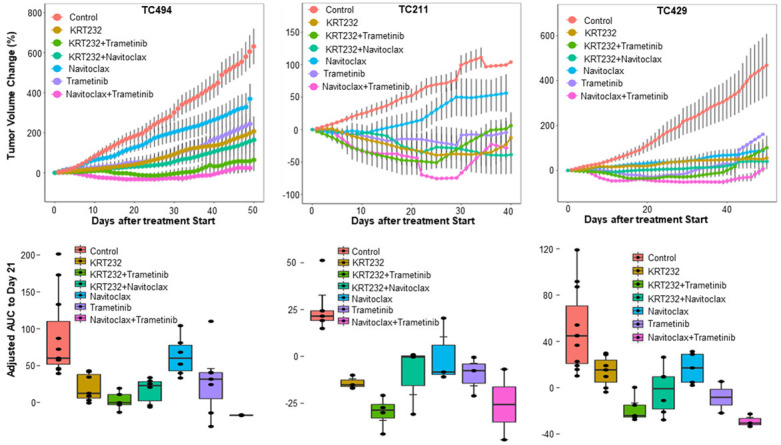
The tumor volume changes and aAUC for 3 KRAS mutant PDXs that were tested for all six treatments. The tumor volume changes and aAUC were determined as described in Figures 1 and 2. The combination therapies of trametinib plus KRT-232 and/or trametinib plus navitoclax had enhanced anticancer activity in these 3 PDXs.
The responses in all treatment groups are summarized in Table 1 and Supplementary Table 1. A PR was induced in 11% (2 of 18), 10% (2 of 21), and 0% (0 of 8) of mice after treatment with KRT-232, trametinib, and navitoclax alone, respectively. The PR rate in animals treated with trametinib plus navitoclax was significantly higher than those treated with trametinib alone (P=0.022) or those treated with navitoclax alone (P=0.036). In the KRAS mutant PDXs tested, the PR in the trametinib plus navitoclax group was significantly higher than in the trametinib alone group (P=0.035) but was not significant when compared with the navitoclax alone group (P=0.06). We did not observe significant differences in body weight in any treatment groups, suggesting that the treatments, including combination therapy, were not toxic at the doses tested in this study.
Molecular biomarkers associated with treatment response
We determined whether treatment responses in NSCLC PDX models were associated with genotype in TP53, KRAS, STK11, and EGFR or with protein expression levels by performing a reverse-phase protein microarray (RPPA) analysis. For this purpose, we combined PR and SD as a sensitive group and compared with PD as a resistant group. Fisher exact test revealed that the TP53 and STK11 genotypes were significantly associated with sensitivity to KRT-232 single-agent therapy. Seven of 11 TP53 wt PDXs were sensitive to KRT-232 (PR or SD [64%]), while none of the 7 TP53 mutant PDXs were sensitive (Supplementary Table S2) (P=0.013). Among 11 TP53 WT PDXs, 4/4 with mutated STK11 and 3/7 with STK11 WT were sensitive to KRT-232 treatment. Interestingly, all 4 STK11 mutant PDXs were sensitive to KRT-232 therapy compared with only 3 of 14 STK11 wt PDXs (21%) (P=0.011). Among 10 PDXs treated with trametinib plus navitoclax, all 7 KRAS mutant PDXs were sensitive, compared with 1 of 3 KRAS wt PDXs was sensitive, indicating that trametinib plus navitoclax is more effective in KRAS mutant tumors; however, the difference was not statistically significant (P=0.067).
A comparison of protein expression levels between treatment-sensitive (PR + SD) and -resistant (PD) groups revealed that levels of Bcl-XL, DUPS4, Rb, and BAP1 were significantly associated with sensitivity to KRT-232 as a single agent (P<0.05), when analyzed with either all PDX models tested or with TP53 wt PDXs only. However, in either case, the false discovery rate was greater than 0.3 (Supplementary Table 3). No significant association between KRT-232 and MDM2pS166 expression was observed when analyzed with all tested models or with TP53 wt only.
In the trametinib plus navitoclax treated group, the protein levels of GSK3b, Nrf2, LKB1/pS334, catalase, CTLA4, SMYD3, PCNA, and RAD50 were significantly different between the sensitive and resistant groups (P<0.005 at false discovery rate of 0.07). PDXs that were sensitive to trametinib plus navitoclax expressed higher levels of Nrf2, LKB1/pS334, catalase, and SMYD3 but lower levels of GSK3b, CTLA4, PCNA, and RAD50 than did those that were resistant (Figure 6). For the other treatment groups tested in this study, we did not identify any proteomic biomarkers that were significantly associated with sensitivity at the genomic and proteomic levels.
Figure 6.

Heatmaps of baseline protein levels in sensitive and resistant PDXs. The heatmaps for the top 8 proteins whose levels were significantly different between sensitive and resistant PDXs are presented for trametinib plus navitoclax (A) and KRT-232 monotherapy (B). Each vertical column represents each PDX model tested. Side bars for treatment responses and genotypes of TP53, KRAS, STK11, and EGFR are shown on the top of each graph. WT: wild type; Mut: mutant.
Discussion
We determined treatment responses to single-agent and combination therapies with the MEK inhibitor trametinib, the MDM2 inhibitor KRT-232, and the BCL-XL/BCL-2 inhibitor navitoclax in molecularly annotated NSCLC PDX models. Our results showed that trametinib plus KRT-232 or navitoclax led to greater in vivo anti-cancer activity than did single-agent therapy, whereas KRT-232 plus navitoclax did not result in improved activity. We did not observe obvious weight loss in any of the treatment groups and tested models, suggesting that all treatment regimens were not toxic. Moreover, we identified genomic and proteomic biomarkers that were significantly associated with treatment responses to single-agent KRT-232 and trametinib plus navitoclax.
KRT-232 is a potent MDM2 inhibitor that activates TP53 signaling and inhibits tumor cell proliferation in TP53 wt tumor cells but has no significant effect on TP53 mutant tumor cells [10]. Similar to the published results from cell lines, we found that among 18 NSCLC PDX models tested, none of the TP53 mutated tumors was sensitive to KRT-232 treatment. KRT-232 anti-cancer activity was observed in 7 of 11 TP53 wt PDXs but none of the 7 TP53 mutant PDXs. Interestingly, we also found an association between response to KRT-232 and STK11 mutations: all 4 SKT11 mutant PDXs responded to KRT-232. As these 4 STK11 mutant NSCLC were TP53 wt and KRAS mutant, it is possible that the response to KRT-232 was primarily due to activation of TP53. Nevertheless, this result indicated that STK11 mutations would not have a negative effect on the anti-cancer activity of KRT-232. Because mutations of SKT11 and TP53 are mutually exclusive in lung adenocarcinoma with KRAS mutations [22] and because lung adenocarcinoma with concomitant mutations of KRAS and STK11 is highly resistant to immune checkpoint blockade therapy [23], it will be important to determine whether KRT-232 can sensitize these tumors to immune checkpoint blockade therapy.
Trametinib plus KRT-232 or navitoclax is currently being studied in clinical trials for cancer treatment [1,12]. Both combination therapies have been found to be more effective than single-agent therapy in several models, particularly in KRAS mutant models. All 7 KRAS mutant PDX models were sensitive to trametinib plus navitoclax in our study (5 PR and 2 SD). Ten of the 11 KRAS mutant PDX models were sensitive to trametinib plus KRT-232 (4 PR and 6 SD).
Our study of proteomic biomarkers revealed that the expression levels of several proteins or phosphorylation sites were associated with treatment responses to trametinib plus navitoclax. It is not yet clear whether the levels of these protein markers were associated with KRAS mutations. Increased expression of NRF2 and catalase may indicate the presence of oxidative stress, whereas increased expression of LKB1/pS334 may indicate increased AKT activity in tumors, as phosphorylation of LKB1 at Ser334 by AKT was reported to block the tumor suppressor activity of LKB1 [24]. KRAS mutations are known to increase oxidative stress and activate the PI3K/AKT pathway [25,26]. In addition, SMYD3, a histone lysine methyltransferase, is reported to promote RAS-driven tumorigenesis by methylating MAP3K2 and potentiating activation of the RAS/RAF/MEK/ERK signaling pathway [27]. Thus, several protein biomarkers that are associated with response to trametinib and navitoclax are involved in RAS-induced alterations in cellular metabolism, signal transduction, and cooperation with oncogenic RAS in tumorigenesis.
Nevertheless, this study has some limitations. Because all PDX models tested in this study were established in immune-compromised mice, the effects of host immune responses and the tumor immune microenvironment could not be determined. Although we tested treatment responses in multiple PDX models, the number of models tested was relatively small and may not cover all subtypes of KRAS mutant lung adenocarcinoma. It may be important to note that not all of the models were tested with all of the treatments, so the statistical comparisons between the treatments and between markers of sensitivity/resistance may be underpowered. It is not yet clear whether the molecular biomarkers identified in this study could be used for patient stratification in future clinical trials, in addition to TP53 wild type as the current patient selection marker for treatment with KRT-232. However, because effective therapy for KRAS-mutant NSCLC, one of the most common molecular subtypes of NSCLC, is not yet available clinically, our results support the initiation of expanded clinical trials of combination therapies of trametinib plus KRT-232 or navitoclax for KRAS mutant NSCLC.
Acknowledgements
We thank Ann M Sutton and the Department of Scientific Publications at The University of Texas MD Anderson Cancer Center for editorial review of the manuscript. This work was supported in part by the National Institutes of Health through grant R01 CA190628, The University of Texas PDX Development and Trial Center grant U54 CA224065, Specialized Program of Research Excellence (SPORE) grant CA070907, Experimental Therapeutics Clinical Trials Network grant UM1CA186688, and The University of Texas MD Anderson Cancer Center support grant P30 CA016672 (we used the Characterized Cell Line Core Facility and Research Animal Support Facility); it was also supported by funds from the University Cancer Foundation via the Lung Cancer Moon Shot Program and the Sister Institution Network Fund at MD Anderson.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kim C, Giaccone G. MEK inhibitors under development for treatment of non-small-cell lung cancer. Expert Opin Investig Drugs. 2018;27:17–30. doi: 10.1080/13543784.2018.1415324. [DOI] [PubMed] [Google Scholar]
- 2.Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, Giannone V, D’Amelio AM Jr, Zhang P, Mookerjee B, Johnson BE. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18:1307–1316. doi: 10.1016/S1470-2045(17)30679-4. [DOI] [PubMed] [Google Scholar]
- 3.Odogwu L, Mathieu L, Blumenthal G, Larkins E, Goldberg KB, Griffin N, Bijwaard K, Lee EY, Philip R, Jiang X, Rodriguez L, McKee AE, Keegan P, Pazdur R. FDA approval summary: dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring BRAF V600E mutations. Oncologist. 2018;23:740–745. doi: 10.1634/theoncologist.2017-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crino L, Orlov S, Blackhall F, Wolf J, Garrido P, Poltoratskiy A, Mariani G, Ghiorghiu D, Kilgour E, Smith P, Kohlmann A, Carlile DJ, Lawrence D, Bowen K, Vansteenkiste J. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical trial. JAMA. 2017;317:1844–1853. doi: 10.1001/jama.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melosky B, Bradbury P, Tu D, Florescu M, Reiman A, Nicholas G, Basappa N, Rothenstein J, Goffin JR, Laurie SA, Wheatley-Price P, Leighl N, Goss G, Reaume MN, Butts C, Murray N, Card C, Ko J, Blais N, Gray S, Lui H, Brown-Walker P, Kaurah P, Prentice LM, Seymour L. Selumetinib in patients receiving standard pemetrexed and platinum-based chemotherapy for advanced or metastatic KRAS wildtype or unknown non-squamous non-small cell lung cancer: a randomized, multicenter, phase II study. Canadian Cancer Trials Group (CCTG) IND.219. Lung Cancer. 2019;133:48–55. doi: 10.1016/j.lungcan.2019.04.027. [DOI] [PubMed] [Google Scholar]
- 6.Greystoke A, Steele N, Arkenau HT, Blackhall F, Md Haris N, Lindsay CR, Califano R, Voskoboynik M, Summers Y, So K, Ghiorghiu D, Dymond AW, Hossack S, Plummer R, Dean E. SELECT-3: a phase I study of selumetinib in combination with platinum-doublet chemotherapy for advanced NSCLC in the first-line setting. Br J Cancer. 2017;117:938–946. doi: 10.1038/bjc.2017.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hellmann MD, Kim TW, Lee CB, Goh BC, Miller WH Jr, Oh DY, Jamal R, Chee CE, Chow LQM, Gainor JF, Desai J, Solomon BJ, Das Thakur M, Pitcher B, Foster P, Hernandez G, Wongchenko MJ, Cha E, Bang YJ, Siu LL, Bendell J. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol. 2019;30:1134–1142. doi: 10.1093/annonc/mdz113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng J, Fang B, Liao Y, Chresta CM, Smith PD, Roth JA. Apoptosis induction by MEK inhibition in human lung cancer cells is mediated by Bim. PLoS One. 2010;5:e13026. doi: 10.1371/journal.pone.0013026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, Deignan J, Duquette J, Eksterowicz J, Fisher B, Fox BM, Fu J, Gonzalez AZ, Gonzalez-Lopez De Turiso F, Houze JB, Huang X, Jiang M, Jin L, Kayser F, Liu JJ, Lo MC, Long AM, Lucas B, McGee LR, McIntosh J, Mihalic J, Oliner JD, Osgood T, Peterson ML, Roveto P, Saiki AY, Shaffer P, Toteva M, Wang Y, Wang YC, Wortman S, Yakowec P, Yan X, Ye Q, Yu D, Yu M, Zhao X, Zhou J, Zhu J, Olson SH, Medina JC. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem. 2014;57:1454–1472. doi: 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- 10.Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, Eksterowicz J, Ye Q, Jin L, Chen A, Zhou J, Cordover D, Kaufman S, Kendall R, Oliner JD, Coxon A, Radinsky R. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol Cancer Ther. 2015;14:649–658. doi: 10.1158/1535-7163.MCT-14-0710. [DOI] [PubMed] [Google Scholar]
- 11.Gluck WL, Gounder MM, Frank R, Eskens F, Blay JY, Cassier PA, Soria JC, Chawla S, de Weger V, Wagner AJ, Siegel D, De Vos F, Rasmussen E, Henary HA. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Invest New Drugs. 2020;38:831–843. doi: 10.1007/s10637-019-00840-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erba HP, Becker PS, Shami PJ, Grunwald MR, Flesher DL, Zhu M, Rasmussen E, Henary HA, Anderson AA, Wang ES. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019;3:1939–1949. doi: 10.1182/bloodadvances.2019030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 14.Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, Ben-Zvi I, Stebbing J, McGuire W, Harris W, Maki R, Gaya A, Bedi A, Zacharoulis S, Ravi R, Wexler LH, Hoque MO, Rodriguez-Galindo C, Pass H, Peled N, Davies A, Morris R, Hidalgo M, Sidransky D. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol. 2017;28:2595–2605. doi: 10.1093/annonc/mdx416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De OE, Rubio-Viqueira B, Strawn S, Wick MJ, Martell J, Sidransky D. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. 2011;10:1311–1316. doi: 10.1158/1535-7163.MCT-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao C, Wang L, Peng S, Cao M, Li H, Hu J, Huang X, Liu W, Zhang H, Wu S, Pataer A, Heymach JV, Eterovic AK, Zhang Q, Shaw KR, Chen K, Futreal A, Wang M, Hofstetter W, Mehran R, Rice D, Roth JA, Sepesi B, Swisher SG, Vaporciyan A, Walsh GL, Johnson FM, Fang B. Gene mutations in primary tumors and corresponding patient-derived xenografts derived from non-small cell lung cancer. Cancer Lett. 2015;357:179–185. doi: 10.1016/j.canlet.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Zhang R, Wang L, Correa AM, Pataer A, Xu Y, Zhang X, Ren C, Wu S, Meng QH, Fujimoto J, Jensen VB, Antonoff MB, Hofstetter WL, Mehran RJ, Pisimisis G, Rice DC, Sepesi B, Vaporciyan AA, Walsh GL, Swisher SG, Roth JA, Heymach JV, Fang B. Tumor characteristics associated with engraftment of patient-derived non-small cell lung cancer xenografts in immunocompromised mice. Cancer. 2019;125:3738–3748. doi: 10.1002/cncr.32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pu X, Zhang R, Wang L, Chen Y, Xu Y, Pataer A, Meraz IM, Zhang X, Wu S, Wu L, Su D, Mao W, Heymach JV, Roth JA, Swisher SG, Fang B. Patient-derived tumor immune microenvironments in patient-derived xenografts of lung cancer. J Transl Med. 2018;16:328. doi: 10.1186/s12967-018-1704-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He Y, Zhou Z, Hofstetter WL, Zhou Y, Hu W, Guo C, Wang L, Guo W, Pataer A, Correa AM, Lu Y, Wang J, Diao L, Byers LA, Wistuba II, Roth JA, Swisher SG, Heymach JV, Fang B. Aberrant expression of proteins involved in signal transduction and DNA repair pathways in lung cancer and their association with clinical parameters. PLoS One. 2012;7:e31087. doi: 10.1371/journal.pone.0031087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang X, Cao M, Wu S, Wang L, Hu J, Mehran RJ, Roth JA, Swisher SG, Wang RY, Kantarjian HM, Andreeff M, Sun X, Fang B. Anti-leukemia activity of NSC-743380 in SULT1A1-expressing acute myeloid leukemia cells is associated with inhibitions of cFLIP expression and PI3K/AKT/mTOR activities. Oncotarget. 2017;8:102150–102160. doi: 10.18632/oncotarget.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 22.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Parra ER, Rodriguez-Canales J, Zhang J, Giri U, Gudikote J, Cortez MA, Yang C, Fan YH, Peyton M, Girard L, Coombes KR, Toniatti C, Heffernan TP, Choi M, Frampton GM, Miller V, Weinstein JN, Herbst RS, Wong KK, Zhang J, Sharma P, Mills GB, Hong WK, Minna JD, Allison JP, Futreal A, Wang J, Wistuba II, Heymach JV. Co-occurring genomic alterations define major subsets of KRAS - mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–77. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, Ali SM, Elvin JA, Singal G, Ross JS, Fabrizio D, Szabo PM, Chang H, Sasson A, Srinivasan S, Kirov S, Szustakowski J, Vitazka P, Edwards R, Bufill JA, Sharma N, Ou SI, Peled N, Spigel DR, Rizvi H, Aguilar EJ, Carter BW, Erasmus J, Halpenny DF, Plodkowski AJ, Long NM, Nishino M, Denning WL, Galan-Cobo A, Hamdi H, Hirz T, Tong P, Wang J, Rodriguez-Canales J, Villalobos PA, Parra ER, Kalhor N, Sholl LM, Sauter JL, Jungbluth AA, Mino-Kenudson M, Azimi R, Elamin YY, Zhang J, Leonardi GC, Jiang F, Wong KK, Lee JJ, Papadimitrakopoulou VA, Wistuba II, Miller VA, Frampton GM, Wolchok JD, Shaw AT, Janne PA, Stephens PJ, Rudin CM, Geese WJ, Albacker LA, Heymach JV. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–835. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, Siu FM, Che CM, Xu A, Wang Y. Akt blocks the tumor suppressor activity of LKB1 by promoting phosphorylation-dependent nuclear retention through 14-3-3 proteins. Am J Transl Res. 2012;4:175–186. [PMC free article] [PubMed] [Google Scholar]
- 25.Storz P. KRas, ROS and the initiation of pancreatic cancer. Small GTPases. 2017;8:38–42. doi: 10.1080/21541248.2016.1192714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang B. RAS signaling and anti-RAS therapy: lessons learned from genetically engineered mouse models, human cancer cells, and patient-related studies. Acta Biochim Biophys Sin. 2016;48:27–38. doi: 10.1093/abbs/gmv090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazur PK, Reynoird N, Khatri P, Jansen PW, Wilkinson AW, Liu S, Barbash O, Van Aller GS, Huddleston M, Dhanak D, Tummino PJ, Kruger RG, Garcia BA, Butte AJ, Vermeulen M, Sage J, Gozani O. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014;510:283–287. doi: 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.