

Abstract
Hepatocellular carcinoma (HCC) is characterized by poor outcome and shows limited drug-response in clinical trials. Tumor immune microenvironment (TIME) exerts a strong selection pressure on HCC, leading to HCC evolvement and recurrence after multiple therapies. T cell-mediated immunoreaction during cancer surveillance and clearance is central in cancer immunity. Heterogenous T cell subsets play multiple roles in HCC development and progression. The re-educated T cells in TIME usually lead to deteriorated T cell response and tumor progression. Investigation into immune system dysregulation during HCC development will shed light on how to turn immune suppressive state to immune activation and induce more efficient immune response. Emerging T cell-based treatment such as cancer vaccines, CAR-T cell therapy, adoptive cell therapy, and immune checkpoint inhibitors (ICIs), have been proved to cause tumor regression in some clinical and preclinical trials. In this review, we focused on recent studies that explored T cells involved in HCC and how they affect the course of disease. We also briefly outlined current T cell-based immunotherapies in HCC.
Keywords: Hepatocellular carcinoma, T lymphocytes, tumor immune microenvironment, prognosis, immunotherapy
Introduction
Liver cancer is the fourth leading cause of cancer-related mortality in the world, and hepatocellular carcinoma (HCC) accounts for approximately 85% to 90% of primary liver cancers [1,2]. While en bloc resection, local ablation and liver transplantation may be curative treatment options for a selective population, high post-operation recurrence makes the survival of HCC patients dismal. A study on natural history of HCC indicated that patients with advanced stage (Barcelona Clinic Liver Cancer, BCLC stage C) had a survival of only 3.4 months if untreated [3]. HCC is characteristic by altered immune microenvironment since patients are usually with chronic liver inflammation such as chronic viral hepatitis and non-alcoholic steatohepatitis (NASH) [4]. Immune cells are re-educated towards immune tolerant type and participate in HCC occurrence and progression. Immune suppressive cells contribute to immune tolerant microenvironment and associate with dismal prognosis of this refractory disease [5]. Targeted therapy and immune checkpoint inhibitors such as nivolumab were approved for treatment of advanced HCC [6,7]. However, the overall survival rates for patients with HCC have been minimally improved. There remains an urgent need for more effective therapies targeting HCC with well tolerant adverse effects.
Immune and immunosuppressive factors are balancing the cancer immunity and participating in HCC development (Figure 1). Immune cells such as lymphocytes, macrophages, neutrophils, myeloid-derived suppressor cells (MDSCs), are abundant in HCC mellitus and play important roles in tumor immune balance (Table 1). Characterizing the heterogeneous populations of immune cells in HCC may help to deepen our understandings of immune activity in tumor progression and recurrence, thus boost the development of effective immunotherapy.
Figure 1.
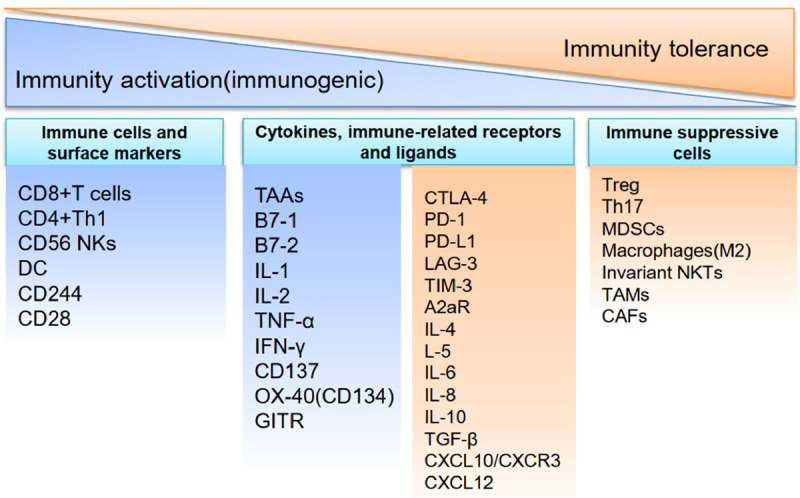
Immune balance conditioning in HCC. The activation and suppression of immunity are associated with anti-tumor response or tumor progression. The balance of immune status is regulated by immune cells, cytokines, and immune-related receptors and ligands. TAAs: tumor associated antigens; TILs: tumor infiltrating lymphocytes; CTLs: cytotoxic lymphocytes; DC: dendritic cell; TNF- α: tumor necrosis factor α; IFN-γ: interferon γ; OX-40R: OX-40 receptor; Treg: T regulatory cell; Th7: T helper 17 cells; MDSCs: myeloid-derived suppressor cells; NKTs: natural killer T cells; TAMs: tumor-associated macrophages TGF-β: transforming growth factor-β; GITR: glucocorticoid induced TNF receptor; CAFs: cancer-associated fibroblasts.
Table 1.
Immune cells in tumor microenvironment
| Cell type | Abbre | Molecular markers |
|---|---|---|
| Tumor-associated macrophages | TAMs | IL-10high, IL-12low; CD68 (pan-macrophage marker); CD163 (M2); c-Maf |
| Dendritic cells | DCs | Human myeloid-derived DCs: CD11c+, CD123- |
| Subgroups: CD16+, CD1c+, CD141+ | ||
| Human plasmacytoid DCs, pDC: CD11c-, MHC-II+, CD123+ | ||
| CD8+ cytotoxic T lymphocytes | CD8+ CTLs | T-cell anergy: direct positive markers are lacking |
| T-cell exhaustion: B7-H1+, PD-1+, CTLA4+, CD160+, LAG-3+, Tim-3+ | ||
| T-cell aging: CD28-, Tim-3high, CD57high, KLRG-1high | ||
| Natural killer T cells | NKTs | CD1d+ |
| Myeloid-derived suppressor cells | MDSCs | Neutrophils: CD15+, CD33+, CD11b+, Lin-, IL-4Rα+, CD80+, CD115+, VEG |
| FR1+, CD62Llow, CD16low (CD66b+ in HCC) | ||
| Monocytes: CD14+, HLA Dr-/low, CD66b+, IL-4Rα+, CD80+, CD115+ | ||
| Regulatory T cells | Tregs | CD4+CD25+FOXP3+ |
| T helper cells | Th1/2, Th17, Th22, Tfh, Th9 | Th1: CD4+, CCR1+, IFN-γ+, IL-2+, IL-22+, TNF-β+ |
| Th2: CD4+, CCR3+, CCR4+, IL-4+, IL-5+, IL-9+, IL-10+ and IL-13+ | ||
| Th17: CD4+, CCR6+, RORγ+, IL-17+ | ||
| Th22: CD4+, CCR4+, CCR6+, IL-22+ | ||
| Tfh: CD4+, IL-21+, IL-4+ and CXCL13+ | ||
| Th9: CD4+, IL-9+ |
CD, cluster of differentiation; IL, interleukin; CCR, C-C motif chemokine receptor; FOXP3, transcription factor forkhead box p3; KLRG1, killer cell lectin-like receptor subfamily G member 1; RORγ, related orphan receptor gamma.
T cells are crucial in surveillance and clearance of tumor cells. Alterations in T cells antigen presenting, trafficking and infiltrating all contribute to tumor formation and recurrence after curative therapy. Single cell RNA sequencing showed distinctive functional composition of infiltrating T cells in HCC, and revealed that exhausted phenotype of cytotoxic T lymphocytes (CTLs) was associated with worse prognosis [8]. Anti-CTLA-4 or anti-PD-1/PD-L1 therapies enhance T cell response to tumor cells, reverse T cell exhaustion, and restore T cell function [9]. Several immune check-point inhibitors have proved their potential in treatment for advanced HCC. However, immune therapies are effective in only a small proportion of patients, implying unknown mechanism underlying the tumor immune escape. With progress in the genomic analyses and gene sequencing, a deeper knowledge of HCC immune contexture and local immune microenvironment in a more comprehensive and multidimensional way will help us to better manage this lethal malignancy. In this review, we mainly discuss different types of T cells in HCC, provide rationales for developing efficient immunotherapies, and briefly describe current immunotherapies based on T cells.
The subtypes of T cells infiltrating and surrounding tumor cells of HCC
CD8+ cytotoxic T lymphocytes (CD8+ CTLs)
HCC immune response highlights the role of CD8+ T cells. CD8+ effector T cells exert antigen specific cytotoxic effects on tumor cells by recognition of specific antigens carried with MHC class I molecules. Tumor antigens are presented to CD8+ T cells by professional antigen-presenting cells (APCs), such as dendritic cells (DCs). CTLs bind to tumor cell surface by TCRs recognition of MHC I. CD8+ T cells exert killing effects by direct lysis of cancer cells and secretion of enzymes and cytotoxic cytokines, such as perforin, granzyme B, interferon γ (IFN-γ), and tumor necrosis factor α (TNF-α). The perforin perforates cell membrane and causes cell lysis, and granzyme B activates caspase cascade. Importantly, the FasL on CD8+ T cells binds to Fas receptors on tumor cells and triggers caspase signal transduction pathways. Those activities together induce tumor cell apoptosis or necrosis. Peripheral CD8+ T cell level were significantly elevated in HCC patients when compared to normal controls [10]. More importantly, tumor infiltrating tumor-associated antigen (TAA) specific CD8+ T lymphocytes are found in the tumor bed or peritumoral tissues, which is recognized as crucial in surveillance and controlling growth of solid tumors [11,12]. T cell infiltration and a high CD4+/CD8+ T cell ratio were found to be significantly associated with decreased recurrence after liver transplantation for HCC [13]. However, a complete tumor immune response requires cancer antigen presentation, T cell priming and trafficking to tumor site. T cell infiltration alone did not predict cancer immune activity. Pathological studies have identified three tumor infiltration phenotypes: the immune-desert phenotype, the immune-excluded phenotype and the inflamed phenotype [14]. Both quantity and quality of infiltrating T cells contribute to anti-tumor efficacy. We mainly discuss studies focusing on CD8+ T cells in HCC from both experimental and clinical aspects.
For decades, cytotoxic T cell infiltration of the tumors has been reported to favor a better survival in different types of human cancers, and CD8+ T cells activation is related to tumor regression. Gabrielson A and colleagues studied surgical specimens from 65 HCC patients and identified three types of tumor infiltration in intratumoral parts and invasive margin: dense nodular clusters of lymphocytes, diffuse, evenly distributed lymphocytes, and rare or scattered lymphocytes. Detailed analysis of T cell subsets showed that CD8+ T lymphocytes infiltration was associated with prolonged recurrence-free survival (RFS) and low rate of recurrence [15]. However, in another study by Kobayashi N and colleges, tumor-infiltrating CD8+ T cells was not predictive for survival of HCC patients, and no significant difference in the survival rate was observed between low and high CD8+ T cell groups [16]. Previous studies have reported that the percentage of infiltrating CD8+ T cells was decreased in HCC compared to normal adjacent tissues [17-20], and tumor antigen-specific CD8+ T cells displayed an exhausted phenotype. In many types of cancers including HCC, quantity of CD8+ T cells alone did not well predict the prognosis [11,19,20]. Apart from immunosuppressive effect of T regulatory cells (Tregs), dysfunction of tumor infiltrating CD8+ T cells usually manifests as exhaustion and tolerance, featured with impaired effector function and expression of immune inhibitory receptors. T cell exhaustion in tumor can be induced by tumor antigen load and immunosuppressive factors. Increased expression of inhibitory receptors such as programmed cell death protein 1 (PD-1), lymphocyte activation gene-3 (LAG-3), 2B4 (CD244), T cell immunoglobulin and mucin domain 3 (TIM-3), cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) are found in exhausted T cells. Those immunoinhibitory receptors are druggable targets for tumor immunotherapy. Moreover, tumor-specific effector T cells are subjected to apoptosis, which was known as activation-induced cell death (AICD) [21] and animal experiment showed that CD8+ T cells undergo high rates of apoptosis within tumor immune microenvironment [22]. These studies indicate tumor microenvironment can educate cytotoxic T cells towards exhausted and impaired function, and may lead to more T cell death through unique mechanism.
To explore the CD8+ T cell responses to tumor-associated antigens (TAAs) in HCC, Flecken and colleagues used overlapping peptides to study tumor antigen-associated CD8+ T cells in a group of 96 HCC patients [23]. They detected TAA-specific CD8+ T cell responses to all four antigens in patients with HCC, while only one individual response in control group. The authors also showed impaired IFN-γ production of tumor infiltrating CD8+ T cells, indicating exhaustion of intratumoral CD8+ T cells. Either the number of TAA-specific CD8+ T cells or the number of TAA targets was positively associated with patient survival. Taken together, the authors suggested the TAA-specific CD8+ T cells were functionally deficient and restoration of CD8+ T cells function would be a promising therapeutic strategy.
CD8+ T cells are dysfunctional and heterogenous in tumor immune microenvironment. Zhou and colleagues studied a population of 59 HCC patients aimed to determine T-cell responses in the HCC microenvironment [24]. They found increased expression of inhibitory receptors, including PD-1, TIM 3 and LAG3, on tumor-infiltrating CD4+ Th cells and CD8+ cytotoxic T cells than that from normal adjacent liver tissue or peripheral blood. They showed PD-1+ infiltrating T cells were significantly more activated using the activation markers HLA-DR, but with no increase or even decrease of percentage of granzyme B, IFN-γ and TNF-α producing cells, indicating impaired functionality of CD8+ T cells. What’s more, in vitro experiment showed PD-L1 blockade alone or in combination with TIM3, LAG3, or CTLA4 blockade enhanced proliferation and cytokine production in CD8+ tumor infiltrating lymphocytes (TILs). In another study by Kim et al., the authors studied the subtypes of CD8+ T cells in HCC [25]. According to PD1 expression on CD8+ TILs, 90 HCC patients who underwent resection were subjected to PD1-high, intermediate and negative subgroups. The authors identified 865 differentially expressed genes between PD1-high and PD1-intermediate subgroups. Based on different enrichment genes, the authors found high-PD1high subgroups had significant poorer overall survival compared to low-PD1high groups in the Cancer Genome Atlas HCC cohort. Notably, LAYN clusters indicating exhaustion were significantly enriched in PD1-high patients, while expression level of LEF1 and CX3CR1 cluster representing memory and effector CD8+ T cells were much lower. Moreover, high PD1 expression on CD8+ T cells was associated with larger tumor sizes, higher AFP levels and larger proportion of microvascular invasion, which may suggest similar heterogeneity of HCC biological features in association with infiltrating CD8+ T cells. Finally, the authors proved combined immune checkpoint blockades restored CD8+ T cells function more efficiently than single use of PD1 blockade in patients with PD1-high TILs, while such effects was absent in PD1-low subgroups. In clinical practice, combination of immune checkpoint inhibitors may be theoretically possible, it should be noted not all HCC patients can benefit. These results suggest immune microenvironment is closely related with HCC phenotypes, and more liable predictive biomarkers for HCC immunotherapy should be further explored.
The presence of T cell infiltration including tumor-specific T cells is not necessarily related with tumor regression, which suggesting a failure of immune surveillance and clearance. Some theories may explain the inadequate immune response to tumors, such as partial antigen masking, failure of antigen processing, inadequate co-stimulation signals, and direct suppression of effector cells. Targeting one or more pathways may provide new insights into HCC treatment. Of note, recent study has revealed that in addition to exhaustion of tumor-specific T cells, tumor-unrelated bystander CD8+ T cells were common and abundant in tumor infiltrates, which shed light on how exhaustion of effector T cells and bystander T cells impact the immune conditioning of HCC [26]. Further investigation may be taken to clarify the characteristics of both exhausted CTLs and bystander CD8+ T cells in HCC, and their roles in HCC development and recurrence.
Natural killer T (NKT) cells
NKT cells are a subset of T lymphocytes expressing both natural killer (NK) and T cell receptors (TCRs), bridging the innate and adaptive immune systems. Two distinct subpopulations are identified as type I (or invariant NKT, iNKT) and type II NKT cells. NKT cells are distributed in blood and lymph tissues, and account for approximately 30% of the T cells in liver [27]. NKT cells produce both pro-inflammatory and anti-inflammatory cytokines, and play critical roles in immune response and tumor surveillance. Dysregulation of NKT cells may lead to immune imbalance and are associated with cancer development. Previous studies showed distinct subsets of NKT cells exerted their positive or negative effects in tumor immunology via distinct mechanisms. For instance, in addition to direct lysis of cancer cells, iNKT cells exert anti-tumor activity mainly through production of cytokines such as INF-γ, IL-2, TNF-α, which subsequently recruit NK cells, dendritic cells and CD8+ T cells. Moreover, iNKT cells were found to restore the function of exhausted NK cells and CD8+ T cells via IL-21, IL-2 and IL-12 [28]. Conversely, type II NKT cells can exert immunosuppressive effect and promote tumor growth by producing IL-13. Apart from the opposite effect in tumor immunity, iNKT and type II NKT cells also cross regulate each other and interact with other immune cells [29]. Elucidating the complex network between NKT cell subpopulations and mechanisms underlying NKT cell immune action will help to improve cancer immunotherapy, especially in development of cancer vaccines.
Enrichment of NKT cells in the liver highlights their roles in liver inflammation and carcinogenesis. Through recruiting macrophages and neutrophils and inducing fat accumulation in hepatocytes, activation of iNKT cells can promote steatosis and steatohepatitis of the liver. Oppositely, type II NKT cells attenuate liver inflammation by inhibiting iNKT cell-mediated inflammatory pathways. As previous studies showed NKT cells might have anti or pro-tumor effects according to cell subtypes and interaction with other immune cells, the roles of NKT cells in HCC remain controversial. Increased NKT cells were observed in both human and mouse HCC samples; iNKT cell frequency was reported to be slightly decreased in blood while significantly increased in tumor tissues [30,31]. Some studies indicated NKT cells suppressed HCC via inducing production of IFN-γ and activation of other effector cells including NK cells and cytotoxic T cells [32,33], while others suggested an oncologic role in HCC development [31]. These studies revealed NKT cells played multiple roles in tumor surveillance and HCC development. More importantly, the association between NKT cells and prognosis in HCC patients has not been fully understood. Since these results are mostly based on animal models, further researches are needed to clarify the exact roles of NKT cells in human HCC. It still needed to explore how heterogenous NKT cell subsets act within liver microenvironment and influence hepatocarcinogenesis.
Regulatory T cells (Tregs)
Tregs are a sub-population of CD4+ T lymphocytes (approximately 5%), and the recruitment of Tregs into tumor microenvironment is involved in tumor cells escaping from immune surveillance and clearance. Two main subsets of Tregs have been identified in cancers as natural Tregs and inducible Tregs. Natural Tregs, which are CD4+CD25+FOXP3+, are major regulatory T cells and maintain peripheral cancer-related inflammation through Fas/FasL pathway. Natural Tregs in tumor can induce cell death of natural killer cells and CD8+ T cells in a granzyme B- and perforin-dependent manner. Inducible Tregs, which are FOXP3+ or not, mediate immune suppression by producing adenosine, IL-10 and TGF-β. Tregs can inhibit effector B and T cells functions after antigen response. In normal condition, Tregs suppress anti-self-immune responses and have important roles in balancing immune tolerance and inflammatory responses.
Tregs exert immunosuppressive effects and hinder cancer immune response through several mechanisms. Those mechanisms include inhibiting the effector cell function by secreting inhibitory cytokines, killing effector cells by granzyme and perforin, disrupting effector cell metabolism, and affecting differentiation and proliferation of dendritic cells which possibly involved signal transduction through CTLA-4 and LAG3. Coordination of TCR stimulation and IL2 receptor signaling is central to stability and immune suppressive function of Tregs. In HCC, Tregs mainly induce immunosuppression by impairing CD8+ T cells. Of note, HCC-derived Tregs could downregulate CD80/CD86 on DCs in a CTLA-4-dependent way, indicating their negative roles on multiple immune cells [34].
Previous studies showed HCC patients had significant increased number of CD4+CD25+ regulatory T cells in peripheral blood and liver tissues [10,35], and increased peripheral Tregs were correlated with disease progression [20]. Fu et al. and Gao et al. demonstrated a correlation of high infiltration of Tregs cells with poor overall survival, and indicated the ratio of Tregs and cytotoxic T cells was a more promising prognostic marker for HCC patients [18,20]. In a study including 19 advanced HCC patients, reduced PD-1+ and Foxp3+ Tregs were shown to be associated with prolonged survival after sorafenib treatment [36]. Accumulating Tregs in liver can also promote HCC recurrence after liver transplantation in a CXCL10/CXCR3 signaling dependent pathway [37]. Further animal experiments demonstrated that Tregs depletion by CD25 monoclonal antibody could hinder HCC development.
The association between Tregs and prognosis indicates Tregs participate in HCC formation and progression. By examining 323 hepatic nodules including precursor lesions, early HCC and advanced HCC, Kobayashi and colleagues demonstrated that infiltrating Tregs increased gradually during the progression of hepatocarcinogenesis in a stepwise manner [16]. In animal experiments, Tregs counts increased in liver after hepatitis B virus (HBV) infection, limiting immune-mediated liver damage but leading to delayed viral clearance [38]. Tregs in patients with HBV-HCC were shown to have increased suppressive effects, featuring decreased expression of PD-1 and increased IL-10 and TGF-β [39]. These studies suggest infiltrating regulatory T cells are associated with HCC development, and Tregs promote hepatocellular carcinoma progression by attenuating viral clearance capacity. The roles of Tregs in balancing immune reaction in HCC development are further validated in preclinical experiments. Studies showed that selective depletion of Tregs led to enhanced antitumor immunity in several tumors including HCC [40]. However, the heterogeneity of Tregs makes it difficult for clinical use of Tregs depletion. It has been noted that regulatory T cell subsets in patients with HCC differ from each other and may represent distinct functions. Kakita and colleagues identified two subtypes of Tregs according to expression of CD25 and CD127 [41]. Both CD25- FOXP3- Tregs and CD25+ FOXP3+ Tregs were detected in HCC tumor tissue, but only CD25- FOXP3- Tregs were significantly higher in tumor than non-tumor tissue. During following up, CD25-FOXP3- Tregs, rather than CD25high+FOXP3+ cells, displayed dynamically change in patients with curative ablation or recurrence. It remains elusive regarding roles of the subpopulations of Tregs in HCC, and the high heterogeneity of Tregs confines their use as prognostic biomarkers and therapeutic targets.
Tregs are attracted to tumor milieu through interaction with other immune cells in the tumor immune contexture, and the latter produce Tregs-recruiting chemokines. Alterations of driver genes such as FAK and EGFR, can recruit Tregs by modulating chemokine transcription, contributing to immunosuppressive microenvironment and tumor progression [42]. MDSCs and tumor associated macrophages also regulate Tregs activity in cancer development. MDSCs are regarded to suppress antigen-specific and nonspecific immune responses in human tumors. Analysis showed Tregs expansion could be induced by MDSCs isolated from HCC patients when cocultured with autologous T cells [43]. Moreover, tumor infiltration of Tregs is positively correlated with density of tumor-associated macrophages (TAMs) in HCC patients [44]. TAMs were proved to produce chemokines CCL17, CCL18, and CCL22, which preferentially induced Tregs and Th2 cells to the tumor tissue, and subsequently impaired CTL activation in ovarian cancer [45]. Tregs can also be regulated by neutrophils to cancer development. A recent study revealed that tumor-associated neutrophils could recruit Tregs to HCC stroma to promote tumor progression in a CCL2+ and CCL17+ dependent way [46]. The crosstalk between Tregs and other tumor infiltrating immune cells indicates a complicated regulatory network in HCC development.
T helper cells
CD4+ T cells are required in the priming of CD8+ T cells and enhance the antitumor effects of CTLs. However, the heterogeneity of CD4+ T helper cells is in accordance with their multiple roles, especially in cancer. Apart from T help 1 (Th1) cells and T help 2 (Th2) cells, IL-17 producing T help 17 (Th17) cells, IL-9 producing T help 9 (Th9) cells, IL-22 producing T help 22 (Th22) cells, and follicular B helper T (Tfh) cells will also be discussed in this review.
Th1/Th2 cells
Based on cytokine secretion profiles, Th1 (IFN-γ, IL-2, IL-22, TNF-β) and Th2 (IL-4, IL-5, IL-9, IL-10 and IL-13) are two major CD4+ T helper lymphocyte subsets. Th1 cells participate in cell-mediated immune responses and delayed-type hypersensitivity reactions, while Th2 cells participate in humoral-mediated immunity. After the dual signal of MHC II/Peptide complex and co-stimulatory molecules, Th1 cells produce cytokines such as IFN-γ to recruit CD8+ T cells into the tumor. Th1/Th2 balance influences the antitumor process. In HCC, the shift of Th1 to Th2 cytokines indicates an immune suppressive tumor microenvironment. Expression of Th1 cytokines (IL-1α, IL-1β, IL-2 and IFN-γ) in tumor tissue is associated with good prognosis, while Th2 cytokines (IL-4, IL-5 and IL-10) are upregulated in HCC with vascular invasion or extrahepatic metastases [47]. Moreover, increased circulating Th2 cells are associated with more advanced tumor stage and poorer treatment response [48]. Patients with low levels of Tregs at baseline before TACE had increased Th1/Th2 ratio compared to those with high Tregs levels, and increased Th1/Th2 ratio was associated better immune response and prolonged overall survival [49]. Cancer vaccines targeting helper T cells showed promising results in some malignancies [50]. However, evidence regarding whether such Th1 response can induce effector T cell action in HCC is lacking and warrants further investigation.
Th17 cells (Th17)
Th17 cells are a subset of CD4+ T helper cells and play complex roles in inflammation and tumor immunity. They have features distinct from Th1 and Th2 cells. Th17 cells express the transcription factor, namely related orphan receptor gamma (RORγ) [51]. Th17 cells secret cytokines including IL-17, IL-17F, and IL-22, IL-21, which act as a proinflammatory mediator and play important roles in inflammatory disease and inflammation against foreign pathogens. Th17 cells are mainly located in gut, lungs and skin, especially in the small intestinal lamina propria. Studies suggested Th17 cells participated in regulating gut mucosal immune responses, maintaining microbial homeostasis, and served both in adaptive and innate immunity.
IL-17 is generally believed to induce chronic inflammation and has pro-tumorigenic effects, and accumulating evidence indicated Th17 cells promoted HCC development and were associated with poor survival [52,53]. Zhang and colleagues studied distribution of Th17 cells in 178 HCC patients. There were increased Th17 cells in tumor tissues when compared to non-tumor regions, and intratumoral IL-17-producing cell density was an independent prognostic factor for significantly shorter overall survival (OS) and disease-free survival (DFS) [54]. It is not clear whether T17 cells could directly promote HCC growth or through a distinct pathway. A positive association between peripheral and intrahepatic Th17 cells and liver cirrhosis was found in patients with HBV-related cirrhosis; in vitro experiment showed that IL-17 promoted cirrhosis progression via activating hepatic stellate cell and inducing production of pro-inflammatory cytokine such as IL-6, IL-8 [55]. It is still under investigation how Th17 cells contribute to the disease progression of liver fibrosis and even HCC in patients with HBV or hepatitis C virus (HCV)-related chronic hepatitis. A recent study by Gomes showed that IL-17 level was elevated in patients with hepatitis, fatty livers, and viral hepatitis-associated HCC. Using mouse model with non-alcoholic steatohepatitis, the study further showed IL-17 could promote chronic inflammation and hepatocarcinogenesis via activating hepatic macrophages [56]. Notably, accumulating evidence has shown microbial dysbiosis contributed to the pathological role of IL-17 in immunity in some cancers such as gastric and colon cancers [57]. Gut microbial translocation and dysbiosis is common in patients with chronic liver disease. It has been established in recent years that dysregulation of intestinal microbiota contributed to hepatocarcinogenesis by endotoxin accumulation, influencing metabolism and promoting cancer-related inflammatory pathways [58]. How Th17 cells are regulated by gut dysbiosis and participate in HCC growth is largely unknown. Unveiling roles of microbial dysbiosis and Th17 cells in cirrhosis or HCC would further our knowledge in liver carcinogenesis.
Th22 cells (Th22)
Th22 cells are a novel subset of CD4+ T cells that were identified recently. Those cells produce IL-22, IL-1β, IL-6, excluding IL-17 or IFN-γ, which are distinct from Th17 cells and Th1 cells [59]. IL-22 are involved in the pathogenesis of autoimmune diseases and different types of cancers. Accumulation of Th22 cells was associated with shorter survival in multiple gastrointestinal cancers [60,61]. Studies showed that IL-22 could both protect hepatocytes from apoptosis and promote HCC development by activation of STAT3 [62,63]. Elevated peripheral and tumor infiltrating Th22 cells were observed in HCC patients and associated with progression of HCC [64,65].
Tfh cells (Tfh)
Tfh cells are derived from the naive CD4+ T cells. These cells secret IL-21, IL-4 and CXCL13, and are involved in B cell function. Defects in Tfh cells can cause the failure of B cell response, resulting in humoral immune deficiency. Tfh cells have been shown to play critical roles in several human disease including autoimmune diseases and cancers. In HCC patients, circulating and tumor infiltrating Tfh cells were significantly decreased when compared to healthy controls and nontumor regions. Tfh cells were found to be exhausted with decreased IL-21 production and impaired function on B cell proliferation and differentiation. Survival analysis showed decreased frequency of circulating Tfh cells was associated with poorer prognosis [66].
Th9 cells (Th9)
Th9 cells are a subset of CD4+ T cells that mainly express IL-9. It has been postulated that Th9 cells could directly exert anti-tumor activity by inducing granzyme B production and promoting dendritic cell recruitment, and IL-9 could enhance cytotoxicity of mast cell and CTL and induce IFN-γ secretion [67]. Studies on the roles of Th9 cells in HCC were limited. In contrast to the anti-tumor role in melanoma and lung adenocarcinoma, a study including 21 HCC patients showed that infiltrating Th9 cells were higher in in peritumor and tumor regions compared with nontumor regions, and higher level of tumor infiltrating Th9 cells were associated with significantly shorter disease-free survival [68].
Mucosal-associated invariant T cells (MAIT)
MAIT cells are innate-like T cells that act at the innate-adaptive interface of immunity. MAIT cells are highly evolutionarily conserved expressing CD161 and a semi-invariant TCR made of an invariant Vα7.2-Jα33. Abundant in peripheral blood and preferably resident in the intestinal mucosa and liver, MAIT cells account for approximately 45% of the liver lymphocytes [69]. MAIT cells secrete multiple cytokines such as IL-17, granzyme B, IFN-γ, and TNF, indicating a defensive role of MAIT cells against pathogens.
MAIT cells are enriched in liver and involved in liver inflammation and cirrhosis. Hegde and colleagues studied MAIT cells in patients with alcoholic or non-alcoholic fatty liver disease-related cirrhosis [70]. They found MAIT cells were reduced in the peripheral blood and increased in cirrhotic liver tissue. In vitro experiments showed MAIT cells could induce myofibroblasts producing IL-6 and IL-8 by secretion of TNF, which contributed to liver fibrosis progression. Another study on autoimmune liver disease indicated MAIT cells contributed to liver fibrosis by activating hepatic stellate cells [71]. In condition of viral infection, peripheral MAIT cells are generally decreased. Study on patients infected with HCV showed MAIT cells were significantly lower in blood and liver. Moreover, infiltrating MAIT cells are activated and inversely correlated with liver inflammation condition, suggesting a protective role of MAIT cells against HCV infection [72]. Significant decreased circulating MAIT cells with exhausted and dysfunctional phenotypes were also found in patients with HBV infection [73]. Though MAIT cells seemed to be associated with liver chronic inflammation and fibrosis, direct evidence regarding the roles of MAIT cells in the development of HCC is lacking. Duan and colleagues studied 50 patients with HCC and found significant lower level of MAIT cells both in peripheral and tumor tissues. Compared to healthy controls, infiltrating MAIT cells of HCC expressed higher level of immune checkpoint molecules such as PD-1, CTLA-4 and TIM-3, indicating an exhausted type of those T cells. Interestingly, when compared to low infiltration group, higher MAIT cells infiltration was associated with poorer prognosis, which indicated MAIT cells were re-routed to a tumor-promoting phenotype [74]. Taken together, MAIT cells may play a pro-inflammatory role in liver chronic inflammation, and partially contribute to fibrosis, cirrhosis and even carcinogenesis. Targeting MAIT cells may shed light on treatment of virus-related cirrhosis and HCC.
Immunotherapy based on T cells in HCC treatment
HCC is characterized by altered immune microenvironment that leads to immune tolerance for tumor cells. Immunotherapy is emerging as a potential treatment option for HCC. We briefly reviewed and highlighted several T cell-based immunotherapies that have been put into clinical practice or under clinical investigation.
Immune checkpoint inhibitors
Immune checkpoint inhibitors reinvigorate T cell function and have shown potential in treating advanced HCC. Nivolumab and pembrolizumab were recently approved by US Food and Drug Administration in HCC patients after treatment of sorafenib. However, the efficacy of single immune checkpoint inhibitor for HCC in subsequent large-scale clinical trials is not satisfying. In the phase III study (KEYNOTE 240) of pembrolizumab as second line therapy for advanced HCC, patients treated with pembrolizumab showed improvement in OS and progression-free survival (PFS) compared with placebo plus best supportive care [median OS 13.9 months vs. 10.6 months, hazard ratio (HR)=0.781, 95% confidence interval (CI): 0.611-0.998, P=0.0238]. Due to P value loss caused by the study design, KEYNOTE 240 did not reach the preset endpoint. Despite the failure of phase III study, objective response rate was 18.3% (95% CI: 14.0%-23.4%) in pembrolizumab group compared to 4.4% (95% CI: 1.6%-9.4%) in control group, and there was significant tendency for longer survival [75]. In another phase III randomized multicenter study (CheckMate 459) which compared nivolumab with sorafenib in unresectable HCC patients, nivolumab also failed to show significant improvement in OS (median OS 16.4 months vs. 14.7 months, HR=0.85, 95% CI: 0.72-1.02, P=0.0752). Despite the effect of single immunotherapy is limited, combining immune checkpoint inhibitors with other therapies are promising. Phase Ib study in unresectable HCC patients has shown that pembrolizumab plus lenvatinib showed good disease control rates and limited adverse effect (KEYNOTE 524, NCT03006926) [76]. A phase III study on lenvatinib plus pembrolizumab for unresectable HCC as the first-line treatment is being conducted (NCT03713593) [77]. Anti-PD-1/PD-L1 combining with anti-angiogenesis agents are also effective in advanced HCC, largely due to inhibiting intratumoral immunosuppression and further producing synergistic effect. The phase I GO30140 study (NCT02715531) indicated that atezolizumab plus bevacizumab showed promising efficacy (36% ORR) and longer progression-free survival (HR 0.55, 80% CI: 0.40-0.74) than atezolizumab alone in patients with advanced HCC [78]. Moreover, IMbrave 150 study (NCT03434379) showed patients treated with atezolizumab and bevacizumab had longer overall survival and progression free survival compared with those with sorafenib alone (median PFS 6.8 months vs. 4.3 months respectively, HR 0.59; 95% CI: 0.47-0.76) [79]. Based on the promising results of the above clinical trials, bevacizumab combined with atezolizumab was approved as first-line therapy for advanced HCC. Main clinical trials of combination therapy based on ICIs are presented in Table 2. Additionally, ICIs are also being explored in neoadjuvant and adjuvant therapy for patients with curative intent. Table 3 shows the main ongoing clinical trials testing ICIs as neoadjuvant and adjuvant therapies. With more emerging combination therapy choices and clinical beneficial data, ICIs seem to play a role through the entire process of HCC treatment. Personalized clinical decisions are needed to achieve better disease control rates and properly manage immune-related adverse events. More knowledge of the roles of the heterogenous T cells in TIME will contribute to better future development of cancer immunotherapies.
Table 2.
Key clinical trials of combination therapy based on immune checkpoint inhibitors in advanced HCC
| ICB | NCT number | Phase/participants | Disease condition | Intervention regimens | Intervention type | Primary endpoints |
|---|---|---|---|---|---|---|
| Anti-PD1 + targeted therapy | ||||||
| Pembrolizumab | 03713593 | Phase III/750 | Advanced HCC | Pembrolizumab + lenvatinib vs. lenvatinib + placebo | Anti-PD-1 antibody + multiple kinases inhibitor | PFS, OS |
| 03347292 | Phase I/40 | Advanced HCC | Pembrolizumab + regorafenib | Anti-PD-1 antibody + multiple kinases inhibitor | TEAEs, DLTs | |
| 03211416 | Phase Ib/II/27 | Locally advanced or metastatic HCC | Pembrolizumab + sorafenib | Anti-PD-1 antibody + multiple kinases inhibitor | ORR-2 | |
| Nivolumab | 03841201 | Phase II/50 | Advanced HCC | Nivolumab + lenvatinib | Anti-PD-1 antibody + multiple kinases inhibitor | ORR-1, AEs |
| 03418922 | Phase Ib/30 | Advanced HCC | Nivolumab + lenvatinib | Anti-PD-1 antibody + multiple kinases inhibitor | DLTs, AEs | |
| 03439891 | Phase II/40 | Advanced, untreated HCC | Nivolumab + sorafenib | Anti-PD-1 antibody + multiple kinases inhibitor | MTD, ORR-2 | |
| 01658878 (CheckMate040) | Phase I/II/1,097 | Advanced HCC | Nivolumab + cabozantinib | Anti-PD-1 antibody + anti-VEGFR2/c-MET | AEs, ORR-1 | |
| 04170556 | Phase I/IIa/60 | Advanced HCC with progression under sorafenib | Nivolumab + Regorafenib | Anti-PD-1 antibody + multiple kinases inhibitor | TEAEs | |
| 04310709 | Phase II/42 | Advanced, chemotherapy-naïve HCC | Nivolumab + Regorafenib | Anti-PD-1 antibody + VEGFR2/TIE2 | Response rate | |
| Camrelizumab | 03764293 | Phase III/510 | Advanced HCC | Camrelizumab + apatinib vs. sorafenib | Anti-PD-1 antibody + VEGFR2 inhibitor | OS, PFS |
| 03463876 | Phase II/190 | Advanced HCC | Camrelizumab + apatinib | Anti-PD-1 antibody + VEGFR2 inhibitor | ORR-1 | |
| 04443309 | Phase I/II/53 | Advanced HCC or progressed following surgical and/or local therapy | Camrelizumab + lenvatinib | Anti-PD-1 antibody + multiple kinases inhibitor | ORR-1 | |
| Tislelizumab | 04183088 | Phase II/125 | Advanced HCC | Tislelizumab + regorafenib vs. placebo + regorafenib | Anti-PD-1 antibody + VEGFR2/TIE2 inhibitor | AEs, ORR-1, PFS |
| 04401800 | Phase II/66 | Locally advanced or metastatic HCC | Tislelizumab + lenvatinib | Anti-PD-1 antibody + multiple kinases inhibitor | ORR-2 | |
| Toripalimab | 04069949 | Phase I/II/39 | Locally advanced HCC with portal vein tumor thrombus | Toripalimab + sorafenib | Anti-PD-1 + multiple kinases inhibitor | PFS, TEAEs |
| 04523493 | Phase III/486 | Advanced HCC | Toripalimab + lenvatinib vs. placebo + lenvatinib | Anti-PD-1 antibody + multiple kinases inhibitor | OS, PFS | |
| 04010071 | Phase II/60 | Advanced hepatobiliary tumors including HCC | Toripalimab + axitinib | Anti-PD-1 antibody + VEGFR1/2/3 inhibitor | ORR-1, PFS | |
| Sintilimab | 03794440 (ORIENT-32) | Phase III/566 | Advanced HCC | Sintilimab + IBI305 vs. sorafenib | Anti-PD-1 antibody + VEGFR inhibitor | OS, PFS |
| 04411706 | Phase II/46 | Advanced HCC | Sintilimab + apatinib and capecitabine | Anti-PD-1 antibody + VEGFR2 inhibitor + chemotherapy | ORR-1 | |
| 04042805 | Phase II/56 | Locally advanced HCC ineligible for curative surgery | Sintilimab + lenvatinib | Anti-PD-1 antibody + multiple kinases inhibitor | ORR-1 | |
| Anti-PD-L1 + targeted therapy | ||||||
| Atezolizumab | 03434379 (IMbrave150) | Phase III/480 | Untreated locally advanced or metastatic HCC | Atezolizumab + bevacizumab vs. sorafenib | Anti-PD-L1 + anti-VEGF antibody | PFS; OS |
| 03755791 | Phase III/740 | Advanced, untreated HCC | Atezolizumab + cabozantinib vs. sorafenib vs. cabozantini | Anti-PD-L1 antibody + VEGFR2/c-MET inhibitor | PFS; OS | |
| Durvalumab | 02519348 | Phase II/433 | Advanced HCC, immunotherapy-naive | Durvalumab +Bevacizumab | Anti-PD-L1 antibody + anti-VEGF antibody | AEs; DLTs |
| 03970616 | Phase Ib/II/42 | Advanced, untreated HCC | Durvalumab + tivozanib | Anti-PD-L1 antibody + VEGFR1/2/3 inhibitor | TEAEs | |
| 03539822 | Phase Ib/30 | Advanced or locally unresectable gastrointestinal cancer including HCC | Durvalumab + cabozantinib | Anti-PD-L1 antibody + VEGFR2/c-MET inhibitor | MTD | |
| Avelumab | 03289533 | Phase Ib/22 | Locally advanced or metastatic HCC | Avelumab + axitinib | Anti-PD-L1 antibody + VEGFR1/2/3 inhibitor | TEAEs |
| Dual ICBs | ||||||
| 04039607 | Phase III/1,084 | Advanced, treatment-naïve HCC | Nivolumab + ipilimumab vs. sorafenib or lenvatinib | Anti-PD-1 antibody + anti-CTLA-4 antibody | OS | |
| 01968109 | Phase I/IIa/1,500 | Advanced, untreated solid tumors including HCC | Nivolumab + relatlimab | Anti-PD-1 antibody + anti-LAG-3 antibody | AEs, ORR-1, DCR, DOR | |
| 01658878 | Phase I/II/1,097 | Advanced HCC not eligible for curative surgery and locoregional therapies | Nivolumab + ipilimumab | Anti-PD-1 antibody + anti-CTLA-4 antibody | AEs, ORR-1 | |
| 03298451 (HIMALAYA) | Phase III/1,324 | Advanced, untreated HCC | Durvalumab + tremelimumab vs. sorafenib | Anti-PD-L1 antibody + anti-CTLA-4 antibody | OS | |
| 02519348 | Phase II/433 | Advanced HCC | Durvalumab + tremelimumab | Anti-PD-L1 antibody + anti-CTLA-4 antibody | AEs, dose limiting toxicities | |
| 03638141 | Phase II/30 | Intermediate stage HCC without vascular invasion and metastasis | Durvalumab + Tremelimumab + TACE | Anti-PD-L1 antibody + anti-CTLA-4 antibody + TACE | ORR-1 | |
| ICB + cytokines inhibitor | ||||||
| 02423343 | Phase Ib/II/75 | Advanced solid tumors including HCC | Nivolumab + galunisertib | Anti-PD-1 antibody + TGFβR1 inhibitor | MTD | |
| 03695250 | Phase I/II/23 | Advanced HCC | Nivolumab + BMS-986205 | Anti-PD-1 antibody + IDO1 inhibitor | AEs, ORR-1 | |
| 04050462 | Phase II/74 | Advanced HCC | Nivolumab + cabiralizumab/BMS-986253 vs. nivolumab | Anti-PD-1 antibody + CSF1R inhibitor/IL-8 inhibitor | ORR-1 | |
| ICB + locoregional therapy | ||||||
| 03033446 | Phase II/40 | Advanced HCC | Nivolumab + Yttrium-90 radioembolization | Anti-PD-1 antibody + radioembolization | ORR-1 | |
| 03380130 | Phase II/40 | Advanced HCC | Nivolumab + Yttrium-90 radioembolization | Anti-PD-1 antibody + radioembolization | AEs | |
| 04268888 | Phase II/III/522 | Intermediate stage HCC | Nivolumab + TACE vs. TACE | Anti-PD-1 antibody + TACE | OS, TTP | |
| 03572582 | Phase II/49 | Intermediate stage HCC | Nivolumab + TACE | Anti-PD-1 antibody + TACE | ORR-1 | |
| 04340193 (CheckMate74W) | Phase III/765 | Intermediate stage HCC | Nivolumab + ipilimumab + TACE vs. Nivolumab + TACE vs. TACE | Anti-PD-1 antibody + anti-PD-L1 antibody + TACE | OS, TTP | |
| 03753659 | Phase II/30 | Early HCC without portal vein invasion and extrahepatic metastasis | Pembrolizumab + RFA/MWA/TACE | Anti-PD-1 antibody + ablation or TACE | ORR-1 | |
| 04246177 | Phase III/950 | Locally advanced HCC | Pembrolizumab + lenvatinib + TACE vs. TACE + placebo | Anti-PD-1 antibody + multiple kinases inhibitor + TACE | PFS, OS | |
| 03316872 | Phase II/30 | Advanced HCC with disease progression after sorafenib | Pembrolizumab + stereotactic body radiotherapy | Anti-PD-1 antibody + radiotherapy | ORR-2 | |
| 03638141 | Phase II/30 | Intermediate stage of HCC | Durvalumab tremelimumab + TACE | Anti-PD-L1 antibody + anti-CTLA-4 antibody + TACE | ORR-1 | |
| 03778957 | Phase III/600 | HCC ineligible for resection or transplantation | Durvalumab + bevacizumab + TACE vs. Durvalumab + TACE vs. TACE | Anti-PD-L1 antibody + anti-VEGF antibody + TACE | PFS | |
| 03482102 | Phase II/70 | Advanced hepatobiliary tumors Including HCC | Durvalumab + tremelimumab + radiation Therapy | Anti-PD-L1 antibody + anti-CTLA-4 antibody + radiation | ORR-2 | |
| 04167293 | Phase II/III/116 | HCC with portal vein invasion | Sintilimab + stereotactic body radiotherapy vs. radiotherapy | Anti-PD-1 antibody + radiotherapy | PFS | |
| 04220944 | Phase I/45 | Advanced HCC ineligible for curative therapy | Sintilimab + TACE + MVA | Anti-PD-1 antibody + TACE + ablation | PFS | |
| 03864211 | Phase I/II/120 | Advanced HCC ineligible for curative therapy | Toriplimab + RFA/WMA vs. toriplimab | Anti-PD-1 antibody + TACE/ablation | AEs, ORR-2 | |
| 04483284 | Phase II/60 | Advanced HCC ineligible for curative therapy | Camrelizumab + TACE | Anti-PD-1 antibody + TACE | PFS | |
| 04479527 | Phase II/56 | Advanced HCC ineligible for curative therapy | Camrelizumab + apatinib + TACE + HAIC | Anti-PD-1 antibody + VEGFR2 inhibitor + embolization and chemotherapy | PFS | |
AEs, adverse events; CSF1R, colony stimulating factor-1 receptor; DCR, disease control rate; DOR, duration of response; DLTs, dose limiting toxicities; HAIC, hepatic arterial infusion chemotherapy; IDO1, indoleamine-2,3-dioxygenase 1; MTD, maximum tolerated dose; MVA, microwave ablation; ORR-1, objective response rate; ORR-2, overall response rate; OS, overall survival; PFS, progression-free survival; RFA, radiofrequency ablation; TACE, transcatheter arterial chemoembolization; TEAE, treatment-emergent adverse event; TGFβR1, transforming growth factor-beta receptor type 1; TRAEs, treatment-related adverse events; TTP, time to progression.
Table 3.
Clinical trials of immune checkpoint inhibitors as neoadjuvant and adjuvant therapy
| NCT number | Phase/participants | Disease condition | Intervention regimens | Intervention type | Primary endpoints |
|---|---|---|---|---|---|
| ICBs as neoadjuvant therapy | |||||
| 03869034 | Phase II/40 | Locally advanced HCC with curative potential for resection | Sintilimab + TAI vs. TAI | Anti-PD-1 antibody + chemotherapy | PFS |
| 04174781 | Phase II/61 | HCC with BCLC stage A/B; beyond Milan criteria | Sintilimab + TACE | Anti-PD-1 antibody + chemotherapy | PFS |
| 03337841 | Phase II/50 | HCC eligible for resection or ablation | Pembrolizumab | Anti-PD-1 antibody | RFS |
| 04224480 | Phase I/45 | HCC eligible for resection | Pembrolizumab | Anti-PD-1 antibody | Recurrence rate, CD8+Ki67+ T cells ratio |
| 04425226 | Phase II/192 | HCC beyond Milan Criteria before transplant | Pembrolizumab + lenvatinib vs. no intervention | Anti-PD-1 antibody + multiple kinases inhibitor | RFS |
| 03510871 | Phase II/40 | Locally advanced HCC with curative potential for resection | Nivolumab + ipilimumab | Anti-PD-1 antibody + anti-CTLA-4 antibody | Percentage of subjects with tumor shrinkage |
| 04123379 | Phase II/50 | Resectable tumors including HCC | Nivolumab + BMS-813160/BMS-986253 | Anti-PD-1 antibody + CCR2/5-inhibitor or IL-8 inhibitor | Major pathologic response, Significant tumor necrosis |
| 04035876 | Phase I/II/120 | HCC patients before transplantation | Camrelizumab + apatinib | Anti-PD-1 antibody + anti-VEGFR2 inhibitor | ORR, RFS |
| ICBs as adjuvant therapy | |||||
| 03383458 (CheckMate9DX) | Phase III/530 | HCC with high recurrence risk after curative therapy | Nivolumab vs. placebo | Anti-PD-1 antibody | RFS |
| 03867084 (KEYNOTE-937) | Phase III/950 | HCC after curative resection or ablation | Pembrolizumab vs. placebo | Anti-PD-1 antibody | RFS, OS |
| 03859128 | Phase II/III/402 | Locally advanced HCC with high recurrence risk after curative resection | Toripalimab vs. placebo | Anti-PD-1 antibody | RFS |
| 03847428 | Phase III/888 | HCC with high recurrence risk after curative therapy | Durvalumab + bevacizumab vs. placebo | Anti-PD-L1 antibody + anti-VEGF antibody | RFS |
| 04102098 (IMbrave050) | Phase III/662 | HCC with high risk of recurrence after curative therapy | Atezolizumab + bevacizumab vs. active surveillance | Anti-PD-L1 antibody + anti-VEGF antibody | RFS |
AEs, adverse events; BCLC, Barcelona clinic liver cancer; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; RFA, radiofrequency ablation; RFS, recurrence-free survival; TACE, transcatheter arterial chemoembolization; TAI, transarterial infusion chemotherapy.
Cancer vaccines
HCC TAAs are potential markers for cancer vaccine development. Vaccines can induce memory T cells and has been investigated in different types of cancers. TAAs such as AFP, GPC-3, MAGE-1 and NY-ESO-1, human telomerase reverse transcriptase (hTERT), are expressed by most HCC cells, but not specific to HCC. Those TAAs-based cancer vaccines were tested in a relatively small population. In a phase I trial of GPC-3 peptide vaccine on 33 HCC patients, only one patient showed a partial response [80]. In another study, 40 patients with advanced HCC vaccinated with a telomerase-derived peptide showed no specific immune responses [81]. In another hand, DC-based cancer vaccine seemed to be theoretically competitive with antigen-presenting cells plus tumor antigens. However, a phase II study using DCs-based HCC vaccines (DCs combined with tumor lysate) were testified and a limited response was observed [82]. Another phase I/II study using DCs plus a combination of α-fetoprotein, glypican-3 and MAGE-1 also showed only one patient (a total of 5 advanced HCC patients) had clinical response [83]. TAAs are usually not limited to tumor cells but also expressed in normal tissues, which deteriorate the immunogenic efficacy and induce possible severe side effects. In such case, identifying neoantigens individually should be taken to improve cell specificity and foster anti-cancer immunotherapy more precisely. Neoantigens-related cancer vaccines have been tested in a few human cancers and showed encouraging preliminary results [84]. Relative high costs and complex vaccine preparation make neoantigen-based vaccine currently not feasible for a large population. Studies on efficacy of neoantigen vaccine in HCC patients are now lacking, probably due to a relatively low tumor mutation burden and great intratumoral heterogeneity. Though results from cancer vaccine are not satisfying, combination immune therapy may lead to better tumor control. In vivo experiment showed combination of ICIs and cancer vaccine resulted in enhanced antitumor effects [85]. In a phase II clinical trial, tumor-specific vaccine combined with immune checkpoint blockades seemed to prolong overall survival in patients with human papillomavirus-related cancers yet warrant further evidence from randomized clinical trials [86]. Cancer vaccines as adjuvant therapy after HCC curative resection are also being explored and only show promising results in some selected patients [87,88]. Development of effective tumor vaccines still warrants further preclinical and clinical studies.
Adoptive cell therapy
Adoptive T-cell therapy (ACT) has been widely investigated in several types of tumors including HCC. Cytokine-induced killer cells (CIKs) and TILs are now commonly used in ACT. It involves isolation and ex vivo expansion of tumor antigen-specific T cells, those cells are then infused into patients with cancer to generate antitumor activity. A study involved 150 HCC patients underwent curative resections showed autologous tumor-infiltrating lymphocytes infusion significantly improved recurrence free time compared to the control group, though overall survival did not differ significantly [89]. Due to requirement of IL-2 and pretreatment of lymphodepletion, TILs therapy has apparent toxicity and has limited application. CIKs can be generated from peripheral blood mononuclear cells and cultured in the absence of IL2. More evidence has yielded from studies using CIKs as adjuvant treatment in postoperative patients. A study on 410 postoperative patients showed that adjuvant CIKs-based ACT prolonged overall survival in patients with tumors larger than 5 cm, while no survival benefit was observed in patients with tumors less than 5 cm [90]. In a phase III randomized trial including 230 HCC patients who received curative therapy, survival benefit and safety of CIKs were evaluated. Compared to controls, patients treated with CIK had markedly increased RFS (median RFS: 44.0 months vs. 30.0 months, P=0.01). There was higher frequency of adverse effects in ACT group, while no significant difference in grade 3 or 4 adverse events was observed between groups [91]. Overall, the safety and efficacy of ACT as adjuvant therapy for HCC still need to be further investigated in an expanded population.
CAR-T cells therapy
Chimeric antigen receptor-engineered T cells (CAR-T) therapy has produced promising results in human cancers especially in hematologic malignancies. For example, CD19-targeted CAR T therapy has yielded remarkable potency in leukemia and lymphoma [92]. CAR comprises a single-chain variable fragment (scFv) of antibody that targets tumor associated antigens, attached to intracellular T-cell signaling domains of the T-cell receptor. Autologous T cells which express CARs are then infused into patients and specifically recognize tumor cells expressing targeted tumor antigen. On-target off-tumor toxicity is the main problem due to low expression of tumor-associated antigen on normal cells. These side-effects were reported to significantly decrease using a co-stimulatory CAR [93] or by optimizing CAR affinity by light-chain exchange method [94].
CAR T therapy has recently been studied in HCC. In mouse models, T cells with a CAR specific for HBV envelope proteins can remarkably suppress liver HBV replication with limited side effects [95]. Among HCC tumor-associated antigens, AFP, CEA, Glypican-3 (GPC3), CD133 are potential candidates for CAR T therapy. Glypican-3 (GPC3) is widely expressed by HCC, and GPC3-targeted CAR T cells therapy has yielded promising anti-tumor activity both in vitro and in vivo [96,97]. In a phase I study including thirteen HCC patients, Zhai and colleagues tested safety and efficacy of GPC3-based CAR-T therapy [98]. All patients but one tolerated well with the therapy. Notably, only GPC3 CAR-T combined with lymphodepletion showed antitumor potential, while GPC3 CAR-T alone did not stop disease progression. Another phase I trial studied CD133-targeted CAR T cells for treatment of metastatic HCC [99]. One of the fourteen HCC patients achieved partial response, with nine participants remained stable disease for 3 months. In general, CAR T therapy has displayed well tolerated adverse effects but limited clinical benefits for HCC. Ongoing clinical trials using CAR T cells therapy are conducting in HCC patients (NCT02723942, NCT02395250, NCT03130712, NCT03084380, NCT03146234).
Prospective and conclusions
TIME exerts a strong selection pressure on HCC, largely leading to HCC evolvement and recurrence after multiple therapies. Crosstalk among tumor cells, immune cells, fibroblasts, endothelial cells, extracellular matrix, and cytokines and their receptors, contributes to tumorigenesis and are impacting tumor progression. Notably, HCC is a disease with a great variety of T cells. Various T cells can inhibit or promote HCC and associate with different survival (Table 4). T cell mediated immunoreaction is central in cancer surveillance and clearance. The mechanism that tips the balance between immune response and immune tolerance to foreign antigens are still elusive. Studies are needed to reveal the landscape of HCC immune cells, neo-antigens, and highlight the heterogeneity of tumor cells.
Table 4.
Roles of various T cell subsets in HCC
| T cell subtypes | Cell frequency | Cell phenotype | Effect on disease | Possible mechanisms | Refs | ||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Blood | Tissue | Blood | Tissue | ||||
| CD8+ T cell | ↑ | ↑↓ | Exhausted | Exhausted | Anti-tumor | PD-1highCD8+ T cells are increased in tumor tissue; High cell infiltration associates with prolonged survival and lower recurrence | [10,15,18] |
| NKT cell | ↓ | ↑ | - | - | Tumor-promoting/anti-tumor | CD4+ iNKT cells inhibit tumor antigen-specific CD8+ T cells expansion; releasing IFN-γ and activate other effector cells | [30,33] |
| Treg | ↑ | ↑ | Immunosuppressive | Immunosuppressive | Tumor-promoting | Increased Tregs associate with tumor invasiveness and poor survival | [16,20] |
| Th17 cell | ↓in early HCC | ↑ | IFN-γ lacking | IFN-γ producing | Tumor-promoting | Increased intratumoral Th17 cells associate with poor survival | [54,56] |
| MAIT cell | ↓ | ↓ | Exhausted | Exhausted | Tumor-promoting | Decreased MAIT cells in tumor tissue correlate with more IL-8; High MAIT cell frequency associates with poor survival | [74] |
| Th1 | - | - | IL-1, IL-2, IL-12, TNF-α, and IFN-γ | - | Anti-tumor | Th1 cells produce IL-2 and IFN-γ and favor cellular immunity | [47,49] |
| Th2 | - | - | IL-4, IL-5, IL-10 | - | Tumor-promoting | Immunosuppressive effect via production of IL-10; Th2 cells were higher in advanced HCC | [47-49] |
| Th22 | ↑ | ↑ | IL-22, IL-6 and IL-1β producing; IL-17 (-) and IFN-γ (-) | IFN-γ (-) IL-17 (+) dominating | Protective/Tumor-promoting | Activation of STAT3 by IL-22 promotes HCC growth; Increased Th22 cells associate with disease progression | [54,64,65] |
| Tfh | ↓ | ↓ | Exhausted; decreased IL-21 production | - | Anti-tumor | Impaired function of Tfh cell promotes HCC progression | [66] |
| Th9 | ↑ | ↑ | - | - | Tumor-promoting | Increased tumor infiltrating Th9 cell frequency associates with shorter disease-free survival | [68] |
↑, increased; ↓, decreased.
Recently, T cell-based immunotherapies have rapidly changed the treatment prospective for HCC. Among these strategies, immune checkpoint inhibitors are emerging as potential therapeutic option for advanced HCC. However, the survival benefit of immunotherapies for HCC patients is still limited. Several reasons may account for the limited efficacy of immunotherapy in HCC. First, there are multiple immune regulators that tip the immune balance between cancer cells and host immune cells, and different pathways are implicated in tumor immunity. The effect of single immunotherapy may not be enough to result in effective host response. Secondly, the liver is a relatively immune-exempt organ that has immunosuppressive properties. It receives and processes a large amount of foreign substances via portal system and is prone to induce immune tolerance. This unique feature of liver partially explained why the efficacy of CIK cells and CAR-T cells in HCC is not satisfying, and single anti-PD-1 or anti-CTLA-4 therapy only produce minimal effect. In fact, studies have demonstrated that the efficacy of PD-1/PD-L1 inhibitors were often decreased in cancers with liver metastasis, indicating the threshold of liver for immune activation was relatively higher than other organs [100,101]. Lastly, the conventional biomarkers were not satisfactory when identifying HCC patients who may benefit from the immunotherapy. For example, nivolumab and pembrolizumab were both found to exert greater efficacy in a prespecified PD-L1-positive population with non-small cell lung cancer. However, there are currently no effective biomarkers for predicting the effect of immunotherapy in HCC. Accordingly, several measures can be taken to improve the potential of therapy that modulating cancer immunity in HCC. First, combination of immunotherapy with traditional therapy such as targeted drugs, radiation therapy or systematic chemotherapy, may be promising for improving patients’ response rates. For example, radiation therapy could stimulate neoantigen release and improve immunotherapy in many cancers. Moreover, boosting neoantigen-specific T cells has been possible, and has great potential in improving efficacy of T cell-based immunotherapy [102]. Combing immunotherapy with other treatments that target distinct biological pathways can produce synergistic effects, and may be promising in improving efficacy of immunotherapy for HCC. Second, novel biomarkers for HCC immunotherapy should be explored. Tumor mutation burden, tumor PD-L1 expression profiles, and circulating tumor DNA should be further explored to evaluate the association with efficacy and the potential in identifying subpopulations who will benefit from immunotherapy. Comprehensive analysis of different biomarkers for HCC is a future direction. High-quality clinical trials are needed to test and validate the predictive power of biomarkers. Lastly, a deeper understanding of HCC immune microenvironment is needed for developing more efficient immunotherapies, especially focusing on the distinct roles and trajectories of heterogenous T cells.
HCC is a malignancy with abundant genetic and epigenetic alterations, leading to great tumor heterogeneity. The distinct phenotypes of HCC result in different therapeutic response and made it difficult for achieving curative intent with single therapy. Moreover, tumor evolution that driven by selective stress from systemic therapies also contributes to tumor recurrence. Novel immunotherapy regimens may play positive roles in neoadjuvant and adjuvant treatment, aiming to achieve curative therapy and reduce recurrence. Of note, the immune-related adverse effects are common and can be serious in some patients, which usually lead to discontinuation of immunotherapy. Since immune-related adverse effects have been shown to be associated with prolonged survival in several cancers, discontinuation of immunotherapy would substantially reduce the anti-tumor effects. The side effects of immunotherapy should be closely monitored and properly handled, in order to avoid life-threatening conditions and intolerance to the treatment.
In conclusion, different subtypes of T lymphocytes can be either friends or foes in HCC treatment. Advancing our understandings of the roles of heterogenous T lymphocytes will help elucidate the underlying molecular mechanisms and pathogenesis of HCC. Progress of genome sequencing techniques such as single cell sequencing will assist in identifying novel therapeutic targets. Immunotherapies based on T cells have shown limited but encouraging efficacy for HCC. More studies should be conducted to evaluate the effects of different combinations and identify the optimal doses and timing of administration. In addition to enhancing synergistic effects, combination strategies should put more emphasis on targets that lead to immunotherapy resistance. Moreover, exploring the potential biomarkers for predicting immunotherapy response will help to improve individualized therapy.
Acknowledgements
This study was supported by International Science and Technology Cooperation Projects, No. 2016YFE0107100; the Beijing Natural Science Foundation, No. L172055 and No. 7192158; the National Ten-thousand Talent Program, the Fundamental Research Funds for the Central Universities, No. 3332018032; and the CAMS Innovation Fund for Medical Science (CIFMS), No. 2017-I2M-4-003 and No. 2018-I2M-3-001.
Disclosure of conflict of interest
None.
Abbreviations
- AEs
adverse events
- AFP
α-fetoprotein
- APC
antigen-presenting cell
- BCLC
Barcelona clinic liver cancer
- CAFs
cancer-associated fibroblasts
- CAR-T
chimeric antigen receptor-engineered T cells
- CCR
C-C motif chemokine receptor
- CD
cluster of differentiation
- CI
confidence interval
- CSF1R
colony stimulating factor-1 receptor
- CTLs
cytotoxic T lymphocytes
- CTLA-4
cytotoxic T lymphocyte-associated antigen 4
- DC
dendritic cell
- DFS
disease-free survival
- DCR
disease control rate
- DOR
duration of response
- DLTs
dose limiting toxicities
- FOXP3
transcription factor forkhead box p3
- GITR
glucocorticoid induced TNF receptor
- HAIC
hepatic arterial infusion chemotherapy
- GPC-3
glypican-3
- HR
hazard ratio
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- ICI
immune checkpoint inhibitor
- IDO1
indoleamine-2,3-dioxygenase 1
- IFN-γ
interferon γ
- IL
interleukin
- KLRG1
killer cell lectin-like receptor subfamily G member 1
- LAG3
lymphocyte activation gene-3
- MAGE-1
melanoma-associated antigen-1
- MDSCs
myeloid-derived suppressor cells
- MTD
maximum tolerated dose
- MVA
microwave ablation
- NASH
non-alcoholic steatohepatitis
- NKTs
natural killer T cells
- NY-ESO-1
New York-esophageal squamous cell carcinoma-1
- ORR
objective response rate/overall response rate
- OS
overall survival
- OX-40R
OX-40 receptor
- PD-1
programmed cell death protein 1
- PD-L1
programmed death-ligand 1
- PFS
progression-free survival
- RFA
radiofrequency ablation
- RFS
recurrence-free survival
- RORγ
related orphan receptor gamma
- TAAs
tumor-associated antigens
- TACE
transcatheter arterial chemoembolization
- TAI
transarterial infusion chemotherapy
- TAMs
tumor-associated macrophages
- TEAE
treatment-emergent adverse event
- TGF-β
transforming growth factor-β
- TGFβR1
transforming growth factor-beta receptor type 1
- Th1/2
T helper 1/2
- Th17
T helper 17
- Th9
T helper 9
- TILs
tumor infiltrating lymphocytes
- TIM-3
T cell immunoglobulin and mucin domain 3
- TIME
tumor immune microenvironment
- TNF
tumor necrosis factor
- TRAEs
treatment-related adverse events
- Tregs
T regulatory cells
- TTP
time to progression
References
- 1.Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, Gores G. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi: 10.1038/nrdp.2016.18. [DOI] [PubMed] [Google Scholar]
- 2.Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–1462. doi: 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 3.Khalaf N, Ying J, Mittal S, Temple S, Kanwal F, Davila J, El-Serag HB. Natural history of untreated hepatocellular carcinoma in a US cohort and the role of cancer surveillance. Clin Gastroenterol Hepatol. 2017;15:273–281. e271. doi: 10.1016/j.cgh.2016.07.033. [DOI] [PubMed] [Google Scholar]
- 4.Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology. 2019;156:477–491. e471. doi: 10.1053/j.gastro.2018.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12:681–700. doi: 10.1038/nrgastro.2015.173. [DOI] [PubMed] [Google Scholar]
- 6.Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, Baron A, Park JW, Han G, Jassem J, Blanc JF, Vogel A, Komov D, Evans TRJ, Lopez C, Dutcus C, Guo M, Saito K, Kraljevic S, Tamai T, Ren M, Cheng AL. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–1173. doi: 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- 7.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling THR, Meyer T, Kang YK, Yeo W, Chopra A, Anderson J, Dela Cruz C, Lang L, Neely J, Tang H, Dastani HB, Melero I. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, Liu Z, Dong M, Hu X, Ouyang W, Peng J, Zhang Z. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342–1356. e1316. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 9.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo CL, Yang XH, Cheng W, Xu Y, Li JB, Sun YX, Bi YM, Zhang L, Wang QC. Expression of Fas/FasL in CD8+ T and CD3+ Foxp3+ Treg cells--relationship with apoptosis of circulating CD8+ T cells in hepatocellular carcinoma patients. Asian Pac J Cancer Prev. 2014;15:2613–2618. doi: 10.7314/apjcp.2014.15.6.2613. [DOI] [PubMed] [Google Scholar]
- 11.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 12.Fatourou EM, Koskinas JS. Adaptive immunity in hepatocellular carcinoma: prognostic and therapeutic implications. Expert Rev Anticancer Ther. 2009;9:1499–1510. doi: 10.1586/era.09.103. [DOI] [PubMed] [Google Scholar]
- 13.Unitt E, Marshall A, Gelson W, Rushbrook SM, Davies S, Vowler SL, Morris LS, Coleman N, Alexander GJ. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J Hepatol. 2006;45:246–253. doi: 10.1016/j.jhep.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 14.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 15.Gabrielson A, Wu Y, Wang H, Jiang J, Kallakury B, Gatalica Z, Reddy S, Kleiner D, Fishbein T, Johnson L, Island E, Satoskar R, Banovac F, Jha R, Kachhela J, Feng P, Zhang T, Tesfaye A, Prins P, Loffredo C, Marshall J, Weiner L, Atkins M, He AR. Intratumoral CD3 and CD8 T-cell densities associated with relapse-free survival in HCC. Cancer Immunol Res. 2016;4:419–430. doi: 10.1158/2326-6066.CIR-15-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi N, Hiraoka N, Yamagami W, Ojima H, Kanai Y, Kosuge T, Nakajima A, Hirohashi S. FOXP3+ regulatory T cells affect the development and progression of hepatocarcinogenesis. Clin Cancer Res. 2007;13:902–911. doi: 10.1158/1078-0432.CCR-06-2363. [DOI] [PubMed] [Google Scholar]
- 17.Shirabe K, Motomura T, Muto J, Toshima T, Matono R, Mano Y, Takeishi K, Ijichi H, Harada N, Uchiyama H, Yoshizumi T, Taketomi A, Maehara Y. Tumor-infiltrating lymphocytes and hepatocellular carcinoma: pathology and clinical management. Int J Clin Oncol. 2010;15:552–558. doi: 10.1007/s10147-010-0131-0. [DOI] [PubMed] [Google Scholar]
- 18.Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J. Clin. Oncol. 2007;25:2586–2593. doi: 10.1200/JCO.2006.09.4565. [DOI] [PubMed] [Google Scholar]
- 19.Unitt E, Rushbrook SM, Marshall A, Davies S, Gibbs P, Morris LS, Coleman N, Alexander GJ. Compromised lymphocytes infiltrate hepatocellular carcinoma: the role of T-regulatory cells. Hepatology. 2005;41:722–730. doi: 10.1002/hep.20644. [DOI] [PubMed] [Google Scholar]
- 20.Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, Zhang Z, Yang H, Zhang H, Zhou C, Yao J, Jin L, Wang H, Yang Y, Fu YX, Wang FS. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132:2328–2339. doi: 10.1053/j.gastro.2007.03.102. [DOI] [PubMed] [Google Scholar]
- 21.Huang D, Chen J, Yang L, Ouyang Q, Li J, Lao L, Zhao J, Liu J, Lu Y, Xing Y, Chen F, Su F, Yao H, Liu Q, Su S, Song E. NKILA lncRNA promotes tumor immune evasion by sensitizing T cells to activation-induced cell death. Nat Immunol. 2018;19:1112–1125. doi: 10.1038/s41590-018-0207-y. [DOI] [PubMed] [Google Scholar]
- 22.Horton BL, Williams JB, Cabanov A, Spranger S, Gajewski TF. Intratumoral CD8(+) T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol Res. 2018;6:14–24. doi: 10.1158/2326-6066.CIR-17-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, Schemmer P, Bruns H, Eiermann T, Price DA, Blum HE, Neumann-Haefelin C, Thimme R. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology. 2014;59:1415–1426. doi: 10.1002/hep.26731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, Pedroza-Gonzalez A, Polak WG, de Jonge J, Gaspersz M, Dong H, Thielemans K, Pan Q, JNM IJ, Bruno MJ, Kwekkeboom J. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology. 2017;153:1107–1119. e1110. doi: 10.1053/j.gastro.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 25.Kim HD, Song GW, Park S, Jung MK, Kim MH, Kang HJ, Yoo C, Yi K, Kim KH, Eo S, Moon DB, Hong SM, Ju YS, Shin EC, Hwang S, Park SH. Association between expression level of PD1 by tumor-infiltrating CD8(+) T cells and features of hepatocellular carcinoma. Gastroenterology. 2018;155:1936–1950. e1917. doi: 10.1053/j.gastro.2018.08.030. [DOI] [PubMed] [Google Scholar]
- 26.Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, Duan K, Ang N, Poidinger M, Lee YY, Larbi A, Khng AJ, Tan E, Fu C, Mathew R, Teo M, Lim WT, Toh CK, Ong BH, Koh T, Hillmer AM, Takano A, Lim TKH, Tan EH, Zhai W, Tan DSW, Tan IB, Newell EW. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 27.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 28.Seo H, Jeon I, Kim BS, Park M, Bae EA, Song B, Koh CH, Shin KS, Kim IK, Choi K, Oh T, Min J, Min BS, Han YD, Kang SJ, Shin SJ, Chung Y, Kang CY. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun. 2017;8:15776. doi: 10.1038/ncomms15776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berzofsky JA, Terabe M. NKT cells in tumor immunity: opposing subsets define a new immunoregulatory axis. J Immunol. 2008;180:3627–3635. doi: 10.4049/jimmunol.180.6.3627. [DOI] [PubMed] [Google Scholar]
- 30.Bricard G, Cesson V, Devevre E, Bouzourene H, Barbey C, Rufer N, Im JS, Alves PM, Martinet O, Halkic N, Cerottini JC, Romero P, Porcelli SA, Macdonald HR, Speiser DE. Enrichment of human CD4+ V(alpha)24/Vbeta11 invariant NKT cells in intrahepatic malignant tumors. J Immunol. 2009;182:5140–5151. doi: 10.4049/jimmunol.0711086. [DOI] [PubMed] [Google Scholar]
- 31.Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, Lorentzen A, Einer C, Schulz S, Clavel T, Protzer U, Thiele C, Zischka H, Moch H, Tschöp M, Tumanov AV, Haller D, Unger K, Karin M, Kopf M, Knolle P, Weber A, Heikenwalder M. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549–564. doi: 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, Ritz T, Longerich T, Theriot CM, McCulloch JA, Roy S, Yuan W, Thovarai V, Sen SK, Ruchirawat M, Korangy F, Wang XW, Trinchieri G, Greten TF. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360:eaan5931. doi: 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mossanen JC, Kohlhepp M, Wehr A, Krenkel O, Liepelt A, Roeth AA, Möckel D, Heymann F, Lammers T, Gassler N, Hermann J, Jankowski J, Neumann UP, Luedde T, Trautwein C, Tacke F. CXCR6 inhibits hepatocarcinogenesis by promoting natural killer T- and CD4(+) T-cell-dependent control of senescence. Gastroenterology. 2019;156:1877–1889. e1874. doi: 10.1053/j.gastro.2019.01.247. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Du Y, Hu Q, Huang Z. Tumor-derived CD4+CD25+regulatory T cells inhibit dendritic cells function by CTLA-4. Pathol Res Pract. 2017;213:245–249. doi: 10.1016/j.prp.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005;65:2457–2464. doi: 10.1158/0008-5472.CAN-04-3232. [DOI] [PubMed] [Google Scholar]
- 36.Kalathil SG, Lugade AA, Miller A, Iyer R, Thanavala Y. PD-1(+) and Foxp3(+) T cell reduction correlates with survival of HCC patients after sorafenib therapy. JCI Insight. 2016;1:e86182. doi: 10.1172/jci.insight.86182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li CX, Ling CC, Shao Y, Xu A, Li XC, Ng KT, Liu XB, Ma YY, Qi X, Liu H, Liu J, Yeung OW, Yang XX, Liu QS, Lam YF, Zhai Y, Lo CM, Man K. CXCL10/CXCR3 signaling mobilized-regulatory T cells promote liver tumor recurrence after transplantation. J Hepatol. 2016;65:944–952. doi: 10.1016/j.jhep.2016.05.032. [DOI] [PubMed] [Google Scholar]
- 38.Stross L, Gunther J, Gasteiger G, Asen T, Graf S, Aichler M, Esposito I, Busch DH, Knolle P, Sparwasser T, Protzer U. Foxp3+ regulatory T cells protect the liver from immune damage and compromise virus control during acute experimental hepatitis B virus infection in mice. Hepatology. 2012;56:873–883. doi: 10.1002/hep.25765. [DOI] [PubMed] [Google Scholar]
- 39.Sharma S, Khosla R, David P, Rastogi A, Vyas A, Singh D, Bhardwaj A, Sahney A, Maiwall R, Sarin SK, Trehanpati N. CD4+CD25+CD127(low) regulatory T cells play predominant anti-tumor suppressive role in hepatitis B virus-associated hepatocellular carcinoma. Front Immunol. 2015;6:49. doi: 10.3389/fimmu.2015.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cany J, Tran L, Gauttier V, Judor JP, Vassaux G, Ferry N, Conchon S. Immunotherapy of hepatocellular carcinoma: is there a place for regulatory T-lymphocyte depletion? Immunotherapy. 2011;3:32–34. doi: 10.2217/imt.11.29. [DOI] [PubMed] [Google Scholar]
- 41.Kakita N, Kanto T, Itose I, Kuroda S, Inoue M, Matsubara T, Higashitani K, Miyazaki M, Sakakibara M, Hiramatsu N, Takehara T, Kasahara A, Hayashi N. Comparative analyses of regulatory T cell subsets in patients with hepatocellular carcinoma: a crucial role of CD25(-) FOXP3(-) T cells. Int J Cancer. 2012;131:2573–2583. doi: 10.1002/ijc.27535. [DOI] [PubMed] [Google Scholar]
- 42.Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16:356–371. doi: 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- 43.Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, Greten TF, Korangy F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135:234–243. doi: 10.1053/j.gastro.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 44.Zhou J, Ding T, Pan W, Zhu LY, Li L, Zheng L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int J Cancer. 2009;125:1640–1648. doi: 10.1002/ijc.24556. [DOI] [PubMed] [Google Scholar]
- 45.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 46.Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, Fan J, Cao Y, Dai Z, Zhou J. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150:1646–1658. e1617. doi: 10.1053/j.gastro.2016.02.040. [DOI] [PubMed] [Google Scholar]
- 47.Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, Kammula US, Chen Y, Qin LX, Tang ZY, Wang XW. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 48.Momiyama K, Nagai H, Sumino Y. Changes of host immunity in relation to efficacy in liver cirrhosis patients with advanced hepatocellular carcinoma treated by intra-arterial chemotherapy. Cancer Chemother Pharmacol. 2009;64:271–277. doi: 10.1007/s00280-008-0866-8. [DOI] [PubMed] [Google Scholar]
- 49.Lee HL, Jang JW, Lee SW, Yoo SH, Kwon JH, Nam SW, Bae SH, Choi JY, Han NI, Yoon SK. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci Rep. 2019;9:3260. doi: 10.1038/s41598-019-40078-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melssen M, Slingluff CL Jr. Vaccines targeting helper T cells for cancer immunotherapy. Curr Opin Immunol. 2017;47:85–92. doi: 10.1016/j.coi.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 52.Lafdil F, Miller AM, Ki SH, Gao B. Th17 cells and their associated cytokines in liver diseases. Cell Mol Immunol. 2010;7:250–254. doi: 10.1038/cmi.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ji Y, Zhang W. Th17 cells: positive or negative role in tumor? Cancer Immunol Immunother. 2010;59:979–987. doi: 10.1007/s00262-010-0849-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang JP, Yan J, Xu J, Pang XH, Chen MS, Li L, Wu C, Li SP, Zheng L. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol. 2009;50:980–989. doi: 10.1016/j.jhep.2008.12.033. [DOI] [PubMed] [Google Scholar]
- 55.Sun HQ, Zhang JY, Zhang H, Zou ZS, Wang FS, Jia JH. Increased Th17 cells contribute to disease progression in patients with HBV-associated liver cirrhosis. J Viral Hepat. 2012;19:396–403. doi: 10.1111/j.1365-2893.2011.01561.x. [DOI] [PubMed] [Google Scholar]
- 56.Gomes AL, Teijeiro A, Buren S, Tummala KS, Yilmaz M, Waisman A, Theurillat JP, Perna C, Djouder N. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2016;30:161–175. doi: 10.1016/j.ccell.2016.05.020. [DOI] [PubMed] [Google Scholar]
- 57.McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. 2019;50:892–906. doi: 10.1016/j.immuni.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, Tang L, Lin Y, He YQ, Zou SS, Wang C, Zhang HL, Cao GW, Wu MC, Wang HY. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology. 2010;52:1322–1333. doi: 10.1002/hep.23845. [DOI] [PubMed] [Google Scholar]
- 59.Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol. 2009;10:864–871. doi: 10.1038/ni.1770. [DOI] [PubMed] [Google Scholar]
- 60.Huang YH, Cao YF, Jiang ZY, Zhang S, Gao F. Th22 cell accumulation is associated with colorectal cancer development. World J Gastroenterol. 2015;21:4216–4224. doi: 10.3748/wjg.v21.i14.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niccolai E, Taddei A, Ricci F, Rolla S, D’Elios MM, Benagiano M, Bechi P, Bencini L, Ringressi MN, Pini A, Castiglione F, Giordano D, Satolli MA, Coratti A, Cianchi F, Bani D, Prisco D, Novelli F, Amedei A. Intra-tumoral IFN-γ-producing Th22 cells correlate with TNM staging and the worst outcomes in pancreatic cancer. Clin Sci (Lond) 2016;130:247–258. doi: 10.1042/CS20150437. [DOI] [PubMed] [Google Scholar]
- 62.Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–1342. doi: 10.1002/hep.20184. [DOI] [PubMed] [Google Scholar]
- 63.Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y, Wang X, Sun B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900–909. doi: 10.1002/hep.24486. [DOI] [PubMed] [Google Scholar]
- 64.Qin S, Ma S, Huang X, Lu D, Zhou Y, Jiang H. Th22 cells are associated with hepatocellular carcinoma development and progression. Chin J Cancer Res. 2014;26:135–141. doi: 10.3978/j.issn.1000-9604.2014.02.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuang DM, Xiao X, Zhao Q, Chen MM, Li XF, Liu RX, Wei Y, Ouyang FZ, Chen DP, Wu Y, Lao XM, Deng H, Zheng L. B7-H1-expressing antigen-presenting cells mediate polarization of protumorigenic Th22 subsets. J Clin Invest. 2014;124:4657–4667. doi: 10.1172/JCI74381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jia Y, Zeng Z, Li Y, Li Z, Jin L, Zhang Z, Wang L, Wang FS. Impaired function of CD4+ T follicular helper (Tfh) cells associated with hepatocellular carcinoma progression. PLoS One. 2015;10:e0117458. doi: 10.1371/journal.pone.0117458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chandwaskar R, Awasthi A. Emerging roles of Th9 cells as an anti-tumor helper T cells. Int Rev Immunol. 2019;38:204–211. doi: 10.1080/08830185.2019.1648453. [DOI] [PubMed] [Google Scholar]
- 68.Tan H, Wang S, Zhao L. A tumour-promoting role of Th9 cells in hepatocellular carcinoma through CCL20 and STAT3 pathways. Clin Exp Pharmacol Physiol. 2017;44:213–221. doi: 10.1111/1440-1681.12689. [DOI] [PubMed] [Google Scholar]
- 69.Dusseaux M, Martin E, Serriari N, Peguillet I, Premel V, Louis D, Milder M, Le Bourhis L, Soudais C, Treiner E, Lantz O. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood. 2011;117:1250–1259. doi: 10.1182/blood-2010-08-303339. [DOI] [PubMed] [Google Scholar]
- 70.Hegde P, Weiss E, Paradis V, Wan J, Mabire M, Sukriti S, Rautou PE, Albuquerque M, Picq O, Gupta AC, Ferrere G, Gilgenkrantz H, Kiaf B, Toubal A, Beaudoin L, Letteron P, Moreau R, Lehuen A, Lotersztajn S. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat Commun. 2018;9:2146. doi: 10.1038/s41467-018-04450-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bottcher K, Rombouts K, Saffioti F, Roccarina D, Rosselli M, Hall A, Luong T, Tsochatzis EA, Thorburn D, Pinzani M. MAIT cells are chronically activated in patients with autoimmune liver disease and promote profibrogenic hepatic stellate cell activation. Hepatology. 2018;68:172–186. doi: 10.1002/hep.29782. [DOI] [PubMed] [Google Scholar]
- 72.Bolte FJ, O’Keefe AC, Webb LM, Serti E, Rivera E, Liang TJ, Ghany M, Rehermann B. Intra-hepatic depletion of mucosal-associated invariant T cells in hepatitis C virus-induced liver inflammation. Gastroenterology. 2017;153:1392–1403. e1392. doi: 10.1053/j.gastro.2017.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yong YK, Saeidi A, Tan HY, Rosmawati M, Enstrom PF, Batran RA, Vasuki V, Chattopadhyay I, Murugesan A, Vignesh R, Kamarulzaman A, Rajarajeswaran J, Ansari AW, Vadivelu J, Ussher JE, Velu V, Larsson M, Shankar EM. Hyper-expression of PD-1 is associated with the levels of exhausted and dysfunctional phenotypes of circulating CD161(++)TCR iValpha7.2(+) mucosal-associated invariant T cells in chronic hepatitis B virus infection. Front Immunol. 2018;9:472. doi: 10.3389/fimmu.2018.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Duan M, Goswami S, Shi JY, Wu LJ, Wang XY, Ma JQ, Zhang Z, Shi Y, Ma LJ, Zhang S, Xi RB, Cao Y, Zhou J, Fan J, Zhang XM, Gao Q. Activated and exhausted MAIT cells foster disease progression and indicate poor outcome in hepatocellular carcinoma. Clin Cancer Res. 2019;25:3304–3316. doi: 10.1158/1078-0432.CCR-18-3040. [DOI] [PubMed] [Google Scholar]
- 75.Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, Breder V, Edeline J, Chao Y, Ogasawara S, Yau T, Garrido M, Chan SL, Knox J, Daniele B, Ebbinghaus SW, Chen E, Siegel AB, Zhu AX, Cheng AL. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J. Clin. Oncol. 2020;38:193–202. doi: 10.1200/JCO.19.01307. [DOI] [PubMed] [Google Scholar]
- 76.Ikeda M, Sung MW, Kudo M, Kobayashi M, Baron AD, Finn RS, Kaneko S, Zhu AX, Kubota T, Kraljevic S, Ishikawa K, Siegel AB, Kumada H, Okusaka T. A phase 1b trial of lenvatinib (LEN) plus pembrolizumab (PEM) in patients (pts) with unresectable hepatocellular carcinoma (uHCC) J. Clin. Oncol. 2018;36:4076–4076. [Google Scholar]
- 77.Llovet JM, Kudo M, Cheng AL, Finn RS, Galle PR, Kaneko S, Meyer T, Qin S, Dutcus CE, Chen E, Dubrovsky L, Zhu AX. Lenvatinib (len) plus pembrolizumab (pembro) for the first-line treatment of patients (pts) with advanced hepatocellular carcinoma (HCC): phase 3 LEAP-002 study. J. Clin. Oncol. 2019;37:TPS4152–TPS4152. [Google Scholar]
- 78.Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, Hack SP, Spahn J, Liu B, Abdullah H, Wang Y, He AR, Lee KH. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol. 2020;21:808–820. doi: 10.1016/S1470-2045(20)30156-X. [DOI] [PubMed] [Google Scholar]
- 79.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, Li D, Verret W, Xu DZ, Hernandez S, Liu J, Huang C, Mulla S, Wang Y, Lim HY, Zhu AX, Cheng AL. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 80.Sawada Y, Yoshikawa T, Nobuoka D, Shirakawa H, Kuronuma T, Motomura Y, Mizuno S, Ishii H, Nakachi K, Konishi M, Nakagohri T, Takahashi S, Gotohda N, Takayama T, Yamao K, Uesaka K, Furuse J, Kinoshita T, Nakatsura T. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18:3686–3696. doi: 10.1158/1078-0432.CCR-11-3044. [DOI] [PubMed] [Google Scholar]
- 81.Greten TF, Forner A, Korangy F, N’Kontchou G, Barget N, Ayuso C, Ormandy LA, Manns MP, Beaugrand M, Bruix J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer. 2010;10:209. doi: 10.1186/1471-2407-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Palmer DH, Midgley RS, Mirza N, Torr EE, Ahmed F, Steele JC, Steven NM, Kerr DJ, Young LS, Adams DH. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology. 2009;49:124–132. doi: 10.1002/hep.22626. [DOI] [PubMed] [Google Scholar]
- 83.Tada F, Abe M, Hirooka M, Ikeda Y, Hiasa Y, Lee Y, Jung NC, Lee WB, Lee HS, Bae YS, Onji M. Phase I/II study of immunotherapy using tumor antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Int J Oncol. 2012;41:1601–1609. doi: 10.3892/ijo.2012.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, Ren S, Zhou C. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. 2019;12:93. doi: 10.1186/s13045-019-0787-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines combined with immune checkpoint antibodies promote cytotoxic T-cell activity and tumor eradication. Cancer Immunol Res. 2016;4:95–100. doi: 10.1158/2326-6066.CIR-14-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, Feng L, Lee JJ, Tran H, Kim YU, Haymaker C, Bernatchez C, Curran M, Zecchini Barrese T, Rodriguez Canales J, Wistuba I, Li L, Wang J, van der Burg SH, Melief CJ, Glisson B. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5:67–73. doi: 10.1001/jamaoncol.2018.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee JH, Tak WY, Lee Y, Heo MK, Song JS, Kim HY, Park SY, Bae SH, Lee JH, Heo J, Kim KH, Bae YS, Kim YJ. Adjuvant immunotherapy with autologous dendritic cells for hepatocellular carcinoma, randomized phase II study. Oncoimmunology. 2017;6:e1328335. doi: 10.1080/2162402X.2017.1328335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sawada Y, Yoshikawa T, Ofuji K, Yoshimura M, Tsuchiya N, Takahashi M, Nobuoka D, Gotohda N, Takahashi S, Kato Y, Konishi M, Kinoshita T, Ikeda M, Nakachi K, Yamazaki N, Mizuno S, Takayama T, Yamao K, Uesaka K, Furuse J, Endo I, Nakatsura T. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology. 2016;5:e1129483. doi: 10.1080/2162402X.2015.1129483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Takayama T, Sekine T, Makuuchi M, Yamasaki S, Kosuge T, Yamamoto J, Shimada K, Sakamoto M, Hirohashi S, Ohashi Y, Kakizoe T. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet. 2000;356:802–807. doi: 10.1016/S0140-6736(00)02654-4. [DOI] [PubMed] [Google Scholar]
- 90.Pan K, Li YQ, Wang W, Xu L, Zhang YJ, Zheng HX, Zhao JJ, Qiu HJ, Weng DS, Li JJ, Wang QJ, Huang LX, He J, Chen SP, Ke ML, Wu PH, Chen MS, Li SP, Xia JC, Zeng YX. The efficacy of cytokine-induced killer cell infusion as an adjuvant therapy for postoperative hepatocellular carcinoma patients. Ann Surg Oncol. 2013;20:4305–4311. doi: 10.1245/s10434-013-3144-x. [DOI] [PubMed] [Google Scholar]
- 91.Lee JH, Lee JH, Lim YS, Yeon JE, Song TJ, Yu SJ, Gwak GY, Kim KM, Kim YJ, Lee JW, Yoon JH. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology. 2015;148:1383–1391. e1386. doi: 10.1053/j.gastro.2015.02.055. [DOI] [PubMed] [Google Scholar]
- 92.Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fisher J, Abramowski P, Wisidagamage Don ND, Flutter B, Capsomidis A, Cheung GW, Gustafsson K, Anderson J. Avoidance of on-target off-tumor activation using a co-stimulation-only chimeric antigen receptor. Mol Ther. 2017;25:1234–1247. doi: 10.1016/j.ymthe.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, Martens ACM, Zweegman S, van de Donk N, Groen RWJ, Lokhorst HM, Mutis T. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol Ther. 2017;25:1946–1958. doi: 10.1016/j.ymthe.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krebs K, Bottinger N, Huang LR, Chmielewski M, Arzberger S, Gasteiger G, Jager C, Schmitt E, Bohne F, Aichler M, Uckert W, Abken H, Heikenwalder M, Knolle P, Protzer U. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145:456–465. doi: 10.1053/j.gastro.2013.04.047. [DOI] [PubMed] [Google Scholar]
- 96.Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, Kong J, Wang H, Yang S, Gu J, Li Z. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20:6418–6428. doi: 10.1158/1078-0432.CCR-14-1170. [DOI] [PubMed] [Google Scholar]
- 97.Dargel C, Bassani-Sternberg M, Hasreiter J, Zani F, Bockmann JH, Thiele F, Bohne F, Wisskirchen K, Wilde S, Sprinzl MF, Schendel DJ, Krackhardt AM, Uckert W, Wohlleber D, Schiemann M, Stemmer K, Heikenwalder M, Busch DH, Richter G, Mann M, Protzer U. T cells engineered to express a T-cell receptor specific for glypican-3 to recognize and kill hepatoma cells in vitro and in mice. Gastroenterology. 2015;149:1042–1052. doi: 10.1053/j.gastro.2015.05.055. [DOI] [PubMed] [Google Scholar]
- 98.Zhai B, Shi D, Gao H, Qi X, Jiang H, Zhang Y, Chi J, Ruan H, Wang H, Ru QC, Li Z. A phase I study of anti-GPC3 chimeric antigen receptor modified T cells (GPC3 CAR-T) in Chinese patients with refractory or relapsed GPC3+ hepatocellular carcinoma (r/r GPC3+ HCC) J. Clin. Oncol. 2017;35:3049–3049. [Google Scholar]
- 99.Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, Liu Y, Huang J, Lv H, Luo C, Feng KC, Yang QM, Li XL, Han W. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology. 2018;7:e1440169. doi: 10.1080/2162402X.2018.1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmid S, Diem S, Li Q, Krapf M, Flatz L, Leschka S, Desbiolles L, Klingbiel D, Jochum W, Früh M. Organ-specific response to nivolumab in patients with non-small cell lung cancer (NSCLC) Cancer Immunol Immunother. 2018;67:1825–1832. doi: 10.1007/s00262-018-2239-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lu LC, Hsu C, Shao YY, Chao Y, Yen CJ, Shih IL, Hung YP, Chang CJ, Shen YC, Guo JC, Liu TH, Hsu CH, Cheng AL. Differential organ-specific tumor response to immune checkpoint inhibitors in hepatocellular carcinoma. Liver Cancer. 2019;8:480–490. doi: 10.1159/000501275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ali M, Foldvari Z, Giannakopoulou E, Böschen ML, Strønen E, Yang W, Toebes M, Schubert B, Kohlbacher O, Schumacher TN, Olweus J. Induction of neoantigen-reactive T cells from healthy donors. Nat Protoc. 2019;14:1926–1943. doi: 10.1038/s41596-019-0170-6. [DOI] [PubMed] [Google Scholar]