

Abstract
Current diagnostic criteria for the neuropathological evaluation of the traumatic brain injury-associated neurodegeneration, chronic traumatic encephalopathy, define the pathognomonic lesion as hyperphosphorylated tau-immunoreactive neuronal and astroglial profiles in a patchy cortical distribution, clustered around small vessels and showing preferential localization to the depths of sulci. However, despite adoption into diagnostic criteria, there has been no formal assessment of the cortical distribution of the specific cellular components defining chronic traumatic encephalopathy neuropathologic change. To address this, we performed comprehensive mapping of hyperphosphorylated tau-immunoreactive neurofibrillary tangles and thorn-shaped astrocytes contributing to chronic traumatic encephalopathy neuropathologic change. From the Glasgow Traumatic Brain Injury Archive and the University of Pennsylvania Center for Neurodegenerative Disease Research Brain Bank, material was selected from patients with known chronic traumatic encephalopathy neuropathologic change, either following exposure to repetitive mild (athletes n = 17; non-athletes n = 1) or to single moderate or severe traumatic brain injury (n = 4), together with material from patients with previously confirmed Alzheimer’s disease neuropathologic changes (n = 6) and no known exposure to traumatic brain injury. Representative sections were stained for hyperphosphorylated or Alzheimer’s disease conformation-selective tau, after which stereotypical neurofibrillary tangles and thorn-shaped astrocytes were identified and mapped. Thorn-shaped astrocytes in chronic traumatic encephalopathy neuropathologic change were preferentially distributed towards sulcal depths [sulcal depth to gyral crest ratio of thorn-shaped astrocytes 12.84 ± 15.47 (mean ± standard deviation)], with this pathology more evident in material from patients with a history of survival from non-sport injury than those exposed to sport-associated traumatic brain injury (P = 0.009). In contrast, neurofibrillary tangles in chronic traumatic encephalopathy neuropathologic change showed a more uniform distribution across the cortex in sections stained for either hyperphosphorylated (sulcal depth to gyral crest ratio of neurofibrillary tangles 1.40 ± 0.74) or Alzheimer’s disease conformation tau (sulcal depth to gyral crest ratio 1.64 ± 1.05), which was comparable to that seen in material from patients with known Alzheimer’s disease neuropathologic changes (P = 0.82 and P = 0.91, respectively). Our data demonstrate that in chronic traumatic encephalopathy neuropathologic change the astroglial component alone shows preferential distribution to the depths of cortical sulci. In contrast, the neuronal pathology of chronic traumatic encephalopathy neuropathologic change is distributed more uniformly from gyral crest to sulcal depth and echoes that of Alzheimer’s disease. These observations provide new insight into the neuropathological features of chronic traumatic encephalopathy that distinguish it from other tau pathologies and suggest that current diagnostic criteria should perhaps be reviewed and refined.
Keywords: chronic traumatic encephalopathy, Alzheimer’s disease, tau, ageing-related tau astrogliopathy, traumatic brain injury
Mapping hyper-phosphorylated tau pathologies contributing to the pathognomonic cortical lesion of chronic traumatic encephalopathy neuropathologic change reveals immunoreactive astrocytes preferentially concentrate to sulcal depths. In contrast, neurofibrillary tangles show limited sulcal concentration, comparable with Alzheimer’s disease. These data provide new insight into the specific pathology of chronic traumatic encephalopathy.
Graphical Abstract
Graphical Abstract.
Introduction
Traumatic brain injury (TBI) is recognized as a major risk factor for a range of neurodegenerative diseases, including Alzheimer’s disease and chronic traumatic encephalopathy (CTE) (Johnson et al., 2017; Smith et al., 2019). Although first described, several decades ago, as dementia pugilistica of former boxers (Millspaugh, 1937; Critchley, 1949; Critchley, 1957; Corsellis et al., 1973), consensus criteria for the neuropathological assessment of what is now recognized as CTE only emerged in the last decade (McKee et al., 2016). These criteria define the pathognomonic lesion of CTE neuropathologic change (CTE-NC) as p-tau aggregates in neurons, astrocytes and cell processes around small vessels in an irregular pattern at the depths of cortical sulci (McKee et al., 2016). However, despite incorporation into diagnostic criteria, the neocortical features currently defining CTE-NC have not been formally evaluated using rigorous and objective measures.
Earliest accounts of the neuropathology of CTE (then dementia pugilistica) described abundant neuronal profiles in the form of neurofibrillary tangles, with a noted preferential involvement of superficial cortical layers and patchy distribution of pathology. Also documented in these early reports was the subjective impression of an apparent concentration of tangles to the depths of cortical sulci (Hof et al., 1992; Geddes et al., 1996; Geddes et al., 1999), although this was never formally quantified. In addition to neurofibrillary tangle pathology, tau-immunoreactive, thorn-shaped astrocytes (TSAs) are increasingly recognized as a prominent feature of CTE (Ikeda et al., 1995; Ikeda et al., 1998; McKee et al., 2009; McKee et al., 2013; McKee et al., 2016), with the combination of these glial and neuronal pathologies defining the pathognomonic lesion of CTE-NC (McKee et al., 2016); a pathology that has now been documented not only in boxers, but in non-boxer athletes (Omalu et al., 2005; McKee et al., 2009; McKee et al., 2013; Smith et al., 2013; Stewart et al., 2016; Ling et al., 2017; Mez et al., 2017; Lee et al., 2019), military veterans (Goldstein et al., 2012; Stein et al., 2014) and individuals exposed to a single moderate or severe TBI (Johnson et al., 2012; Zanier et al., 2018; Tribett et al., 2019; Arena et al., 2020).
While neurofibrillary tangles have long been described in association with a variety of neurodegenerative pathologies, only more recently attention has been paid to astrocytic tau pathologies in ageing and wider neurodegeneration. Ageing-related tau astrogliopathy (ARTAG) is increasingly recognized in the brains of older individuals and as a co-morbidity in a variety of neurodegenerative diseases (Kovacs et al., 2016; Kovacs et al., 2017), with its presence suggested to correlate with a greater degree of cognitive impairment (Robinson et al., 2018). Comprising morphologically distinct phospho-tau immunoreactive astrocytes, ARTAG typically localizes to interface sites, such as sub-pial, perivascular or sub-ependymal locations, or around the boundary between grey and white matter (Kovacs et al., 2016). Notably, one morphological variant of ARTAG, TSA, shows comparable morphology and immunophenotype to the astroglial pathology of CTE-NC (Kovacs et al., 2016; Liu et al., 2016; Kovacs et al., 2017; Kovacs et al., 2018; Forrest et al., 2019; Arena et al., 2020).
There is, therefore, a need to critically appraise the pathology of CTE-NC and, where required, refine neuropathological criteria for its recognition and differentiation from the pathologies of ageing and wider neurodegenerative disease. To this end, we pursued quantitative mapping of the distribution of cortical neurofibrillary tangle and astroglial pathologies in cases with previously documented CTE-NC and in cases with Alzheimer’s disease neuropathologic changes and no known history of TBI. Contrary to understanding reflected in current diagnostic criteria, our observations demonstrate that the astroglial pathology alone shows marked concentration to the depths of cortical sulci in CTE-NC. In contrast, neurofibrillary tangles show only limited concentration towards sulcal depths, which echoes that seen in Alzheimer’s disease.
Materials and methods
All tissue samples were obtained from the Glasgow TBI Archive, Department of Neuropathology, Queen Elizabeth University Hospital, Glasgow, UK, or the University of Pennsylvania Center for Neurodegenerative Disease Research (CNDR) Brain Bank, Philadelphia, PA, USA. Brain tissue was acquired by means of planned donation after multi-year longitudinal follow-up, or at routine diagnostic autopsy. Ethical approval for use of tissue in this study was provided by the West of Scotland Research Ethics Committee (Project ID 225271), the Greater Glasgow and Clyde Biorepository (Application Number 340) and the Institutional Review Board of the University of Pennsylvania.
Cases for inclusion were identified as all available cases from each archive with documented pathology of CTE-NC, or a random sample of cases from the CNDR archive with known Alzheimer’s disease neuropathologic changes and no known history of exposure to TBI. All CTE-NC cases (n = 22) had a history of previous exposure to head trauma, had been subject to comprehensive and standardized neuropathological assessment for neurodegenerative disease pathologies at autopsy and had documented cortical tau pathology consistent with the pathognomonic pathology of CTE-NC defined in diagnostic criteria (McKee et al., 2016). In each case where CTE-NC was present, the extent and distribution of pathology was further dichotomized as ‘low’ or ‘high’; corresponding to stages I/II or III/IV, respectively, of a widely used protocol (McKee et al., 2013). For CTE-NC cases, histories included a remote history of exposure to repetitive mild TBI (soccer, n = 8; American football, n = 4; rugby union, n = 3; boxing, n = 1; mixed sports, n = 1; non-sport, n = 1) or a single moderate or severe TBI (fall, n = 2; assault, n = 1; motor vehicle collision, n = 1). Detailed reports from the original diagnostic autopsies were available for all cases; supplemented, where necessary, by forensic and clinical records. All Alzheimer’s disease cases had no documented history of TBI or participation in contact sport, and met neuropathological criteria for the diagnosis of Alzheimer’s disease [n = 6; 4 high and 2 intermediate Alzhemier’s disease neuropathologic changes (ADNC) (Montine et al., 2012)]. A single representative cortical sulcus containing pathognomonic hyperphosphorylated tau (p-tau) pathology was selected for analysis in each case. Regionally matched tissue sections from the frontal, angular and temporal cortices were selected from ADNC cases for comparison with CTE-NC. Clinical, demographic and neuropathologic information for each case, including integrated clinicopathologic diagnosis (Lee, 2018; Lee et al., 2019), is summarized in Table 1.
Table 1.
Case demographics
| Case no. | Age at death | Sex | TBI/sport exposure | Integrated clinicopathologic diagnosis | CTE-NC stage | PMI |
|---|---|---|---|---|---|---|
| C1 | 40s | M | Football | CBD | Low | 7 h |
| C2 | 50s | M | sTBI, Fall | No NDD | Low | 108 h |
| C3 | 60s | M | Football | CBD | Low | 3 h |
| C4 | 60s | M | Multiple non-sport mTBI | DLB | Low | 21 h |
| C5 | 70s | M | Football | DLB | Low | 18 h |
| C6 | 70s | M | Rugby | AD | Low | 48 h |
| C7 | 70s | M | Rugby | Mixed AD/VaD | Low | 48 h |
| C8 | 70s | M | sTBI, MVC | CTE | Low | 24 h |
| C9 | 70s | M | sTBI, Fall | PDD | Low | 7.5 h |
| C10 | 80s | M | Soccer | AD | Low | 24 h |
| C11 | 80s | M | Soccer | AD | Low | 20 h |
| C12 | 50s | M | Soccer | CTE | High | Unknown |
| C13 | 60s | M | sTBI, Assault | No NDD | High | 24 h |
| C14 | 60s | M | Boxing | CTE | High | 24 h |
| C15 | 60s | M | Soccer | VaD | High | 11 days |
| C16 | 70s | M | Rugby | CTE | High | 12 h |
| C17 | 70s | M | Soccer | DLB | High | 4 days |
| C18 | 70s | M | Soccer | CTE | High | 3 days |
| C19 | 80s | M | Football | FTLD-TDP | High | 7 h |
| C20 | 80s | M | Soccer | NPH | High | 3 days |
| C21 | 80s | M | Soccer | CTE and PD | High | 3 days |
| C22 | 80s | M | Boxing, Rugby, Soccer | PDD | High | 4 days |
| A1 | 60s | M | No | AD | n/a | 13.5 h |
| A2 | 60s | M | No | AD | n/a | 5 h |
| A3 | 70s | M | No | AD | n/a | 4 h |
| A4 | 70s | M | No | AD | n/a | 8.5 h |
| A5 | 70s | F | No | AD | n/a | 11 h |
| A6 | 80s | F | No | AD | n/a | 6 h |
AD, Alzheimer’s disease; ADNC, Alzheimer’s disease neuropathologic changes; CBD, corticobasal degeneration; CTE-NC, chronic traumatic encephalopathy neuropathologic change; DLB, dementia with Lewy bodies; FTLD-TDP, frontotemporal lobar degeneration with TDP-43 inclusions; MVC, motor vehicle collision; n/a, not applicable; NDD, neurodegenerative disease; NPH, normal pressure hydrocephalus; PD, Parkinson’s disease; PDD, Parkinson’s disease dementia; PMI, post-mortem interval; sTBI, single moderate or severe traumatic brain injury; TBI, traumatic brain injury; VaD, vascular dementia.
Brain tissue handling and immunohistochemistry
Whole brains from the Glasgow TBI Archive were fixed in 10% formol saline at autopsy for a minimum of 2 weeks prior to dissection, standardized anatomical sampling, tissue processing and paraffin embedding as described previously (Graham et al., 1995). Tissue blocks sampled from fresh brains at the University of Pennsylvania CNDR were fixed overnight in 70% ethanol and 150 mmol sodium chloride or 10% neutral-buffered formalin and processed to paraffin as described previously (Toledo et al., 2014). For each case, a single representative cortical block was chosen to include the defining pathognomonic pathology of CTE-NC or, for AD cases, a high burden of ADNC. From each tissue block, 8 μm tissue sections were prepared and subjected to deparaffinization and rehydration to H2O before immersion in 3% aqueous H2O2 to quench endogenous peroxidase activity. Antigen retrieval was performed via microwave pressure cooker in either Tris/EDTA (CP13) or citrate buffer following formic acid pre-treatment (GT-38), as optimized for each antibody. Sections were blocked using normal horse serum (Vector Labs, Burlingame, CA) in Optimax buffer (BioGenex, Fremont, CA) for 30 min followed by incubation in the primary antibody overnight at 4°C. Specifically, tau antibodies CP13 (specific for phosphoepitope S202, 1:1000 dilution, courtesy Dr Peter Davies) (Jicha et al., 1999) and GT-38 (1:1000 dilution, UPenn CNDR) were applied. GT-38 has been shown to detect a conformation-dependent epitope present in tau within the inclusions of Alzheimer’s disease requiring both 3R and 4R tau, but not the 3R or 4R-only tau of other primary tauopathies, and in a phosphorylation-independent manner (Gibbons et al., 2018; Gibbons et al., 2019). After rinsing, sections were incubated in a biotinylated universal secondary antibody (Vector Labs) for 30 min, followed by the avidin–biotin complex for 30 min (Vector Labs). Visualization was achieved using the DAB peroxidase substrate kit (Vector Labs). Sections were counterstained with haematoxylin, followed by rinsing, dehydration and coverslipping. Sections from a known positive control were stained in parallel with test sections, with omission of the primary antibody for one section to control for non-specific staining.
Mapping of pathologies
Stained sections from the Glasgow TBI Archive were scanned at a magnification of 20× with a Hamamatsu NanoZoomer 2.0-HT slide scanner, saved as NDPI files and evaluated using Aperio ImageScope Viewer 12.3.3 software (Leica Biosystems, Wetzlar, Germany). Sections from the University of Pennsylvania CNDR were scanned at a magnification of 20× with a Lamina Slide Scanner (Perkin Elmer, Waltham, MA) and saved as MSRX files, exported to ImageJ (National Institutes of Health, Bethesda, MD) using QuPath Open Source Digital Pathology software and then saved as TIFF file format and evaluated using Aperio ImageScope Viewer 12.3.3 (Leica Biosystems, Wetzlar, Germany).
On each scanned image, the region of interest was defined as the cortical grey matter of an entire involved sulcus and the adjacent gyral crests in which the pathologies of either CTE-NC or ADNC were present. This region of interest was then further sub-divided into sulcal depth and gyral crest (non-depth cortex) regions; the sulcal depth region defined as extending 1 mm from the deepest aspect of the sulcus towards the gyral crest and to the underlying grey-white junction (Holleran et al., 2017). In accordance with standard diagnostic and research practices, neuronal or astroglial tau-immunoreactive pathologies were identified as classical neurofibrillary tangles or thorn-shaped astrocytes by their defining and stereotypical cellular morphologies (Montine et al., 2012; Crary et al., 2014; Kovacs, 2015; Kovacs et al., 2016; McKee et al., 2016; Kovacs et al., 2017). CP13 immunostaining revealed both neurofibrillary tangles and thorn-shaped astrocytes, whereas GT-38 exclusively identified neurofibrillary tangles, as described previously (Arena et al., 2020). The location of each immunoreactive profile was then annotated within the region of interest (Fig. 1). All assessments were performed blind to clinical information and neuropathological diagnoses.
Figure 1.

Neuronal and astroglial tau pathologies of CTE neuropathologic change. (A) Low power view of cortical material from a former American football player who died in his 70s with a diagnosis of dementia with Lewy bodies (Case C5) reveals a patchy distribution of p-tau-immunoreactive (CP13) profiles, with apparent concentration of pathology to the sulcal depth (outlined in green) compared to the gyral crest (outlined in black). (B, C) There is clustering of p-tau immunoreactive profiles around cortical vessels, (D, E) which on higher power are revealed as typical neurofibrillary tangles (blue circles) and thorn-shaped astrocytes (red circles). Scale bars: (A) 1 mm, (B, C) 500 μm and (D, E) 50 μm
Quantification of pathology and statistical analysis
Densities of tau-immunoreactive profiles in each region were calculated as the number of profiles per mm2 assessed. From these data, a depth to crest ratio was calculated for neurofibrillary tangles or thorn-shaped astrocytes by dividing the density of the pathology in the depth sub-region by the density in the adjacent crest. A subset of cortical regions was independently annotated and assessed by two reviewers, resulting in a good-to-excellent inter-rater reliability, with intra-class correlation coefficient 0.923 (95% confidence interval, 0.718–0.980; two-way random effects, absolute agreement and single measurement) (Koo and Li, 2016).
Statistical analyses
Statistical analyses were performed using STATA v15.1 statistics software (College Station, TX) and GraphPad Prism v8.2.1 (San Diego, CA). Non-parametric tests were used in comparisons of depth to crest ratio, as assumptions of normality were not met after tests of skewness and kurtosis. Unpaired data samples were compared using the Mann–Whitney test, and paired samples were compared using the Wilcoxon signed-rank test. Associations between pathology and age were calculated by linear regression. Statistical significance was determined using an alpha level of 0.05.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
Results
Thorn-shaped astrocytes show concentration at sulcal depths in CTE-neuropathologic change
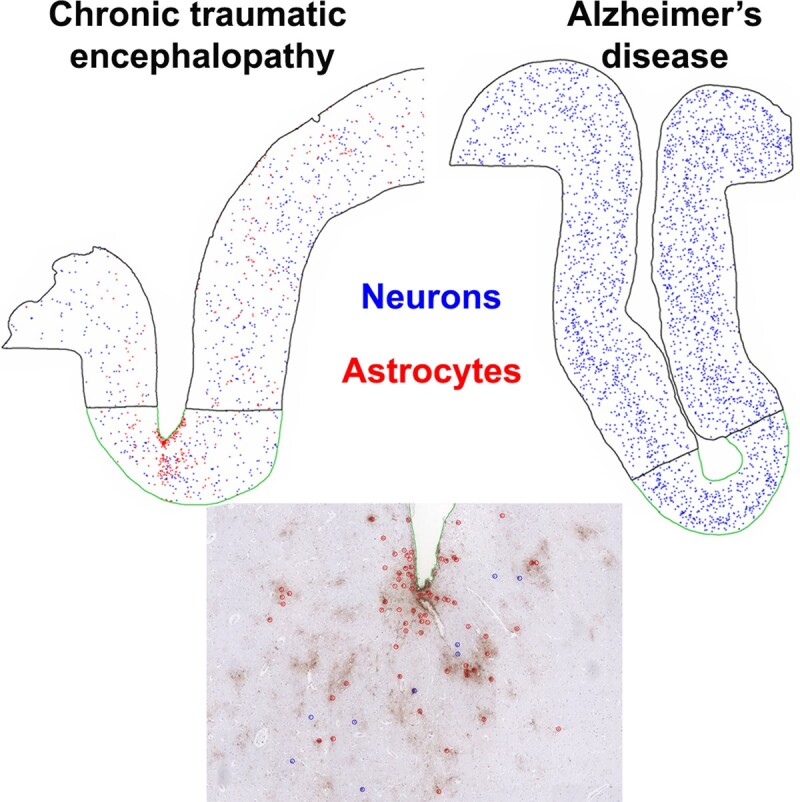
In keeping with previous descriptions, low power examination of sections from cases with known CTE-NC showed a patchy deposition of p-tau immunoreactivity, with apparent concentration to the depths of cortical sulci and distribution to superficial cortical layers (Fig. 1). While p-tau-immunoreactive thorn-shaped astrocytes were not observed in sections with ADNC, these were abundant in cases with CTE-NC (Fig. 1). Typically, p-tau immunoreactive thorn-shaped astrocytes in cases with CTE-NC were present in sub-pial locations and extending into deeper cortical layers (Fig. 2), with clustering around intracortical vessels. Formal mapping and quantitative assessment revealed thorn-shaped astrocytes in CTE-NC heavily concentrated to the sulcal depths (sulcal depth to gyral crest ratio of thorn-shaped astrocytes 12.84 ± 15.47, mean ± SD) (Figs. 3A and 4).
Figure 2.

Maps of neurofibrillary tangles and thorn-shaped astrocytes in CTE-NC and ADNC. (A) Reviewing the annotated map of p-tau-immunoreactive (CP13) profiles in CTE-NC reveals numerous neurofibrillary tangles (blue) and thorn-shaped astrocytes (red), (C) with the former showing relatively uniform distribution across the sulcal depth (outlined in green) and gyral crest (outlined in black). (E) In contrast, thorn-shaped astrocytes (red) show marked clustering and concentration to the sulcal depth. (B) In parallel, analysis of ADNC (D) reveals a heavy burden of neurofibrillary tangles arranged in a classical bilaminar distribution across the cortex, with limited concentration to the sulcal depth. (F) No thorn-shaped astrocytes were present in ADNC. (A, C, E) Case C10, a former soccer player age 80s. (B, D, F) Case A6, a patient with known Alzheimer’s disease (age, 80 years)
Figure 3.

Sulcal depth to gyral crest distributions of neuronal and astroglial pathologies in each case. (A) In cases with known CTE-NC, the ratio of sulcal depth to gyral crest CP13-positive thorn-shaped astrocyte pathology was typically high, in contrast to neurofibrillary tangles, which showed only limited sulcal concentration. (B) There was a close correlation between density of neurofibrillary tangles immunoreactive for CP13 and neuronal profiles identified using the antibody GT-38, which detects a conformation-dependent epitope of tau present in Alzheimer’s disease. The sulcal depth concentration of these pathologies in cases with CTE-NC was similar to that seen in cases with ADNC. NFT, neurofibrillary tangles; TSA, thorn-shaped astrocytes
Limited sulcal neurofibrillary tangle concentration in CTE-neuropathologic change echoes Alzheimer’s disease
Mapping of neuronal profiles demonstrated a mild increase in density of p-tau immunoreactive (CP13) neurofibrillary tangles and GT-38 immunoreactive profiles at the depths of cortical sulci compared to adjacent gyral crests in cases with CTE-NC and also in cases with ADNC (Figs. 2 and 3B). Notably, the mild sulcal depth concentration of neuronal profiles in CTE-NC was similar to that observed in cases with ADNC, whether assessed in sections stained for CP13 [sulcal depth to gyral crest ratio of neurofibrillary tangles in CTE-NC 1.40 ± 0.74 (mean ± standard deviation); ADNC 1.20 ± 0.10; P = 0.82; Mann–Whitney test] or for GT-38 (1.64 ± 1.05, CTE-NC; 1.29 ± 0.15, ADNC; P = 0.91) (Fig. 4). Furthermore, the sulcal depth to gyral crest concentration of neurofibrillary tangles in CTE-NC was considerably lower than that of thorn-shaped astrocytes in matched sections (P < 0.001, Wilcoxon signed-rank test) (Fig. 4).
Figure 4.

Depth to crest ratio of tau pathologies in CTE-NC and ADNC. Quantitative assessment of thorn-shaped astrocytes and neuronal profiles in each case reveals astrocytes with a higher ratio of sulcal depth to gyral crest density (sulcal depth to gyral crest ratio 12.84 ± 15.47; mean ± SD) than co-existing neurofibrillary tangles (1.40 ± 0.74; P < 0.001). There was mild concentration of both CP13- and GT-38-immunoreactive neurofibrillary tangles in CTE-NC, which was similar to that seen in material from patients with ADNC (all analyses not significant). TSA, thorn-shaped astrocytes; NFT, neurofibrillary tangles; open circles, individual data points; crossed circles, outliers
Association between injury and patient variables and distribution of thorn-shaped astrocytes in CTE
Sub-dividing cases with known CTE-NC by history of injury exposure demonstrated greater sulcal concentration of thorn-shaped astrocytes in cases with a history of survival from non-sport TBI than in cases with history of exposure to repetitive mild TBI through contact sports participation (sulcal depth to gyral crest ratio of thorn-shaped astrocytes 31.21 ± 22.79 versus 7.44 ± 6.92; P = 0.009; Fig. 5A). No difference in concentration of CP13 or GT-38 neurofibrillary tangles was noted between sport and non-sport TBI cases. There was no significant difference in sulcal concentration of thorn-shaped astrocytes in low-stage compared to high-stage CTE-NC (18.90 ± 19.80 versus 6.78 ± 5.48; p = 0.12) (Fig. 5A). Sucal depth to gyral crest ratios of both neurofibrillary tangles and thorn-shaped astrocytes (Fig. 5B) was independent of patient age.
Figure 5.

Association of injury exposure, CTE-NC stage and age at death with thorn-shaped astrocyte distribution. (A) Concentration of thorn-shaped astrocytes to the sulcal depth was greater where there was a history of non-sports TBI (depth to crest ratio, 31.21 ± 22.79) than when exposure to TBI was through participation in contact sports (7.44 ± 6.92: P = 0.009). Although there was a greater concentration of thorn-shaped astrocytes to sulcal depths in cases with low (18.90 ± 19.80) versus high (6.78 ± 5.48) stage CTE-NC, this was not significant (P = 0.12). (B) There was no association between age at death and distribution of thorn-shaped astrocytes (R2 = 0.063, P = 0.26). TSA, thorn-shaped astrocytes; open circles, individual data points; crossed circles, outliers
Discussion
Herein, we present formal, quantitative evaluation of the distribution of neocortical neuronal and astroglial tau pathologies contributing to CTE neuropathologic change. Specifically, our data demonstrate that the typical p-tau immunoreactive, thorn-shaped astrocytes of CTE-NC showed considerable concentration to the depths of cortical sulci compared to adjacent gyral crests. In contrast, p-tau immunoreactive neurofibrillary tangles showed only limited concentration at the depths of cortical sulci in CTE-NC, which was not different from that observed in material from patients with known Alzheimer’s disease neuropathologic changes. Notably, the concentration of p-tau-immunoreactive astroglial pathology to the depths of cortical sulci in CTE-NC was greater in material from patients with a history of survival from non-sport TBI than in cases with a history of exposure to repetitive mild TBI through participation in contact sports. In contrast, we found no clear association between cortical distribution of these pathologies and neuropathological stage of CTE-NC or with patients’ age at death.
Prior studies have assessed the distribution of individual components of p-tau pathology in CTE-NC (Armstrong et al., 2017, 2019). In contrast to our data demonstrating that the astroglial pathologies alone show distinctive concentration to the depths of cortical sulci, previous quantitative analysis suggested both astrocytic and neuronal pathologies concentrated to the sulcal depths (Armstrong et al., 2017). However, unlike this study, no comparison with equivalent cortical p-tau pathologies in wider neurodegenerative disease, including Alzheimer’s disease, was pursued. In this context, we observed sulcal concentration of neurofibrillary tangles in CTE-NC when compared to gyral crests; however, this was mild in comparison to the equivalent assessment of astroglial pathology. Furthermore, the mild concentration of neurofibrillary tangles we observed in cases with CTE-NC was of the same degree as that recorded in our cases with ADNC which, in turn, was consistent with the previous quantitative assessments of cortical distribution of pathologies in ADNC (Gentleman et al., 1992; Clinton et al., 1993; Armstrong, 1994). Preferential distribution of neurofibrillary tangle pathology to sulcal depths, therefore, appears neither a marked nor a specific finding in CTE-NC.
Although finding only limited concentration of neurofibrillary tangle pathology at sulcal depths in CTE-NC, thorn-shaped astrocytes showed marked sulcal concentration. Early descriptions of thorn-shaped astrocytes in ARTAG reported distribution to the depths of cortical sulci in the temporal lobe (Ikeda et al., 1995; Ikeda, 1996). Furthermore, reported patterns of subpial ARTAG across the cerebral convexities share a striking resemblance to CTE-NC and, in part, are hypothesized might result from prior exposure to trauma (Kovacs et al., 2018). Elsewhere, re-evaluation of Corsellis’ original cohort revealed ARTAG pathology in 10 of 14 ex-boxers, including all cases with sulcal CTE-NC (Goldfinger et al., 2018). Sub-pial and perivascular thorn-shaped astrocytes have also been described in chronic survivors of military blast injury (Shively et al., 2016) and spinal cord thorn-shaped astrocytes develop in the setting of chronic compressive forces in cervical spondylosis (Shimizu et al., 2008). Therefore, the possibility exists that, in the context of neurodegeneration, distribution of astroglial tau pathology towards sulcal depths might serve as evidence of prior exposure to TBI (Forrest et al., 2019).
Prior characterization has revealed that the neuronal and astroglial pathologies of CTE-NC contain differing tau isoform compositions and immunophenotypes, echoing tau phenotypes of comparable pathologies in ageing and Alzheimer’s disease (Arena et al., 2020). As such, in addition to shared cellular morphologies and anatomic distributions, thorn-shaped astrocytes of CTE-NC and ARTAG are composed of 4R tau, with similar post-translational modifications (Arena et al., 2020). Furthermore, assessment of the distribution of ARTAG has implicated a possible role for chronic disruption of CSF–brain and blood–brain barriers in its development (Kovacs et al., 2017; Kovacs et al., 2018). In this context, persisting and widespread blood–brain barrier disruption has been shown following both single moderate or severe (Hay et al., 2015) and repetitive mild TBI in humans (Doherty et al., 2016), and as a pathological consequence of mild TBI in experimental models (Johnson et al., 2018). As such, the possibility that the astroglial pathologies of CTE-NC and ARTAG share similar aetiologies might be considered.
Current consensus criteria for the neuropathological assessment and diagnosis of CTE define the pathognomonic lesion based on the apparent presence of both neuronal and astroglial pathologies concentrated at the depths of cortical sulci (McKee et al., 2016). These preliminary criteria were generated following standardized qualitative methodologies for consensus reporting in which 10 cases of known CTE with ‘late’ stage pathology were selected from a single archive and reviewed by a panel of neuropathologists blind to the original diagnosis. These known CTE cases were assessed alongside cases of multiple other tauopathies and, based on subjective assessments, a consensus agreed upon a so-called defining pathology of CTE. Recognized limitations of this process are the small number of presumed CTE cases selected, their selection as displaying already defined stereotypical pathology and the subjective nature of assessment and review. Hence, these criteria remain preliminary. In keeping with experiences in wider neurodegenerative disease, there is a need for continual reappraisal and refinement of consensus criteria (Montine et al., 2012, 2016; McKeith et al., 2017).
Conclusion
Our quantitative assessment considered neocortical p-tau pathologies in 22 cases with varying exposures to TBI and known CTE-NC spanning ‘low’- and ‘high’-stage pathology. Furthermore, we compared these pathologies with those observed in material from patients with clinically diagnosed Alzheimer’s disease and confirmed Alzheimer’s disease neuropathologic changes. Our observations suggest that, contrary to the description of the pathognomonic pathology incorporated into current consensus criteria for the neuropathologic assessment and diagnosis of CTE, the distribution of p-tau immunoreactive astroglial pathology, alone, might represent the specific pathology of CTE-NC. As such, current consensus criteria for the identification of CTE-NC might require review and refinement.
Acknowledgements
The authors thank Dr Peter Davies of the Albert Einstein College of Medicine for generously providing the tau antibody CP13 and Josephine Atkinson of the University of Glasgow for technical assistance with immunohistochemical studies.
Funding
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award numbers U54NS115322, R01NS092398, R01NS094003 and R01NS038104; National Institute on Aging of the National Institutes of Health grants AG010124, AG017586, AG054991, AG09215 and AG17586; National Health Service Research Scotland; Clinical and Translational Science Awards training grant TL1TR001880; National Institutes of Health award F32AG053036 and by The Football Association and the Professional Footballers Association.
Competing interests
The authors report no competing interests.
Glossary
- AD =
Alzheimer’s disease
- ADNC =
Alzheimer’s disease neuropathologic changes
- ARTAG =
ageing-related tau astrogliopathy
- CNDR =
Penn Center for Neurodegenerative Disease Research
- CTE-NC =
Chronic traumatic encephalopathy neuropathologic change
- NFT =
neurofibrillary tangle
- sTBI =
single moderate or severe traumatic brain injury
- TBI=
traumatic brain injury
- TSA =
thorn-shaped astrocytes
References
- Arena JD, Smith DH, Lee EB, Gibbons GS, Irwin DJ, Robinson JL, et al. Tau immunophenotypes in chronic traumatic encephalopathy recapitulate those of ageing and Alzheimer’s disease. Brain 2020; 143: 1572–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RA. Quantitative differences in beta/A4 protein subtypes in the parahippocampal gyrus and frontal cortex in Alzheimer’s disease. Dementia 1994; 5: 1–5. [DOI] [PubMed] [Google Scholar]
- Armstrong RA, McKee AC, Alvarez VE, Cairns NJ.. Clustering of tau-immunoreactive pathology in chronic traumatic encephalopathy. J Neural Transm 2017; 124: 185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RA, McKee AC, Stein TD, Alvarez VE, Cairns NJ.. A quantitative study of tau pathology in 11 cases of chronic traumatic encephalopathy. Neuropathol Appl Neurobiol 2017; 43: 154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RA, McKee AC, Stein TD, Alvarez VE, Cairns NJ.. Cortical degeneration in chronic traumatic encephalopathy and Alzheimer’s disease neuropathologic change. Neurol Sci 2019; 40: 529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton J, Roberts GW, Gentleman SM, Royston MC.. Differential pattern of beta-amyloid protein deposition within cortical sulci and gyri in Alzheimer’s disease. Neuropathol Appl Neurobiol 1993; 19: 277–81. [DOI] [PubMed] [Google Scholar]
- Corsellis JA, Bruton CJ, Freeman-Browne D.. The aftermath of boxing. Psychol Med 1973; 3: 270–303. [DOI] [PubMed] [Google Scholar]
- Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128: 755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley M, Punch-drunk syndromes: the chronic traumatic encephalopathy of boxers. Hommage a` Clovis Vincent Paris: Maloine; 1949. [Google Scholar]
- Critchley M. Medical aspects of boxing, particularly from a neurological standpoint. Br Med J 1957; 1: 357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty CP, O’Keefe E, Wallace E, Loftus T, Keaney J, Kealy J, et al. Blood-brain barrier dysfunction as a hallmark pathology in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 2016; 75: 656–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest SL, Kril JJ, Wagner S, Honigschnabl S, Reiner A, Fischer P, et al. Chronic traumatic encephalopathy (CTE) is absent from a European community-based aging cohort while cortical aging-related tau astrogliopathy (ARTAG) is highly prevalent. J Neuropathol Exp Neurol 2019; 78: 398–405. [DOI] [PubMed] [Google Scholar]
- Geddes JF, Vowles GH, Nicoll JA, Revesz T.. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 1999; 98: 171–8. [DOI] [PubMed] [Google Scholar]
- Geddes JF, Vowles GH, Robinson SF, Sutcliffe JC.. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol 1996; 22: 12–6. [PubMed] [Google Scholar]
- Gentleman SM, Allsop D, Bruton CJ, Jagoe R, Polak JM, Roberts GW.. Quantitative differences in the deposition of beta A4 protein in the sulci and gyri of frontal and temporal isocortex in Alzheimer’s disease. Neurosci Lett 1992; 136: 27–30. [DOI] [PubMed] [Google Scholar]
- Gibbons GS, Banks RA, Kim B, Changolkar L, Riddle DM, Leight SN, et al. Detection of Alzheimer disease (AD)-specific tau pathology in AD and nonAD tauopathies by immunohistochemistry with novel conformation-selective tau antibodies. J Neuropathol Exp Neurol 2018; 77: 216–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons GS, Kim S-J, Robinson JL, Changolkar L, Irwin DJ, Shaw LM, et al. Detection of Alzheimer’s disease (AD) specific tau pathology with conformation-selective anti-tau monoclonal antibody in co-morbid frontotemporal lobar degeneration-tau (FTLD-tau). Acta Neuropathol Comm 2019; 7: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 2012; 4: 134ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfinger MH, Ling H, Tilley BS, Liu AKL, Davey K, Holton JL, et al. The aftermath of boxing revisited: identifying chronic traumatic encephalopathy pathology in the original Corsellis boxer series. Acta Neuropathol 2018; 136: 973–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DI, Gentleman SM, Lynch A, Roberts GW.. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol 1995; 21: 27–34. [DOI] [PubMed] [Google Scholar]
- Hay JR, Johnson VE, Young AM, Smith DH, Stewart W.. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol 2015; 74: 1147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Bouras C, Bu�e L, Delacourte A, Perl DP, Morrison JH.. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol 1992; 85: 23–30. [DOI] [PubMed] [Google Scholar]
- Holleran L, Kim JH, Gangolli M, Stein T, Alvarez V, McKee A, et al. Axonal disruption in white matter underlying cortical sulcus tau pathology in chronic traumatic encephalopathy. Acta Neuropathol 2017; 133: 367–80. [DOI] [PubMed] [Google Scholar]
- Ikeda K. Glial fibrillary tangles and argyrophilic threads: classification and disease specificity. Neuropathology 1996; 16: 71–7. [Google Scholar]
- Ikeda K, Akiyama H, Arai T, Nishimura T.. Glial tau pathology in neurodegenerative diseases: their nature and comparison with neuronal tangles. Neurobiol Aging 1998; 19: S85–91. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Akiyama H, Kondo H, Haga C, Tanno E, Tokuda T, et al. Thorn-shaped astrocytes: possibly secondarily induced tau-positive glial fibrillary tangles. Acta Neuropathol 1995; 90: 620–5. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, et al. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J Neurosci 1999; 19: 7486–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Arena JD, Smith DH.. Traumatic brain injury as a trigger of neurodegeneration. Adv Neurobiol 2017; 15: 383–400. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH.. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012; 22: 142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Weber MT, Xiao R, Cullen DK, Meaney DF, Stewart W, et al. Mechanical disruption of the blood-brain barrier following experimental concussion. Acta Neuropathol 2018; 135: 711–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo TK, Li MY.. A guideline of selecting and reporting intraclass correlation coefficients for reliability research. J Chiropr Med 2016; 15: 155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 2015; 41: 3–23. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 2016; 131: 87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Lee VM, Trojanowski JQ.. Protein astrogliopathies in human neurodegenerative diseases and aging. Brain Pathol 2017; 27: 675–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Robinson JL, Xie SX, Lee EB, Grossman M, Wolk DA, et al. Evaluating the patterns of aging-related tau astrogliopathy unravels novel insights into brain aging and neurodegenerative diseases. J Neuropathol Exp Neurol 2017; 76: 270–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Xie SX, Robinson JL, Lee EB, Smith DH, Schuck T, et al. Sequential stages and distribution patterns of aging-related tau astrogliopathy (ARTAG) in the human brain. Acta Neuropathol Comm 2018; 6: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Yousef A, Kaindl S, Lee VM, Trojanowski JQ.. Connexin-43 and aquaporin-4 are markers of ageing-related tau astrogliopathy (ARTAG)-related astroglial response. Neuropathol Appl Neurobiol 2018; 44: 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB. Integrated neurodegenerative disease autopsy diagnosis. Acta Neuropathol 2018; 135: 643–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Kinch K, Johnson VE, Trojanowski JQ, Smith DH, Stewart W.. Chronic traumatic encephalopathy is a common co-morbidity, but less frequent primary dementia in former soccer and rugby players. Acta Neuropathol 2019; 138: 389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Morris HR, Neal JW, Lees AJ, Hardy J, Holton JL, et al. Mixed pathologies including chronic traumatic encephalopathy account for dementia in retired association football (soccer) players. Acta Neuropathol 2017; 133: 337–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AK, Goldfinger MH, Questari HE, Pearce RK, Gentleman SM.. ARTAG in the basal forebrain: widening the constellation of astrocytic tau pathology. Acta Neuropathol Comm 2016; 4: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. ; the TBI/CTE Group. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016; 131: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 2009; 68: 709–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Stein TD, Nowinski CJ, Stern RA, Daneshvar DH, Alvarez VE, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013; 136: 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology 2017; 89: 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 2017; 318: 360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millspaugh JA. Dementia pugilistica. United States Naval Medicine. Bulletin 1937; 35: 297–303. [Google Scholar]
- Montine TJ, Monsell SE, Beach TG, Bigio EH, Bu Y, Cairns NJ, et al. Multisite assessment of NIA-AA guidelines for the neuropathologic evaluation of Alzheimer’s disease. Alzheimers Dement 2016; 12: 164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH.. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005; 57: 128–34. [DOI] [PubMed] [Google Scholar]
- Robinson JL, Corrada MM, Kovacs GG, Dominique M, Caswell C, Xie SX, et al. Non-Alzheimer’s contributions to dementia and cognitive resilience in The 90+ Study. Acta Neuropathol 2018; 136: 377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Kakita A, Takahashi H.. Spinal cord tau pathology in cervical spondylotic myelopathy. Acta Neuropathol 2008; 115: 185–92. [DOI] [PubMed] [Google Scholar]
- Shively SB, Horkayne-Szakaly I, Jones RV, Kelly JP, Armstrong RC, Perl DP.. Characterisation of interface astroglial scarring in the human brain after blast exposure: a post-mortem case series. Lancet Neurol 2016; 15: 944–53. [DOI] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Stewart W.. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 2013; 9: 211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Trojanowski JQ, Stewart W.. Chronic traumatic encephalopathy—confusion and controversies. Nat Rev Neurol 2019; 15: 179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Alvarez VE, McKee AC.. Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther 2014; 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart W, McNamara PH, Lawlor B, Hutchinson S, Farrell M.. Chronic traumatic encephalopathy: a potential late and under recognized consequence of rugby union? QJM 2016; 109: 11–5. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL, et al. A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 2014; 10: 477–84.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribett T, Erskine B, Bailey K, Brown T, Castellani RJ.. Chronic traumatic encephalopathy pathology after shotgun injury to the brain. J Forensic Sci 2019; 64: 1248–52. [DOI] [PubMed] [Google Scholar]
- Zanier ER, Bertani I, Sammali E, Pischiutta F, Chiaravalloti MA, Vegliante G, et al. Induction of a transmissible tau pathology by traumatic brain injury. Brain 2018; 141: 2685–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article.