

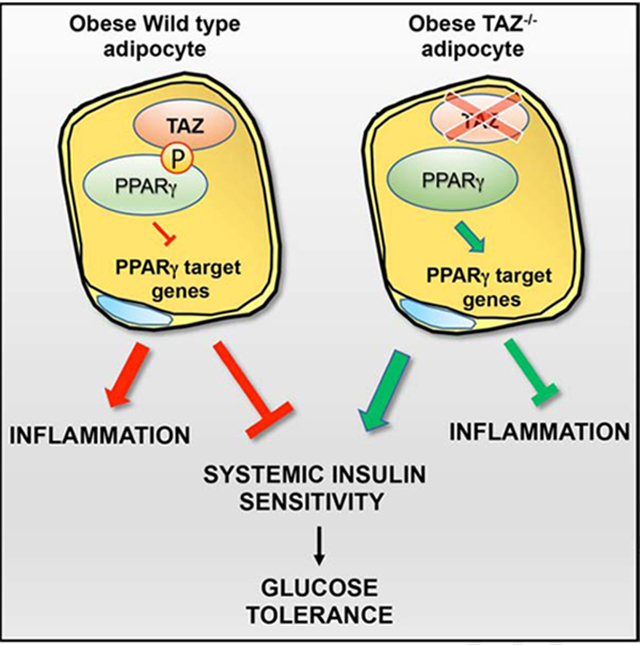
Summary
Insulin resistance is a major factor in obesity-linked type 2 diabetes. PPARγ is a master regulator of adipogenesis and small molecule agonists, termed thiazolidinediones, are potent therapeutic insulin sensitizers. Here, we studied the role of TAZ (Transcriptional Co-Activator with PDZ-binding motif) as a transcriptional co-repressor of PPARγ. We found that adipocyte-specific TAZ knockout (TAZ AKO) mice demonstrate a constitutively active PPARγ state. Obese TAZ AKO show improved glucose tolerance and insulin sensitivity compared to littermate controls. PPARγ response genes are up-regulated in adipose tissue from TAZ AKO mice and adipose tissue inflammation was also decreased. In vitro and in vivo mechanistic studies revealed that the TAZ-PPARγ interaction is partially dependent on ERK-mediated Ser112 PPARγ phosphorylation. As adipocyte PPARγ Ser112 phosphorylation is increased in obesity, repression of PPARγ activity by TAZ could contribute to insulin resistance. These results identify TAZ as a new factor in the development of obesity-induced insulin resistance.
eTOC blurb
El Ouarrat & Isaac et. al show that TAZ, a member of the Hippo pathway, functions as a PPARγ co-repressor in adipocytes. Adipocyte-specific TAZ KO increases PPARγ activity, reduces adipose inflammation, improves insulin sensitivity and glucose tolerance in obese mice.
Graphical Abstract
Introduction
Obesity is a major global public health problem and underlies several human diseases connected to insulin resistance such as Type 2 Diabetes Mellitus (T2DM), Polycystic Ovary Syndrome (PCOS) and metabolic syndrome. Adipose tissue is a critical regulator of systemic energy homeostasis by acting both as an energy reservoir in the form of triglyceride storage and as an endocrine organ. Adipose tissue dysfunction is a hallmark of obesity. For example, failure to properly store excess energy can lead to ectopic lipid deposition in other metabolically sensitive organs, which can promote insulin resistance. In addition, adipose tissue inflammation in obesity can lead to systemic insulin resistance and various factors released by adipose tissue, such as adipokines, Galectin-3, exosomes etc., have been implicated in this process (Rosen and Spiegelman, 2006).
The PPARγ nuclear receptor is a master transcriptional regulator of adipogenesis and modulates the role of adipose tissue in whole-body glucose metabolism. PPARγ is the receptor for the thiazolidinedione class of insulin sensitizing anti-diabetic drugs (Evans et al., 2004; Lehrke and Lazar, 2005; Rangwala and Lazar, 2004; Tontonoz and Spiegelman, 2008). Transcriptional control of PPARγ depends on multiprotein complexes containing coregulators, which have various effects on metabolic dysfunction and insulin resistance (Glass and Rosenfeld, 2000; Koppen and Kalkhoven, 2010; Powell et al., 2007). Several co-activators such as PGC1α and CBP, and co-repressors like SMRT and NCoR, have important roles in regulating PPARγ transcriptional activity (Kleiner et al., 2012; Li et al., 2011; Sutanto et al., 2010). In addition to ligands and co-regulators, the transcriptional activity of PPARγ is also modulated by various posttranslational modifications, such as phosphorylation at serine 112 and 273. (Ahmadian et al., 2013; Choi et al., 2010; Hu et al., 1996; van Beekum et al., 2009). Thus, PPARγ S112 and S273 can have significant effects on target gene expression and can affect whole-body insulin sensitivity (Banks et al., 2015; Burns and Vanden Heuvel, 2007; Rangwala and Lazar, 2004).
In recent years, the Hippo pathway has emerged as an important signaling network coordinating cell-proliferation and differentiation to regulate tissue growth (Wang et al., 2017). Similarly, several studies have demonstrated the importance of Hippo signaling in adipocyte proliferation and differentiation (An et al., 2013; Kamura et al., 2018). Originally identified in Drosophila melanogaster, Transcriptional Co-Activator with PDZ-binding motif (TAZ) and Yes-associated protein (YAP) are the major effectors of this pathway, regulating genes important for cell proliferation and survival (Huang et al., 2005). TAZ, also called WWTR1, contains a WW domain and functions by binding to PPXY motifs on transcription factors (Kanai et al., 2000; Salah and Aqeilan, 2011; Sudol, 2010). As a transcriptional co-regulator, TAZ interacts with and regulates multiple transcription factors, such as TEAD, TBX5, RUNX2 and PPARγ (Hong et al., 2005; Murakami et al., 2005; Zhang et al., 2009). A direct interaction between TAZ and the AF-1 domain of PPARγ has been shown, which requires the TAZ WW domain. TAZ can promote osteoblast differentiation by enhancing RUNX2-dependent transcriptional activation, and can repress PPARγ dependent transcription, thereby inhibiting adipocyte differentiation (Hong et al., 2005). Studies of knockout mice and of clinical samples have demonstrated the significance of TAZ in human-associated diseases such as cancer, kidney and liver disease (Anakk et al., 2013; Higashi et al., 2015; Varelas et al., 2010). However, the possible role of TAZ in adipocyte dysfunction, glucose tolerance and insulin resistance in vivo remains unexplored.
To assess the role of TAZ in glucose metabolism and insulin sensitivity, we generated adipocyte-specific TAZ knockout (TAZ AKO) mice. We demonstrate that TAZ AKO mice exhibit improved glucose tolerance, enhanced insulin sensitivity and decreased adipose tissue inflammation consistent with a derepressed actively PPARγ phenotype. Consistent with a derepressed, activated, PPARγ phenotype, these studies reveal a new role of TAZ in the development of obesity-linked insulin resistance.
Results
Generation of adipocyte-specific TAZ knockout mice.
To assess the potential role of Hippo pathway cofactors in metabolic disease, we first examined gene expression patterns in metabolism-related cell types from WT mice by performing RNA-seq experiments. Quantitative mRNA levels of TAZ were much higher in adipocytes compared to adipose stromal vascular cells (SVC), hepatocytes, non-parenchymal liver cells and muscle. Notably, in adipocytes the expression levels of the TAZ paralog, YAP, was similar compared to hepatocytes, and remained approximately 2-fold lower relative to TAZ expression in adipocytes (Figure 1A). A comparable trend for the YAP/TAZ protein expression ratio in adipocytes was also observed (Figure S1A), and TAZ protein levels are the same in adipose tissue from normal chow diet (NCD) vs. high-fat diet (HFD) (Figure S1B). To investigate the specific role of TAZ in adipogenesis and insulin sensitivity, we generated adipocyte-specific knockout mice using the adiponectin Cre-lox system. We bred mice carrying the floxed Wwtr1 allele, containing two loxP sites flanking exon 2, with transgenic mice expressing Cre recombinase driven by the Adiponectin-promoter in order to generate TAZ f/f;Adiponectin-Cre+/− mice (TAZ AKO). Please note, to conform to the literature, from here on we use the common name TAZ rather than Wwtr1. Mice with the floxed TAZ alleles, that do not express Cre recombinase (TAZ f/f; Adiponectin-Cre−/−), were used as controls, referred to hereafter as TAZ f/f mice (Figures 1B and 1C). As expected, quantitative PCR (qPCR) analysis revealed an ~80% reduction of TAZ expression in epididymal white adipose tissue (eWAT) and subcutaneous white adipose tissue (sqWAT) in TAZ AKO compared to TAZ f/f mice (Figure 1D). mRNA levels of TAZ in brown adipose tissue (BAT) were also substantially diminished, while there was no change in mRNA levels in other tissues, verifying the specificity of the adipocyte knockout mice. Furthermore, we confirmed by Western blot analysis that TAZ protein levels were greatly reduced in eWAT and sqWAT of TAZ AKO mice compared to TAZ f/f (Figure 1E) but not in other tissues (Figure S1C). Since adipose tissue contains many non-adipocyte cell types, it is likely that much of the residual TAZ expression detected in total TAZ AKO adipose tissue comes from cells other than adipocytes.
Figure 1. Creation of TAZ adipocyte-specific knockout mice.

(a) Expression profile of TAZ and YAP in RNAseq experiments. (b) Schematic representation of wildtype TAZ, conditional floxed and deleted TAZ gene loci. Primers used to distinguish WT and floxed alleles are indicated. (c) Genotyping results of wildtype (+/+), (F/+) and (F/F) mice. (d) Relative TAZ mRNA levels in various tissues. (e) Immunoblot of TAZ in Epididymal white adipose tissue (eWAT) and subcutaneous white adipose tissue (sqWAT), with HSP90 as loading control. (f) Body weight of TAZ adipocyte knock out (TAZ ako) and TAZ f/f mice on standard normal chow diet (NCD) for 12 weeks. (g) Weight of eWAT at 12 weeks of NCD. (h) Food intake of TAZ f/f and ako at 5 weeks of NCD. (i) Body weight of TAZ f/f and ako mice on high fat diet (HFD) for 12 weeks. (j) Weight of eWAT at 12 weeks of HFD. (k) Food intake of TAZ f/f and ako mice at 5 weeks of HFD. Values are fold induction of gene expression normalized to the housekeeping gene Gapdh and expressed as mean ± SEM, n=8–10 mice/group in (e-k) *p<0.05 for TAZ AKO vs TAZ f/f. See also figure S1.
To investigate the functional significance of adipocyte-specific TAZ deletion, both TAZ AKO and TAZ f/f mice were fed normal chow diet (NCD) or 60% high fat diet (HFD) for 12 weeks, starting at 8 weeks of age. Both TAZ AKO and TAZ f/f mice showed a similar age-dependent increase in body weight on either diet (Figures 1F and I). Moreover, fat mass weight was the same in NCD f/f vs TAZ AKO mice as was the HFD-induced increase in adipose tissue mass (eWAT, sqWAT and BAT) (Figures 1G, J and S1D-I). No significant differences were found in food intake in TAZ f/f vs. TAZ AKO mice on HFD (Figures 1H and K).
Adipocyte-specific TAZ deletion protects against HFD-induced systemic insulin resistance.
We assessed glucose homeostasis in NCD and HFD-fed TAZ f/f and TAZ AKO mice. No changes in fasting insulin, glucose or insulin tolerance were observed in the lean NCD-fed mice between genotypes (Figures 2B-C, S2A-B). On the other hand, on HFD, basal glucose and insulin levels were reduced in TAZ AKO mice (Figures 2A,D). Furthermore, the HFD TAZ AKO mice showed improved glucose tolerance and an enhanced hypoglycemic response to injected insulin compared to TAZ f/f littermates (Figures 2B-C). These improved values were now comparable to the results in chow-fed mice. To gain a better understanding of the role of TAZ in the pathophysiology of insulin resistance, hyperinsulinemic-euglycemic clamp studies were performed. As seen in Figures 2E-G, the glucose infusion rate (GIR) required to maintain euglycemia, and the ability of insulin to suppress hepatic glucose production (HGP), were both significantly higher in TAZ AKO mice compared to f/f controls. There was no impact on the basal or insulin-stimulated glucose disposal rate (GDR and IS-GDR), indicating that muscle insulin sensitivity is not altered between the two groups (Figures 2H,I). Finally, the ability of insulin to suppress plasma free fatty acid (FFA) levels provides a good indication of adipose tissue insulin sensitivity, and FFA suppression was nearly two-fold higher in TAZ AKO vs TAZ f/f mice (Figures 2J). This increased suppressibility of FFAs in the TAZ AKO mice could also contribute to the enhanced insulin effects to inhibit HGP.
Figure 2. Improved glucose metabolism and insulin sensitivity in TAZ AKO mice.

(a) Fasting blood glucose levels. (b) Glucose tolerance tests (GTT) in NCD and after 12 weeks HFD feeding. (c) Insulin tolerance test (ITT) in NCD and after 12 weeks HFD feeding (8–10 mice per group). (d) Fasting blood insulin levels. (e) Glucose infusion rate (GIR) during hyperinsulinmic euglycemic clamp. (f) Basal hepatic glucose production (HGP) rate. (g) Percent suppression of HGP by insulin. (h) Glucose disposal rate (GDR). (i) Insulin-stimulated glucose disposal rate (IS-GDR). (j) Percent suppression of free fatty-acid levels (FFA). (k) Western blot analysis of insulin-stimulated phospho-Akt Ser 473 (pSer473AKT) in adipose tissue. (l) Insulin stimulated pSer473 AKT in liver. (m) Insulin-stimulated pSer473AKT in muscle. Values are expressed as mean ± SEM, n=8–10 mice/group in (a-d) and n=6–7 mice/group *p< 0.05, p< 0,01 for TAZ AKO vs TAZ f/f. See also figure S2 and S3.
To measure ex vivo insulin action we harvested adipose, liver and muscle tissue. Consistent with the glucose clamp studies, insulin-stimulated Akt phosphorylation was significantly higher in liver and adipose, but not in muscle tissue of TAZ AKO mice when compared to f/f mice (Figures 2K-M).
Liver mass of TAZ AKO was the same as in f/f mice (Figures S3B). However, when examined by H&E staining, the livers of the obese TAZ AKO mice were markedly less steatotic than controls (Figure S3A). Triglyceride (TAG) measurements revealed significantly lower levels of TAG in liver from TAZ AKO mice, consistent with the lower FFA levels, reduced expression of lipogenic genes and higher expression of FA β-oxidation genes (Figures S3C-E). In addition, mRNA levels of inflammatory genes in liver from TAZ AKO were significantly lower compared to liver from f/f mice (Figure S3F). Taken together, these results show that deletion of TAZ in adipocytes leads to a marked improvement in systemic insulin resistance due to enhanced adipose and liver insulin sensitivity.
Improved adipocyte function in TAZ AKO mice.
In obesity, adipose tissue enlarges in order to store excess energy, and this is mainly driven by adipocyte hypertrophy. We studied the adipocyte architecture of HFD obese TAZ AKO and f/f mice. Interestingly, adipocytes from TAZ AKO mice were smaller with a lower average cell size (Figures 3A-C). On the other hand, an increase in adipocyte number was observed in HFD AKO compared to WT mice (Figure 3B). To explain this phenotype, we measured adipocyte differentiation rate in isolated stromal vascular cells (SVCs) from TAZ AKO and f/f mice. As shown in Figure S4A-C, SVCs from TAZ AKO mice differentiated into adipocytes faster than SVCs from f/f mice, as evident by Oil-red O staining and the expression of both adipogenic and lipogenic genes. Importantly, the expression of PPARγ genes, including FABP4, PEPCK, LIPE and Adiponectin were significantly higher in TAZ AKO compared to f/f cells. In addition, caspase activity as well as TUNEL staining was reduced by ~50% in eWAT from TAZ AKO compared to f/f mice (Figure 3D-E). Taken together, these results suggest that TAZ deletion can protect against the adipocyte hypertrophy and apoptosis seen in obese TAZ f/f eWAT and accelerate adipocyte differentiation. Since adipocyte hypertrophy affects circulating levels of adiponectin, we measured blood levels of adiponectin, as well as other adipokines, in both genotypes. As shown in Figure 3F, TAZ AKO mice exhibited higher adiponectin levels and lower leptin levels, favoring increased insulin sensitivity.
Figure 3. Increased adipogenesis and improved adipocyte function in the absence of TAZ.

(a) Hematoxylin & Eosin (H&E) staining of eWAT; scale bar =100 μM. (b) Quantification of adipocyte number per image and area (μM). (c) Histogram showing distribution of adipocyte frequency, diameter and cell mean from TAZ AKO and TAZ f/f mice. Values are expressed in ± SEM (n=5 mice/group and 3 images/mouse were analyzed). (d) Caspase activity 3/7 of eWAT lysate; n=6 mice. (e) TUNEL staining of eWAT; n=5 mice; scale bar =50 μM (f) Plasma circulating adipokine levels. (g) Plasma free fatty-acid (FFA) levels. (h) Basal and insulin-stimulated 2-DOG glucose uptake in primary adipocytes. Values are expressed as mean ± SEM, n=7–9 mice/group in (f-h) *p< 0.05 for TAZ AKO vs TAZ f/f. See also figure S4.
Insulin promotes fatty acid uptake and storage in the form of triglycerides (TG) and inhibits adipocyte TG lipolysis. In TAZ AKO mice, while basal levels of phospho-perilipin and hormone-sensitive lipase (HSL) were the same as in f/f mice (Figure S4D), the circulating levels of free fatty acids (FFA) were lower, suggesting a lower rate of lipolysis in eWAT (Figure 3G). To extend these findings, insulin stimulated glucose uptake in primary adipocytes isolated from TAZ AKO and f/f mice was measured. As seen in Figure 3H, the ability of insulin to enhance glucose transport was enhanced in the TAZ AKO versus f/f mice. This is consistent with the results in Figure 2K, which demonstrate higher adipocyte insulin sensitivity in TAZ AKO adipose tissue compared to control.
Decreased macrophage infiltration and inflammation in eWAT of TAZ AKO mice.
Adipose tissue macrophages (ATMs) accumulate in large quantities in obese eWAT, leading to the development of chronic tissue inflammation and contributing to decreased insulin sensitivity. Accordingly, flow cytometry analysis from isolated SVCs showed less accumulation of F4/80 and CD11b double-positive macrophages in TAZ AKO adipose tissue compared to f/f mice (Figure 4A). This was consistent with lower mRNA expression of F4/80, and a lower number of crown-like structures as measured by F4/80 immunohistochemical staining in eWAT from TAZ AKO compared to TAZ f/f mice (Figures 4B,C).
Figure 4. Decreased adipose tissue inflammation in TAZ AKO mice.

(a) FACS analysis of F4/80+/cd11b+ cells in stromal vascular fraction (SVF). (b) Relative mRNA levels of macrophage marker F4/80 in eWAT. (c) F4/80 immuno-staining in eWAT; scale bar = 100 μM. (d) FACS analysis of F4/80+/cd11b+/cd11c+ cells in SVF. (e) Relative mRNA levels of pro inflammatory M1-like marker CD11c in eWAT. (f) Relative mRNA levels of pro inflammatory cytokines in eWAT. (g) Plasma circulating levels of inflammatory cytokines. (h) Effect of conditioned medium (CM) from TAZ AKO and TAZ f/f primary adipocytes on macrophage chemotaxis. Values are expressed as mean ± SEM, n=5–6 mice/group in (a-b,de) and n=8 mice/group in (f-h) *p< 0,05, **p< 0,01 for TAZ AKO vs TAZ f/f.
Adipose tissue contains different subpopulations of ATMs, including M1-like macrophages (F4/80+,CD11b+,CD11c+) that are highly pro-inflammatory. We found a reduction in these triple-positive infiltrating macrophages, as well as reduced CD11c mRNA levels, in TAZ AKO eWAT compared to WT littermates (Figures 4D,E). Furthermore, there was reduced expression of pro-inflammatory genes in eWAT from TAZ AKO mice as measured by qPCR analysis (Figure 4F). On a systemic basis, HFD TAZ AKO mice also exhibited lower circulating cytokine levels (IL-1β, MCP-1 and TNFα) compared to HFD TAZ f/f mice (Figure 4G).
To assess the mechanisms underlying these differences in ATM content, we measured the ability of conditioned medium (CM) collected from TAZ AKO vs f/f primary adipocytes to induce macrophage chemotaxis. In vitro macrophage chemotaxis towards CM from TAZ AKO adipocytes was markedly reduced compared to TAZ f/f CM (Figure 4H). Taken together, these results suggest that adipocyte specific TAZ deletion leads to decreased chemokine secretion from adipocytes, causing reduced ATM content, decreased inflammation, contributing to improved insulin sensitivity.
Effects of TAZ AKO on PPARγ expression and activity.
Given the central role of PPARγ in adipogenesis, we examined whether TAZ affects PPARγ expression and activity. To address this question, we used siRNA mediated depletion of TAZ in differentiated 3T3-L1 adipocytes. TAZ knockdown induced an increase in mRNA levels of PPARγ (Figure 5A). In addition, silencing the TAZ gene led to increased mRNA levels of PPARγ target genes (Figure 5A). Notably, TAZ did not affect binding of PPARγ to the PEPCK promoter as seen in PPARγ ChIP experiments in control and TAZ siRNA knockdown 3T3-L1 adipocytes (Figure S5A). PPARγ levels were also several fold higher in adipose tissue from obese TAZ AKO mice, inducing several adipogenic PPARγ response genes compared to WT (Figure 5B). These effects were observed both in eWAT and scWAT (S5B). As expected, there was higher PPARγ protein levels in adipose tissue from HFD-fed TAZ AKO mice compared to HFD TAZ f/f mice (Figure 5C). Interestingly, along with the increase in total PPARγ expression, a marked reduction in phosphorylation of the inhibitory Ser sites, S273 and S112 of PPARγ was observed in these mice (Figure 5C,D).
Figure 5. Effects of TAZ AKO on PPARγ expression and activity.

(a) mRNA levels of PPARγ target genes in siRNA TAZ and non-targeting siRNA transfected differentiated 3T3-L1 adipocytes. (b) Adipogenic gene and PPARγ target genes expression levels in eWAT. (c) Phospho-PPARγ Ser273 (p273PPARγ) and PPARγ levels in eWAT. (d) Phospho-PPARγ Ser112 (p112PPARγ) and PPARγ levels in eWAT. (e) GTT of TAZ f/f and TAZ ako mice on HFD with or without rosiglitazone treatment for 3 weeks (* TAZ f/f HFD vs Rosi) and (# TAZ AKO HFD vs TAZ f/f HFD). (f) p273PPARγ and PPARγ levels in eWAT with or without rosiglitazone treatment. (g) mRNA levels of PEPCK, ACC and RGS2, which are responsive to PPARγ agonist rosiglitazone in eWAT. (h,i) Transcriptional repression effect of TAZ and NCoR in the basal (h) and with rosiglitazone treatment (i) on PPARγ-driven gene transcription in dual-luciferase reporter assay in U2OS cells. Values are expressed as mean ± SEM, n=5–6 mice/group in (e) # p< 0.05, for TAZ f/f vs ako. n=6–9 mice/group in (a-b, g) *p< 0.05, **p< 0.01. n=3 **p< 0.01 in (h). See also figure S5.
Insulin sensitizing thiazolidinediones (TZDs) inhibit PPARγ S273 phosphorylation, which confers an active PPARγ transcriptional program with improved whole-body insulin sensitivity. To further assess the impact of reduced S273 phosphorylation, TAZ AKO and f/f mice fed HFD for 12 weeks were treated with rosiglitazone for an additional 3 weeks. As expected, HFD TAZ f/f mice displayed improved glucose tolerance after rosiglitazone treatment compared to untreated TAZ f/f HFD mice (Figure 5E). However, HFD TAZ AKO mice showed a greater improvement in glucose tolerance and rosiglitazone treatment in this group had no additional effect. (Figure 5E). Rosiglitazone treatment led to the expected reduction of p-S273 PPARγ levels in TAZ f/f but did not further reduce PPARγ S273 phosphorylation in TAZ AKO eWAT (Figure 5F). TAZ KO leads to a substantial increase in PPARγ mRNA and protein expression both in vitro and in vivo. It is very likely that this contributes to the increased PPARγ signaling and the overall insulin sensitive, glucose tolerant phenotype. This finding can be explained by the fact that deletion of TAZ derepresses PPARγ, placing it in a relatively constitutively active transcriptional state, and since PPARγ itself is a PPARγ target gene, this would lead to the increase in PPARγ expression which we have observed.
To assess the transcriptional basis for the resistance to PPARγ agonism in TAZ AKO mice, qPCR analysis of PPARγ responsive genes was performed. As shown in Figures 5G and S5C, these genes were upregulated in TAZ AKO mice compared to f/f littermates. Rosiglitazone treatment enhanced gene expression in TAZ f/f mice but did not further increase expression in TAZ AKO mice. Similarly, RGS2 a gene known to be downregulated by rosiglitazone showed lower expression in TAZ AKO mice and was not further down-regulated by rosiglitazone treatment (Figure 5G). We also show that treatment with rosiglitazone did not affect protein levels of TAZ in adipose tissue from f/f HFD mice (Figure S5D).
NCoR is a well-known PPARγ co-repressor, and adipocyte-specific knockout of NCoR leads to constitutively active PPARγ stimulation with increased insulin sensitivity (Li et al., 2011). As TAZ is also a PPARγ co-repressor, we investigated the functional interaction between these two co-repressors in relation to PPARγ. As shown in Figure 5H, NCoR inhibits basal PPARγ-driven transcriptional activity. However, while TAZ led to ~50% decrease in rosi-stimulated PPARγ-mediated transcription, the stimulatory effects of rosiglitazone on PPARγ transcription were not repressed by NCoR (5I). These results suggest that TAZ and NCoR inhibit PPARγ activity by different pathways and stimulation of PPARγ activity by rosiglitazone is affected differentially by these two co-repressors.
S112 phosphorylation contributes to inhibition of PPARγ activity by TAZ protein.
To investigate the functional interaction between endogenous TAZ and PPARγ, we performed co-immunoprecipitation (Co-IP) experiments in adipose tissue lysates. As shown in Figure 6A, PPARγ protein co-precipitated with TAZ. Furthermore, in “reverse” experiments, TAZ protein was co-precipitated by PPARγ antibody in f/f, but not TAZ AKO mice (Figure S6A). We next explored potential mechanisms underlying TAZ and PPARγ interactions. TAZ contains a WW domain that can bind to Pro-Pro-X-Tyr (PPXY) motifs and PPARγ contains a PPXY motif within its AF1 activation domain. Interestingly, PPARγ serine 112 (S112) is an ERK-dependent inhibitory phosphorylation site (Adams et al., 1997), which is located adjacent to this motif (Figure 6B). We also find increased levels of S112 phosphorylation in adipose tissue samples from HFD obese compared to chow fed mice (Figure S6B). Indeed, upon treatment of differentiated 3T3-L1 adipocytes with the MEK inhibitor PD98059, both p-Erk1/2 and p-S112PPARγ levels were reduced. We also observed that the TAZ-PPARγ interaction was decreased upon MEK inhibitor treatment (Figure 6C).
Figure 6. TAZ interaction with PPARγ is regulated by Serine112 phosphorylation.

(a) Co-immunoprecipitation of TAZ with PPARγ in eWAT. (b) Schematic representation of PPARγ depicting several protein domains, including PPYY motif and neighboring serine S112 phosphorylation site. (c) Inhibition of PPARγ S112 phosphorylation by MEK inhibitor PD98059 treatment decreases protein interaction with TAZ in 3T3-L1 adipocytes. (d) S112A phosphorylation of PPARγ by constitutively active (CA) ERK potentiates TAZ and PPARγ interaction in HEK293T cells. (e,f) Co-immunoprecipitation of TAZ and PPARγ WT, mutant S112D (e) or S112A (f) in HEK293T cells. (g) Effect of rosiglitazone treatment on interaction TAZ with PPARγ in differentiated 3T3-L1 adipocytes. (h) Transcriptional repression effect of TAZ on WT and S112A mutant PPARγ-driven gene transcription in dual-luciferase reporter assay in U2OS cells. Values are expressed as mean ± SEM. (e-g) n=3–4 *p< 0.05, **p< 0.01. See also figure S6.
Therefore, we hypothesized that the TAZ-PPARγ interaction could be regulated by S112 phosphorylation. To address this idea, we examined the interaction between TAZ and PPARγ, with and without constitutively active (CA) ERK in Co-IP experiments. Some TAZ and PPARγ co-immunoprecipitated under basal conditions, even though PPARγ S112 is unphosphorylated.
Interestingly, CA-ERK-induced S112 phosphorylation of PPARγ increased the association with TAZ (Figure 6D). The same effect was observed when the phospho-mimic S112 mutant of PPARγ (S112 to aspartate - S112D) was expressed in 293 HEK cells. Thus, as seen in Figure 6E, the association of PPARγ S112D with TAZ protein was increased by ~2-fold compared to WT PPARγ. Conversely, mutation of PPARγ S112 to Ala112 led to decreased TAZ-PPARγ association by 40% when compared to WT PPARγ (Figure 6F, S5C). To assess any potential effects of PPARγ Ser 273 de-phosphorylation on the TAZ-PPARγ interaction, we performed Co-IP experiments. As shown in Figure 6G, p-S273 PPARγ levels were reduced upon rosiglitazone treatment, but no difference was observed in the association between TAZ and PPARγ. This indicates that, unlike Ser 112, PPARγ S273 is not directly involved in TAZ-PPARγ binding. Taken together, these results show that PPARγ S112, but not S273 phosphorylation, contributes to the TAZ-PPARγ interaction.
To further assess the role of S112A on the TAZ/PPARγ corepressor effect, we performed transcriptional assays. Luciferase reporter assays show that the activity of WT PPARγ and the mutant S112A are the same (Figure 6H). Overexpression of TAZ completely inhibited WT PPARγ driven transcriptional activity, while S112A PPARγ mutant activity was only partially repressed. Likewise, the repression of WT PPARγ by mutant TAZ lacking the WW domain (TAZΔWW), was also substantially less than the full repression conferred by full length TAZ. While the effects of the TAZΔWW were somewhat less than seen in the PPARγ S112A/TAZ experiment, these differences were not statistically significant (p=0.18). Taken together, these results suggest that PPARγ S112A and the TAZΔWW domain are at least partially responsible for the TAZ/ PPARγ interaction, but that other points of interaction likely exist. In addition, unlike TAZ, the other PPARγ co-repressor, NCoR, was fully able to inhibit transcriptional activity of the S112A mutant PPARγ comparable to its inhibitory effect on WT PPARγ transcription.This finding adds strength to the concept that PPARγ S112 phosphorylation participates in TAZ mediated PPARγ corepression but not NCoR corepression (Figure S6D).
Discussion
Very little is known about the function of TAZ, a Hippo pathway component, in adipocyte biology and nothing has been reported, to the best of our knowledge, in the setting of insulin resistance and diabetes. Here, we have generated adipocyte-specific TAZ knockout mice and have performed detailed studies revealing that TAZ is a critical physiological regulator of PPARγ activity, insulin sensitivity and glucose tolerance. We find that deletion of TAZ from adipocytes protects mice from HFD-induced insulin resistance and glucose intolerance in vivo. This was accompanied by a reduction in adipocyte hypertrophy, reduced ATM content and decreased inflammatory signaling and apoptosis. Although TAZ has established functions as a co-activator in many systems, our data clearly show that in adipocytes TAZ functions to co-repress PPARγ by inhibiting its transcriptional output. Therefore, deletion of TAZ from adipocytes in vivo leads to a constitutively active PPARγ state, consistent with the enhanced insulin sensitivity and improved glucose tolerance seen in TAZ AKO mice.
Based on hyperinsulinemic-euglycemic clamp studies, increased insulin sensitivity was found not only in adipose tissue but also in liver, as measured by increased HGP suppression. These in vivo findings were strongly supported by tissue measurements of insulin signaling, which showed improved insulin-stimulated Akt phosphorylation in adipose tissue and liver, but not muscle.
We explored the physiological mechanisms underlying the improvement of insulin resistance in HFD TAZ AKO mice. Adipocyte size correlates inversely with insulin sensitivity (Yang et al., 2012), and TAZ AKO adipocytes were quantitatively smaller than WT. We also observed an increase in adipocyte number in AKO adipose tissue which can explain why overall adipose tissue mass was not different between the two genotypes. The increase in adipocyte number in AKO adipose tissue could be due to both: 1) A higher rate of adipocyte differentiation as was observed in isolated SVCs from TAZ AKO and f/f mice; and 2) A decrease in HFD-induced apoptosis (Li et al. 2011; Feng et al., 2011) as measured by caspase activity and TUNEL staining.
Interestingly, we also observed a marked improvement of hepatic steatosis in the TAZ AKO mice. The TAZ AKO mice displayed a reduction in plasma free fatty acid levels, which most likely represent slower rates of lipolysis in adipocytes, and this could contribute to the decreased liver triglyceride content of obese TAZ AKO mice. In addition, we found decreased expression of genes involved in hepatic de novo lipogenesis in HFD TAZ AKO vs TAZ f/f littermates. Interestingly, expression of inflammatory genes, such as IL-1β, MCP1 and TNFα were also decreased, a likely contribution to the improved hepatic steatosis. These results highlight the physiological significance of adipose specific TAZ function and its systemic effects on liver function and whole-body insulin resistance.
Chronic tissue inflammation and increased ATM infiltration is a well-known mechanism of insulin resistance in obesity (McArdle et al., 2013), and we found that ATM accumulation and expression of proinflammatory genes was reduced in TAZ AKO adipose tissue. Furthermore, we found that CM harvested from TAZ AKO primary adipocytes displayed a reduced ability to stimulate macrophage migration compared to CM from TAZ f/f controls. The expression and secretion of proinflammatory cytokines and chemokines (TNFα and MCP1) are known to be inhibited by PPARγ activation (Chen et al., 2008). These data suggest that adipocyte TAZ deletion leads to decreased chemokine secretion from adipocytes, causing reduced ATM content and decreased inflammation. In conclusion, we attribute the improved insulin sensitivity observed in TAZ AKO mice to the ability of TAZ to act as a PPARγ co-repressor. TAZ knockout leads to increasing PPARγ expression and a constitutively active PPARγ phenotype, thereby enhancing insulin sensitivity and glucose tolerance.
These findings prompted us to investigate the effects of TZD treatment in our in vivo studies. We found the predicted improvement of insulin sensitivity in rosiglitazone treated HFD WT mice. However, the HFD TAZ AKO mice did not show further improvement in glucose tolerance with rosiglitazone treatment. This is consistent with the gene expression results, which show an activated PPARγ transcriptional program in TAZ AKO adipose tissue with no additional effect of rosiglitazone. Although TAZ AKO mice resemble the TZD treated state with respect to improved systemic insulin sensitivity, they did not phenocopy the increased weight gain associated with TZD treatment. This suggests that even though the set of constitutively upregulated genes in TAZ AKO mice overlaps with genes involved in TZD-mediated improvement of insulin sensitivity, both modes of PPARγ activity may result in expression of qualitatively different genes.
Several studies have shown that Erk1/2 can phosphorylate PPARγ within the AF-1 region at serine 112, and this represses PPARγ transcriptional activity (Adams et al., 1997; Rangwala et al., 2003). However, the mechanism by which S112 phosphorylation reduces PPARγ activity has remained unknown. The adjacency of S112 to the PPARγ PPYY motif suggested that this site plays a role in the recruitment of TAZ to the PPARγ transcriptional complex. Indeed, in the current studies, we demonstrated that S112 phosphorylation contributes to the association between PPARγ and TAZ. Thus, Erk-induced S112 phosphorylation enhances association of PPARγ with TAZ, whereas, inhibition of S112 phosphorylation reduced this interaction. Furthermore, TAZ association with the PPARγ S112D mutant was increased. While mutation of S112 to alanine or deletion of the TAZ WW domain do not completely ablate the TAZ-PPARγ interaction, they do decrease it by 50–75%, confirming the contribution of these sites to the protein:protein interaction. These findings suggest that de-repression of PPARγ activity in adipose tissue, by either TAZ deletion or prevention of S112 phosphorylation (thereby inhibiting TAZ-PPARγ association), would have comparable in vivo consequences. Consistent with this idea, S112A PPARγ mutant mice showed no differences in bodyweight on NCD or HFD, compared to WT, but did show improved glucose tolerance, insulin sensitivity, reduced FFA levels and smaller fat cells in the setting of diet-induced obesity (Rangwala et al., 2003) Together, this strengthens the notion that TAZ’s role in insulin sensitivity is mediated through PPARγ association and co repressor activity.
Erk can phosphorylate both S112 and S273 on PPARγ (Banks et al., 2015), but it is unlikely that S273 phosphorylation plays a direct role in TAZ association. For example, rosiglitazone treatment of 3T3L-1 adipocytes (Figure 6F) led to a large decrease in S273 phosphorylation with no change in the TAZ-PPARγ interaction as measured by Co-IP. Given the inverse correlation of PPARγ S273 phosphorylation with insulin sensitivity (Choi et al., 2010), we examined the status of PPARγ S273 phosphorylation in our TAZ AKO mice studies. Interestingly, we found that S273 phosphorylation was decreased in whole adipose tissue from HFD TAZ AKO compared to WT mice. S112 phosphorylation is known to inhibit PPARγ activity as does S273 phosphorylation, and rosiglitazone treatment has no effect on S112 phosphorylation. This could explain why rosiglitazone treatment still had a partial effect to increase PPARγ transcriptional activity.
NCoR is a well-known PPARγ co-repressor, and knockout of NCoR leads to constitutively active PPARγ stimulation with an increase in insulin sensitivity and improved glucose tolerance (Korman et al. 2018, Li et al., 2011). Here, we show that TAZ also acts as a PPARγ co-repressor and TAZ KO leads to a similar insulin sensitive phenotype, resistant to further stimulation by rosiglitazone. However, there are several important mechanistic differences between NCoR and TAZ. Firstly, rosiglitazone causes dissociation of NCoR from PPARγ, whereas, it does not affect the TAZ-PPARγ association. The ability of PPARγ ligands to dismiss NCoR from PPARγ allows increased TZD-induced transcription of target genes. Since rosiglitazone does not decrease TAZ/PPARγ association, this observation could explain why TAZ inhibits both basal and rosiglitazone-stimulated PPARγ transcriptional activity, whereas, NCoR only inhibits basal transcription activity. Secondly, TAZ and NCoR bind to different sites on PPARγ. Specifically, PPARγ S112 participates in the TAZ/PPARγ association, but does not influence the NCoR/PPARγ interaction. This could underlie the differential phenotypes seen between TAZ and NCoR KO, such as an increase in body weight, food intake and insulin sensitivity in muscle of obese AKO NCoR mice (Li, 2011). This suggests there may be selective PPARγ pathways for insulin sensitization differentially affected by these co-repressors.
It is of interest that S112 PPARγ phosphorylation is increased in obese adipose tissue, most likely due to the increased ERK activation, as reported previously (Lee and Ge, 2014) and as shown in Figure S6B. It is also known that adipose tissue PPARγ expression levels are lower in obese adipose tissue, consistent with a reduced effect of TZDs to induce target genes in obesity. Since PPARγ induces its own expression in adipocytes (Lee and Ge, 2014), the increased S112 PPARγ phosphorylation in obesity would lead to greater TAZ-PPARγ association, providing, a mechanism for the decreased PPARγ expression. This would also provide an explanation for restored adipose tissue PPARγ expression in TAZ AKO mice. This increases in PPARγ expression is likely to contribute to the increased insulin sensitization caused by TAZ KO and constitutively activated PPARγ activity. This increased S112 phosphorylation, with down regulated PPARγ signaling, suggests that the TAZ PPARγ association could be a cause of insulin resistance.
In these studies, we have shown the TAZ functions as a PPARγ co-repressor in adipocytes and that deletion of TAZ leads to a constitutively active PPARγ state, both in vivo and in vitro. This is manifested by improved glucose tolerance, increased insulin sensitivity, and resistance to any additional effects of rosiglitazone treatment to further improve insulin resistance. PPARγ S112 phosphorylation and the TAZ WW domain contribute to the TAZ-PPARγ association, and any method that inhibits this interaction should lead to improved insulin sensitivity.
Limitations of the studies
It should be noted that while our TAZ KO was tissue selective for adipocytes, it was not an inducible KO model, which leaves open the possibility of developmental effects within adipocytes. However, this seems unlikely, since we were able to recapitulate the essential constitutively active PPARγ phenotype using in vitro studies. While the mechanism of the TAZ-PPARγ interaction involves the TAZ WW domain binding to PPARγ S112P, our studies indicate that this is not the complete mechanism, since deletion of the WW domain or mutation of S112 only partially (50–75%) blocked the PPARγ TAZ interaction. This suggests that there are, yet to be identified, interaction points between these two molecules. Finally, while PPARγ S112 phosphorylation is increased in human obesity, the degree to which the TAZ PPARγ interaction affects insulin resistance in clinical situations remains to be defined with further translational studies.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE
Further information and requests for reagents may be directed to the corresponding author Jerrold M Olefsky (University of California San Diego, jolefsky@ucsd.edu ).
EXPERIMENTAL MODEL DETAILS
Animals
TAZfl/fl mice were kindly provided by Dr. Jeff Wrana from University of Toronto. These mice were backcrossed to the C57BL/6J strain for 5 generations. Mice were bred with transgenic mice harboring Cre recombinase driven by adiponectin promotor (Adipoq-Cre) to generated the following genotypes: TAZ flox/flox (TAZ f/f), TAZ flox/flox; adipoq-Cre (TAZ ako) Animals were housed in an animal facility on a 12 h/12 h light/dark cycle at the room temperature of 20–22°C with free access to food and water. Beside obesity, mice were in generally good health. All animal procedures were in accordance with UC San Diego and Institutional Animal Care and Use Committee-approved protocols, and conformed to the Guide for Care and Use of Laboratory Animals of the National Institutes of Health.
Eight-week-old mice male TAZ AKO and f/f were fed with high fat diet (HFD, 60% kcal from fat; D12492, Research Diets) for 12 weeks. Male control and AKO mice were fed with 60% high fat diet (HFD) for 12 weeks, with or without 3 weeks of TZD treatment (Rosiglitazone 3 mg per kg per d). Serum leptin, resistin, and PAI-1 levels were measured using a multiplex Luminex assay (Millipore /Linco research). Plasma insulin and HMW-adiponectin levels were measured with ELISA kits from ALPCO. Plasma FFA levels were measured enzymatically using a kit from WAKO Chemicals. Serum leptin, resistin, and PAI- 1 levels were measured using a multiplex Luminex assay (Millipore /Linco research).
Cell lines
HEK and U2OS cells were cultured in DMEM supplemented with 10% FBS, Pen/strep, and glutamine). To generate adipocytes, 3T3-L1 cells were differentiated in induction medium (DMEM/F12 medium) containing 10% FBS, penicillin-streptomycin, and glutamine and then induced with a differentiation cocktail consisting of 0.5 mM 3-isobutyl-1-methylxanthine, 1 mM dexamethasone, 10 mg/mL insulin, 0.2 mM indomethacin, and 1 mM rosiglitazone in DMEM supplemented with 10% FBS, PS, and glutamine) for 7–10 days (Li et al., 2016).
METHOD DETAILS
ITTs, GTTs, and hyperinsulinemic-euglycemic clamp
Male TAZ AKO and TAZ f/f mice (n = 8–10 each group) were fed with 60% high fat diet (HFD) for 8 weeks, followed by glucose tolerance tests (GTTs), insulin tolerance tests (ITTs) and hyperinsulinemic-euglycemic clamps. For GTTs, animals were IP injected with dextrose (1 g/kg) after 6 hours of fasting, and blood was drawn to measure glucose levels at 0, 15, 30, 60, 90, and 120 minutes after dextrose injection. For ITTs, 0.4–0.5 units/kg of insulin was IP injected after 6 hours of fasting, and blood was drawn at 0, 15, 30, 60, 90, and 120 minutes after insulin injection.
Hyperinsulinemic-euglycemic clamps were performed as previously described (Li et al., 2010). In brief, dual catheters (MRE-025, Braintree Scientific) were implanted in the right jugular vein and tunneled subcutaneously and exteriorized at the back of the neck. Only mice losing <6% of their pre-cannulation weight after 4–5 d of recovery were used. The clamp experiments began with a constant infusion (5 μCi h−1) of D-[3–3H] glucose (Du Pont–NEN) in 6-h-fasted mice. After 90 min of tracer equilibration and basal sampling, glucose (50% dextrose; Abbott) and tracer (5 μCi h−1) plus insulin (6 mU kg−1 min−1) were infused into the jugular vein. Small blood samples were drawn from the tail vein at 10-min intervals and achievement of steady-state conditions (120 mg dl−1 ± 5 mg dl−1) was confirmed at the end of the clamp by maintaining glucose infusion and plasma glucose concentration for a minimum of 20 min. Blood samples at t = −10, 0 (basal), 110 and 120 (end of experiment) min were taken to determine glucose-specific activity and insulin concentration. Tracer-determined rates were quantified by using the Steele equation for steady-state conditions (Steele, 1959). At steady state, the rate of glucose disappearance, or total GDR, is equal to the sum of the rate of endogenous glucose productions (HGP) plus the exogenous (cold) GIR. The IS- GDR is equal to the total GDR minus the basal glucose turnover rate.
Adipocyte, SVCs isolation, and FACS analysis
Briefly, epididymal fat pads from male mice were weighed, rinsed three times in PBS, and then minced in FACS buffer (PBS + 1% low endotoxin BSA). Tissue suspensions were treated with collagenase (1 mg/ml, Sigma-Aldrich) for 30 minutes, and then were filtered through a 100 μm filter (BD Biosciences). After centrifugation at 500 g for 5 min, the supernatant containing adipocytes was removed and the pellet containing the SVC fraction was incubated with RBC lysis buffer (eBioscience) for 5 min followed by another centrifugation (300 g, 5 min) and resuspension in FACS buffer. SVCs were incubated with Fc block for 20 min at 4°C before staining with fluorescence labeled primary antibodies or control IgGs for 30 min at 4°C. F4/80-APC FACS antibody was purchased from AbD Serotec (Raleigh, NC); CD11b-fluorescein isothiocyanate and CD11c-PE FACS antibodies were from BD Biosciences. Cells were gently washed twice and resuspended in FACS buffer with propidium iodide (PI) (Sigma-Aldrich). SVCs were analyzed using a FACS Aria flow cytometer (BD Biosciences). Unstained, single stains and FMO (fluorescence minus one) controls were used to set compensation and gates. In addition, bead compensation was used to verify negative and positive fluorescence signals using FlowJo software. To gate out negative/positive signals for each color used, compensation beads were prepared (each fluorescence antibody + compensation beads) and data applied to the + FMO compensation in the control samples.
Primary pre-adipocyte differentiation
For adipocyte differentiation, pre-adipocytes from male mice were first cultured to confluence in DMEM/F12 medium containing 10% FBS, penicillin-streptomycin (PS), and glutamine and then induced with a differentiation cocktail consisting of 0.5 mM 3-isobutyl-1-methylxanthine, 1 μM dexamethasone, 10 μg/mL insulin, 0.2 mM indomethacin, and 1 μM rosiglitazone in DMEM supplemented with 10% FBS, PS, and glutamine. After 2 days, the medium was replaced to DMEM containing 10% FBS, PS, glutamine,10 μg/mL insulin, and 1 μM rosiglitazone every other day.
Immunohistochemistry of adipose tissue and cell size
Epi-WAT and liver were fixed and embedded in paraffin and sectioned. Liver sections were stained with hematoxylin and eosin (H&E). Adipose tissue sections from at least 4 mice per group were stained with H&E. Using Adiposoft software, adipocyte (n = 100 adipocytes) diameter was measured, and an average diameter was recorded for each animal. The method for IHC study of mouse adipose tissue was carried out as previously described (Lumeng et al., 2008). Anti-F4/80 antibody was purchased from BD Biosciences. TUNEL staining was performed using apoptosis detection kit (Chemicon) according to the manufacturer’s instructions.
In vitro chemotaxis assay
In vitro chemotaxis assay was performed as previously described (Oh et al., 2010). Briefly, primary adipocytes from TAZ AKO and TAZ f/f mice were used for preparation of conditioned media. For the migration per se, 100,000 intraperitoneal-macrophages (IPMacs) from WT mice were used per condition. The IPMacs were placed in the upper chamber of an 8 μm polycarbonate filter (24-transwell format; Corning, Lowell, MA), whereas primary adipocyte conditioned medium was placed in the lower chamber. After 3 hr of migration, cells were fixed in formalin and stained with 4′, 6-diamidino-2-phenylindole. Images taken on an Olympus MXV10 Macro-view microscope and counted using Image J software (NIH).
Glucose uptake assay
The glucose uptake assay in TAZ AKO and TAZ f/f primary adipocytes was performed as previously described (Li et al, 2010). In brief, after 8 hr serum starvation, cells were stimulated with 100 nM insulin for 30 min in KRH buffer (137 nM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, 0.2% BSA, 16 mM HEPES) at 37°C. Then 3H-2-deoxy-D-glucose (3H-2-DOG, 0.1 mM, 0.4 μCi/ml) was supplemented to cells. After 10 min incubation at 37°C, cells were washed with ice-cold PBS twice. NaOH (1 N) was then added and incubated for 20 min to efficiently dissolve cells. An aliquot was used for protein concentration measurement. After neutralizing NaOH by adding HCl (1 N), the extracts were transferred to scintillation vials, and scintillation fluid was added, and the radioactivity was counted. Results were normalized with protein concentration of cell lysates.
RNA isolation and qPCR
Total RNA was extracted from adipose tissue using an RNA purification kit (Qiagen). First-strand cDNA was synthesized using SuperScript III and random hexamers. Quantitative PCR was carried out in 25-μl reactions using iTaq SYBR Green supermix on an MJ Research Chromo4 Real Time PCR system (Bio-Rad). Relative Gene expression was calculated as mRNA level normalized to that of a standard housekeeping gene (36B4) using the ΔΔCT method. The specificity of the PCR amplification was verified by melting curve analysis of the final products using Opticon 3 software. Primer sequences were provided in Table S1.
Immunoprecipitation assay and Western blot analysis
Primary adipocyte, liver, muscle and Epi-WAT were homogenized in RIPA buffer supplemented with protease and phosphatase inhibitors. Tissue lysates were subjected to Western blotting and proteins were detected by corresponding antibodies. The protein bands were analyzed using densitometry and Image J image analysis software, normalizing phosphorylated protein to total protein bands. Arbitrary densitometry units were quantified by ImageJ analysis.
For immunoprecipitation, HEK293T cells and 3T3-L1 adipocytes were homogenized in lysis buffer (150 mM NaCl, 50 mM HEPES (pH 7.4), 1mM EDTA, 1% NP-40, 0.2 mM PMSF, with protease and phosphatase inhibitors) and 2 mg of cell lysate protein was incubated with 2μg of mouse anti-TAZ antibody. Presence of PPARγ in the precipitates was detected by a rabbit anti-PPARγ antibody. Amount of phosphorylated PPARγ was quantified by ImageJ and normalized to amount of total PPARγ.
Plasmids and cell culture
The inserts of mouse pSV sport-TAZ and PpSV sport-PPARgamma 2 from Addgene were cloned in HA-pCDNA3 and GFP-pCDNA3. The S112A and S112D mutations of PPARγ was introduced using Quickchange XL site directd mutagenesis kit. PPREx3-TK-luc from Addgene, and Flag-YAP, myc- TEAD was provided by Guan lab.
U2OS cells in a 24-well plate were co-transfected with a DNA mixture consisting of 10 ng of PPREx3-TK-Luc and pGL4.75, 100 ng of Ha-TAZ WT or HA-TAZΔWW, 100 ng of PPARγ WT or mutant PPARγ S112A/S112D as indicated prior to the luciferase assay. The level of promoter activity was evaluated by measuring firefly luciferase activity relative to renilla luciferase activity using the Dual Luciferase Assay System, as described by the manufacturer. siRNA sequences against mouse Taz and scrambled RNA (ON-TARGETplus Wwtr1 siRNA) were purchased from Dharmacon. siRNA and scrambled control RNA were transfected into 3T3l-1 (ATCC) using RNAiMAX according to the manufacturer’s instruction.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as the means ± s.e.m. The significance of differences between groups was evaluated using analysis of variance. The P value < 0.05 was considered significant.
Statistical parameters including the exact value of n, the definition of center, dispersion and precision measures (mean ± s.e.m) and statistical significance are reported in the Figures and the Figure Legends. Data is judged to be statistically significant when P < 0.05 by two-tailed Student’s t-Test or 2-way ANOVA, where appropriate. Outliers were excluded based upon analysis by the ROUT Test with Q = 0.2% (selecting for only definitive outliers). In Fasting blood insulin levels experiment (Figure 2D), one outlier was excluded from each group (TAZ f/f and AKO mice). In figures, asterisks denote statistical significance (*, P < 0.05; **, P < 0.01). Pearson correlation coefficients were calculated with PRISM and statistical significance is based upon the assumption that values exhibit a Gaussian distribution. Statistical analysis was performed in Graph Pad PRISM 6.
No blinding experiments were performed. Mice were randomly allocated to groups (e.g. NCD vs HFD groups). Sample size was estimated by pilot experiments that showed trends of effects and their sizes as well as previous experience for similar experiments.
DATA AND SOFTWARE AVAILABILITY
Software Availability
The software is available on the web sites linked in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HSP90 | Cell Signaling Technology | Cat#4874;RRID:AB_2121214 |
| Phospho AKT(Ser473) | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| AKT | Cell Signaling Technology | Cat#9272;RRID:AB_329827 |
| Phospho-Erk1/2 | Cell Signaling Technology | Cat#9101;RRID:AB_331646 |
| Erk | Cell Signaling Technology | Cat#4695;RRID:AB_390779 |
| YAP/TAZ | Cell Signaling Technology | Cat#8418;RRID:AB_10950494 |
| HSL | Cell Signaling Technology | Cat#4107;RRID:AB_2296900 |
| Phospho-HSL (Ser563) | Cell Signaling Technology | Cat#4139;RRID:AB_2135495 |
| Actin | Cell Signaling Technology | Cat#3700;RRID:AB_2242334 |
| GAPDH | Cell Signaling Technology | Cat#5174;RRID:AB_10622025 |
| Adiponectin | Invitrogen | Cat#MA1-054;RRID:AB_557516 |
| PPARg | Santa Cruz | Cat#7196;RRID:AB_654710 |
| IgG | Santa cruz | Cat#2027;RRID:AB_737197 |
| Phospho-PPARg (Ser 112) | Millipore | Cat# MAB3632; RRID:AB_2166194 |
| Phospho-PPARg (Ser 273) | LS-Bio | Cat#LS-B7391;RRID:AB_11203925 |
| TAZ | BD sciences | Cat# 560235;RRID:AB_1645338 |
| Perilipin | Affinity Bioreagents | Cat#PA1-1052;RRID:AB_2268558 |
| Phospho-perilipin (S522) | Vala sciences | Cat#4856 |
| Plasmids and Chemicals | ||
| Novolin R regular human insulin in ITTs | Novo-Nordisk | NDC0169-1833-11 |
| Insulin in glucose uptake and hepatic glucose production assays | Sigma-Aldrich | I9278 |
| SuperScript III | Thermo Fisher Scientific | 18080051 |
| iTaq SYBR Green supermix | Bio-Rad Laboratories |
1725121 |
| 2-deoxy-D-glucose | Sigma | D8375 |
| Dextrose | Hospira, Inc | 0409-6648-02 |
| pSV Sport TAZ | Addgene | Cat# 31795 |
| CA-Erk | Addgene | Cat# 39197 |
| Psv Sport PPARgamma 2 | Addgene | Cat# 8862 |
| PPREx3-TK-Luc | Addgene | Cat#1015 |
| PPARg-S112A | This paper | N/A |
| PPARg-S112D | This paper | N/A |
| TAZAWW | This paper | N/A |
| Pgl4.75 [hRluc/CMV] | Promega | E6931 |
| Ha-Pcdna3 | This paper | N/A |
| Flag-NcoR | This paper | N/A |
| RNAi-MAX | Life Technologies | (#13778150) |
| ON-TARGETplus Wwtr1 siRNA | Dharmacon | L-041057-01 |
| 3H-deoxyglucose | Perkin Elmer | NET328001MC |
| 3H-glucose | Perkin Elmer | NET331C001MC |
| RBC lysis buffer | eBioscience | # 00-4333-57 |
| 4’, 6-diamidino-2-phenylindole | Molecular Probes | Cat: #D1306 |
| 60% HFD | Research Diets Inc. | D12492 |
| Critical Commercial Assays | ||
| Quickchange XL site directed mutagenesis kit | Agilent | N/A |
| Plasma FFA enzymatical kit | WAKO Chemicals | 999–34691 |
| RNA purification kit | Qiagen | Cat: 74104 |
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1910 |
| Experimental Models: Cell Lines | ||
| 3T3-L1 adipocytes | ATCC | N/A |
| Primary adipocytes (TAZ WT, TAZ AKO) knockout) | This paper | N/A |
| HEK293T cells | ATCC | N/A |
| U2OS cells | ATCC | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: TAZ floxed/floxed | Dr. Jeff Wrana | N/A |
| Mouse: WT C57Bl6/J | Jackson Laboratories | N/A |
| Mouse:Adipoq-Cre | Jackson Laboratories | N/A |
| Mouse:TAZ ako mice | This paper | N/A |
| Sequence-Based Reagents | ||
| Primers, see Table S1 | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo | http://www.flowjo.com/ |
| Image J | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Adiposoft | Image J | http://imagej.net/Adiposoft |
| Other | ||
| 8 μm polycarbonate filter, 24-transwell Format | Corning | Cat#3464 |
| Mini-Osmotic pump | Alzet | Model 2002 |
Supplementary Material
Highlights
TAZ binds to PPARγ and functions as a corepressor of PPARγ in adipose tissue
TAZ deletion leads to increased PPARγ activity and target gene expression
TAZ KO leads to increased insulin sensitivity and improved glucose tolerance
TAZ KO leads to decreased adipose tissue inflammation
Context and Significance
Insulin resistance is a common metabolic abnormality in obese individuals and can directly lead to the development of Type 2 Diabetes Mellitus (T2DM). Activation of the nuclear hormone receptor PPARγ is an established method for treating insulin resistance. This work shows that the protein TAZ is an endogenous inhibitor of PPARγ activity and that genetic deletion of TAZ from fat cells locks PPARγ into an activated state. In obese mice, TAZ deletion results in increased systemic insulin sensitivity with an improvement in glucose tolerance. Taken together, these results identify TAZ as a new factor in the development of obesity-induced insulin resistance and suggests that inhibition of TAZ could have beneficial therapeutic effects in the treatment of T2DM and other disorders marked by insulin resistance.
ACKNOWLEDGEMENTS
We would like to thank Dr. Jeff Wrana from University of Toronto for providing the TAZ floxed mice. We also thank Jachelle Pimentel and Jan Pferdekamper for technical assistance and Angela Tyler for administrative support. This study was funded by grants (JMO) from the National Institutes of Health (DK033651, DK074868, DK063491, DK09062) and a grant from Merck & Co., Inc. D.Y.O. has been supported by grants from the NIH (R01 DK108773) and the American Heart Association (14SDG19880020).
Footnotes
Declaration of Interests
The authors have no conflicts of interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams M, Reginato MJ, Shao D, Lazar MA, and Chatterjee VK (1997). Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. The Journal of biological chemistry 272, 5128–5132. [DOI] [PubMed] [Google Scholar]
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, and Evans RM (2013). PPARgamma signaling and metabolism: the good, the bad and the future. Nature medicine 19, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Y, Kang Q, Zhao Y, Hu X, Li N. (2013). Lats2 modulates adipocyte proliferation and differentiation via hippo signaling. PLoS One. 8(8):e72042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anakk S, Bhosale M, Schmidt VA, Johnson RL, Finegold MJ, and Moore DD (2013). Bile acids activate YAP to promote liver carcinogenesis. Cell reports 5, 1060–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, and Spiegelman BM (2015). An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 517, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KA, and Vanden Heuvel JP (2007). Modulation of PPAR activity via phosphorylation. Biochimica et biophysica acta 1771, 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FL, Yang ZH, Liu Y, Li LX, Liang WC, Wang XC, Zhou WB, Yang YH, Hu RM (2008). Berberine inhibits the expression of TNFalpha, MCP-1, and IL-6 in AcLDL-stimulated macrophages through PPARgamma pathway. Endocrine. 2008 Jun;33(3):331–7. [DOI] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, et al. (2010). Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RM, Barish GD, and Wang YX (2004). PPARs and the complex journey to obesity. Nature medicine 10, 355–361. [DOI] [PubMed] [Google Scholar]
- Glass CK, and Rosenfeld MG (2000). The coregulator exchange in transcriptional functions of nuclear receptors. Genes & development 14, 121–141. [PubMed] [Google Scholar]
- Higashi T, Hayashi H, Ishimoto T, Takeyama H, Kaida T, Arima K, Taki K, Sakamoto K, Kuroki H, Okabe H, et al. (2015). miR-9–3p plays a tumour-suppressor role by targeting TAZ (WWTR1) in hepatocellular carcinoma cells. British journal of cancer 113, 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, Mueller E, Benjamin T, Spiegelman BM, Sharp PA, et al. (2005). TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309, 1074–1078. [DOI] [PubMed] [Google Scholar]
- Hu E, Kim JB, Sarraf P, and Spiegelman BM (1996). Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 274, 2100–2103. [DOI] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, and Pan D. (2005). The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122, 421–434. [DOI] [PubMed] [Google Scholar]
- Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, Hisaminato A, Fujiwara T, Ito Y, Cantley LC, et al. (2000). TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. The EMBO journal 19, 6778–6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner S, Mepani RJ, Laznik D, Ye L, Jurczak MJ, Jornayvaz FR, Estall JL, Chatterjee Bhowmick D, Shulman GI, and Spiegelman BM (2012). Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proceedings of the National Academy of Sciences of the United States of America 109, 9635–9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamura K, Shin J, Kiyonari H, Abe T, Shioi G, Fukuhara A, Sasaki H. (2018). Obesity in Yap transgenic mice is associated with TAZ downregulation. Biochem Biophys Res Commun. 505(3):951–957. [DOI] [PubMed] [Google Scholar]
- Koppen A, and Kalkhoven E. (2010). Brown vs white adipocytes: the PPARgamma coregulator story. FEBS letters 584, 3250–3259. [DOI] [PubMed] [Google Scholar]
- Korman B, Marangoni RG, Lord G, Olefsky J, Tourtellotte W, Varga J. (2018). Adipocyte-specific Repression of PPAR-gamma by NCoR Contributes to Scleroderma Skin Fibrosis. Arthritis Research & Therapy. 11;20(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, and Ge K. (2014). Transcriptional and epigenetic regulation of PPARgamma expression during adipogenesis. Cell & bioscience 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrke M, and Lazar MA (2005). The many faces of PPARgamma. Cell 123, 993–999. [DOI] [PubMed] [Google Scholar]
- Li P, Lu M, Nguyen MT, Bae EJ, Chapman J, Feng D, Hawkins M, Pessin JE, Sears DD, Nguyen AK, Amidi A, Watkins SM, Nguyen U, Olefsky JM. (2010). Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. JBC. 14;285(20):15333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, Sears DD, Talukdar S, Oh D, Chen A, et al. (2011). Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 147, 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Liu S, Lu M, Bandyopadhyay G, Oh D, Imamura T, Johnson AMF, Sears D, Shen Z, Cui B, Kong L, Hou S, Liang X, Iovino S, Watkins SM, Ying W, Osborn O, Wollam J, Brenner M, Olefsky JM. (2016). Hematopoietic-Derived Galectin-3 Causes Cellular and Systemic Insulin Resistance. Cell 167(4):973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. (2008). Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008 57(12):3239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM, (2013). Mechanisms of Obesity-Induced Inflammation and Insulin Resistance: Insights into the Emerging Role of Nutritional Strategies. Frontiers in Endocrinology 4: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Nakagawa M, Olson EN, and Nakagawa O. (2005). A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proceedings of the National Academy of Sciences of the United States of America 102, 18034–18039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell E, Kuhn P, and Xu W. (2007). Nuclear Receptor Cofactors in PPARgamma-Mediated Adipogenesis and Adipocyte Energy Metabolism. PPAR research 2007, 53843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala SM, and Lazar MA (2004). Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends in pharmacological sciences 25, 331–336. [DOI] [PubMed] [Google Scholar]
- Rangwala SM, Rhoades B, Shapiro JS, Rich AS, Kim JK, Shulman GI, Kaestner KH, and Lazar MA (2003). Genetic modulation of PPARgamma phosphorylation regulates insulin sensitivity. Developmental cell 5, 657–663. [DOI] [PubMed] [Google Scholar]
- Rosen ED, and Spiegelman BM (2006). Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444, 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah Z, and Aqeilan RI (2011). WW domain interactions regulate the Hippo tumor suppressor pathway. Cell death & disease 2, e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudol M. (2010). Newcomers to the WW Domain-Mediated Network of the Hippo Tumor Suppressor Pathway. Genes & cancer 1, 1115–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutanto MM, Ferguson KK, Sakuma H, Ye H, Brady MJ, and Cohen RN (2010). The silencing mediator of retinoid and thyroid hormone receptors (SMRT) regulates adipose tissue accumulation and adipocyte insulin sensitivity in vivo. The Journal of biological chemistry 285, 18485–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, and Spiegelman BM (2008). Fat and beyond: the diverse biology of PPARgamma. Annual review of biochemistry 77, 289–312. [DOI] [PubMed] [Google Scholar]
- van Beekum O, Fleskens V, and Kalkhoven E. (2009). Posttranslational modifications of PPAR-gamma: fine-tuning the metabolic master regulator. Obesity 17, 213–219. [DOI] [PubMed] [Google Scholar]
- Varelas X, Miller BW, Sopko R, Song S, Gregorieff A, Fellouse FA, Sakuma R, Pawson T, Hunziker W, McNeill H, et al. (2010). The Hippo pathway regulates Wnt/betacatenin signaling. Developmental cell 18, 579–591. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yu A, and Yu FX (2017). The Hippo pathway in tissue homeostasis and regeneration. Protein & cell 8, 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Eliasson B, Smith U, Cushman SW, and Sherman A. (2012). The size of large adipose cells is a predictor of insulin resistance in first-degree relatives of type 2 diabetics. Obesity 20(5): 932–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, Xiong Y, Lei QY, and Guan KL (2009). TEAD transcription factors mediate the function of TAZ in cell growth and epithelial mesenchymal transition. The Journal of biological chemistry 284, 13355–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Software Availability
The software is available on the web sites linked in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HSP90 | Cell Signaling Technology | Cat#4874;RRID:AB_2121214 |
| Phospho AKT(Ser473) | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| AKT | Cell Signaling Technology | Cat#9272;RRID:AB_329827 |
| Phospho-Erk1/2 | Cell Signaling Technology | Cat#9101;RRID:AB_331646 |
| Erk | Cell Signaling Technology | Cat#4695;RRID:AB_390779 |
| YAP/TAZ | Cell Signaling Technology | Cat#8418;RRID:AB_10950494 |
| HSL | Cell Signaling Technology | Cat#4107;RRID:AB_2296900 |
| Phospho-HSL (Ser563) | Cell Signaling Technology | Cat#4139;RRID:AB_2135495 |
| Actin | Cell Signaling Technology | Cat#3700;RRID:AB_2242334 |
| GAPDH | Cell Signaling Technology | Cat#5174;RRID:AB_10622025 |
| Adiponectin | Invitrogen | Cat#MA1-054;RRID:AB_557516 |
| PPARg | Santa Cruz | Cat#7196;RRID:AB_654710 |
| IgG | Santa cruz | Cat#2027;RRID:AB_737197 |
| Phospho-PPARg (Ser 112) | Millipore | Cat# MAB3632; RRID:AB_2166194 |
| Phospho-PPARg (Ser 273) | LS-Bio | Cat#LS-B7391;RRID:AB_11203925 |
| TAZ | BD sciences | Cat# 560235;RRID:AB_1645338 |
| Perilipin | Affinity Bioreagents | Cat#PA1-1052;RRID:AB_2268558 |
| Phospho-perilipin (S522) | Vala sciences | Cat#4856 |
| Plasmids and Chemicals | ||
| Novolin R regular human insulin in ITTs | Novo-Nordisk | NDC0169-1833-11 |
| Insulin in glucose uptake and hepatic glucose production assays | Sigma-Aldrich | I9278 |
| SuperScript III | Thermo Fisher Scientific | 18080051 |
| iTaq SYBR Green supermix | Bio-Rad Laboratories |
1725121 |
| 2-deoxy-D-glucose | Sigma | D8375 |
| Dextrose | Hospira, Inc | 0409-6648-02 |
| pSV Sport TAZ | Addgene | Cat# 31795 |
| CA-Erk | Addgene | Cat# 39197 |
| Psv Sport PPARgamma 2 | Addgene | Cat# 8862 |
| PPREx3-TK-Luc | Addgene | Cat#1015 |
| PPARg-S112A | This paper | N/A |
| PPARg-S112D | This paper | N/A |
| TAZAWW | This paper | N/A |
| Pgl4.75 [hRluc/CMV] | Promega | E6931 |
| Ha-Pcdna3 | This paper | N/A |
| Flag-NcoR | This paper | N/A |
| RNAi-MAX | Life Technologies | (#13778150) |
| ON-TARGETplus Wwtr1 siRNA | Dharmacon | L-041057-01 |
| 3H-deoxyglucose | Perkin Elmer | NET328001MC |
| 3H-glucose | Perkin Elmer | NET331C001MC |
| RBC lysis buffer | eBioscience | # 00-4333-57 |
| 4’, 6-diamidino-2-phenylindole | Molecular Probes | Cat: #D1306 |
| 60% HFD | Research Diets Inc. | D12492 |
| Critical Commercial Assays | ||
| Quickchange XL site directed mutagenesis kit | Agilent | N/A |
| Plasma FFA enzymatical kit | WAKO Chemicals | 999–34691 |
| RNA purification kit | Qiagen | Cat: 74104 |
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1910 |
| Experimental Models: Cell Lines | ||
| 3T3-L1 adipocytes | ATCC | N/A |
| Primary adipocytes (TAZ WT, TAZ AKO) knockout) | This paper | N/A |
| HEK293T cells | ATCC | N/A |
| U2OS cells | ATCC | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: TAZ floxed/floxed | Dr. Jeff Wrana | N/A |
| Mouse: WT C57Bl6/J | Jackson Laboratories | N/A |
| Mouse:Adipoq-Cre | Jackson Laboratories | N/A |
| Mouse:TAZ ako mice | This paper | N/A |
| Sequence-Based Reagents | ||
| Primers, see Table S1 | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo | http://www.flowjo.com/ |
| Image J | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Adiposoft | Image J | http://imagej.net/Adiposoft |
| Other | ||
| 8 μm polycarbonate filter, 24-transwell Format | Corning | Cat#3464 |
| Mini-Osmotic pump | Alzet | Model 2002 |