

Abstract
Integrating MS-based metabolomics approaches, LC-MS-PCA and molecular networking enabled the targeted isolation of five new pyrrole-derived alkaloids phallusialides A−E (1−5), from a marine-derived Micromonospora sp. bacterium. The structures of 1−5 were elucidated by analysis of their HRMS, MS/MS, and NMR spectroscopic data. The absolute configuration of phallusialide A (1) was determined on the basis of comparisons of experimental and theoretically calculated ECD spectra. Compounds 1 and 2 exhibited antibacterial activity against methicillin resistant S. aureus (MRSA) and E. coli, with MIC values of 32 and 64 µg/mL, respectively, whereas 3−5 showed no antibacterial activity even at 256 µg/mL, yielding important SAR insights for this class of compounds.
TOC Graphic
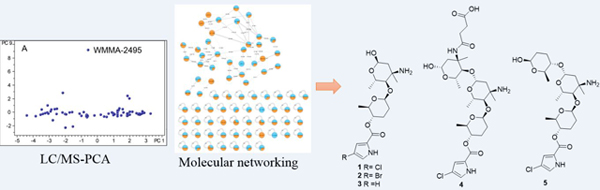
Despite the fact that bacterially-derived natural products (NPs) have been a successful source of drug leads, most pharmaceutical companies have reduced or shut down their NP-based drug discovery programs due to historically high rates of rediscovery. To address this problem, we have developed strain prioritization by metabolomics (SPM) to help identify bacterial strains most likely to yield previously unknown agents. LC-MS-based principal component analysis (PCA)1,2 has been a useful tool for SPM by helping to distinguish unique and common metabolites. Nonetheless, reliable dereplication is often very challenging when solely using MS-1 data and available databases, especially at m/z values < 600. In contrast, MS/MS fragmentation represents the ultimate mass spectral fingerprint of a molecule, and thus can be effectively used in dereplication. Molecular networking,3,4 which visualizes and organizes MS/MS data based on the observation that structurally similar molecules share similar MS/MS fragmentation patterns, has emerged as a promising technique for dereplication. Particularly, it is available on the Global Natural Products Social Molecular Networking (GNPS; http://gnps.ucsd.edu) open-access Web-based platform.5 Since molecular networking groups secondary metabolites into categories of similar scaffolds, a major strength of the method is the ability to identify putative analogues of both known and unknown compounds. Indeed, by employing metabolomics approaches LC-MS-PCA and molecular networking, it is possible to prioritize strains and dereplicate large numbers of compounds without complete knowledge of their structures. This tactic provides many opportunities to identify new molecular families in a rapid and efficient manner.
In our efforts to discover novel antibiotics, 72 marine-derived Micromonospora sp. bacterial extracts were analyzed by PCA leading to the identification of strain WMMA-2495 as a metabolic outlier (Figure 1A and 1B). The bucket statistics (Figure 1C) plot indicated that a compound at m/z 403.1634 ([M+H]+) was detected only in strain WMMA-2495, indicating the unique chemistry specific to WMMA-2495. The isotopic pattern in the mass spectrum indicated that this m/z contained Cl. To further investigate the secondary metabolite produced by WMMA-2495, we employed molecular networking in tandem with Br supplementation during WMMA-2495 fermentations. We hypothesized that molecular networking would greatly illuminate possible connections between the Cl and Br families for these putatively novel natural products. Therefore, we used two media; one contained our typically used artificial seawater (ASW-A), and the other contained excess KBr and no NaCl (a component of ASW-A). After fermentation of WMMA-2495 in the two media, extracts were subjected to LC-MS/MS analysis, and the resultant data were used to generate the molecular network using the GNPS Web site, providing 27 nodes connected with 43 edges (Figure 2). The molecular network quickly allowed us to establish the structural relationships of these unknown molecules. For example, a difference of 18 Da suggested a putative H2O increment, a difference of 34 Da inferred the substitution of a proton by a chlorine atom, and a difference of 44 Da invoked a Br for Cl substitution. Notably, the same compound was found to yield different nodes due to in source fragmentation; thus, it is important to note that the number of nodes in the network does not correspond exactly to the number of metabolites. The molecular network for WMMA-2495 indicated the presence of several analogues of the most abundant ion at 403 ([M+H]+), clear differences in the production of the secondary metabolites depending on the culture media, and no hits in the GNPS public dataset. Thereafter, from a careful analysis of the molecular networking of this strain, five new pyrrole-derived alkaloids were discovered.
Figure 1.

PCA scores and loadings plots for strain WMMA-2495. (A) The scores plot for PC1 vs. PC9 shows that WMMA-2495 separates from the other strains. (B) The loadings plot shows molecular features that are unique for WMMA-2495. (C) The bucket statistics plot indicated that the compound at m/z 403.1634 ([M+H]+) was only detected in the strain WMMA-2495.
Figure 2.

Cluster of pyrrole-derived analogues generated by GNPS. Thickness of the edges between nodes indicated the degree of similarity between their respective MS/MS spectra. Cytoscape 4.3.0 was used for visualizing the corresponding molecular networks.
Results and Discussion
The molecular formula of phallusialide A (1) was established as C18H27ClN2O6 based on HRESIMS data that also showed an isotopic pattern consistent with one chlorine atom. Evaluation of the 1H and 13C NMR and HSQC spectra made clear the presence of 18 carbons, including one α,β-unsaturated carbonyl carbon within the amide/ester range, three methyl groups, three methylenes, six oxygenated methines (including two anomeric carbons at δC 103.4 and 91.5, representing two sugar moieties), four aromatic/olefinic carbons with two protonated, one sp3 tetrasubstituted carbon and four exchangeable protons. The COSY NMR data for 1 (Table 1) showed a series of correlations establishing three spin systems, H1’−H6’, H1’’−H2’’, and H4’’−H5’’−H7’’. In addition, the HMBC cross-peaks from H-2’’, H-4’’ and H-5’’ to C-3’’ corroborated the C1’’−C5’’−C7’’ backbone. A methyl group (δH 1.30) was attached to C-3’’ on the basis of HMBC correlations from H3-6’’ to C-2’’, C-3’’, and C-4’’. Moreover, the 13C NMR chemical shift of C-3’’ (δC 56.3) indicated a nitrogen attachment to C-3’’, which was also confirmed by the 1H-15N HMBC correlations from H2-2’’, H-4’’ and H3-6’’ to a nitrogen at δN 43.9 (Figure S7). The HMBC correlations from H-3 to C-2 and C-5, and from H-5 to C-2 and C-4, together with the small coupling constant (1.5 Hz) between H-3 and H-5, suggested the presence of a 2, 4-substituted pyrrole moiety. The HMBC correlations from H-3, H-5 and H-4’ to C-6 indicated connection of the pyrrole moiety unit to sugar A via an ester bond, whereas a chlorine was the other substituent attached to the pyrrole moiety. MS/MS fragmentation returned a mass fragment at m/z 242, consistent with the presence of a chloro-1H-pyrrole-carboxylic acid. There were two possible arrangements with respect to regiochemistry of the 2, 4-substituted pyrrole moiety, 4-chloro-1H-pyrrole-2-carboxylic acid or 2-chloro-1H-pyrrole-4-carboxylic acid (Figure S51). In order to distinguish between these two possibilities, complementary approaches were utilized. A strong HMBC correlation from a proton at δH 6.96 to C-6, as well as a weaker HMBC correlation from a proton at δH 6.74 to C-6 were observed, which suggested the pyrrole moiety as 2-chloro-1H-pyrrole-4-carboxylic acid, but could not entirely rule out the possibility of 4-chloro-1H-pyrrole-2-carboxylic acid due to the unique features of pyrrole ring system. To explore the possibility of using the intensity of the HMBC correlations, coupling constants between H-5 to C-6 (4JH-2,C-6) and between H-3 to C-6 (3JH-4,C-6) of 4-chloro-1H-pyrrole-2-carboxylic acid were calculated using Gaussian 096 by the density functional theory (DFT) method at the B3LYP/6-311+G(2d,p) level.7 We hypothesized that the 4J coupling constant would be substantially less than that of 3J. However, DFT calculations for 4-chloro-1H-pyrrole-2-carboxylic acid revealed the 4JH-5,C-6 and 3JH-3,C-6 values as 1.6 and 1.3 Hz, respectively, suggesting that a structural hypothesis based on linking HMBC intensity to coupling constants was not appropriate. However, the location of the chlorine atom was ultimately confirmed by comparing the 1H and 13C chemical shifts of compound 1 and the corresponding data of 4-chloro-1H-pyrrole-2-carboxylic acid in the same solvent (CD3OD) (Table S1); signals for the pyrrole moiety in 1 proved nearly identical to those of 4-chloro-1H-pyrrole-2-carboxylic acid. To satisfy the molecular formula, there were four remaining exchangeable protons that were assigned by default as 1-NH, an hydroxy group attached to C-1’’ and an amine attached to C-3’’ to complete the planar structure of 1 as depicted.
Table 1.
1H and 13C NMR Data for Phallusialide A (1) (600 MHz for 1H, 125 MHz for 13C, CD3OD).
| Positon | δC, mult. | δH (J in Hz) | COSY | HMBCa | ROESY |
|---|---|---|---|---|---|
| 2 | 123.2, C | ||||
| 3 | 115.5, CH | 6.74, d (1.5) | 5 | 2, 5, 6 | |
| 4 | 114.5, C | ||||
| 5 | 122.6, CH | 6.96, d (1.5) | 3 | 2, 4, 6 | |
| 6 | 161.2, C | ||||
| Sugar A, β-amicetose | |||||
| 1’ | 103.4, CH | 4.72, dd (9.0, 1.7) | 2’ | 2’, 5’, 4’’ | 5’, 4’’, 7’’ |
| 2’ | 31.4, CH2 | ax: 2.01, m eq: 1.72, m |
1’, 3’ | 1’, 3’, 4’ | |
| 3’ | 28.8, CH2 | 2.19, m 1.69, m |
2’, 4’ | 1’, 2’, 4’ | |
| 4’ | 74.2, CH | 4.56, ddd (10.0, 10.0, 4.8) | 3’, 5’ | 6, 5’, 6’ | 2’ax, 6’ |
| 5’ | 74.9, CH | 3.69, dq (10.0, 6.1) | 4’, 6’ | 1’, 3’, 4’, 6’ | 1’, 2’eq |
| 6’ | 18.5, CH3 | 1.22, d (6.1) | 5’ | 4’, 5’ | 4’ |
| Sugar B, α−4-epi-vancosamine | |||||
| 1’’ | 91.5, CH | 5.22, d (2.8) | 2’’ | 3’’, 5’’ | |
| 2’’ | 40.4, CH2 | 2.06, d (14.8); 1.89, dd (14.8, 3.5) |
1’’ | 1’’, 3’’, 4’’, 6’’ | |
| 3’’ | 56.3, C | ||||
| 4’’ | 82.3, CH | 3.44, d (10.0) | 5’’ | 1’, 5’’, 6’’, 7’’ | 1’, 2’’b, 6’’, 7’’ |
| 5’’ | 64.4, CH | 4.13, dq (10.0, 6.1) | 4’’, 7’’ | 1’’, 3’’,4’’, 7’’ | |
| 6’’ | 25.7, CH3 | 1.44, s | 2’’, 3’’, 4’’ | 4’’ | |
| 7’’ | 18.7, CH3 | 1.30, d (6.1) | 5’’ | 4’’, 5’’ | 1’, 4’’ |
The relative configuration of phallusialide A (1) was determined by a combination of NOE studies, coupling constant analyses, extensive molecular modeling, and DFT calculations. The H-1’ was assigned to be axial based on its multiplicity, which displayed a large coupling constant (9.0 Hz) between H-1’ and H-2’ax, and a small coupling constant (1.7 Hz) between H-1’ and H-2’eq. The ROESY correlations from H-5’ to H-1’ and H-2’eq implied that H-5’ was in the axial position (Figure 3). The large coupling constant between H-5’ and H-4’ (10 Hz), as well as the ROESY correlation between H-4’ and H-2’aq indicated that H-4’ was axial. Therefore, sugar A was established as β-amicetose.6 The large coupling constant (10 Hz) observed between H-4’’ and H-5’’ confirmed the axial configuration of H-4’’ and H-5’’. The anomeric proton of sugar B (H-1’’) was assigned to be in an α-configuration based on the small coupling constant (2.8 Hz) between H-1’’ and H2-2’’. The ROESY correlation between H-4’’ and H3-6’’ was not able to unambiguously validate the configuration of C-6’’. However, the amino sugar in 1 (sugar B) was determined to be 4-epi-vancosamine based on comparisons of its 1H and 13C NMR data with those of α-avidinosaminide,9,10 vancosamine,11 and vancosamine related epimers.12,13 The only stereochemical information linking the two sugar units was the NOESY correlation between H-1’ and H-4’’. Nevertheless, this correlation was insufficient to determine the relative configuration of the two sugar moieties (1a and 1b; Figure 4). Therefore, DFT 13C NMR chemical shift calculations were undertaken and analyzed with the DP4 probability method.14 Low-energy conformers of the two possible stereoisomers 1a and 1b were determined with Spartan 14 followed by DFT NMR calculations using Gaussian 09 with the B3LYP method at the 6–31G (d,p) level. The diastereoisomer 1a was deduced to be the correct relative configuration with a 100% DP4 probability (Table S2).
Figure 3.

Key ROESY correlations of 1.
Figure 4.

Possible diastereoisomers of 1 based on ROESY correlations.
The electronic circular dichroism (ECD) approach by comparing experimental and calculated electronic ECD spectra generated by time-dependent functional theory (TDDFT)15,16 is only effective for establishing the absolute configuration at C-4’ (and possibly C-5’), since the chromophore is remote from ring B. While the relative configuration of 1 was firmly established, the absolute configuration of the whole molecule can be established by relaying the absolute configuration at C-4’. Conformational analysis was carried out employing Spartan 14 software and MMFF molecular mechanics calculations. MMFF conformational search was further optimized using TDDFT at the B3LYP/6-31G(d) basis set level, to render the nine lowest energy conformers. The overall calculated ECD spectra of 1a and 1c (Figure 5) were generated by Boltzmann distribution of the nine lowest energy conformers with 46.1%, 16.7%, 8.0%, 8.0%, 5.9%, 3.5%, 3.1%, 1.9%, and 1.9% population. The calculated ECD spectrum of enantiomer 1a agreed well with the experimental data of 1, therefore establishing the absolute configuration of C-4’ as S. Accordingly, the two sugar residues were determined as d-β-amicetose and l-α−4-epi-vancosamine.
Figure 5.

Experimental ECD spectrum of compound 1 in CH3OH and calculated ECD spectra of enantiomers 1a and 1c.
Biosynthetically driven halide incorporation is often integrally dependent on relative halogen concentrations within fermentation media.17–19 Accordingly, the KBr concentration was increased to 10 g/L (from 0.1 g/L) and NaCl was cut from 20 g/L to 0 g/L in fermentation medium ASW-A; these halide alterations enabled the production of brominated analogue, phallusialide B (2), with a molecular formula of C18H27BrN2O6 as determined by HRESIMS. A comparison of the 1H and 13C NMR shifts of phallusialides A (1) and B (2) showed that the chemical shifts of H-3, H-5 and C-2 shifted deshielded, and C-4 shifted shielded in phallusialide B (2), together with the prominent product ion at m/z 286 observed in the MS/MS data (Figure S17), suggesting that the chlorine atom was replaced by a bromine atom in compound 2. No other significant changes in chemical shifts were noted between 1 and 2, indicating that both 1 and 2 have the same relative configuration. The absolute configuration of 2 was deduced by comparing the Cotton effects seen in the ECD spectrum of 2 with those of 1, which showed a positive Cotton effect at 270 nm.
Phallusialide C (3) was isolated as a colorless, amorphous powder, with molecular formula of C18H28N2O6 on the basis of its HRESIMS data. A comparison of the 1H and 13C NMR chemical shifts (Table S4) of phallusialide C (3) with phallusialide A (1) and phallusialide B (2) confirmed that the only difference in the structures was the lack of a halogen atom at C-4. An additional aromatic/olefinic proton was observed in 3, indicating 3 as a nonhalogenated analogue of phallusialides A and B. Comparisons of the ECD spectra for compounds 3 and 1 revealed that both have the same absolute configuration; the configuration of 3 is identical to that of 1.
The elemental composition of phallusialide D (4) was established as C29H44ClN3O11 on the basis of HRESIMS. Analysis of the 1H, 13C, and HMQC NMR data of 4 (Table 2) revealed the presence of 29 carbons, including five methyl groups, six methylene units, nine oxygenated methines, two sp3 tetrasubstituted carbons (two oxygenated), four aromatic/olefinic carbons (two of which were protonated), and three carbonyl carbons including one α,β-unsaturated carbonyl carbon. These data, together with six unobserved exchangeable protons, accounted for all of the 1H and 13C NMR resonances for compound 4. NMR data for 4 revealed that it possessed a core scaffold similar to that of 1; this was further confirmed by our observation of a fragment ion at m/z 403, which was equivalent to the molecular weight of compound 1. In addition, another daughter ion at m/z 244 in the ESI-MS/MS data (Figure 6) accounted for the remaining partial structure of the molecule, with the molecular formula C11H17NO5. The determination of the remaining partial structure was carried out as follows. The COSY NMR data of 4 (Table 2) established the three spin systems, H-1’’’−H-2’’’, H-4’’’−H-5’’’−H3-7’’’, and H2-9’’’−H2-10’’’. HMBC correlations from H2-2’’’, H-5’’’ and H3-6’’’ to C-3’’’, combined with the chemical shift of C-3’’’ (δC 57.6), suggested the presence of an additional amino sugar (sugar C) in 4. Furthermore, the HMBC correlation from H-4’’’ to C-1’’, and the ROESY correlations between H-4’’’ to H-1’’ revealed the linkage of sugars C and B via C-4’’’ to C-1’’. The HMBC correlations from H2-9’’’ and H2-10’’’ to C-10’’’ and C-11’’’, along with the m/z 244 daughter ion, established that a succinic acid moiety was linked to the sugar C, satisfying the molecular formula of the remaining substructure. The chemical shift of the nitrogen attached to C-3’’’ (δN 122.7) was found to be reasonably far deshielded compared to that of the nitrogen attached to C-3’’ (δN 38.9). As a result, it was apparent that the succinic acid was attached to C-3’’’ via an amide linkage.
Table 2.
1H and 13C NMR Data for Phallusialide D (4) (600 MHz for 1H, 125 MHz for 13C, CD3OD).
| Positon | δC, mult. | δH (J in Hz) | COSY | HMBCa | ROESY |
|---|---|---|---|---|---|
| 2 | 123.2, C | ||||
| 3 | 115.5, CH | 6.74, d (1.5) | 5 | 2, 5, 6 | |
| 4 | 114.5, C | ||||
| 5 | 122.6, CH | 6.96, d (1.5) | 3 | 2, 4, 6 | |
| 6 | 161.2, C | ||||
| Sugar A, β-amicetose | |||||
| 1’ | 103.4, CH | 4.71, dd (9.0, 1.7) | 2’ | 2’, 4’’ | 5’, 4’’ |
| 2’ | 31.5, CH2 | ax: 2.00, m eq: 1.71, m |
1’, 2’eq, 3’ 1’, 2’ax, 3’ |
1’, 3’, 4’ | |
| 3’ | 28.8, CH2 | 2.19, m 1.69, m |
2’, 4’ |
1’, 4’, 5’ | |
| 4’ | 74.2, CH | 4.56, ddd (10, 10, 4.8) | 3’, 5’ | 6, 5’, 6’ | 6’ |
| 5’ | 74.9, CH | 3.69, dq (10.0, 6.0) | 4’, 6’ | 1’, 3’, 4’, 6’ | 1’ |
| 6’ | 18.5, CH3 | 1.22, d (6.0) | 4’, 5’ | 4’ | |
| Sugar B, β−4-epi-vancosamine | |||||
| 1’’ | 99.0, CH | 4.89, d (10.0) | 2’’ | 2’’, 4’’’ | |
| 2’’ | 42.4, CH2 | eq: 2.19, d (14.8); ax: 1.88, dd (14.8, 10.0) |
1’, 2’eq 1’, 2’ax |
1’’, 3’’, 4’’, 6’’ | |
| 3’’ | 56.9, C | ||||
| 4’’ | 82.4, CH | 3.43, d (10.0) | 5’’ | 1’, 5’’,6’’, 7’’ | 6’’, 7’’ |
| 5’’ | 71.0, CH | 3.69, dq (10.0, 6.0) | 4’’, 7’’ | 1’’, 3’’, 4’’, 7’’ | 7’’ |
| 6’’ | 26.6, CH3 | 1.47, s | 1’’, 2’’, 3’’, 4’’ | 4’’ | |
| 7’’ | 19.1, CH3 | 1.33, d (6.0) | 5’’ | 4’’, 5’’ | 5’’ |
| Sugar C, β−4-epi-vancosamine | |||||
| 1’’’ | 93.7, CH | 4.82, dd (9.6) | 2’’’ | 2’’’ | 5’’’ |
| 2’’’ | 41.7, CH2 | eq: 3.25, dd (13.8); ax: 1.26, dd (13.8, 9.6) |
1’, 2’eq 1’, 2’ax |
1’’’, 3’’’, 4’’’, 6’’’ | 6’’’ |
| 3’’’ | 57.6, C | ||||
| 4’’’ | 85.9, CH | 3.15, d (9.5) | 5’’’ | 1’’, 5’’’, 6’’’, 7’’’, 8’’’ | 6’’’, 7’’’ |
| 5’’’ | 71.0, CH | 3.84, dq (9.5, 6.1) | 4’’’, 7’’’ | 1’’’, 3’’’, 4’’’, 7’’’ | |
| 6’’’ | 25.3, CH3 | 1.42, s | 1’’’, 2’’’, 3’’’, 4’’’ | 2’’’, 4’’’ | |
| 7’’’ | 18.7, CH3 | 1.27, d (6.1) | 5’’’ | 1’’’, 4’’’, 5’’’ | 4’’’ |
| 8’’’ | 176.0, C | ||||
| 9’’’ | 34.1, CH2 | 2.47, t (7.0) | 10’’’ | 8’’’, 10’’’, 11’’’ | |
| 10’’’ | 34.9, CH2 | 2.36, t (7.0) | 9’’’ | 8’’’, 9’’’, 10’’’ | |
| 11’’’ | 178.9. C | ||||
Figure 6.

MS/MS fragmentation of 4 and 5.
The coupling constants (9.0, 1.7 Hz) of H-1’, the large coupling constant between H-5’ and H-4’ (10 Hz), as well as the ROESY correlation between H-1’ and H-5’ indicated the same relative configuration of sugar A (β-amicetose) in compound 4 as had been found for compound 1. The 13C NMR chemical shifts of C-1’ and C-4’’ of compound 4 were almost identical to those of compound 1, suggesting the cis relationship between C-1’ and C-4’’. H-4’’ and H-5’’ were assigned in axial positions on the basis of the large coupling constant (10 Hz) observed between H-4’’ and H-5’’. Comparison of the 13C NMR shifts of C-1’ and C-4’’ of compound 4 with those of compound 1 suggested the methyl group attached to C-3 was axial. Moreover, the large coupling constants of between H-1’’ and H-2’’ax (J = 10.0 Hz) and between H-1’’’ and H-2’’’ax (J = 9.6 Hz) allowed both the anomeric protons H-1’’ and H-1’’’ to be assigned as axial. The other amino sugar (sugar C), was assigned the same relative configuration as sugar B in 4, based on comparisons of the 1H and 13C NMR data. Consequently, both amino sugars were assigned as β−4-epi-vancosamine. The positive CE at 270 nm from the ECD data of 4 suggested the absolute configuration of C-4’ was the same as that of 1. On the basis of the relative configuration established for 4, the absolute configurations of the sugar moieties in 4 were proposed as d-β-amicetose and l-β−4-epi-vancosamine, which was also in consistent with the biosynthetic considerations.
Phallusialide E (5) was found to have the molecular formula C24H37ClN2O8, as deduced from HRESIMS data. Three characteristic fragment ions at m/z 385, 276 and 242 were observed in the ESI- MS/MS data (Figure 5). The daughter ions at m/z 385 and 242 indicated that 5 has the same partial structure (2-chloro-1H-pyrrole-4-carboxylic acid attached to sugars A and B) as that of compound 4, which was further confirmed by comparisons of NMR data for 4 and 5. The remaining unit of 5 with the molecular formula C6H12O3 was deduced by COSY and HMBC data, which revealed the remaining moiety to be amicetose. The HMBC correlations from H-4’’’ to C-1’’, and the ROESY correlation between H-4’’’ and H-1’’ strongly suggested attachment of sugar C unit to C-1’’, thus completing the planar structure of compound 5.
The 13C NMR chemical shifts of the sugar units A and B of 5 were almost identical to those of 4, suggesting that these two units have the same relative configurations as those in 4. The ROESY correlation between H-1’’’ and H-5’’’, together with the large coupling constant observed between H-4’’’ and H-5’’’, indicated that sugar C in 5 possesses the same relative configuration as the sugar A unit of 4. Moreover, the configuration of C-4’ in 5 was determined on the basis of ECD data comparisons, therefore establishing the sugars in 5 as d-β-amicetose (sugar C) and l-β−4-epi-vancosamine (sugar B).
Compounds 1−5 were tested for antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) and Escherichia coli. Both compounds 1 and 2 showed antibacterial activity against MRSA with the MIC value of 32 μg/mL, and against E. coli with an MIC value of 64 μg/mL; compounds 3−5 failed to show any detectable antibacterial activities against either organism even at concentrations up to 256 μg/mL. The absence of antimicrobial activity observed for nonhalogenated 3 suggests that halogenation (Cl or Br) at the pyrrole C-4 (of 1−3) is crucial to the antibacterial activities of 1 and 2. Despite their chlorinated pyrrole moieties, neither compound 4 or 5 demonstrated any antibacterial activity, suggesting that the extra sugar moieties modulate the biological activities. Alternatively, it may be the case that the extra sugars of 4 and 5 prevent their uptake by bacterial cells. Taken together, these findings provide early glimpses into the SAR for these new natural products.
In conclusion, employing a combination of MS-based metabolomics approaches, five new pyrrole-derived alkaloids phallusialides A−E (1−5) were identified from a marine-derived bacterium Micromonospora sp. bacterium; compounds 1 and 2 were found to have moderate antibacterial activity. Compounds 1−5 contain a 2-carboxy-pyrrole aglycone linked to 2−3 unusual sugars, which is rare in secondary metabolites. The most structurally related compounds reported thus far include 2,4-dimethyl-3-pyrrolycarbonyl α-L-rhamnopyranoside, 2-pyrrolycarbonyl α-l-rhamnopyranoside,20 salaciaglycoside A,21 β-d-glucopyranos-1-yl N-methylpyrrole-2-carboxylate,22 and pyrrol-2-ylcarbonyl 6-deoxy-α-l-talopyranoside.23 Notably, in all of these known cases, the linkage between the sugar moiety and the aglycone was via an anomeric carbon. In contrast, the sugar moieties in 1−5 are attached to the respective aglycones via a C-4 linkage. An LC-MS-PCA approach was extremely useful in allowing us to distinguish unique compounds among large number of metabolites, whereas MS/MS molecular networking proved efficient for dereplicating known molecules and identifying new compound classes. This study demonstrates that merging LC-MS-PCA and MS/MS molecular networking provides a promising tool in the search for novel natural molecules.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were measured on a Perkin–Elmer 241 Polarimeter. UV spectra were recorded on an Aminco/OLIS UV-Vis Spectrophotometer. ECD spectra were recorded on an AVIV Model 420 Circular Dichroism Spectrometer. IR spectra were measured with a Bruker Equinox 55/S FT–IR Spectrophotometer. Both 1D and 2D NMR spectra were obtained using a Bruker Avance 600 MHz spectrometer with 1H{13C/15N} cryoprobe and a 500 MHz spectrometer with 13C/15N{1H} cryoprobe; chemical shifts were referenced to the residual solvent peaks (CD3OD: δH = 3.31, δC = 49.15). HRMS and MS/MS data were acquired with a Bruker MaXis 4G QTOF mass spectrometer. RP HPLC was performed using a Shimadzu Prominence HPLC system and a Phenomenex Luna C18 column (250 × 10 mm, 5 µm).
Biological Material.
WMMA-2495 was isolated from the tunicate Phallusia nigra, which was collected on 02/17/15, in the Florida Keys (24° 39’ 390” N, 81° 26’ 265”W). A voucher specimen is housed at the University of Wisconsin-Madison. For cultivation, a sample of tunicate (1 cm3) was rinsed with sterile seawater, macerated using a sterile pestle in a micro-centrifuge tube, and dilutions were made in sterile seawater, with vortexing between steps to separate bacteria from heavier tissues. Strain WMMA-2495 was purified from a Gauze 1 medium isolation plate.24
Sequencing.
Genomic DNA was extracted using the UltraClean Microbial DNA Isolation kit (Mo Bio Laboratories, Inc.). 16S rDNA genes were amplified using 100–200 ng genomic DNA template with the primers 8–27F (5’ to 3’ GAGTTTGATCCTGGCTCAG) and 1492R (5’ to 3’ GGTTACCTTGTTACGACTT). The following PCR conditions were used: 94 °C for 5 min, followed by 30 cycles of 94 °C for 30 s, 55 °C for 1 min, 72 °C for 1.5 min, with a final step of 72 °C for 5 min. The PCR bands were excised from the gel and purified using the QIAquick Gel Extraction kit (QIAGEN). A 1 μL sample of purified product was sequenced. Sequencing reactions were performed by the UW Biotechnology Center and reactions were sequenced with an ABI 3730xl DNA Analyzer. WMMA-2495 was identified as a Micromonospora sp., and its 16s gene sequence was deposited to GenBank and assigned the accession number MK765852.
Fermentation, Extraction, and Isolation.
Three 10 mL seed cultures (25 × 150 mm tubes) in medium ASW-A (20 g soluble starch, 10 g glucose, 5 g peptone, 5 g yeast extract, 5 g CaCO3 per liter of artificial seawater) were inoculated with strain WMMA-2495 and agitated at 200 RPM for 14 d at 28 °C. For making artificial seawater, solution I (415.2 g NaCl, 69.54 g Na2SO4, 11.74 g KCl, 3.40 g NaHCO3, 1.7 g KBr, 0.45 g H3BO3, 0.054 g NaF) and II (187.9 g MgCl2•6H2O, 22.72 g CaCl2•2H2O, 0.428 SrCl2•6H2O) were made up separately, and combine to give a total volume of 20 L. Two-liter flasks (6 × 500 mL) containing ASW-A medium with Diaion HP20 (7% by weight) were inoculated with 10 mL from the culture tube and shaken at 200 rpm at 28 °C for 14 days. Filtered HP20 was washed with distilled H2O and extracted with acetone. The acetone extract (10 g) was subjected to a liquid-liquid partitioning using 30% aqueous CH3OH and CHCl3 (1:1). The CHCl3-soluble partition (0.8 g) was fractionated by Sephadex LH20 column (3 cm diam. x 45 cm tall) chromatography (CHCl3:CH3OH, 1:1). The fractions containing 1 and 3 were further subjected to RP HPLC (10–70% CH3OH-H2O with H2O containing 0.1% acetic acid over 30 min, 4.0 mg/mL) using a Phenomenex Luna C18 column (250 × 10 mm, 5 µm), yielding 1 (14 mg, tR 23.1 min) and 3 (1.0 mg, tR 19.3 min). The fractions containing 4 and 5 were further subjected to RP HPLC (20–75% CH3OH-H2O with H2O containing 0.1% acetic acid over 30 min, 4.0 mg/mL) using a Phenomenex Luna C18 column (250 × 10 mm, 5 µm), yielding 4 (4.6 mg, tR 24.9 min) and 5 (2.0 mg, tR 25.9 min). For the incorporation of bromine and the production of 2, the same procedure was used with 2 L flasks (2 × 500 mL) containing medium ASW-A (with an increase of KBr from 0.1 g/L to 10 g/L and elimination of 20 g/L NaCl). The LH20 fractions containing 2 were further subjected to RP HPLC (10–70% CH3OH-H2O with H2O containing 0.1% acetic acid over 30 min, 4.0 mg/mL) using a Phenomenex Luna C18 column (250 × 10 mm, 5 µm), yielding 2 (4.6 mg, tR 22.3 min).
Phallusialide A (1):
colorless oil; [α]25D +21 (c 0.14, CH3OH); UV (CH3OH) λmax (log ε) 205 (3.63), 232 (3.56), 271 (4.86) nm; ECD (c 0.1, CH3OH), λmax (Δε) 271 nm (6.7); IR (ATR) υmax 3356.9, 2979.3, 2950.9, 2835.9, 1701.7, 1642.8, 1564.3, 1448.9, 1394.9, 1328.2, 1163.6, 1124.9, 1067.1, 1021.0 cm−1; 1H and 13C NMR, Table 1; HRESIMS m/z 403.1635 [M + H]+ (calcd for C18H28ClN2O6+, 403.1630).
Phallusialide B (2):
colorless oil; [α]25D +13 (c 0.4, CH3OH); UV (CH3OH ) λmax (log ε) 204 (3.12), 233 (3.20), 272 (3.39) nm; ECD (c 0.17, CH3OH), λmax (Δε) 269 nm (13.3); IR (ATR) υmax 3348.9, 2971.9, 2935.3, 2881.8, 1693.9, 1564.1, 1409.9, 1391.6, 1324.8, 1162.4, 1129.1, 1066.3, 1029.0, 951.3 cm−1; 1H and 13C NMR, Table S3; HRESIMS m/z 447.1123 [M + H]+ (calcd for C18H28BrN2O6+, 447.1125).
Phallusialide C (3):
colorless oil; [α]25D +16 (c 0.1, CH3OH); UV (CH3OH) λmax (log ε) 206 (3.42), 229 (3.33), 271 (3.62) nm; ECD (c 0.11, CH3OH), λmax (Δε) 270 nm (7.7); IR (ATR) υmax 3349.4, 2980.7, 2953.4, 2838.2, 1658.0, 1563.4, 1409.7, 1162.0, 1020.0 cm−1; 1H and 13C NMR, Table S4; HRESIMS m/z 369.2021 [M + H]+ (calcd for C18H29N2O6+, 369.2020).
Phallusialide D (4):
colorless oil; [α]25D +8 (c 0.4, CH3OH); UV (CH3OH) λmax (log ε) 205 (3.29), 233 (3.27), 271 (3.49) nm; ECD (c 0.09, CH3OH), λmax (Δε) 233 (4.7), 269 nm (3.2); IR (ATR) υmax 3345.5, 2947.3, 2946.3, 2835.9, 1650.7, 1562.8, 1450.3, 1393.1, 1329.1, 1194.1, 1163.4, 1127.3, 1019.1 cm−1; 1H and 13C NMR, Table 2; HRESIMS m/z 646.2740 [M + H]+ (calcd for C29H45ClN3O11+, 646.2737).
Phallusialide E (5):
colorless oil; [α]25D +12 (c 0.1, CH3OH); UV (CH3OH) λmax (log ε) 204 (3.22), 232 (3.22), 271 (3.46) nm; ECD (c 0.12, CH3OH), λmax (Δε) 269 nm (8.3); IR (ATR) υmax 3347.6, 2971.5, 2935.1, 2883.0, 1702.8, 1659.2, 1573.3, 1410.6, 1380.1, 1310.0, 1161.9, 1129.7, 1066.5, 952.1 cm−1; 1H and 13C NMR, Table S5; HRESIMS m/z 517.2320 [M + H]+ (calcd for C24H38ClN2O8+, 517.2311).
Conformational Analysis and ECD Calculations.
Systematic conformational analysis for compound 1 was performed using the Spartan 14 software package using the MMFF molecular mechanics force field calculation. Conformers occurring within a 10 kcal/mol energy window from the global minimum were chosen for geometry optimization and energy calculation using DFT with the B3LYP functional and the 6–31G(d) basis set with the Gaussian09 program. TD-DFT with the basis set B3LYP/6–31G(d) was used to calculate the spin-allowed excitation energies and rotatory (Rn) and oscillator strengths (fn) of the lowest 50 excited states. The overall ECD spectra were then generated according to Boltzmann weighting of each conformer. All quantum computations were performed using the Gaussian09 package, performed on a Dell Precision T5500 Linux workstation with a Xeon processor (3.3 GHz, 6-core).
Molecular Modeling and DFT calculations.
Molecular modeling calculations were performed on a Dell Precision T5500 Linux workstation with a Xeon processor (3.3 GHz, 6-core). Equilibrium conformers of the 1a and 1b using Spartan 14 software (MMFF, 10000 conformers examined). The conformer for each compound was analyzed using Gaussian 09 for geometry optimization and NMR calculations (B3LYP/6-31 G(d,p)). NMR shifts were referenced to TMS and benzene using the multi-standard approach.25 Molecules were modeled in the gas phase. The chemical shifts were compared using the DP4 probability method.26
Antibacterial Assay.
Compounds 1−5 were tested for antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) (ATCC #33591), and E. coli (ATCC #25922), and MICs were determined using a dilution antimicrobial susceptibility test for aerobic bacteria.27 Compounds 1−5 were dissolved in DMSO, and serially diluted to 10 concentrations (0.5–256 μg/mL) in a 96-well plate. Vancomycin was used as a positive control and exhibited an MIC of 0.25 μg/mL against MRSA. Gentamicin was used as a positive control and exhibited an MIC of 4 μg/mL against E. coli. Compounds 1−5 and positive controls were tested in triplicate. On each plate, there were six untreated media controls. The plates were incubated at 35 °C for 18 h. The MIC was defined as the lowest concentration that inhibited visible growth of bacteria.
Molecular Networking.
The acetone extract of WMMA-2495 was analyzed with LC-MS and LC-MS/MS. A molecular network was created using the online workflow at GNPS (gnps.ucsd.edu). The spectra in the network were searched against all GNPS spectral libraries. The library spectra were filtered in the same manner as the input data. Results were visualized in Cytoscape 3.4.0 (www.cytoscape.org).
Supplementary Material
ACKNOWLEDGMENT
This work was supported by funding from the University of Wisconsin-Madison School of Pharmacy. This work was also funded by the NIH, grant R01 GM104192. We thank the Analytical Instrumentation Center (AIC) at the University of Wisconsin-Madison School of Pharmacy for the facilities to acquire spectroscopic data, especially MS data. This study made use of the National Magnetic Resonance Facility at Madison (NMRFAM), which is supported by NIH grants P41RR02301 (BRTP/NCRR) and P41GM66326 (NIGMS).
Footnotes
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: Spectroscopic data including 1D and 2D NMR, HRMS, MS/MS and ECD spectra of compounds 1−5.
References:
- (1).Hou Y; Braun DR; Michel CR; Klassen JL; Adnani N; Wyche TP; Bugni TS Anal. Chem 2012, 84, 4277–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chanana S; Thomas CS; Braun DR; Hou Y; Wyche TP; Bugni TS Metabolites 2017, 7, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kersten RD; van der Voort M; Pogliano K; Gross H; Raaijmakers JM; Moore BS; Laskin J; Bandeira N; Dorrestein PC Proc. Natl. Acad. Sci. U. S. A 2012, 109, E1743− E1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bouslimani A; Sanchez LM; Garg N; Dorrestein PC Nat. Prod. Rep 2014, 31, 718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan T; Porto C; Bouslimani A; Melnik AV; Meehan MJ; Liu W-T; Crüsemann M; Boudreau PD; Esquenazi E; Sandoval-Calderon M; Kersten RD; Pace LA; Quinn RA; Duncan KR; Hsu C-C; Floros DJ; Gavilan RG; Kleigrewe K; Northen T; Dutton RJ; Parrot D; Carlson EE; Aigle B; Michelsen CF; Jelsbak L; Sohlenkamp C; Pevzner P; Edlund A; McLean J; Piel J; Murphy BT; Gerwick L; Liaw C-C; Yang Y-L; Humpf H-U; Maansson M; Keyzers RA; Sims AC; Johnson AR; Sidebottom AM; Sedio BE; Klitgaard A; Larson CB; Boya P CA; TorresMendoza D; Gonzalez DJ; Silva DB; Marques LM; Demarque DP; Pociute E; O’Neill EC; Briand E; Helfrich EJN; Granatosky EA; Glukhov E; Ryffel F; Houson H; Mohimani H; Kharbush JJ; Zeng Y; Vorholt JA; Kurita KL; Charusanti P; McPhail KL; Nielsen KF; Vuong L; Elfeki M; Traxler MF; Engene N; Koyama N; Vining OB; Baric R; Silva RR; Mascuch SJ; Tomasi S; Jenkins S; Macherla V; Hoffman T; Agarwal V; Williams PG; Dai J; Neupane R; Gurr J; Rodríguez AMC; Lamsa A; Zhang C; Dorrestein K; Duggan BM; Almaliti J; Allard P-M; Phapale P; Nothias L-F; Alexandrov T; Litaudon M; Wolfender J-L; Kyle JE; Metz TO; Peryea T; Nguyen D-T; VanLeer D; Shinn P; Jadhav A; Müller R; Waters KM; Shi W; Liu X; Zhang L; Knight R; Jensen PR; Palsson BØ; Pogliano K; Linington RG; Gutierrez M; Lopes NP; Gerwick WH; Moore BS; Dorrestein PC; Bandeira N Nat. Biotechnol 2016, 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr., Peralta JE; Ogliaro F; Bearpark M, Heyd JJ, Brothers E, Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam NJ; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision A.1. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- (7).Katritzky AR; Akhmedov NG; Doskocz J; Mohapatra PP; Hall CD; Güven A Magn. Reson. Chem 2007, 45, 532–545. [DOI] [PubMed] [Google Scholar]
- (8).Helay SE; Kulik A; Zinecker H; Ramachandaran K; Tan GYA; Imhoff JF; Sussmuth RD; Fiedler H; Sabaratnam V J. Nat. Prod 2012, 75, 1018–1024. [DOI] [PubMed] [Google Scholar]
- (9).Aoki M; Shirai H; Nakayama N; Itezono Y; Mori M; Satou T; Ohshima S; Watanabe J; Yokose K J. Antibiot 1991, 44, 635–645. [DOI] [PubMed] [Google Scholar]
- (10).Martin GDA; Tan LT; Jensen PR; Dimayuga RE; Fairchild CR; Raventos-Suarez C; Fenical W J. Nat. Prod 2007, 70, 1406–1409. [DOI] [PubMed] [Google Scholar]
- (11).Johnson AW; Smithand RM; Guthrie RD J. Chem. Soc., Perkin Trans. I 1972, 2153–2159. [DOI] [PubMed] [Google Scholar]
- (12).Debono M; Molloy M J. Org. Chem 1980, 45, 4685–4687. [Google Scholar]
- (13).Hunt AH; Molly RM; Debono M; Occolowitz JL Tetrahedron Lett. 1988, 29, 1223–1226. [Google Scholar]
- (14).Smith SG; Goodman JM J. Org. Chem 2009, 74, 4597–4607. [DOI] [PubMed] [Google Scholar]
- (15).Furche F; Ahlrichs R; Wachsmann C; Weber E; Sobanski A; Vogtle F; Grimme S J. Am. Chem. Soc 2000, 122, 1717–1724. [Google Scholar]
- (16).Ding Y; Li XC; Ferreira D J. Org. Chem 2007, 72, 9010–9017. [DOI] [PubMed] [Google Scholar]
- (17).Eustaquio AS; Pojer F; Noel JP; Moore BS Nat. Chem. Biol 2008, 4, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wagner C; EI Omari M; König GM J. Nat. Prod 2009, 72, 540–553. [DOI] [PubMed] [Google Scholar]
- (19).Wyche TP; Standiford M; Hou Y; Braun D; Johnson DA; Johnson JA; Bugni TS Mar. Drugs 2013, 11, 5089–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Grond S; Langer H; Henne P; Sattler I; Thiericke R; Grabley S; Zähner H; Zeeck A Eur. J. Org. Chem 2000, 929–937. [Google Scholar]
- (21).Yao F; Chen Y; Zou Y; Ye Q; Lv S; Chen W; Liang L; Sun L Phytochem. Lett 2017, 19, 263–265. [Google Scholar]
- (22).Pailee P; Prachyawarakorn V; Mahidol C; Ruchirawat S; Kittakoop P Eur. J. Org. Chem 2011, 3809–3814. [Google Scholar]
- (23).Bitzer J; Zeeck A Eur. J. Org. Chem 2006, 3661–3666. [Google Scholar]
- (24).Gauze GF, Preobrazhenskaya JP, Kudrina EE, Blinov N0, Ryabova ID, and Sveshnikova MA. 1957. Problems in the classification of antagonistic actinomycetes. State Publishing House for Medical Literature, Medgiz, Moscow. [Google Scholar]
- (25).Sarotti AM; Pellegrinet SC J. Org. Chem 2009, 74, 7254–7260. [DOI] [PubMed] [Google Scholar]
- (26).Smith SG; Goodman JM J. Am. Chem. Soc 2010, 132, 12946–12959. [DOI] [PubMed] [Google Scholar]
- (27).National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 7th ed; NCCLS: Villanova, PA, USA, 2006; Approved standard M7–A7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.