

Abstract
Chemical modifications of histone tails influence genome accessibility and the transcriptional state of eukaryotic cells. Lysine acetylation is one of the most common modifications and acetyllysine‐binding bromodomains (BDs) provide a means for acetyllysine marks to be translated into meaningful cellular responses. Here, we have investigated the mechanism underlying the reported association between the Bromodomain and Extra Terminal (BET) family of BD proteins and the essential histone variant H2A.Z. We use NMR spectroscopy to demonstrate a physical interaction between the N‐terminal tail of H2A.Z and the BDs of BRD2, BRD3, and BRD4, and show that the interaction is dependent on lysine acetylation in H2A.Z. The BDs preferentially engage a diacetylated H2A.Z‐K4acK7ac motif that is reminiscent of sequences found in other biologically important BET BD target proteins, including histones and transcription factors. A H2A.Z‐K7acK11ac motif can also bind BET BDs—with a preference for the second BD of each protein. Chemical shift perturbation mapping of the interactions, together with an X‐ray crystal structure of BRD2‐BD1 bound to H2A.Z‐K4acK7ac, shows that H2A.Z binds the canonical AcK binding pocket of the BDs. This mechanism mirrors the conserved binding mode that is unique to the BET BDs, in which two acetylation marks are read simultaneously by a single BD. Our findings provide structural corroboration of biochemical and cell biological data that link H2A.Z and BET‐family proteins, suggesting that the function of H2A.Z is enacted through interactions with these chromatin readers.
Keywords: acetylation, BET family proteins, BRD2, BRD3, BRD4, epigenetics, H2A.Z, histone
Short abstract
PDB Code(s): 7JX7;
1. INTRODUCTION
The reversible acetylation of lysine sidechains in histones and other transcriptional regulators is an important aspect of transcriptional control in eukaryotic organisms. Histone lysine acetylation is predominantly associated with gene activation and, conversely, silenced areas of the genome carry low levels of histone acetylation. 1 , 2 It is thought that the mechanism by which acetylated lysines impact gene expression relies to a large extent on the recognition of these marks by “reader” domains on transcriptional regulatory proteins. Bromodomains (BDs) are the most abundant domains that are capable of specifically engaging acetyllysine (AcK) residues. 3 BDs are widespread in proteins that control the expression of essential developmental and regulatory genes. 4 Not surprisingly, dysregulation of BD function and mutation of BD‐containing proteins have been associated with several chronic disease states such as cancer and inflammation. 5
The Bromodomain and Extra Terminal (BET) family of BD‐containing proteins has received much attention for their promise as drug targets for a wide range of human diseases. 6 , 7 The BET family in eukaryotes comprises four members; BRD2, BRD3, and BRD4 are widely expressed (Figure 1a), whereas BRDT displays testes‐specific expression. Each of the BET proteins contains two tandem BDs separated by a ~100–150‐residue linker, and both domains have been shown to bind AcK in a deep pocket. A characteristic feature of these domains is that they preferentially bind diacetylated motifs—sequences that contain two AcK residues, most commonly in an AcK‐X‐X‐AcK arrangement. For example, diacetylation of the N‐terminal tail of histone H4 (K5acK8ac) drives the recruitment of BRD4 and BRDT to chromatin, resulting in the compaction of chromatin. 8 Lysine acetylation is also emerging as a common feature of many DNA‐binding transcription factors, including GATA‐family transcription factors, TWIST1, and E2F1. 9 , 10 , 11 , 12 , 13 Diacetylation of GATA1 (K312acK315ac) creates a motif that binds BRD3‐BD1, an interaction that is crucial for GATA1 chromatin occupancy and erythroid maturation. 9 , 10 , 14
Histone variants are an important additional layer of regulatory control for the eukaryotic genome. Approximately 20 variants of core histone genes are known, and these proteins play diverse roles in genome biology. 15 Although these variants can have preferential distributions in the genome (e.g., H2A.X is central to the DNA damage response) and their deposition into nucleosomes can dramatically alter higher order chromatin structure and in turn gene transcription, relatively little is known about the biochemical basis for their activity. 16
H2A.Z is one of the most studied histone variants and is upregulated in several human diseases such as cancer. 17 , 18 , 19 It is found across Eukarya and shares 60% sequence identity with the canonical H2A. 20 , 21 Interestingly, H2A.Z is the only histone variant that is obligate for viability and development in numerous organisms. 22 Two H2A.Z paralogues exist—H2A.Z.1 and H2A.Z.2—that differ by three amino acids in humans and emerging data suggests that these paralogues might have non‐redundant functions. 23 , 24 We will refer to the two paralogues together as H2A.Z, unless otherwise indicated. H2A.Z is found in nucleosomes at both promoter and enhancer regions and has also been connected with both activation and repression of gene expression in different contexts. 17 Recently, it has been realized that these contexts can be distinguished, at least in part, by post‐translational modifications (PTMs) of H2A.Z. Acetylation of the H2A.Z N‐terminal tail is correlated with the occupation of promoters and enhancers of active genes. 25 , 26 In contrast, ubiquitination of H2A.Z in gene bodies is correlated with transcriptional repression. 27 The mechanisms through which these modifications signal for gene activation are, however, unclear.
Several reports describe a connection between H2A.Z, specifically the H2A.Z.1 isoform, and the BET‐family protein BRD2. BRD2 co‐purifies with and is enriched at H2A.Z nucleosomes, particularly those at promoters that display H2A.Z acetylation. 18 Accordingly, it has been shown that BRD2 does not colocalize with an acetylation‐deficient H2A.Z mutant, suggesting an acetylation dependent interaction. 23 Similarly, Draker et al. demonstrated that BRD2 cooperates with H2A.Z to favor activation of androgen‐receptor (AR) related genes, and that interaction of H2A.Z nucleosomes with BRD2 is dependent on the AcK‐binding properties of the BRD2 BDs. 22 However, they were unable to detect a direct interaction between acetylated H2A.Z and BRD2.
Thus, unresolved questions remain regarding whether acetylated H2A.Z directly interacts with BRD2. To address these questions, we examined the ability of acetylated H2A.Z motifs (K4acK7ac and K7acK11ac) to bind the BDs of BRD2, as well as BRD3 and BRD4. Using 15N‐HSQC NMR spectroscopy, we demonstrate that H2A.Z binds the BET BDs in an acetylation dependent manner. The BET BDs preferentially bind the H2A.Z‐K4acK7ac acetylation pattern with weak affinities in the single digit mM range—affinities that are comparable to those measured for some other important BET BD‐acetylated histone interactions. H2A.Z‐K7acK11ac on the other hand selectively interacts with the second BD of each BET protein, although the interactions are weaker than that for the corresponding interactions with H2A.Z‐K4acK7ac. NMR chemical shift perturbation analysis demonstrates a conserved binding mode for both acetylation states to the canonical AcK binding pocket of the BET BDs. These data, together with an X‐ray crystal structure of BRD2‐BD2 in complex with H2A.Z‐K4acK7ac, confirm that the interactions closely mimic the mechanism of binding that has been characterized for BET‐BD interactions with histones and transcription factors. Our work supports the idea that H2A.Z activity might be mediated by direct interactions with BET BDs.
2. RESULTS
2.1. The bromodomains of BRD2 selectively bind acetylated H2A.Z peptides with a preference for a K4acK7ac modification pattern
To investigate the possibility of a direct interaction between the BDs of BRD2 and H2A.Z, we searched the literature for reports of H2A.Z acetylation sites. Residues K4, K7, and K11 of H2A.Z were found to be the most frequently reported positions of acetylation and, furthermore, doubly acetylated species (K4acK7ac and K7acK11ac) were present at significant abundance in vivo. 25 , 28 Given the known preference of BET‐BDs for Kac‐X‐X‐Kac motifs, we therefore decided to assess the ability of BRD2‐BD1 and BRD2‐BD2 to bind a H2A.Z (1–15) peptide bearing (a) no acetylation (H2A.Z‐unacetylated), (b) acetylation on K4 and K7 (H2A.Z‐K4acK7ac), or (c) acetylation on K7 and K11 (H2A.Z‐K7acK11ac).
Figure 2a,b show sections of 15N‐HSQC spectra of BRD2‐BD1 and BRD2‐BD2 during titrations with the indicated H2A.Z peptides. No chemical shift perturbations (CSPs) were observed upon addition of up to 10 molar equivalents of the unacetylated H2A.Z peptide (H2A.Z‐unacetylated) to either BD. In contrast, several signals in the spectra of both BD1 and BD2 showed clear CSPs upon addition of comparable quantities of H2A.Z‐K4acK7ac or H2A.Z‐K7acK11ac. For each BD, both peptides induced CSPs in the same subset of peaks and, furthermore, the directions of movement were conserved, suggesting a shared binding mode. All signals were in fast exchange and the CSPs for the peaks displaying the greatest movement were quantified and fit to a simple 1:1 binding model to calculate the affinities for each interaction (Table 1). Representative CSP data and fits for a single peak for each titration performed for BRD2‐BD1 and BRD2‐BD2 are shown in Figure 2c,d. K D values of 1.4 mM and 1.5 mM were obtained for the interaction of H2A.Z‐K4acK7ac with BRD2‐BD1 and BRD2‐BD2, respectively. A weaker affinity of 2.2 mM was measured for the interaction of H2A.Z‐K7acK11ac with BRD2‐BD2, whereas the K D for the same peptide with BRD2‐BD1 was estimated to be >10 mM.
FIGURE 2.
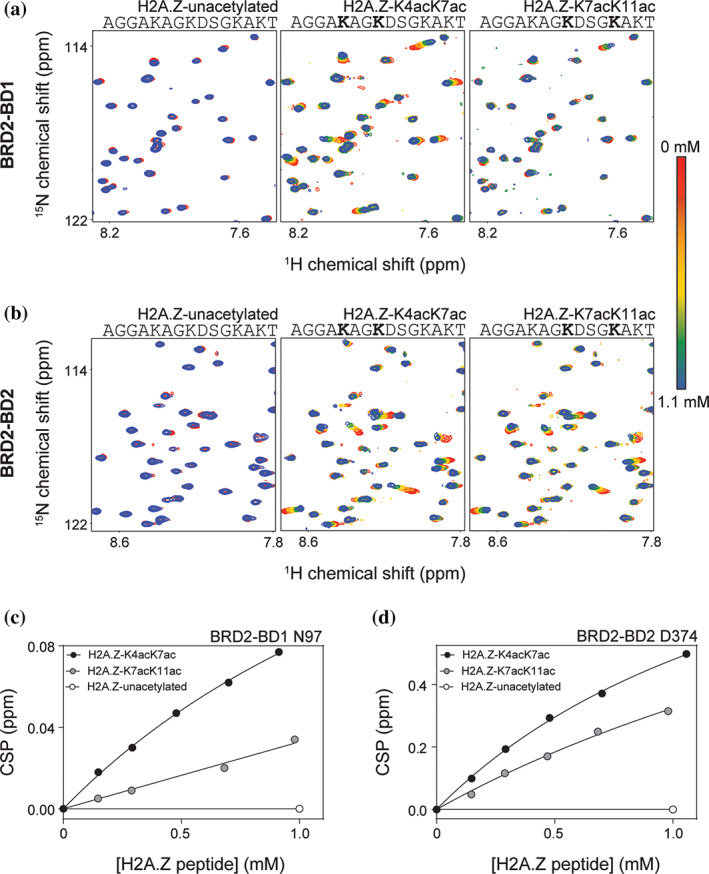
15N‐HSQC titration data for BRD2 BD‐H2A.Z peptide interactions. (a) 15N‐HSQC titrations monitoring the addition of the indicated H2A.Z peptides (up to 1.1 mM peptide—~10 molar equivalents) to BRD2‐BD1 (~100 μM). (b) 15N‐HSQC titrations monitoring the addition of the indicated H2A.Z peptides (up to 1.1 mM peptide—~10 molar equivalents) to BRD2‐BD2 (~100 μM). (c) Binding curves derived by tracking the 15N CSP in BRD2‐BD1 N97 in response to titration with the indicated H2A.Z peptide variants. The data were fitted to a simple 1:1 binding isotherm using GraphPad. (d). Binding curves derived by tracking the 15NCSP induced in BRD2‐BD2 D374 in response to titration with the indicated H2A.Z peptide variants. The data were fitted to a simple 1:1 binding isotherm using GraphPad
TABLE 1.
Affinities of H2A.Z I peptides for BET BDs as determined by 15N‐HSQC titration
| KD (mM) | |||||||
|---|---|---|---|---|---|---|---|
| Peptide | BRD2‐BD1 | BRD3‐BD1 | BRD4‐BD1 | BRD2‐BD2 | BRD3‐BD2 | BRD4‐BD2 | |
| H2A.Z I‐unacetylated | AGGAKAGKDSGKAKT | No binding observed | |||||
| H2A.Z I‐K4acK7ac | AGGAKAGKDSGKAKT | 1.4 | 2.1 | 1.2 | 1.5 | 1.6 | 1.2 |
| H2A.Z I‐K7acK11ac | AGGAKAGKDSGKAKT | >10 | 2.2 | 2.1 | 1.5 | ||
We note that the binding that we observed for all titrations performed did not reach saturation at the concentrations that we used in our experiments. As a result, it is difficult to precisely quantify the differences in affinity observed between H2A.Z‐K4acK7ac and H2A.Z‐K7acK11ac and also between H2A.Z‐K7acK11ac binding to BRD2‐BD1 and BRD2‐BD2. However, it is clear that H2A.Z‐K4acK7ac triggers greater CSPs in both BRD2‐BD1 and BRD2‐BD2 in comparison to H2A.Z‐K7acK11ac, consistent with a preference of the BRD2 BDs for the H2A.Z‐K4acK7ac motif.
Similarly, if we compare the CSPs that BRD2‐BD1 and BRD2‐BD2 undergo in response to addition of H2A.Z‐K7acK11ac, it is notable that the CSPs induced by the peptide are ~55% smaller than the CSPs induced by the H2A.Z‐K4acK7ac peptide for the BRD2‐BD1 titration (Figure 2c). On the other hand, the CSPs induced by H2A.Z‐K7acK11ac are only ~40% smaller than the CSPs induced by H2A.Z‐K4acK7ac in the BRD2‐BD2 titration, hinting at the possibility that the H2A.Z‐K7acK11ac peptide weakly prefers BRD2‐BD2 over BRD2‐BD1 (Figure 2d).
Together our data confirm that BRD2 BDs can bind directly to the N‐terminal tail of H2A.Z in a lysine‐acetylation dependent manner and further demonstrate a preference of these BDs for the H2A.Z‐K4acK7ac acetylation pattern.
2.2. The BDs from other BET‐family proteins display similar H2A.Z‐binding properties to BRD2
To assess the H2A.Z‐binding behavior of BRD3 and BRD4, the other broadly expressed BET family members, we performed the corresponding 15N‐HSQC titrations. Figure 3a–d show sections of 15N‐HSQC spectra for each titration and Figure 3e–h shows representative CSP data and fits for a single peak from each titration. The affinities calculated from the CSP data are given in Table 1. Both H2A.Z‐K4acK7ac and H2A.Z‐K7acK11ac induced sizeable CSPs to the same set of signals in each titration, whereas no significant CSPs were observed in any of the BD spectra upon addition of up to ~10 molar equivalents of the unacetylated peptide. Overall, the H2A.Z‐K4acK7ac peptide induced larger CSPs in all BDs. The K D values for the interaction between the peptide and BDs (~1.2–2.1 mM) were comparable to the affinities measured for the same peptide with the BRD2 BDs, indicating a family‐wide preference of the BET BDs for the H2A.Z‐K4acK7ac acetylation pattern.
FIGURE 3.
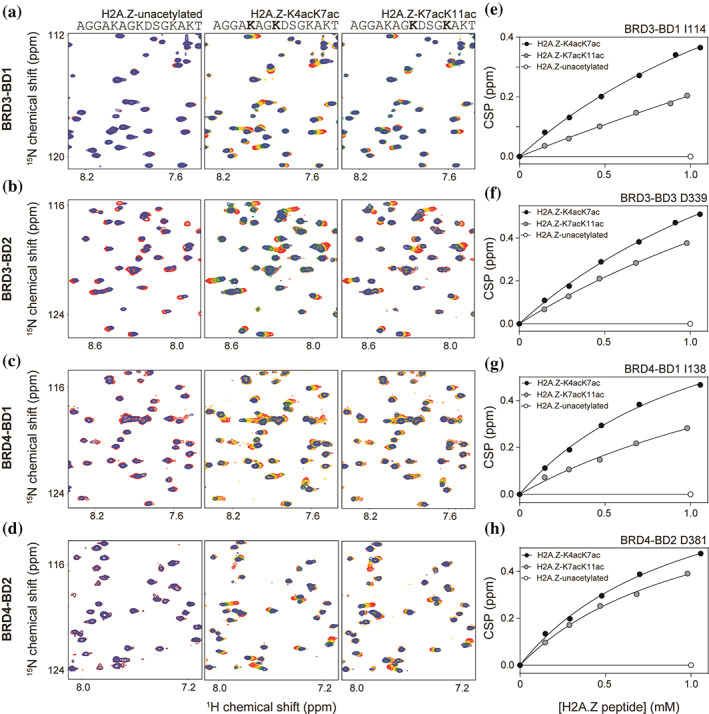
15N‐HSQC titration data for BRD3 and BRD4 BD‐H2A.Z interactions. (a–d) 15N‐HSQC titrations monitoring the addition of the indicated H2A.Z peptides (up to 1.1 mM peptide—~10 molar equivalents) to BRD3‐BD1 (a), BRD3‐BD2 (b), BRD4‐BD1 (c), and BRD4‐BD2 (d). The concentrations of BDs in each experiment are ~100 μM. (e‐h) Binding curves derived by tracking the 15NCSPs induced in BRD3‐BD1 I114 (e), BRD3‐BD2 D339 (f), BRD4‐BD1 I138 (g), and BRD4‐BD2 D381 (h), in response to titration with the indicated H2A.Z peptide variants. The data were fitted to a simple 1:1 binding isotherm using GraphPad
Interestingly, as observed for the BRD2 BD experiments, H2A.Z‐K7acK11ac displayed selectivity for BD2 domains over BD1 domains. K Ds of ~1.5–2.5 mM were measured for the interactions between H2A.Z‐K7acK11ac and the second bromodomains (BD2s) of BRD3 and BRD4, whereas the affinity for the peptide for the first bromodomains (BD1s) of BRD3 and BRD4 was estimated to be >10 mM. Consistent with these KD differences, the magnitudes of the CSPs induced by H2A.Z‐K7acK11ac for the BD1s were ~40–45% smaller than the CSPs induced for the same BDs in response to titration with H2A.Z‐K4acK7ac (Figure 3e,g). In contrast, the magnitudes of the CSPs induced by H2A.Z‐K7acK11ac for the BD2s were only ~20–30% smaller than the CSPs induced for the same BDs in response to titration with H2A.Z‐K4acK7ac (Figure 3f,g). Overall, the acetylated H2A.Z peptides demonstrate the same pattern of binding as observed for the BRD2 BDs.
2.3. Structural mapping indicates that H2A.Z‐K4acK7ac And H2A.Z‐K7acK11ac Bind BET BDs in the canonical AcK binding pocket
To delineate the mechanism of acetylated H2A.Z recognition, we made backbone assignments for each BET BD by collecting and analyzing triple resonance NMR spectra (HNCACB, CBCA[CO]NH, HNCO, and HN[CA]CO) using 15N/13C‐labeled BDs. 1H and 15N CSPs were then calculated and plotted for each BD (Figures S1 and S2) and mapped onto the structure of that domain (Figure 4). For the H2A.Z‐K4acK7ac titrations, the residues that experience the greatest CSPs are localized to the same section of each protein, suggesting a conserved binding mode of the peptide to the BET BDs (Figure 4a). These residues overlap closely with the canonical AcK binding pocket (Figure S3). Other surfaces of each BD show little to no CSP.
FIGURE 4.
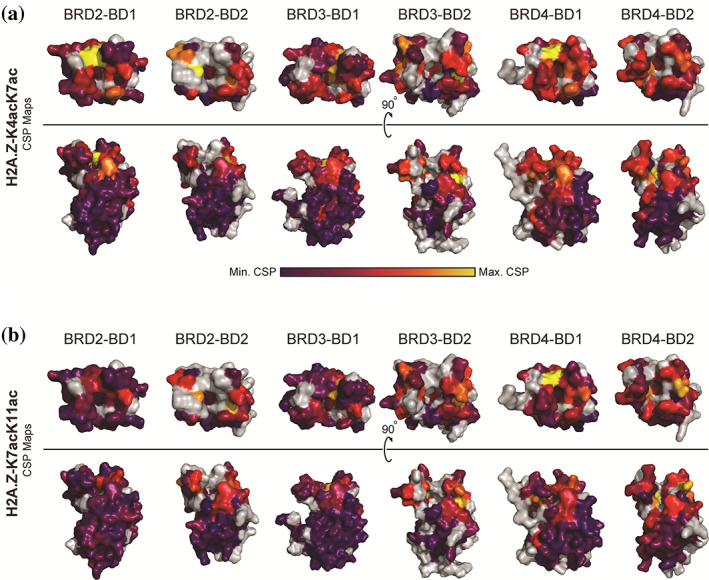
Structural mapping of the 1H and 15N CSP data for the interactions of BET BDs with H2A.Z‐K4acK7ac and H2A.Z‐K7acK11ac. (a) CSPs observed upon the formation of a complex with H2A.Z‐K4acK7ac are mapped onto the structures of the BET BDs (PDB IDs: 4UYF for BRD2‐BD1, 3ONI for BRD2‐BD2, 3S91 for BRD3BD1, 3S92 for BRD3‐BD2, 4LYI for BRD4‐BD1, and 5UVV for BRD4‐BD2). 29 , 30 , 31 CSP magnitude is displayed through a purple–red–yellow gradient in which purple indicates no CSP and yellow indicates the maximum CSPs. Unassigned residues are gray. (b) CSPs observed upon the formation of a complex with H2A.Z‐K7acK11ac are mapped onto the structures of the BET BDs (PDB IDs: 4UYF for BRD2‐BD1, 3ONI for BRD2‐BD2, 3S91 for BRD3BD1, 3S92 for BRD3‐BD2, 4LYI for BRD4‐BD1, and 5UVV for BRD4‐BD2). 29 , 30 , 31 CSP magnitude is displayed through a purple–red–yellow gradient in which purple indicates no CSP and yellow indicates the maximum CSPs. Unassigned residues are gray
Figure 4b shows that, although the CSPs are smaller for the H2A.Z‐K7acK11ac titrations, the pattern of changes closely mimics those observed for H2A.Z‐K4acK7ac. These data show that the canonical AcK pocket is the preferred binding site for both peptides.
2.4. The crystal structure of a BRD2‐BD2:H2A.Z‐K4acK7ac complex demonstrates that the interaction mechanism is conserved with other BD‐diacetylated peptide complexes
Lastly, to corroborate the binding mode of the preferred H2A.Z‐K4acK7ac acetylation state with the BET BDs, we used X‐ray crystallography to determine the structure of a complex formed between BRD2‐BD2 and H2A.Z‐K4acK7ac (Figure 5a, Table S1, PDB ID 7JX7). Molecular replacement using a structure of BRD2‐BD2 (PDB ID: 3ONI) as a search model revealed significant electron density in the canonical AcK binding site. 29 We were able to build the H2A.Z‐K4acK7ac peptide into this density iteratively through rounds of manual building and refinement and the final refined structure showed continuous density for residues 2–7 (Figure 5b). The electron density for the two AcK residues present in the structure was clear, allowing us to unambiguously determine the orientation of the peptide and identities of the two AcK residues.
FIGURE 5.
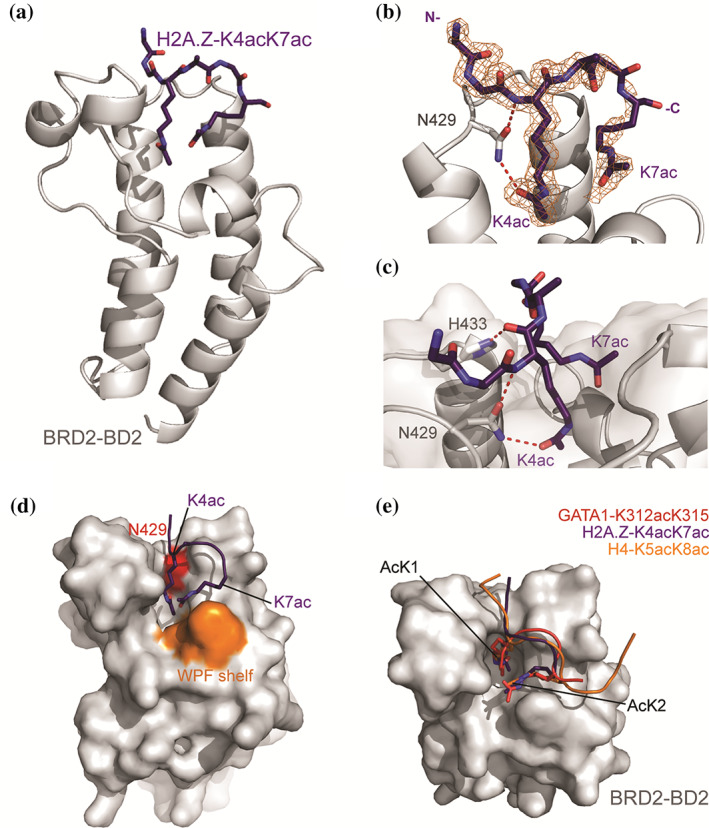
Crystal structure of BRD2‐BD2 in complex with H2A.Z.1‐K4acK7ac (1.75 Å resolution, PDB ID: 7JX7). (a) Ribbon diagram representation of BRD2‐BD2 (gray) in complex with H2A.Z.1‐K4acK7ac (purple, shown as sticks). (b) Close up of the interaction between H2A.Z.1‐K4acK7ac and BRD2‐BD2. The 2Fo‐Fc map for the H2A.Z.1‐K4acK7ac density is shown as an orange mesh. The conserved asparagine (N429) that forms a hydrogen bond with the AcK is shown as sticks, with the hydrogen bonds as red dashed lines. (c) Depiction of all hydrogen bonds formed between BRD2‐BD2 and the H2A.Z.1‐K4acK7ac. Hydrogen bonds are shown as red dashed lines. (d) Surface representation of BRD2‐BD2 highlighting the WPF shelf and conserved N429 residue in orange and red, respectively. The H2A.Z.1‐K4acK7ac peptide is shown as a cartoon with the two AcK residues shown as sticks. The C‐terminal AcK (K7ac) contacts the WPF shelf. (e) Overlay of H2A.Z.1‐K4acK7ac with the BD‐bound conformations of diacetylated GATA1 (K312acK315ac, PDB ID: 2L5E, red) and diacetylated H4 (K5acK8ac, PDB ID: 3UVW, orange)
The structure of BRD2‐BD2 in this complex is indistinguishable from previous structures of the same domain (RMSD over ordered Cα atoms = 0.4 Å with PDB 3ONI). The N‐terminal AcK (K4ac) of the H2A.Z‐K4acK7ac peptide occupies the canonical AcK binding pocket and the side chain acetyl group of K4ac forms the canonical hydrogen bond with the conserved BRD2‐BD2 N429 side chain amide (Figure 5b,c). The backbone amide of K4ac additionally forms a hydrogen bond with the sidechain carbonyl group of N429 (Figure 5c). Finally, H433 of BRD2‐BD2 participates in a hydrogen bond with the backbone carbonyl group of K4ac (Figure 5c). These interactions likely stabilize the position of K4ac in the AcK binding pocket. In turn, the C‐terminal AcK (K7ac) forms van der Waals interactions with the BET BD “WPF shelf” that is well known to mediate interactions with the second AcKs of diacetylated motifs from other proteins (Figure 5d).
Overlay of the conformations adopted by the diacetylated GATA1 (K312acK315ac) motif bound to BRD3‐BD1 and the diacetylated H4 (K5acK8ac) motif bound to BRD4‐BD1 shows that the AcK residues from each peptide take up near identical locations (Figure 5e). 9 , 32 The peptide backbones likewise share similar trajectories, diverging mainly at the less organized termini of the peptides. Overall, the interaction between H2A.Z‐K4acK7ac and BRD2‐BD2 displays many of the conserved contacts formed during several biologically relevant BET BD‐diacetylated histone and transcription factor motif interactions.
3. DISCUSSION
There has been considerable focus on the BET‐family proteins for their promise as therapeutic targets. Despite this focus, much remains unresolved regarding their biochemical activity. In this study, we have demonstrated that these proteins can recognize a specific pattern of diacetylation on the essential histone variant H2A.Z—a protein whose mechanism of action has also proven somewhat elusive.
3.1. The balance of H4 vs H2A.Z acetylation for BRD2 recruitment to H2A.Z nucleosomes
Although the binding that we measured for H2A.Z‐K4acK7ac and H2A.Z‐K7acK11ac was weak, the K D values are comparable to measured affinities for several important BET BD‐acetylated histone interactions that have been determined using NMR. 33 , 34 For example, 15N‐HSQC data reveal a K D of 2.9 mM for the interaction between BRD2‐BD2 and H4‐K12ac; this is the only acetylation mark on H4 that is critical for BRD2 engagement in vivo. 34 , 35 More recently, a study investigating the binding of H2A.Z.1 and H2A.Z.2 to BDs from diverse bromodomain‐containing protein families reports similarly K Ds (3 mM and 0.5 mM for the interaction between a H2A.Z‐K7acK13ac peptide and BRD4‐BD1 and BRD4‐BD2, respectively). 36
It has been suggested that H4 acetylation is the main determinant of BRD2 recruitment to H2A.Z nucleosomes, an idea that is in line with the known ability of the BET protein BRD4 to bind to H4‐K12ac and H4‐K5acK8ac. 8 , 22 , 35 However, a recent study demonstrated that recruitment of BRD2 to the H2A.Z‐containing promoters of genes that are upregulated during DNA repair is abrogated following the expression of a H2A.Z mutant that cannot be acetylated, arguing that a direct interaction of the type observed in the current study could play a role in BRD2 recruitment. 23 It is therefore possible that BRD2 recognizes additional properties on H2A.Z that augment its interaction with H2A.Z nucleosomes, though the relevant properties have not been identified.
It is worth noting that the presence of two BDs with AcK‐binding potential in BET proteins allows for both H4 and H2A.Z binding to occur simultaneously, and indeed there is evidence that combinations of multiple low‐affinity interactions can synergistically improve specificity, affinity and dynamics in the regulation of chromatin biology. 37 , 38 It is known that H2A.Z nucleosomes are enriched for H4 acetylation relative to H2A nucleosomes, and our data support the idea that BRD2 might therefore preferentially be recruited to nucleosomes in which both H2A.Z and H4 are acetylated. Furthermore, the interactions between BET family BDs and acetylated nucleosomes have recently been reported to be enhanced by direct interactions between the BDs and nucleosomal DNA itself. 39 In such a situation, BRD2 could effectively read out multiple nucleosome “signals” at the same time: acetylation of K4 and K7 or K7 and K11 on H2A.Z, acetylation of K5 and K8 or K12 of H4, nucleosomal DNA, and the very presence of the histone variant H2A.Z. 8 Interrogation of multivalent binding of this type represents an important direction for future analysis.
3.2. A conserved target recognition mode for BET‐family BDs
There are now 12 published examples of BET‐family BDs recognizing target proteins through a diacetylated AcK‐X‐X‐AcK motif (~40% of all published BET BD‐native ligand structures). 8 , 9 , 11 , 32 , 40 , 41 The preference for BET BDs to bind diacetylated sequences highlights the distinctive role played by BET‐family proteins in chromatin biology for their ability to recognise two PTMs via a single domain—and therefore read the acetylation of four lysines through its two BDs.
We found H2A.Z‐K4acK7ac diacetylation to be the preferred target motif for each of the tested BET BDs. It has been demonstrated that the sequence context of the AcK residues is important for BET‐BD recognition—the first position between the lysines is most often Ala or Gly, whereas the second position most prominently features Arg and Gly. H2A.Z conforms to this consensus (Figure 1b). 32 , 41 Given the high level of conservation of histone proteins, this observation further supports our proposal that recognition of the characteristic H2A.Z‐K4acK7ac diacetylation motif by BET‐family proteins is functionally relevant.
FIGURE 1.

Domain architecture of the BET family proteins used in this study and H2A.Z acetylation marks. (a) Domain topologies of the ubiquitously expressed human BET proteins BRD2, BRD3, and BRD4. The bromodomains (BDs), extra terminal domain (ET), and the C‐terminal domain of each protein are indicated. (b) Sequence comparison of the N‐terminal residues of human H2A.Z, H2A, and H4. The acetylation sites investigated in this study are boxed in gray. The AcK residues of the H4 diacetylation motif that is known to mediate an interaction with the BET BDs is also shown
Complexes have also been isolated for BET BDs bound to diacetylated sequences with more than two residues between the AcK moieties. In histone H4, two distinct diacetylation motifs with three residues between the AcKs (K12acK16ac and K16acK20ac) have been demonstrated to bind BRD4‐BD1 in a manner that positions the two AcK residues in the same locations (the first AcK hydrogen bonding with the conserved BD Asn and the second forming contacts near the WPF shelf) that are observed for AcK‐X‐X‐AcK type interactions (Figure S4). 32 This distinct diacetylation pattern resembles the H2A.Z‐K7acK11ac acetylation state investigated in this study. Although the binding of the H2A.Z‐K7acK11ac peptide was weaker than H2A.Z‐K4acK7ac, the affinities measured for H2A.Z‐K7acK11ac binding to BD2 domains are within the range of the K D values calculated for H2A.Z‐K4acK7ac. Taken together with our 15N‐HSQC CSP analysis, these data suggest that the H2A.Z‐K7acK11ac peptide might interact with the BET BD2s using a diacetylation‐dependent mechanism.
3.3. Specificity for H2A.Z in BET‐family proteins
BRD2, but not BRD3 or BRD4, was identified by mass spectrometry (MS) following the immunoprecipitation (IP) of FLAG‐tagged H2A.Z nucleosomes isolated from 293 T cells. 22 Related IP‐MS approaches in SKmel147 and HeLa Kyoto cells yielded the same outcome, strongly hinting at H2A.Z exhibiting a preference for BRD2 over BRD3 or BRD4. 18 , 42 On the other hand, a recent interactome analysis of BET‐family proteins identified H2A.Z as an interaction partner for both BRD2 and BRD3 (though not BRD4 and BRDT). 41 Our biophysical data indicate that the isolated BDs of all three of these BET proteins have a similar capacity to bind H2A.Z‐K4acK7ac, suggesting that BRD2 (and perhaps BRD3) might harbor additional sequence features that give rise to the observed specificity—possibly through the recognition of a third partner protein or stabilizing interactions of peripheral BET protein sequences with chromatin. It is notable that only BRD2 and BRD3 share a predicted coiled‐coil domain and that this domain contributes to BRD2 activity in cells that may aid in such interactions. 43 Additionally, BRD2 is unique in that both of its BDs are capable of interacting with nucleosomal DNA whereas only the BD2s of the remaining BET family proteins can bind DNA. 39 It is possible that the increased capacity of BRD2 to engage nucleosomal DNA compared to the other BET proteins many contribute to the specificity of the BRD2‐H2A.Z interaction observed in vivo.
Although H2A.Z‐K4acK7ac displayed no specificity for the BDs of BRD2, BRD3, BRD4, H2A.Z‐K7acK11ac bound preferentially to the BD2s of each BET BD we studied; K Ds with BD1s were weaker than 10 mM. Our data suggest that H2A.Z‐K7acK11ac might be a native BD2‐specific recognition motif. This finding is somewhat surprising given the high degree of structure and sequence conservation of the AcK binding pocket of the BET BDs. This similarity has been a notorious challenge for the development of paralogue selective BET BD inhibitors and very few BET protein or BD1 and BD2 selective inhibitors and native binding partners have been reported.
As both diacetylated H2A.Z‐K4acK7ac and H2A.Z‐K7acK11ac species are present at comparable abundances in vitro, it is possible that both modification patterns may play distinct but important roles in mediating an interaction with BRD2. 25 The selectivity we observe for H2A.Z‐K7acK11ac and the functional implications of both diacetylation patterns we studied here require further investigation at a cellular level.
Overall, our findings provide evidence for the direct interaction between acetylation H2A.Z motifs and the BD modules of the BET family proteins. While our data are confined to biophysical analysis of individual BDs and the N‐terminal tail of H2A.Z, they corroborate and extend a number of previous studies that have identified a connection between BRD2 and H2A.Z in a cellular context.
4. MATERIALS AND METHODS
4.1. Protein expression and purification
The BDs from human BRD2 (BD1: 65–194; BD2: 347–455), rat BRD3 (BD1: 25–147; BD2: 307–419), and human BRD4 (BD1: 42–168; BD2: 348–464) were expressed recombinantly using BL21(DE3) Escherichia coli (E. coli) cell as N‐terminal GST‐tagged fusion proteins. LB expression cultures were inoculated with starter cultures (1:100 dilution, grown overnight using colonies from fresh transformation plates). Expression cultures were incubated with shaking at 150 rpm at 37°C until an OD600 of ~0.6–0.8 was reached. The cultures were then cooled to room temperature and expression was induced with 0.25 mM IPTG. The cultures were subsequently transferred to 18°C for expression for a further ~20–24 h with shaking at 150 rpm before being harvested via centrifugation at 5,000g for 20 min. Cell pellets were stored at −20°C.
15N‐labeled and 15N/13C‐labeled proteins were expressed as described above until an OD600 of ~0.6–0.8 was reached. Cultures were then harvested and gently washed with M9 minimal media salts. The cultures were resuspended in minimal medium (half the volume of the original LB culture) supplemented with 15NH4Cl or 15NH4Cl + 13C‐glucose. Cultures were incubated at 18°C for ~1 h before expression was induced with 0.25 mM IPTG. Expression was allowed for a further ~20–24 h before harvesting and storage as described above.
A purification procedure consisting of GSH‐affinity chromatography, followed by HRV‐3C cleavage for GST‐tag removal, and size exclusion chromatography (SEC) was used to purify the BET BDs. Cells were lysed using sonication in a buffer composed of 50 mM Tris pH 7.2, 500 mM NaCl, 5 mM β‐mercaptoethanol, 0.1% v/v Triton X‐100, 1× complete EDTA‐free protease inhibitor, 10 μg/ml DNase I, 10 μg/ml RNase A, and 100 μg/ml lysozyme. Lysates were clarified via centrifugation at 18,000g for 30–60 min. The soluble fraction of the cell lysate was incubated with GSH‐Sepharose for ~3–4 h to isolate GST‐tagged proteins. Proteins bound to the GSH‐Sepharose were eluted using a buffer comprising 50 mM Tris pH 7.2, 150 mM NaCl, 10 mM reduced glutathione, and 5 mM β‐mercaptoethanol. The eluate was incubated with HRV‐3C protease overnight at 4°C to remove the GST tag. The cleaved protein sample was then concentrated to a volume below 5 ml and injected onto a HiLoad 16/600 Superdex 75 SEC column. The protein was eluted from the column in a buffer comprising 10 mM Tris pH 7.2, 100 mM NaCl, and 1 mM DTT. Protein containing fractions were pooled and concentrated before aliquoting. 15N‐labeled and 15N/13C‐labeled protein was concentrated to 5–10 mg/ml and unlabeled BRD2‐BD2 expressed for crystallography was concentrated to 10–15 mg/ml. Protein aliquots were snap frozen in liquid nitrogen and stored at −80°C.
The quality of protein purification was assessed using SDS‐PAGE analysis and monitoring UV absorbance at 280 nm.
4.2. Peptide synthesis
All peptides used in this study were synthesized at a nominal percentage purity of >80% by Ontores Biotechnologies (Hangzhou, China) with acetylation at the N‐termini and amidation at the C‐termini. The identity and purity of the proteins were assessed by Ontores Biotechnologies using RP‐HPLC and mass spectrometry, respectively.
4.3. NMR spectroscopy
BET BDs were diluted to a concentration of ~100 μM in preparation for titration with H2A.Z peptides. NMR spectra were acquired at 298 K using Bruker Avance III 600‐ or 800‐MHz NMR spectrometers fitted with TCI probe heads and using standard pulse sequences from the Bruker library. TOPSPIN3 (Bruker) and NMRFAM‐SPARKY were used for analysis of spectra. 44 Spectra were internally referenced to 10 μM 4,4‐dimethyl‐4‐silapentane‐1‐sulfonic acid.
Chemical shift assignments were made for all BET BDs using the standard triple resonance approach. CSP experiments were performed by collecting 15N‐HSQC spectra of 15N‐labeled BDs before and after addition of unlabeled peptide to the labeled BDs. Interactions were assessed by tracking the CSPs induced upon titration with H2A.Z peptides. CSP plots were generated by using the APES algorithm in NMRFAM‐SPARKY to generate 1H and 15N peak lists for 15N‐labeled BDs before and after addition of peptide. 44 The CSP tracking data were fit to a simple 1:1 binding model using GraphPad to calculate K D values for each interaction. The 1H and 15N CSPs were also plotted against BET BD residue number and mapped to the structures of each BD.
4.4. X‐ray crystallography
The BRD2‐BD2:H2A.Z‐K4acK7ac complex was crystalized via vapor‐diffusion. BRD2‐BD2 was mixed with 10 M equivalents of H2A.Z‐K4acK7ac and incubated on ice for 30 min prior to dispensing into crystallization trays. Crystallization was performed using various commercially available sparse matrix screens. The BRD2‐BD2:H2A.Z‐K4acK7ac mixture was dispensed into an MRC two‐drop, sitting drop 96‐well crystallization plate, using a Mosquito crystallization robot. Each condition was screen at a 1:1 and 2:1 protein to precipitant ratio (maintaining a final 300‐nl drop volume). The experiments were conducted at 18°C. Crystals took several weeks to grow in a solution containing 24% w/v PEG 1500, 20% w/v glycerol. Crystals of the complex were frozen by plunge‐freezing in liquid nitrogen.
X‐ray diffraction data were collected at the Australian Synchrotron using the Macromolecular Crystallography MX1 beamline (bending magnet) at 100 K and a wavelength of 0.9537 Å. 45 Data were collected using an ADSC QUANTUM 210r detector. XDS was used to index and integrate the data and the data were processed further using the CCP4i suite. 46 The data were scaled and merged using AIMLESS and the molecular replacement program PhaserMR was used to calculate the initial phases using an existing X‐ray structures of BRD2‐BD2 as the molecular replacement models (PDB ID: 3ONI). 47 COOT was used for manual model building and refinement was performed by iterative rounds of manual building in COOT followed by refinement using Phenix. 48 , 49 The quality of the final model was validated with the wwPDB server and submitted to the PDB (PDB ID: 7JX7). Structure diagrams were generated using PyMol (Schrödinger, Inc.). The data collection and refinement statistics for this structure are outlined in Table S1.
AUTHOR CONTRIBUTIONS
Karishma Patel: Formal analysis; investigation; methodology; visualization; writing‐original draft; writing‐review and editing. Paul Solomon: Formal analysis; visualization. James Walshe: Investigation; methodology. Jason Low: Methodology; supervision; writing‐review and editing. Joel Mackay: Conceptualization; project administration; resources; supervision; writing‐original draft; writing‐review and editing.
Supporting information
Data S1: The CSPs induced in the 15N‐HSQC titration of the BET BDs upon addition of ~10 M equivalents of the H2A.Z‐K4acK7ac peptide. The data have been normalized to the peak that displays the greatest CSP. Unassigned residues are indicated by red stars.
Figure S2: Plots of the CSPs induced in the 15N‐HSQC titration of each BET BD upon addition of ~10 M equivalents of the H2A.Z‐K7acK11ac peptide. The data have been normalized to the peak that displays the greatest CSP. Unassigned residues are indicated by red stars.
Figure S3: CSP data for the BRD3‐BD1:diacetylated GATA1 interaction highlighting the canonical BET BD binding pocket. The binding mode of a diacetylated GATA1 motif (K312acK315ac) interacting with BRD3‐BD1 (PDB ID: 2L5E)4 is shown. The backbone of the GATA1 peptide is depicted in blue, BRD3‐BD1 is depicted in gray, and the BRD3‐BD1 residues that undergo the greatest CSP upon titration with the diacetylated GATA1 peptide in a 15N‐HSQC titration are highlighted in red. The orientations shown match those shown in Figure 4.
Figure S4: Overlay of the H2A.Z‐K4acK7ac peptide from the structure of the complex with BRD2‐BD2 with the BD‐bound conformations of diacetylated H4 motifs that have the AcK residues separated by three amino acids (K12acK16ac—PDB ID: 3UVX; K16acK20ac—PDB ID: 3UVY).5 The orientation shown is similar to the upper panels of Figure 4a,b.
Table S1 Data collection and refinement statistics for the crystal structure of the BRD2‐BD2:H2A.Z‐K4acK7ac (PDB ID 7JX7) complex solved by molecular replacement.
ACKNOWLEDGEMENTS
This research was undertaken using MX1 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF). We acknowledge the Sydney Analytical Core Facility at the University of Sydney for providing access to NMR infrastructure.
Patel K, Solomon PD, Walshe JL, Low JKK, Mackay JP. The bromodomains of BET family proteins can recognize diacetylated histone H2A.Z. Protein Science. 2021;30:464–476. 10.1002/pro.4006
DATA AVAILABILITY STATEMENT
All data are provided in this manuscript and the supplementary material. The coordinates and structure factors for the BRD2‐BD2:H2A.Z‐K4acK7ac structure has been deposited to the Protein Data Bank (PDB accession code: 7JX7). The NMR backbone assignments for the BET BDs used in this study have been deposited to the Biological Magnetic Resonance Data Bank (BMRB accession codes are as follows: BRD2‐BD1: 50143, BRD2‐BD2: 50149, BRD3‐BD1: 50148, BRD3‐BD2: 50147, BRD4‐BD1: 50145, and BRD4‐BD2: 50146).
REFERENCES
- 1. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. [DOI] [PubMed] [Google Scholar]
- 2. You L, Nie J, Sun W‐J, Zheng Z‐Q, Yang X‐J. Lysine acetylation: Enzymes, bromodomains and links to different diseases. Essays Biochem. 2012;52:1–12. [DOI] [PubMed] [Google Scholar]
- 3. Mujtaba S, Zeng L, Zhou MM. Structure and acetyl‐lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. [DOI] [PubMed] [Google Scholar]
- 4. Fujisawa T, Filippakopoulos P. Functions of bromodomain‐containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol. 2017;18:246–262. [DOI] [PubMed] [Google Scholar]
- 5. Pervaiz M, Mishra P, Günther S. Bromodomain drug discovery – The past, the present, and the future. Chem Rec. 2018;18:1808–1817. [DOI] [PubMed] [Google Scholar]
- 6. Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hsu SC, Blobel GA. The role of bromodomain and extraterminal motif (BET) proteins in chromatin structure. Cold Spring Harb Symp Quant Biol. 2017;82:37–43. [DOI] [PubMed] [Google Scholar]
- 8. Morinière J, Rousseaux S, Steuerwald U, et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature. 2009;461:664–668. [DOI] [PubMed] [Google Scholar]
- 9. Gamsjaeger R, Webb SR, Lamonica JM, Billin A, Blobel GA, Mackay JP. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol Cell Biol. 2011;31:2632–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hung HL, Lau J, Kim AY, Weiss MJ, Blobel GA. CREB‐binding protein acetylates hematopoietic transcription factor GATA‐1 at functionally important sites. Mol Cell Biol. 1999;19:3496–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi J, Wang Y, Zeng L, et al. Disrupting the interaction of BRD4 with diacetylated twist suppresses tumorigenesis in basal‐like breast cancer. Cancer Cell. 2014;25:210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kouzarides T, Martínez‐Balbás MA, Bauer U‐M, Brehm A, Nielsen SJ. Regulation of E2F1 activity by acetylation. EMBO J. 2002;19:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu L, Chen Y, Mayakonda A, et al. Targetable BET proteins‐ and E2F1‐dependent transcriptional program maintains the malignancy of glioblastoma. Proc Natl Acad Sci U S A. 2018;115:E5086–E5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lamonica JM, Vakoc CR, Blobel GA. Acetylation of GATA‐1 is required for chromatin occupancy. Blood. 2006;108:3736–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martire S, Banaszynski LA. The roles of histone variants in fine‐tuning chromatin organization and function. Nat Rev Mol Cell Biol. 2020;21:522–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double‐stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. [DOI] [PubMed] [Google Scholar]
- 17. Giaimo BD, Ferrante F, Herchenröther A, Hake SB, Borggrefe T. The histone variant H2A.Z in gene regulation. Epigenet Chromatin. 2019;12:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vardabasso C, Gaspar‐Maia A, Hasson D, et al. Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol Cell. 2015;59:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Domaschenz R, Kurscheid S, Nekrasov M, Han S, Tremethick DJ. The histone variant H2A.Z is a master regulator of the epithelial‐mesenchymal transition. Cell Rep. 2017;1:943–952. [DOI] [PubMed] [Google Scholar]
- 20. Van Daal A, Elgin SCR. Histone variant, H2AvD, is essential Drosophila melanogaster . Mol Biol Cell. 1992;3:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Faast R, Thonglairoam, V , & Schulz, TC , et al. Histone variant H2A.Z is required for early mammalian development. Curr Biol. 2001;1:1183–1187. [DOI] [PubMed] [Google Scholar]
- 22. Draker R, Ng MK, Sarcinella E, Ignatchenko V, Kislinger T, Cheung P. A combination of H2A.Z and H4 acetylation recruits Brd2 to chromatin during transcriptional activation. PLoS Genet. 2012;8:e1003047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Semer M, Bidon B, Larnicol A, et al. DNA repair complex licenses acetylation of H2A.Z.1 by KAT2A during transcription. Nat Chem Biol. 2019;15:992–1000. [DOI] [PubMed] [Google Scholar]
- 24. Greenberg RS, Long HK, Swigut T, Wysocka J. Single amino acid change underlies distinct roles of H2A.Z subtypes in human syndrome. Cell. 2019;178:1421–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishibashi T, Dryhurst D, Rose KL, Shabanowitz J, Hunt DF, Ausió J. Acetylation of vertebrate H2A.Z and its effect on the structure of the nucleosome. Biochemistry. 2009;48:5007–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ren Q, Gorovsky MA. The nonessential H2A N‐terminal tail can function as an essential charge patch on the H2A.Z variant N‐terminal tail. Mol Cell Biol. 2003;23:2778–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gómez‐Zambrano Á, Merini W, Calonje M. The repressive role of Arabidopsis H2A.Z in transcriptional regulation depends on AtBMI1 activity. Nat Commun. 2019;10:2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dryhurst D, Ishibashi T, Rose KL, et al. Characterization of the histone H2A.Z‐1 and H2A.Z‐2 isoforms in vertebrates. BMC Biol. 2009;7:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gosmini R, Nguyen VL, Toum J, et al. The discovery of I‐BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up‐regulator and selective BET bromodomain inhibitor. J Med Chem. 2014;57:8111–8131. [DOI] [PubMed] [Google Scholar]
- 31. Lucas X, Wohlwend, M , & Hügle, M , et al. 4‐Acyl pyrroles: Mimicking acetylated lysines in histone code reading. Angew Chemie Int Ed. 2013;52:14055–14059. [DOI] [PubMed] [Google Scholar]
- 32. Filippakopoulos P, Picaud S, Mangos M, et al. Histone recognition and large‐scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2692–2704. [DOI] [PubMed] [Google Scholar]
- 34. Huang H, Zhang J, Shen W, et al. Solution structure of the second bromodomain of Brd2 and its specific interaction with acetylated histone tails. BMC Struct Biol. 2007;7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanno T, Kanno Y, Siegel RM, Jang MK, Lenardo MJ, Ozato K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol Cell. 2004;13:33–43. [DOI] [PubMed] [Google Scholar]
- 36. Olson NM, Kroc S, Johnson JA, et al. NMR analyses of acetylated H2A.Z isoforms identify differential binding interactions with the bromodomain of the NURF nucleosome remodeling complex. Biochemistry. 2020;59:1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. [DOI] [PubMed] [Google Scholar]
- 38. Nguyen UTT, Bittova L, Müller MM, et al. Accelerated chromatin biochemistry using dnA‐barcoded nucleosome libraries. Nat Methods. 2014;11:834–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miller TCR, Simon, B , & Rybin, V , et al. A bromodomain‐DNA interaction facilitates acetylation‐dependent bivalent nucleosome recognition by the BET protein BRDT. Nat Commun. 2016;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu J, Zhibing, D , & Guo, W , et al. Targeting the BRD4/FOXO3a/CDK6 axis sensitizes AKT inhibition in luminal breast cancer. Nat Commun. 2018;9:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lambert JP, Picaud S, Fujisawa T, et al. Interactome rewiring following pharmacological targeting of BET bromodomains. Mol Cell. 2019;73:621–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pünzeler S, Link S, Wagner G, et al. Multivalent binding of PWWP2A to H2A.Z regulates mitosis and neural crest differentiation. EMBO J. 2017;36:2263–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Werner MT, Wang H, Hamagami N, et al. Comparative structure‐function analysis of bromodomain and extraterminal motif (BET) proteins in a gene‐complementation system. J Biol Chem. 2020;295:1898–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee W, Tonelli M, Markley JL. NMRFAM‐SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2014;31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cowieson NP, Aragao D, Clift M, et al. MX1: A bending‐magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron. J Synchrotron Radiat. 2015;22:187–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Cryst D. 2003;59:1131–1137. [DOI] [PubMed] [Google Scholar]
- 47. McCoy AJ, Grosse‐Kunstleve, R , Adams, P , et al. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst D. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: A comprehensive Python‐based system for macromolecular structure solution. Acta Cryst D. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: The CSPs induced in the 15N‐HSQC titration of the BET BDs upon addition of ~10 M equivalents of the H2A.Z‐K4acK7ac peptide. The data have been normalized to the peak that displays the greatest CSP. Unassigned residues are indicated by red stars.
Figure S2: Plots of the CSPs induced in the 15N‐HSQC titration of each BET BD upon addition of ~10 M equivalents of the H2A.Z‐K7acK11ac peptide. The data have been normalized to the peak that displays the greatest CSP. Unassigned residues are indicated by red stars.
Figure S3: CSP data for the BRD3‐BD1:diacetylated GATA1 interaction highlighting the canonical BET BD binding pocket. The binding mode of a diacetylated GATA1 motif (K312acK315ac) interacting with BRD3‐BD1 (PDB ID: 2L5E)4 is shown. The backbone of the GATA1 peptide is depicted in blue, BRD3‐BD1 is depicted in gray, and the BRD3‐BD1 residues that undergo the greatest CSP upon titration with the diacetylated GATA1 peptide in a 15N‐HSQC titration are highlighted in red. The orientations shown match those shown in Figure 4.
Figure S4: Overlay of the H2A.Z‐K4acK7ac peptide from the structure of the complex with BRD2‐BD2 with the BD‐bound conformations of diacetylated H4 motifs that have the AcK residues separated by three amino acids (K12acK16ac—PDB ID: 3UVX; K16acK20ac—PDB ID: 3UVY).5 The orientation shown is similar to the upper panels of Figure 4a,b.
Table S1 Data collection and refinement statistics for the crystal structure of the BRD2‐BD2:H2A.Z‐K4acK7ac (PDB ID 7JX7) complex solved by molecular replacement.
Data Availability Statement
All data are provided in this manuscript and the supplementary material. The coordinates and structure factors for the BRD2‐BD2:H2A.Z‐K4acK7ac structure has been deposited to the Protein Data Bank (PDB accession code: 7JX7). The NMR backbone assignments for the BET BDs used in this study have been deposited to the Biological Magnetic Resonance Data Bank (BMRB accession codes are as follows: BRD2‐BD1: 50143, BRD2‐BD2: 50149, BRD3‐BD1: 50148, BRD3‐BD2: 50147, BRD4‐BD1: 50145, and BRD4‐BD2: 50146).