

Abstract
Transferrins function in iron sequestration and iron transport by binding iron tightly and reversibly. Vertebrate transferrins coordinate iron through interactions with two tyrosines, an aspartate, a histidine, and a carbonate anion, and conformational changes that occur upon iron binding and release have been described. Much less is known about the structure and functions of insect transferrin‐1 (Tsf1), which is present in hemolymph and influences iron homeostasis mostly by unknown mechanisms. Amino acid sequence and biochemical analyses have suggested that iron coordination by Tsf1 differs from that of the vertebrate transferrins. Here we report the first crystal structure (2.05 Å resolution) of an insect transferrin. Manduca sexta (MsTsf1) in the holo form exhibits a bilobal fold similar to that of vertebrate transferrins, but its carboxyl‐lobe adopts a novel orientation and contacts with the amino‐lobe. The structure revealed coordination of a single Fe3+ ion in the amino‐lobe through Tyr90, Tyr204, and two carbonate anions. One carbonate anion is buried near the ferric ion and is coordinated by four residues, whereas the other carbonate anion is solvent exposed and coordinated by Asn121. Notably, these residues are highly conserved in Tsf1 orthologs. Docking analysis suggested that the solvent exposed carbonate position is capable of binding alternative anions. These findings provide a structural basis for understanding Tsf1 function in iron sequestration and transport in insects as well as insight into the similarities and differences in iron homeostasis between insects and humans.
Keywords: hemolymph, insect, iron coordination, iron homeostasis, metal binding, protein structure, transferrin
Short abstract
PDB Code(s): 6WB6;
1. INTRODUCTION
Members of the transferrin protein superfamily are known for their roles in the iron homeostasis of animals. 1 Their functions are mediated by the ability to bind iron tightly and, in some cases, reversibly. 1 , 2 , 3 The well‐understood transferrins are those found in vertebrates and include mammalian serum transferrin, which sequesters iron in the blood and delivers it into cells via receptor mediated endocytosis 4 ; mammalian lactoferrin, which sequesters iron in secreted fluids as an iron withholding mechanism of innate immunity 5 , 6 ; and avian ovotransferrin that has both an iron transport and immune function. 7 These proteins are typically 70–80 kDa monomeric glycoproteins that form two distinct lobes: an amino‐lobe and carboxyl‐lobe (N‐lobe and C‐lobe, respectively). 1 , 2 , 3 Each lobe has the ability to bind a ferric (Fe3+) ion with high affinity at neutral pH, 8 , 9 , 10 and to release the Fe3+ ion as a function of pH decrease. 11
High resolution crystal structures of lactoferrin 12 and serum transferrin 13 have demonstrated that these mammalian transferrins have remarkably similar iron binding sites and several common structural features that facilitate iron binding. The iron binding site in each of the N‐ and C‐ lobes is in a deep cleft that separates each lobe into two domains: the N1 and N2 domains of the N‐lobe and the C1 and C2 domains of the C‐lobe. 14 Each iron binding site contains an aspartate, a histidine and two tyrosines, which, along with an anion, typically carbonate (CO3 2−), coordinate a ferric ion. 1 , 2 , 3 , 14 Coordination of the CO3 2− occurs through the side chains of a threonine and an arginine, and the amide groups of two N‐terminal helix residues. 14 The binding of CO3 2− at the site is considered synergistic because it is crucial for the formation of a stable Fe3+–CO3 2−‐transferrin complex. 14 , 15 Domain 1 (N1 or C1) contains the ligating aspartate, domain 2 (N2 or C2) contains one of the tyrosines, and a hinge region between domains 1 and 2 contains the histidine and second tyrosine. 16 With the four ligating side chains from the protein and two from the synergistic anion, the site has a distorted octahedral coordination sphere. The iron release characteristic of vertebrate serum transferrins arises from conformational changes that occur during binding of transferrin to its receptor and protonation of residues in the intralobe binding cleft. 2 , 17 As domains 1 and 2 begin to separate and as the coordination of iron at the binding site is abolished, the tertiary structure of the protein changes from a “closed” conformation in the holo‐form to an “open” conformation in the apo‐form 18 (Figure S1).
The roles of insect transferrin‐1 (Tsf1) in iron homeostasis are functionally similar to those of lactoferrin and serum transferrin. 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 Recent in vivo studies in Drosophila melanogaster have provided strong evidence for the function of Tsf1 in iron transport and immunity. 22 , 26 Xiao et al. 26 showed that Tsf1 functions in the transportation of iron from the gut to the fat body (insect liver and adipose equivalent). Iatsenko et al. 22 demonstrated that after infection, Tsf1 functions in relocating iron from the hemolymph to the fat body. However, unlike vertebrates, insects have no known Tsf1 receptor, and the mechanism by which iron bound to Tsf1 enters the cell remains elusive. In addition, Tsf1's iron binding and release mechanisms appear to differ from the well‐studied vertebrate transferrins. 28 Bioinformatic studies have suggested that most Tsf1s have only a single iron binding site, in their N‐lobe, and that this site lacks an iron coordinating histidine. 20 , 28 , 29 , 30 Spectroscopic analysis of the ligand‐to‐metal charge transfer (LMCT) peak, which occurs when transferrins bind iron, 3 was done for Tsf1s from Manduca sexta (MsTsf1) and D. melanogaster (DmTsf1). Both iron saturated MsTsf1 and DmTsf1 showed large LMCT peak shifts compared to the 470 nm peak for serum transferrin and lactoferrin, with MsTsf1 having a peak at 420 nm and DmTsf1 at 434 nm. These results indicate that Tsf1s coordinate iron differently than the two mammalian transferrins. 28 Despite their spectroscopic differences, biochemical analysis of MsTsf1 and DmTsf1 showed that both bind a single Fe3+ with high affinity (log K′ = 18 at pH 7.4), and release Fe3+ under moderately acidic conditions, similar to iron release by serum transferrin.
The goal of this study was to further our understanding of iron coordination in Tsf1s through structural analysis. To this end, the first crystal structure of a Tsf1 (MsTsf1) was obtained using protein purified from M. sexta larval hemolymph in an iron bound and glycosylated form. The structure of MsTsf1 revealed a single Fe3+ binding site in the N‐lobe that surprisingly is coordinated by two tyrosine ligands, Tyr90 and Tyr204, and two CO3 2− anions. Moreover, the positioning of the C‐lobe suggests that it acts as wedge between the N1 and N2 domains leaving the iron bound N‐lobe in a relatively open conformation. These novel findings provide a structural explanation for the differences in the biochemical properties of Tsf1s compared to vertebrate transferrins.
2. RESULTS
2.1. Overall structure of MsTsf1
The final MsTsf1 model contained two molecules in the asymmetric unit and included residues spanning Ser3‐Leu662 (Subunit A) and Tyr5‐Gly661 (Subunit B). Residues Ala1‐Lys2, Pro508‐Pro514, and Ala663 in Subunit A as well as Ala1‐Ser4, Gly445, Asn511‐Ser513, and Leu662‐Ala663 in Subunit B could not be modeled due to disorder. Crystallographic data are provided in Table 1. The overall structure of MsTsf1 Subunit A along with the noncrystallographic dimer is shown in Figure 1a,b respectively. Each subunit contains an Fe3+ ion and a single N‐glycosylation site, and MsTsf1 adopts an overall bilobal fold. The amino‐ and carboxyl‐lobes are labeled N‐lobe (Ser3‐Glu343) and C‐lobe (Val352‐Leu662), respectively, and are covalently connected by a short linker peptide (Arg344‐Leu351). Furthermore, each lobe contains two domains delineated by residues Ser3‐Arg79 and Gly275‐Glu343 (domain N1), Arg89‐Arg271 (domain N2), Val352‐Tyr429 and Thr587‐Leu662 (domain C1), and Tyr437‐Met582 (domain C2). The topology of the secondary structure elements for each domain is depicted in Figure S2. The MsTsf1 structure is composed of 20 α‐helices, 26 β‐strands and twelve disulfide bonds (Figure 2).
TABLE 1.
Crystallographic data for MsTsf1
| MsTsf1 (K2PtCl4) | MsTsf1 (native) | |
|---|---|---|
| Data collection | ||
| Unit‐cell parameters (Å, o) | a = 65.61, b = 141.42, c = 150.40 | a = 64.49, b = 139.06, c = 146.70 |
| Space group | P212121 | P212121 |
| Resolution (Å) a | 49.44–2.85 (2.99–2.85) | 47.28–2.05 (2.09–2.05) |
| Wavelength (Å) | 1.0000 | 1.0000 |
| Temperature (K) | 100 | 100 |
| Observed reflections | 545,842 | 544,369 |
| Unique reflections | 33,530 | 83,612 |
| <I/σ (I)> a | 10.8 (2.1) | 7.6 (2.2) |
| Completeness (%) a | 99.9 (99.6) | 100 (100) |
| Multiplicity a | 16.3 (17.1) | 6.5 (6.6) |
| R merge (%) a , b | 24.1 (187.2) | 16.2 (82.7) |
| R meas (%) a , c | 24.9 (193.0) | 17.6 (89.8) |
| R pim (%) a , c | 6.2 (46.6) | 6.9 (34.8) |
| CC1/2 a , d | 0.997 (0.777) | 0.991 (0.697) |
| DelAnom CC e | 0.481 (0.039) | — |
| Refinement | ||
| Resolution (Å) a | 40.00–2.05 | |
| Reflections (working/test) a | 79,455/4,065 | |
| R factor/R free (%) a , f | 20.9/27.2 | |
| No. of atoms (Protein/Fe3+/CO3/water) | 9,925/2/16/422 | |
| Model quality | ||
| R.m.s deviations | ||
| Bond lengths (Å) | 0.008 | |
| Bond angles (o) | 0.964 | |
| Average B‐factor (Å b ) | ||
| All Atoms | 33.4 | |
| Protein | 33.5 | |
| Fe3+ | 18.7 | |
| CO3 | 22.6 | |
| Water | 49.3 | |
| Coordinate error (maximum likelihood) (Å) | 0.30 | |
| Ramachandran plot | ||
| Most favored (%) | 94.8 | |
| Additionally allowed (%) | 4.6 |
Values in parenthesis are for the highest resolution shell.
R merge = ∑hkl∑i|I i(hkl) − <I(hkl)>|/∑hkl∑i I i(hkl), where I i(hkl) is the intensity measured for the ith reflection and <I(hkl)> is the average intensity of all reflections with indices hkl.
R meas = redundancy‐independent (multiplicity‐weighted) R merge. 52 , 70 R pim = precision‐indicating (multiplicity‐weighted) R merge. 71 , 72
CC1/2 is the correlation coefficient of the mean intensities between two random half‐sets of data. 73 , 74
DelAnom CC is the correlation coefficient between the Bijvoet differences (I (hkl) – I (−h‐k‐l)) from two random half‐sets of data 52 and is used to estimate the anomalous signal strength.
R factor = ∑hkl||F obs (hkl)| − |F calc (hkl)||/∑hkl|F obs (hkl)|; R free is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement.
FIGURE 1.
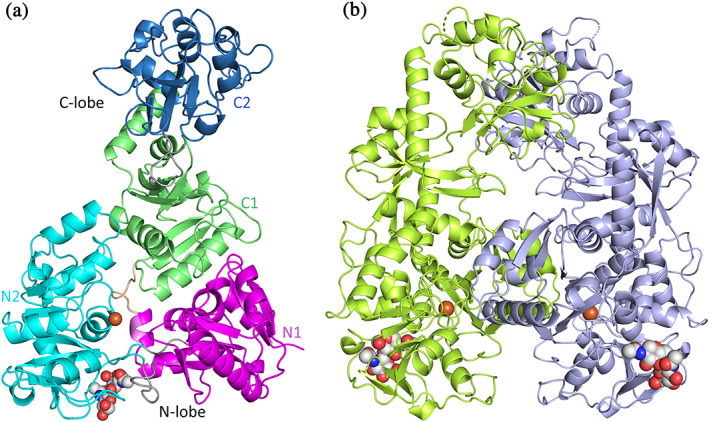
Structure of MsTsf1. (a) Individual domains of Subunit A with N1 in magenta, N2 in cyan, C1 in green, and C2 in blue. Also shown are the interlobe linker peptide in wheat and the intralobe “hinge” segments in grey. (b) Noncrystallographic dimer showing Subunit A (yellow‐green) and B (light blue). The Fe3+ ions (brown) and NAG molecules are modeled as spheres
FIGURE 2.
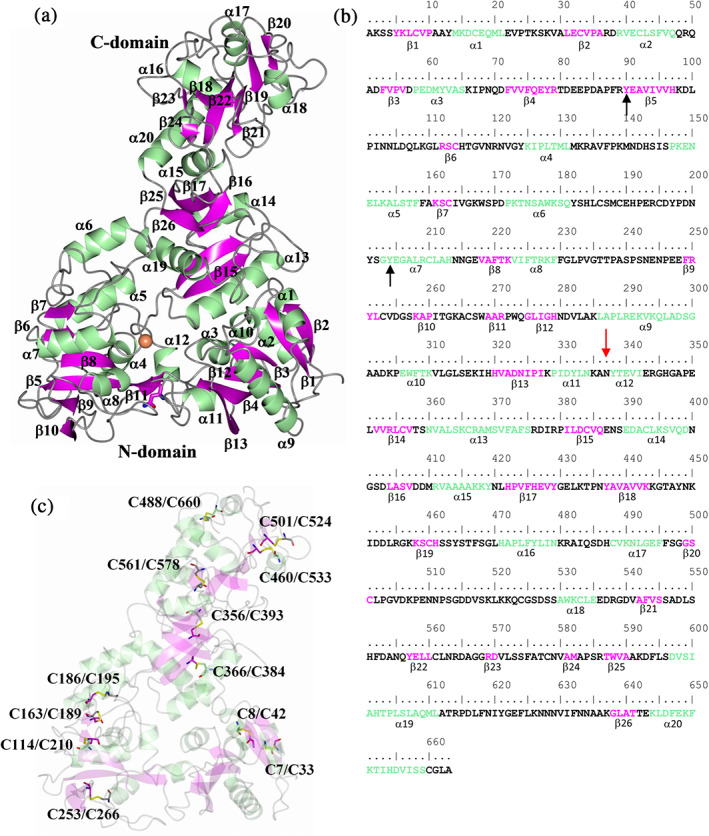
Secondary structure elements relative to the MsTsf1 sequence. (a) MsTsf1 structure showing the secondary structure elements and (b) secondary structure annotation relative to the amino acid sequence. The red arrow and black arrows indicate the glycosylation site and residues that coordinate the Fe3+ ion, respectively. (c) Locations of the disulfide bonds
The structure of MsTsf1 was subjected to a DALI 31 , 32 search in an effort to identify structures that display a similar fold, and the top 100 hits are listed in Table S1. Not surprisingly, the top hits consisted of various transferrin type proteins. However, one interesting observation was that only a small number of residues were aligned (lali). The average number of aligned residues was 269 amongst the top 100 hits even though the MsTsf1 protein contains 663 amino acids and most transferrin proteins are of similar length, which is evident in Table S1 (nres). It should be noted that the hits in Table S1 that contain “nres” in the 300 amino acid range are structures of individual N‐ or C‐lobes of transferrins. To compare MsTsf1 further with other transferrin proteins, superposition of apo‐human serum transferrin in the glycosylated form (PDB 2HAV) was conducted, which yielded an RMSD deviation of 2.46 Å between Cα atoms (282 residues aligned). The N‐lobe domains had the highest sequence similarity and comparatively displayed a similar spatial arrangement of the secondary structure elements (Figure 3a). However, the C‐lobe domains were in completely different orientations relative to the N‐lobe core and would need to rotate approximately 180o about the linker peptide that connects the N1 and C1 domains in order to superimpose (Figure 3b). Superposition of the individual C‐lobe domains of 2HAV and MsTsf1 yielded an RMSD deviation of 3.38 Å between Cα atoms (258 residues aligned, Figure S3a). Although the secondary structure elements in the C‐terminus were similar, their overall spatial arrangements differ, which accounts for the high RMSD deviation.
FIGURE 3.
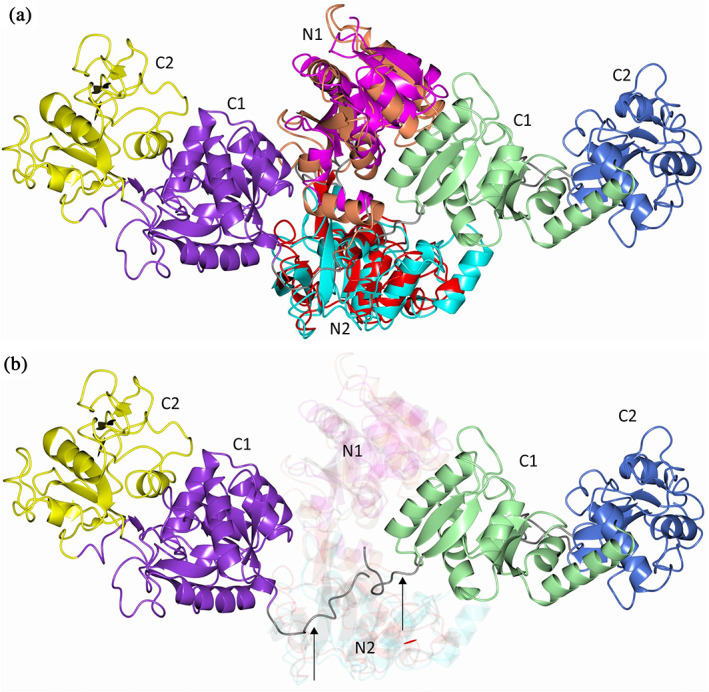
Comparison of MsTsf1 with human serum transferrin 2HAV. (a) The domains are colored as follows. MsTsf1: magenta (N1), cyan (N2), green (C1), and blue (C2); 2HAV: orange (N1), red (N2), purple (C1), and yellow (C2) (b) Same as panel a highlighting the differences in the orientation of the C1 and C2 domains. The linker peptides that connect the N1 and C1 domains are indicated by the arrows and colored as grey
The N‐ and C‐lobes of transferrins are believed to have arisen from a gene duplication event from a single lobed ancestral gene. 33 The N1 and C1 domains and the N2 and C2 domains of MsTsf1 were subjected to a pairwise structure comparison using the DALI server 34 to assess their sequence and structure similarity. Superposition of the N1 and C1 domains yielded an RMSD deviation of 2.8 Å between Cα atoms (135 out of 154 residues aligned). Superposition of the N2 and C2 domains yielded an RMSD deviation of 2.3 Å between Cα atoms (134 out of 139 residues aligned). The structural alignments show that despite having low amino acid sequence identity (24%) and no metal binding ability by the C‐lobe, the lobes do have homologous folding patterns (Figure S3b).
The solvent exposed surface of MsTsf1 does not contain patches of positive charge on the surface as was observed for lactoferrin. 35 However, one notable area in the intralobe cleft of the N‐lobe contains a large mass of positive charge. This positive patch surrounds the Fe3+ binding site (Figure S4) and is formed by residues Arg89, Arg120, Lys125, Lys222, and Arg271.
2.2. Structure of the Fe3+ and CO32− binding sites
The MsTsf1 structure showed difference electron density consistent with the Fe3+ in each subunit and displayed appreciable peak heights (8.6 and 7.4σ) in the phased anomalous difference maps using diffraction data collected at a wavelength of 1.0000 Å. Additionally, electron density for two CO3 2− anions was observed at each Fe3+ ion site. (Note that, the resolution of the structure does not allow us to determine whether these resonance stabilized anions are carbonate or bicarbonate; however, for simplicity and to adhere to past nomenclature in transferrin research, we have used the term carbonate (CO3 −2) to refer to these anions.) The Fe3+ ion is positioned in the N2 domain and adopts an octahedral coordination from six coordinating oxygens: two phenolic oxygens of residues Tyr90 and Tyr204, which are located in strand β5 and helix α7, respectively, and two oxygen atoms from each of the carbonate ions (CO3 2−‐1 and CO3 2−‐2; Figure 4a). Tyr90 and Tyr204 are highly conserved in all iron binding transferrins, including human serum transferrin and lactoferrin (Figure S5), and have been predicted to be involved in iron coordination in Tsf1s. 20 , 28 , 29 , 30 However, the presence of two CO3 2− ions in the coordination of iron is a novel observation amongst all transferrin structures. The bond lengths of the six coordinating ligands to the Fe3+ are within the range of 1.9–2.3 Å, and the bond angles of the octahedral coordination are distorted from their ideal geometry (Figure S6a and Table 2).
FIGURE 4.
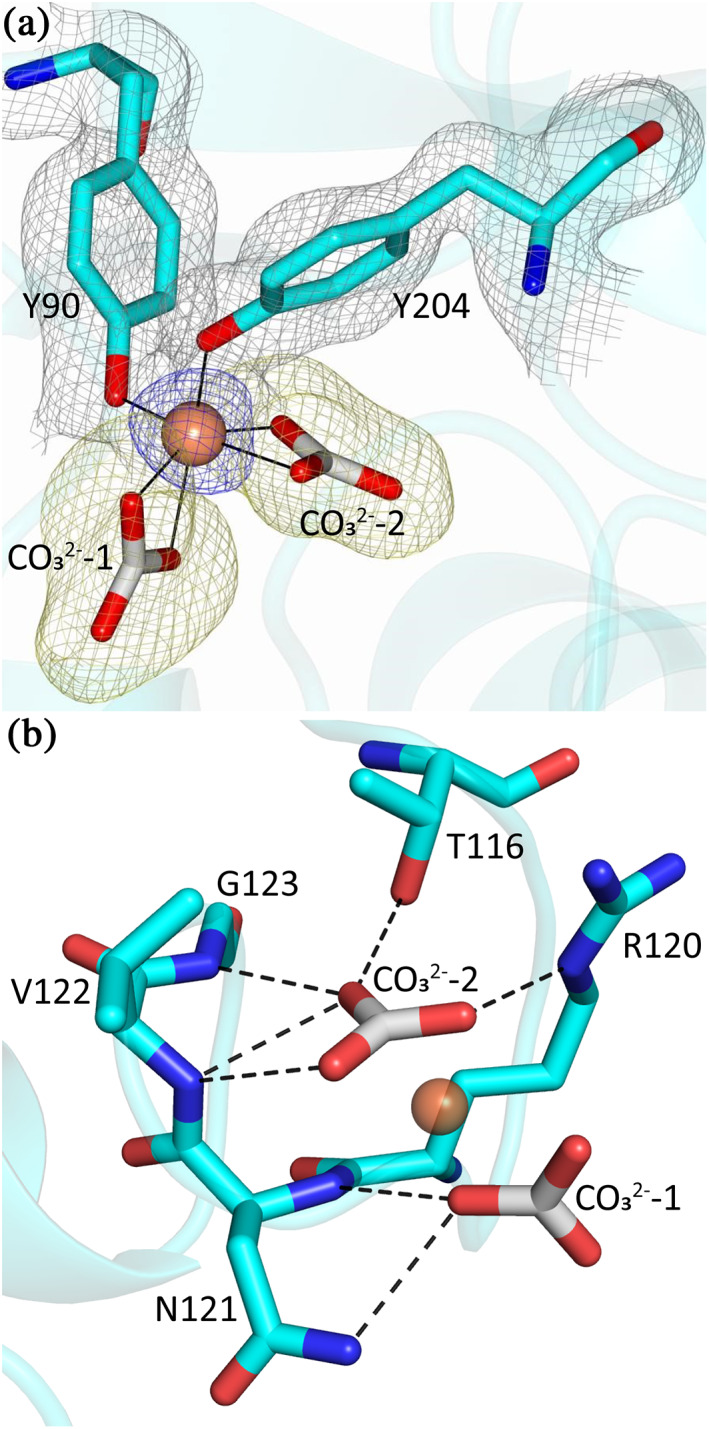
The coordination of Fe3+ and two CO3 2− ions in MsTsf1. (a) The site of Fe3+ coordination. The ligating tyrosines, Tyr90 and Tyr204, and two CO3 2− ions are in cyan, and the Fe3+ is shown as a brown sphere. The blue anomalous difference map and yellow Fo‐Fc are contoured at 3.0 σ and the grey 2Fo‐Fc map is contoured at 1.0 σ. (b) Hydrogen bond interactions (dashed lines) between MsTsf1 residues (cyan) and the CO3 2− ions (grey)
TABLE 2.
Bonds lengths (Å) of Fe3+, CO3 2−‐1, and CO3 2−‐2 to coordinating residues and bond angles (°) of Fe3+ to coordinating residues
| Bond | Length | Bonds | Angle |
|---|---|---|---|
| Fe—Oη (Y90) | 2.1 | Oη (Y90)—Fe—Oη (Y204) | 101.9 |
| Fe—Oη (Y204) | 1.9 | Oη (Y90)—Fe—O3 (CO3 2−‐2) | 89.9 |
| Fe—O3 (CO3 2−‐2) | 2.0 | Oη (Y90)—Fe—O1 (CO3 2−‐2) | 157.6 |
| Fe—O1 (CO3 2−‐2) | 2.1 | Oη (Y90)—Fe—O3 (CO3 2−‐1) | 92.6 |
| Fe—O3 (CO3 2−‐1) | 2.2 | Oη (Y90)—Fe—O1 (CO3 2−‐1) | 111.6 |
| Fe—O1 (CO3 2−‐1) | 2.3 | Oη (Y204)—Fe—O3 (CO3 2−‐2) | 102.2 |
| Oγ1 (T116)—O2 (CO3 2−‐2) | 2.8 | Oη (Y204)—Fe—O1 (CO3 2−‐2) | 85.9 |
| Nε (R120)—O1 (CO3 2−‐2) | 2.7 | Oη (Y204)—Fe—O3 (CO3 2−‐1) | 157.9 |
| N (G123)—O2 (CO3 2−‐2) | 3.1 | Oη (Y204)—Fe—O1 (CO3 2−‐1)) | 95.9 |
| N (V122)—O2 (CO3 2−‐2) | 3.1 | O3 (CO3 2−‐2)—Fe—O1 (CO3 2−‐2) | 67.8 |
| N (V122)—O3 (CO3 2−‐2) | 3.0 | O3 (CO3 2−‐2)—Fe—O3 (CO3 2−‐1) | 94.3 |
| NΔ2 (N121)—O3 (CO3 2−‐1) | 3.0 | O3 (CO3 2−‐2)—Fe—O1 (CO3 2−‐1) | 148.4 |
| O1 (CO3 2−‐2)—Fe—O3 (CO3 2−‐1) | 86.9 | ||
| O1 (CO3 2−‐2)—Fe—O1 (CO3 2−‐1) | 88.1 | ||
| O3 (CO3 2−‐1)—Fe—O1 (CO3 2−‐1) | 63.0 |
The carbonate ions are coordinated by residues Thr116, Arg120, Asn121, Val122, and Gly123, which are located in the loop connecting strand β6 and helix α4 in the N2 domain. CO3 2−‐2 forms hydrogen bonds with the Oγ1 atom of Thr116, the Nε atom of Arg120 and the backbone nitrogen group of Val122 and Gly123. CO3 2−‐1 forms hydrogen bonds with the backbone nitrogen group and the NΔ2 atom of Asn121 (Figure 4b and Table 2). A surface representation of the iron binding site shows that one carbonate ion (CO3 2−‐2) is deeply buried at the site, while the other carbonate ion (CO3 2−‐1) is solvent exposed (Figure S6b). Superposition of MsTsf1 and human serum transferrin (PDB 1D3K) demonstrates that the position of CO3 2−‐1 in MsTsf1 and the position of Asp63 in human serum transferrin are similar (Figure S6c). Moreover, CO3 2−‐2 is very similar in its position and hydrogen bonding network to the carbonate anion bound to serum transferrin (Figure S6d).
By performing an analysis of amino acid sequence alignments of Tsf1 sequences, 28 , 30 we found that all five carbonate binding residues are highly conserved (Table S2); therefore, anion coordination appears to be conserved in Tsf1 orthologs. To predict whether any noninsect transferrins have an N‐lobe that binds two anions, we analyzed transferrin sequences from noninsect species. We identified sequences that have the conserved carbonate‐binding residues in four noninsect arthropod species (Table S2), but we did not find this consensus in other types of animals, including molluscs, annelids, nematodes, and deuterostomes; therefore, the binding of a solvent exposed anion by Asn121 is likely to be specific to arthropods.
2.3. Domain interactions
The N‐ and C‐lobe of the MsTsf1 structure are connected by a linker peptide (Arg344‐Leu351) between the N1 and C1 domains (Figure 1). The MsTsf1 linker peptide adopts a loop conformation, similar to the linker peptides in serum transferrin, 18 which is different from lactoferrin linker peptides, that form an α‐helical structure. 36 The linker peptide further connects the two lobes through several of its residues forming bonds with the N1, N2, and C1 domains (Figure S7). One residue of the linker peptide, Arg344, interacts with all three domains. The Nη1 and Nη2 atoms of Arg344 form a salt bridge with the Oε2 atom of Glu151 as well as the Oδ2 atom of Asp377. The backbone nitrogen of Arg344 forms hydrogen bonds with the backbone oxygen of Val341 and Glu340 while the backbone oxygen of Arg344 forms hydrogen bonds with the Nε and Nη2 atoms of Arg379 (Figure 5a).
FIGURE 5.
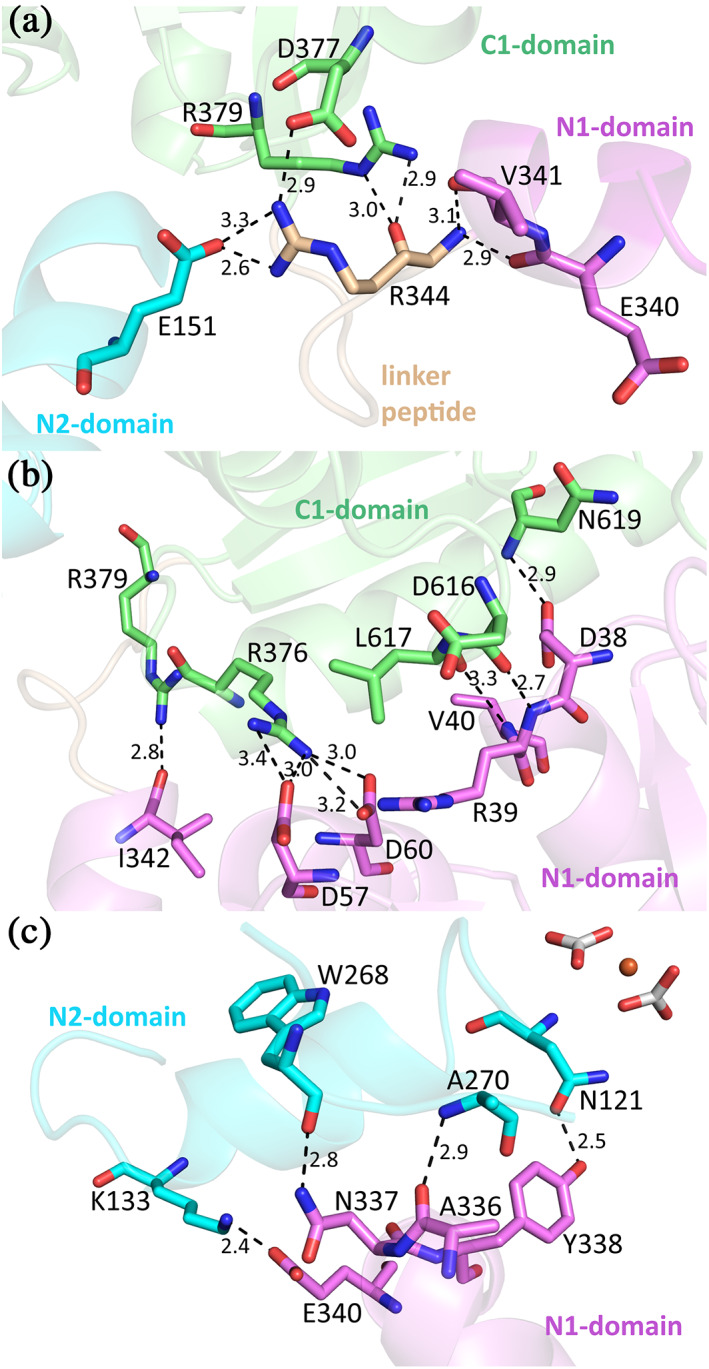
Interactions between the domains of MsTsf1. (a) The interaction of the linker peptide residue, Arg344, with residues of the N1, N2, and C1 domains. (b) The noncovalent interlobe contacts between the N1 and C1 domains. (c) The noncovalent intralobe contacts in the N‐lobe. For clarity, the NAGs were removed from Asn337. All residues and domains are colored according to Figure 1a
Noncovalent interlobe contact in the form of direct hydrogen bonds and salt bridges comes solely from the N1 and C1 domains. The Nη1 and Nη2 atoms of Arg376 form a salt bridge with Oδ2 of Asp57. The Nη2 atom of Arg376 also engages in hydrogen bonds with the Oδ1 and Oδ2 atoms of Asp60. Atom Nη2 of Arg379 forms a hydrogen bond with the backbone oxygen of Ile342. The N‐terminal end of helix α2 in the N1 domain, three hydrogen bonds are formed with a disordered region of the C1‐domain. The Oδ2 atom of Asp38 bonds with the backbone nitrogen of Asn619, the backbone nitrogen of Arg39 with the backbone oxygen of Asp616 and the backbone nitrogen of Val40 with the backbone oxygen of Leu617 (Figure 5b). While there is no direct interaction of the C‐lobe with the N2‐domain, there is a notable hydrogen bond network mediated by several water molecules located in the space between the N2 and C1 domain. This network is composed of side chain and backbone groups of residues from the C1 domain (Asp377, Arg379, Gln609, and Thr641) and the N2 domain (Asn119, Lys148, Lys167, Pro170, and Asp171; Figure S8).
Despite the N1 domain having no direct interaction with the bound Fe3+ or anions, it does make several intralobe contacts with the N2 domain. Two peptide segments covalently connect the two lobes (Thr80‐Phe88 and Pro272‐Gln274). These two segments help to form the back of the cleft between the two lobes (Figures 1a and 2a), similar in position to the hinge regions in the N‐ and C‐lobes of serum transferrin and lactoferrin. 2 , 3 , 16 Because these two covalent segments adopt loop structures, they may provide flexibility to the two domains to facilitate any conformational changes. There are also several noncovalent contacts that occur between the N1 and N2 domain. A salt bridge is formed between the Nζ atom of Lys133 and the Oε1 atom of Glu340. Two residues, Ala336 and Asn337 (Asn337 is also the site of glycosylation), between helix α11 and α12 form hydrogen bonds with the N2 domain residues Trp268 and Ala270. The most interesting contact comes from a hydrogen bond formed between the Oη atom of Tyr338 and the Oδ1 atom of the CO3 2−‐1 coordinating Asn121 (Figure 5c). This 2.5 Å interaction between Tyr338 and Asn121 is not only an intralobe contact connecting the N1 domain to the N2 domain, but also closely links the N1 domain to anion coordination and likely also to iron binding.
The C2 domain of MsTsf1 makes no contact with the N‐lobe. Besides the two covalent peptide linkages made with the C1 domain (Gly430‐Asn436 and Ala583‐Arg586), the C2 domain's only other direct contact comes through a disulfide linkage between its Cys488 and the Cys660 very near to the C‐terminal end in the C1 domain.
2.4. Glycosylation site
Characteristic of the transferrin family, MsTsf1 is glycosylated. MsTsf1 has three potential glycosylation sites at Asn200, Asn337, and Asn400, as predicted from the N‐GlyDE server. 37 However, electron density consistent with glycosylation was only observed at Asn337 in each subunit (Figure 6a,b), which is located in the N1 domain within the loop connecting α11 and α12. Two N‐acetylglucosamine (NAG) glycans could be modeled at each Asn337 residue, although there was weaker electron density present that suggested that more extensive glycan branching was present. Therefore, the glycosylated MsTsf1 was analyzed by mass spectrometry, using a site‐specific glycosylation analysis, in an effort to identify the glycans on each potential N‐glycosylation site. The mass spectrometry results (Figure 6c) confirmed the observations from the crystal structure that glycosylation was only present at Asn337. Additionally, heterogeneous glycosylation of high mannose variants was observed at Asn337 which explains why only the NAG–NAG glycans could only be modeled to the electron density, as the mannose occupancies presumably varied and/or were disordered. This level of glycosylation of MsTsf1 is consistent with previous reports of mannose and NAG being at a 5:1 ratio in MsTsf1. 38
FIGURE 6.
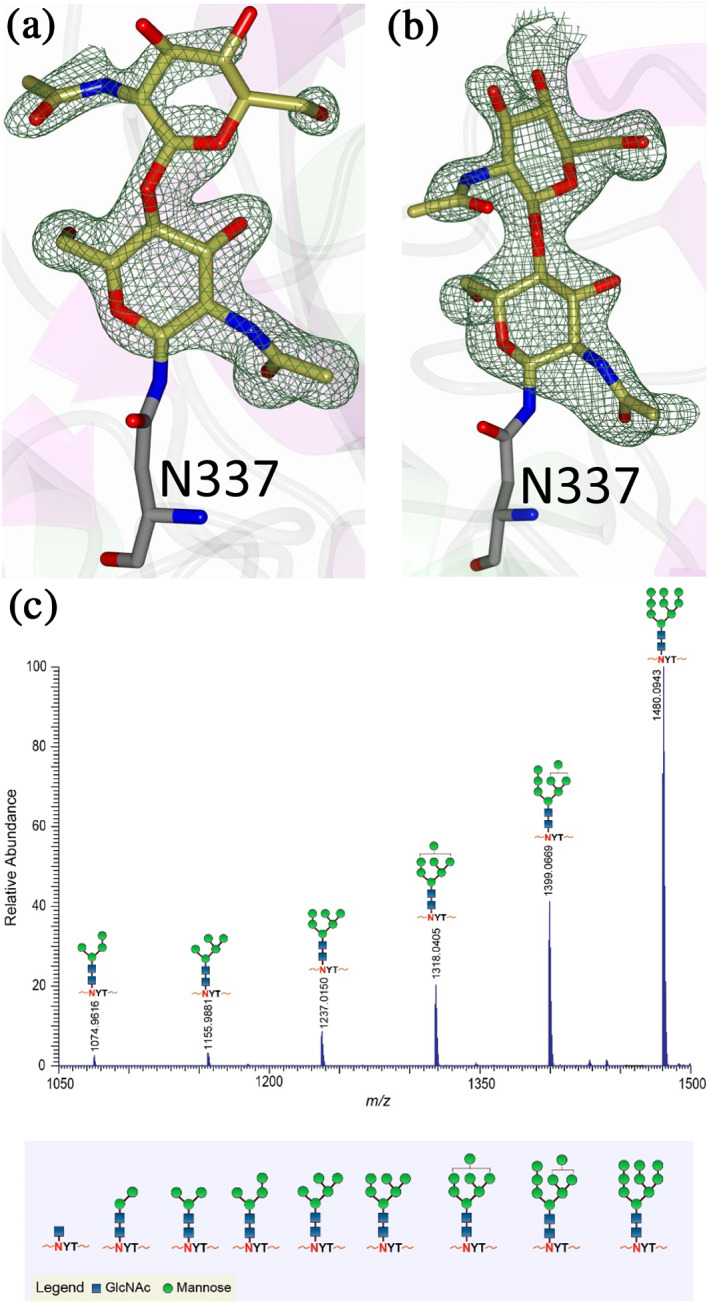
Glycosylation in MsTsf1. Fo‐Fc Polder omit electron density map (green mesh) contoured at 3σ showing the modeled glycans in (a) Subunit A and (b) Subunit B. (c) Mass spectrometry results showing the most abundant glycoforms present at residue Asn337
Studies of vertebrate transferrins have revealed highly variable sites of N‐glycosylation; however, these post‐translational modifications have mostly been found on the protein surface and are not believed to alter iron binding and release. 1 , 39 The glycosylation of Asn337 in the N‐lobe of MsTsf1 is unique because of its location at the intralobe contact points (Figure 5c) and near the hinge‐like region. In vertebrate transferrins, this region would normally be buried in the interlobe interface, but it is exposed due to the difference in the orientation of the MsTsf1 C‐lobe. The positioning of glycosylation at the hinge‐like region of MsTsf1's N‐lobe could influence the flexibility of the domains to form open or closed conformations.
2.5. Molecular docking of organic anions
Substitutes for carbonate as the synergistic anion bound to vertebrate transferrins have been previously studied. 14 , 15 , 40 While many organic anions can form weak Fe‐anion‐transferrin complexes, oxalate is the only substitute that forms a relatively similar stable complex to that of carbonate. 15 , 41 In the case of MsTsf1, CO3 2−‐2 has a similar coordination as the carbonate in vertebrate transferrins, but CO3 2−‐1 is solvent exposed and has less steric restrictions. The concentration of carbonate in insect hemolymph 42 is similar to many other organic acids. 43 Thus, our hypothesis is that the CO3 2−‐1 binding site is susceptible to the binding of alternative anions that could form a stable Fe‐anion‐MsTsf1 complex. As an initial probe of this hypothesis, we performed a molecular docking study. Candidate anions were selected from reported organic anions and acids found in insect hemolymph 43 , 44 : acetoacetate, α‐ketoglutarate, ascorbate, fumarate, glycine, glyoxylate, lactate, malate, oxaloacetate, pyruvate and succinate. Searchable pockets in the cleft between the N1 and N2 domains were identified (Figure S9a) and docking experiments were performed using AutoDock Vina. 45 Nearly all of the candidate anions had poses within an RMSD of 4 Å of CO3 2−‐1; however, not all positions were sterically possible with respect to where the iron is located in the pocket. Analysis of the sterically acceptable docking poses at the iron binding site are found in Table 3. Figure S9b‐l shows the sterically acceptable poses for each anion with the Fe3+ ion and CO3 2−‐2 replaced in the MsTsf1 structure. Citrate was the exception, as no poses were found near the site, which explains its high average RMSD from the CO3 2−‐1 position, >11 Å. The binding of the dicarboxylic acid candidates, α‐ketoglutarate, fumarate, malate, oxaloacetate and succinate, were consistently more energetically favorable because one carboxyl group becomes the oxygen ligands for the iron and the other carboxyl group can interact with nearby positively charged residues Lys125 and Arg271 that protrude into the cleft. Figure 7 shows an example of α‐ketoglutarate's predicted interactions at the iron binding site.
TABLE 3.
Analysis of sterically acceptable organic anions docked at the iron binding site of MsTsf1
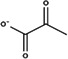
| Anion | Structure | Average RMSD deviation from carbonate (Å) | Average energy (kcal/mol) |
|---|---|---|---|
| Acetoacetate |
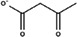
|
1.52 | −4.0 |
| α‐Ketoglutarate |
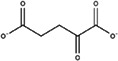
|
1.40 | −4.8 |
| Ascorbate |
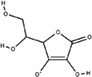
|
2.86 | −5.3 |
| Carbonate a |
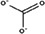
|
1.16/0.27 | −3.2/−3.5 |
| Fumarate |
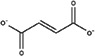
|
2.21 | −4.0 |
| Glycine |
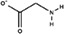
|
0.84 | −3.1 |
| Glyoxylate |
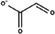
|
1.68 | −3.6 |
| Lactate |
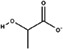
|
1.24 | −3.6 |
| Malate |
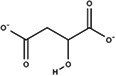
|
3.42 | −4.4 |
| Oxaloacetate |
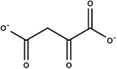
|
1.21 | −4.6 |
| Pyruvate |
|
1.74 | −3.8 |
| Succinate |
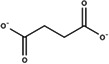
|
2.30 | −4.3 |
The docked carbonate results have two values, the first being the measurements of the docking result at the CO3 2−‐1 position (solvent exposed carbonate) and the second being the CO3 2−‐2 position (buried carbonate).
FIGURE 7.
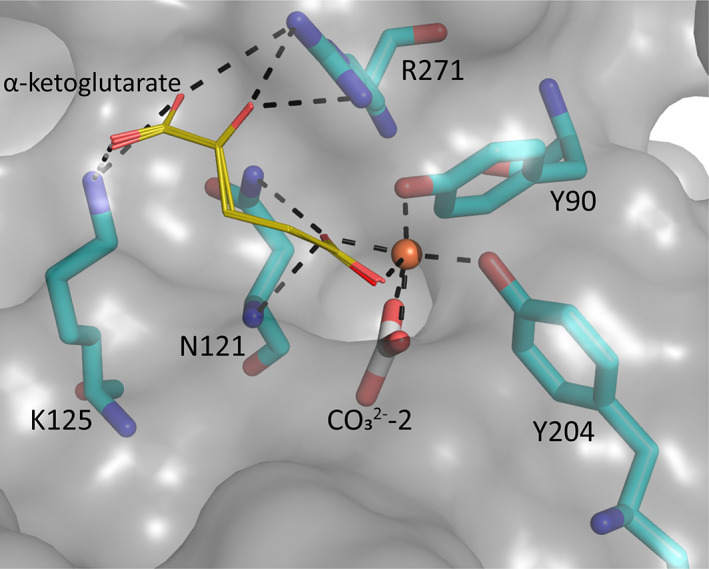
Docking poses of α‐ketoglutarate and the possible interaction in MsTsf1 N‐lobe. A surface representation of the predicted interactions of α‐ketoglutarate (gold stick) with iron and residues Asn121, Lys125, and Arg271 (cyan). Predicted bonds are shown as dashed lines and are within 3.5 Å. The Fe3+ ion (brown sphere) and known coordinating ligands, Tyr90 and Tyr204, and CO3 2−‐2 are also shown
To assess the robustness of the search area and docking parameters, two controls were applied to the study. As a positive control, carbonate was redocked in the structure. This proved to be a good control, as it was the only anion that had poses at the CO3 2−‐1 and CO3 2−‐2 positions (Figure S9m); moreover, carbonate had low RMSD values for both positions (CO3 2−‐1 = 1.16 Å and CO3 2−‐2 = 0.27 Å). The cation phosphonium was used as a negative control because of its positive charge. The average energy of the phosphonium binding poses was considerably higher (−0.5 kcal/mol) than all the anions and had no poses near the iron binding site.
3. DISCUSSION
Previous studies have provided bioinformatic and biochemical evidence that that Tsf1s have different iron binding properties than the well‐characterized vertebrate transferrins. 20 , 28 , 29 , 30 Most Tsf1s have only one iron‐binding site, and it is found in the N‐lobe. 20 , 28 , 29 , 30 Moreover, while Tsf1s are predicted to have two tyrosines at the iron binding site, the histidine and, in many species, the aspartate are substituted with other amino acid residues. In addition, Tsf1 and vertebrate transferrins differ in their spectroscopic properties. 28 Despite these differences between Tsf1s and vertebrate transferrins, DmTsf1 and MsTsf1 bind iron tightly and reversibly. 28
This structural analysis of MsTsf1 verifies that an iron binding site is present in the N‐lobe but not the C‐lobe. It also demonstrates that the iron binding site differs from other transferrin structures. The distorted octahedral coordination of the Fe3+ is mediated by two N2 domain residues, Tyr90 and Tyr204, and two carbonate anions. These two tyrosine ligands and the residues that hydrogen bond with the two carbonates, Thr116, Arg120, Asn121, Val122, and Gly123, are highly conserved in all available Tsf1 sequences, leading us to believe that similar coordination is likely to occur in other species of insects. The use of a second carbonate instead of histidine and aspartate in iron coordination explains the differences in the spectroscopic characteristics of Tsf1s compared to serum transferrin and lactoferrin. 28 One of the carbonate anions, CO3 2−‐2, is buried behind the Fe3+ in a pocket within the N1 domain and adopts a similar position observed for the structures of other transferrins. 12 , 13 , 46 , 47 The other carbonate, CO3 2−‐1, is more solvent exposed and held in place through hydrogen bounds with Asn121, which is also involved in intralobe contacts within the N‐lobe. The intralobe contacts suggest that Asn121 influences the integrity of the iron binding site during expected conformational changes.
The overall structure of MsTsf1 is bilobal, with each lobe separated into two domains, forming folds similar to those of vertebrate transferrin structures. 12 , 13 , 46 , 47 However, the orientation of the MsTsf1 C‐lobe differs from other transferrin structures in that it is rotated approximately 180° from the core of the N‐lobe. Thus, the interaction of the C1 domain with the N1 and N2 domains is at the opening of the N‐lobe cleft. The C1 domain appears to act as a wedge between the N1 and N2 domains, hindering further closing of the N‐lobe around the iron binding site. This most likely explains why the first several hits of the DALI search were vertebrate transferrin structures in the open conformation. The glycosylation site of MsTsf1 at Asn337 makes it unlikely that the C‐lobe orientation is induced by crystal contacts, because the glycosylation at Asn337 is positioned on the backside of the N‐lobe cleft, near the hinge‐like region, thereby sterically inhibiting the C‐lobe from the positioning itself like the vertebrate transferrin structures.
The finding of iron‐bound MsTsf1 in an open conformation was surprising. However, our docking study provides evidence that this conformation results in a solvent exposed anion binding site that is accessible to several different organic anions found in insect hemolymph. The concentration of carbonate (10 mM) in M. sexta hemolymph 42 is similar to other organic anions, many of which fall within the 3–7 mM range in similar lepidopteran insects. 43 With the numerous organic anions found in insect hemolymph and their fluctuating concentrations during deveopment, 42 , 43 , 44 this flexibility to bind alternative anions could be functionally significant.
The functional role of the C‐lobe in Tsf1s is still unknown. In the MsTsf1 structure, the C‐lobe is in a novel orientation that makes contacts with the N‐lobe domains and likely stabilizes the tertiary structure. However, why the C‐lobe adopts a secondary structure similar to vertebrate transferrins but does not bind iron, remains unknown. One hypothesis is that the C‐lobe could act as a decoy for receptors of iron scavenging pathogens. 27 , 48 Future work is needed to elucidate the conformational changes and interplay between domains that takes place in MsTsf1.
In summary, this paper describes the first structure of an insect transferrin and details several novel structural features, including iron coordination, domain interactions, C‐lobe orientation, and glycosylation position. Two tyrosines from the N2 domain and two carbonate anions coordinate iron binding. The C1 domain is wedged at the cleft opening between the N1 and N2 domains. The unique properties of this transferrin structure illustrate the important fact that insect and vertebrate transferrins have evolved different structural features, and suggest that Tsf1s have different mechanisms for carrying out their proposed roles in iron homeostasis. It will be important to consider these differences when evaluating model insect studies of iron homeostasis and iron‐related diseases of humans.
4. MATERIALS AND METHODS
4.1. Crystallization and data collection
MsTsf1 was purified from larval hemolymph following a procedure previously described. 28 Purified MsTsf1 was extensively dialyzed in 50 mM NaCl, 20 mM Tris pH 7.4, 5 mM sodium bicarbonate and concentrated to 9.2 mg/ml for crystallization screening. All crystallization experiments were setup using an NT8 drop setting robot (Formulatrix Inc.) and UVXPO MRC (Molecular Dimensions) sitting drop vapor diffusion plates at 18°C. 100 nl of protein and 100 nl crystallization solution were dispensed and equilibrated against 50 μl of the latter. Initial crystals that formed needle clusters were obtained from the Index HT screen (Hampton Research) condition D10 (20% (w/v) PEG 5,000 MME, 100 mM Bis‐Tris pH 6.5). These crystals were reproduced in a new crystallization plate in order to generate a crystal seed stock and 20 nl of seeds were added to each drop of new crystallization experiments dispensed with the NT8 robot as described above. Crystals that displayed a needle morphology were obtained in 2 weeks from the Proplex HT screen (Molecular Dimensions) condition F6 (15% (w/v) PEG 20,000, 100 mM Hepes pH 7.5). These crystals could be reproducibly obtained overnight by streak seeding new crystallization drops in a sitting drop plate. Heavy atom derivatized crystals were obtained by transferring native crystals to a solution containing crystallant supplemented with 10 mM K2PtCl4 and incubating for 18 hr. Crystals were transferred to a cryoprotectant solution composed of 80% crystallization solution and 20% PEG 200 before storing in liquid nitrogen. X‐ray diffraction data were collected at the Advanced Photon Source beamline 17‐ID using a Dectris Pilatus 6 M pixel array detector.
4.2. Structure solution and refinement
Intensities were integrated using XDS 49 , 50 via Autoproc 51 and the Laue class analysis and data scaling were performed with Aimless. 52 Structure solution, using two data sets that were scaled together from Pt‐derivatized crystals, was conducted by SAD phasing with Crank2 53 using the Shelx, 54 Refmac, 55 Solomon, 56 Parrot, 57 and Buccaneer 58 pipeline via the CCP4 59 interface. Eleven Pt sites were located with occupancies greater than 0.25 and phasing/density modification resulted in a mean figure of merit of 0.44 in the space group P212121. Subsequent model building utilizing density modification and phased refinement yielded R/R free = 0.352/0.425 for the initial model. This model was used for molecular replacement with Phaser 60 against the higher resolution native data set and the top solution was obtained for two molecules of MsTsf1 in the asymmetric unit in the space group P212121 (TFZ = 51.5, LLG = 2,945). The model was further improved by automated model building with Phenix and additional refinement and manual model building were conducted with Phenix and Coot, 61 respectively. Disordered side chains were truncated to the point for which electron density could be observed. Structure validation was conducted with Molprobity 62 and figures were prepared using the CCP4MG package 63 and Pymol (The PyMOL Molecular Graphics System, Version 2.3.2 Schrödinger, LLC). Surface electrostatics were determined using APBS. 64 Crystallographic data are provided in Table 1. The coordinates and structure factors were deposited in the Protein Data Bank with the code 6WB6.
4.3. Mass spectrometry and chromatography
A 25 μg aliquot of MsTsf1 sample at a concentration of 9.25 mg/ml was denatured with 7 M urea in 100 mM Tris buffer (pH 8.5), then reduced with 5 mM TCEP at room temperature for 1 hr, followed by alkylation with 20 mM iodoacetamide for an additional hour in the dark. The reduced and alkylated sample was buffer exchanged with 50 mM ammonium bicarbonate (pH 8) using a 30‐kDa MWCO filter (Millipore) prior to trypsin digestion. The sample was digested with trypsin at a 30:1 protein:enzyme ratio and was incubated overnight at 37°C for 18 hr.
High‐resolution LC/MS experiment was performed using an Orbitrap Fusion Lumos Tribrid (Thermo Scientific) mass spectrometer equipped with ETD that is coupled to an Acquity UPLC M‐Class system (Waters). Mobile phases consisted of solvent A: 99.9% deionized H2O + 0.1% formic acid and solvent B: 99.9% CH3CN + 0.1% formic acid. Three microliters of the sample were injected onto C18 PepMap™ 300 column (300 μm i.d. × 15 cm, 300 Å, Thermo Fisher Scientific) at a flow rate of 3 μl/min. The CH3CN/H2O gradient ramping from 3 to 40% B in 45 min was used to separate the MsTsfl digest. All mass spectrometric analysis was performed in the positive ion mode using data‐dependent acquisition with the instrument set to run in 3‐s cycles for the survey and two consecutive MS/MS scans with CID and ETD. Mass spectrometry data acquisition parameters include: a survey scan in the mass range, 350–1800 m/z at a resolution of 60,000 at m/z 200 with an AGC target of 4 × 105 and a maximum injection time of 50 ms, CID collision energy of 30%, and ETD was performed using the calibrated charge dependent reaction time. Data dependent acquisition was carried out by dynamic selection of ions with intensity greater than 5,000. Resulting fragments were detected using rapid scan rate in the ion trap. Glycopeptide compositions were manually identified in the LC/MS data file and were confirmed by a combination of high resolution MS data, CID, and ETD data.
4.4. Structure preparation and molecular docking
Water molecules and ligands were removed from the MsTsf1 PDB file and the structure was subjected to the fpocket online server. 65 Pockets located in the N‐lobe cleft were identified and grouped as one pocket. The size (x = 15.0 Å, y = 28.5 Å, and z = 15.0 Å) and center (x = −16.2 Å, y = −7.4 Å, and z = 4.2 Å) coordinates of this grouped pocket were determined. The structures of the candidate anions were drawn using MolView (molview.org) and converted to MOL2 files using the program OpenBabel. 66 The anions and MsTsf1 structures were prepared for docking in AutoDockTools version 1.5.6 67 and structural files in PDBQT format were generated for use in AutoDock Vina. 45
AutoDock Vina 1.1.2 was used to carry out the docking simulations of the organic anions in the identified pocket of MsTsf1. The parameters of experiments were carried out under default conditions except that the searchable area was set to the size and center of the pocket in the N‐lobe cleft and the exhaustiveness was set to 100. Four independent trials were conducted. This program predicted the global minimum binding energy (kcal/mol) of the anions in the given pocket dimensions and reported the top nine poses for each trial. The poses that had an RMSD of 4 Å or less from CO3 2−‐1 were further analyzed to determine if they would sterically clash with the Fe3+ in the original structure. The average RMSD from CO3 2−‐1 and the average binding energy of these sterically acceptable poses were then calculated. A carbonate anion structure was generated and used as positive control for the study. It had poses at both the CO3 2−‐1 and CO3 2−‐2 positions; therefore, the average energy and average RMSD from each position was analyzed and reported. A phosphonium cation was generated as a negative control and we analyzed its predicted binding poses. Pockets, coordinates and poses were analyzed using Pymol (The PyMOL Molecular Graphics System, Version 2.3.2 Schrödinger, LLC).
4.5. Alignment analysis
To identify possible anion‐binding residues in transferrin sequences, we analyzed sequence alignments of 107 Tsf1 and Tsf1‐like sequences and 14 noninsect hexapod transferrin sequences. 26 , 28 We evaluated whether these sequences had the five anion‐binding residues identified in the MsTsf1 crystal structure. To identify transferrins from other types of animals that may also have these anion‐binding residues, we used BLASTP at NCBI to search nonredundant protein databases for Crustacea, Arachnida, Myriapoda, Xiphosura, Mollusca, Annelida, Nematoda, and Deuterostomia.
The sequence alignment of MsTsf1, human lactoferrin and human serum transferrin was created by collecting sequences through the UniProt data base. 68 The signal peptide sequences were removed. A sequence alignment was generated with Clustal Omega using the EMBL‐EBI server. 69 The accession numbers used in the alignment are as follows: P22297 for MsTsf1, P02788 for human lactoferrin and P02787 for human serum transferrin.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
AUTHOR CONTRIBUTIONS
Jacob Weber: Conceptualization; formal analysis; investigation; methodology; visualization; writing‐original draft; writing‐review and editing. Maithri Kashipathy: Conceptualization; investigation; methodology; writing‐review and editing. Kevin Battaile: Conceptualization; investigation; methodology; writing‐review and editing. Eden Go: Conceptualization; investigation; methodology; writing‐review and editing. Heather Desaire: Supervision; writing‐review and editing. Michael Kanost: Conceptualization; funding acquisition; resources; writing‐review and editing. Scott Lovell: Conceptualization; investigation; project administration; resources; supervision; writing‐original draft; writing‐review and editing. Maureen Gorman: Conceptualization; funding acquisition; investigation; project administration; resources; supervision; writing‐review and editing.
Supporting information
Figure S1 Comparison of human serum transferrin in the holo‐ and apo‐form.
Figure S2. Topology diagram of MsTsf1.
Figure S3. Structural alignments of transferrin lobes and domains.
Figure S4. Electrostatic surface charge of MsTsf1.
Figure S5. Sequence alignment of MsTsf1, human lactoferrin and human serum transferrin.
Figure S6. Characteristics of Fe3+ and carbonate coordination in MsTsf1
Figure S7. Noncovalent interactions between the linker peptide and the N1, N2 and C1 domains.
Figure S8. The mediated contacts between the C1 and N2 domains.
Figure S9. Results from docking study of organic anions in the N‐lobe cleft of MsTsf1.
Table S1. Top 100 hits from the DALI search.
Table S2. Predicted carbonate binding residues of the N‐lobe of insect Tsf1 orthologs and other arthropod transferrins
ACKNOWLEDGMENTS
The authors thank Brian Geisbrecht for helpful suggestions regarding this work. This work was supported by National Science Foundation Grant 1656388, National Institute of General Medical Sciences grant R37 GM041247 and grant R35 GM130354, and the Johnson Cancer Research Center. This is contribution 21‐023‐J from the Kansas Agricultural Experiment Station. Use of the IMCA‐CAT beamline 17‐ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman‐Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE‐AC02‐06CH11357.
Weber JJ, Kashipathy MM, Battaile KP, et al. Structural insight into the novel iron‐coordination and domain interactions of transferrin‐1 from a model insect, Manduca sexta . Protein Science. 2021;30:408–422. 10.1002/pro.3999
Funding information Basic Energy Sciences, Grant/Award Number: DE‐AC02‐06CH11357; National Institute of General Medical Sciences, Grant/Award Numbers: R35 GM130354, R37 GM041247; National Science Foundation, Grant/Award Number: 1656388
REFERENCES
- 1. Baker EN. Structure and reactivity of transferrins In: Sykes AG, editor. Advances in Inorganic Chemistry. Volume 41 Cambridge, Massachusetts: Academic Press, 1994; p. 389–463. [Google Scholar]
- 2. Mizutani K, Toyoda M, Mikami B. X‐ray structures of transferrins and related proteins. Biochim Biophys Acta, Gen Subj. 2012;1820:203–211. [DOI] [PubMed] [Google Scholar]
- 3. Sun H, Li H, Sadler PJ. Transferrin as a metal ion mediator. Chem Rev. 1999;99:2817–2842. [DOI] [PubMed] [Google Scholar]
- 4. Octave J‐N, Schneider Y‐J, Trouet A, Crichton RR. Iron uptake and utilization by mammalian cells. I: Cellular uptake of transferrin and iron. Trends in Biochem Sci. 1983;8:217–220. [Google Scholar]
- 5. Farnaud S, Evans RW. Lactoferrin—A multifunctional protein with antimicrobial properties. Mol Immunol. 2003;40:395–405. [DOI] [PubMed] [Google Scholar]
- 6. Jenssen H, Hancock REW. Antimicrobial properties of lactoferrin. Biochimie. 2009;91:19–29. [DOI] [PubMed] [Google Scholar]
- 7. Giansanti F, Leboffe L, Pitari G, Ippoliti R, Antonini G. Physiological roles of ovotransferrin. Biochim Biophys Acta. 2012;1820:218–225. [DOI] [PubMed] [Google Scholar]
- 8. Aisen P, Leibman A, Zweier J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J Biol Chem. 1978;253:1930–1937. [PubMed] [Google Scholar]
- 9. Pakdaman R, Petitjean M, Chahine J‐MEH. Transferrins. Eur J Biochem. 1998;254:144–153. [DOI] [PubMed] [Google Scholar]
- 10. Brandts JF, Lin LN. Study of strong to ultratight protein interactions using differential scanning calorimetry. Biochemistry. 1990;29:6927–6940. [DOI] [PubMed] [Google Scholar]
- 11. Day CL, Stowell KM, Baker EN, Tweedie JW. Studies of the N‐terminal half of human lactoferrin produced from the cloned cDNA demonstrate that interlobe interactions modulate iron release. J Biol Chem. 1992;267:13857–13862. [PubMed] [Google Scholar]
- 12. Anderson BF, Baker HM, Dodson EJ, et al. Structure of human lactoferrin at 3.2‐A resolution. Proc Natl Acad Sci U S A. 1987;84:1769–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bailey S, Evans RW, Garratt RC, et al. Molecular structure of serum transferrin at 3.3‐A resolution. Biochemistry. 1988;27:5804–5812. [DOI] [PubMed] [Google Scholar]
- 14. Harris WR. Anion binding properties of the transferrins. Implications for function. Biochim Biophys Acta. 2012;1820:348–361. [DOI] [PubMed] [Google Scholar]
- 15. Schlabach MR, Bates GW. The synergistic binding of anions and Fe3+ by transferrin. Implications for the interlocking sites hypothesis. J Biol Chem. 1975;250:2182–2188. [PubMed] [Google Scholar]
- 16. Mizutani K, Mikami B, Hirose M. Domain closure mechanism in transferrins: New viewpoints about the hinge structure and motion as deduced from high resolution crystal structures of ovotransferrin N‐lobe. J Mol Biol. 2001;309:937–947. [DOI] [PubMed] [Google Scholar]
- 17. Eckenroth BE, Steere AN, Chasteen ND, Everse SJ, Mason AB. How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH. Proc Natl Acad Sci U S A. 2011;108:13089–13094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang N, Zhang H, Wang M, Hao Q, Sun H. Iron and bismuth bound human serum transferrin reveals a partially‐opened conformation in the N‐lobe. Sci Rep. 2012;2:999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brummett LM, Kanost MR, Gorman MJ. The immune properties of Manduca sexta transferrin. Insect Biochem Mol Biol. 2017;81:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geiser DL, Winzerling JJ. Insect transferrins: Multifunctional proteins. Biochim Biophys Acta. 2012;1820:437–451. [DOI] [PubMed] [Google Scholar]
- 21. Huebers HA, Huebers E, Finch CA, et al. Iron binding proteins and their roles in the tobacco hornworm, Manduca sexta (L.). J Comp Physiol B. 1988;158:291–300. [DOI] [PubMed] [Google Scholar]
- 22. Iatsenko I, Marra A, Boquete J‐P, Peña J, Lemaitre B. Iron sequestration by transferrin 1 mediates nutritional immunity in Drosophila melanogaster . Proc Natl Acad Sci U S A. 2020;117:7317–7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim BY, Lee KS, Choo YM, et al. Insect transferrin functions as an antioxidant protein in a beetle larva. Comp Biochem Physiol, Part B: Biochem Mol Biol. 2008;150:161–169. [DOI] [PubMed] [Google Scholar]
- 24. Kurama T, Kurata S, Natori S. Molecular characterization of an insect transferrin and its selective incorporation into eggs during oogenesis. Eur J Biochem. 1995;228:229–235. [PubMed] [Google Scholar]
- 25. Lee KS, Kim BY, Kim HJ, et al. Transferrin inhibits stress‐induced apoptosis in a beetle. Free Radic Biol Med. 2006;41:1151–1161. [DOI] [PubMed] [Google Scholar]
- 26. Xiao G, Liu Z‐H, Zhao M, Wang H‐L, Zhou B. Transferrin 1 functions in iron trafficking and genetically interacts with ferritin in Drosophila melanogaster . Cell Rep. 2019;26:748–758.e5. [DOI] [PubMed] [Google Scholar]
- 27. Yoshiga T, Hernandez VP, Fallon AM, Law JH. Mosquito transferrin, an acute‐phase protein that is up‐regulated upon infection. Proc Natl Acad Sci U S A. 1997;94:12337–12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weber JJ, Kanost MR, Gorman MJ. Iron binding and release properties of transferrin‐1 from Drosophila melanogaster and Manduca sexta: Implications for insect iron homeostasis. Insect Biochem Mol Biol. 2020;125:103438 10.1016/j.ibmb.2020.103438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lambert LA, Perri H, Halbrooks PJ, Mason AB. Evolution of the transferrin family: conservation of residues associated with iron and anion binding. Comp Biochem Physiol, Part B: Biochem Mol Biol. 2005;142:129–141. [DOI] [PubMed] [Google Scholar]
- 30. Najera DG, Dittmer NT, Weber JJ, Kanost MR, Gorman MJ. Phylogenetic and sequence analyses of insect transferrins suggest that only transferrin 1 has a role in iron homeostasis. Insect Science. Cambridge, Massachusetts; 2012. 10.1111/1744-7917.12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holm L, Laakso LM. Dali server update. Nucleic Acids Res. 2016;44:W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holm L, Rosenström P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lambert LA. Molecular evolution of the transferrin family and associated receptors. Biochim Biophys Acta. 2012;1820:244–255. [DOI] [PubMed] [Google Scholar]
- 34. Holm L. DALI and the persistence of protein shape. Protein Sci. 2020;29:128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker HM, Baker EN. A structural perspective on lactoferrin function. Biochem Cell Biol. 2012;90:320–328. [DOI] [PubMed] [Google Scholar]
- 36. Wally J, Buchanan SK. A structural comparison of human serum transferrin and human lactoferrin. Biometals. 2007;20:249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pitti T, Chen CT, Lin HN, Choong WK, Hsu L, Sung TY. N‐GlyDE: A two‐stage N‐linked glycosylation site prediction incorporating gapped dipeptides and pattern‐based encoding. Sci Rep. 2019;9:15975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bartfeld NS, Law JH. Isolation and molecular cloning of transferrin from the tobacco hornworm, Manduca sexta. Sequence similarity to the vertebrate transferrins. J Biol Chem. 1990;265:21684–21691. [PubMed] [Google Scholar]
- 39. Baker EN, Baker HM. A structural framework for understanding the multifunctional character of lactoferrin. Biochimie. 2009;91:3–10. [DOI] [PubMed] [Google Scholar]
- 40. Dubach J, Gaffney BJ, More K, Eaton GR. Effect of the synergistic anion on electron paramagnetic resonance spectra of iron‐transferrin anion complexes is consistent with bidentate binding of the anion. Biophys J. 1991;59:1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Halbrooks PJ, Mason AB, Adams TE, Briggs SK, Everse SJ. The oxalate effect on release of iron from human serum transferrin explained. J Mol Biol. 2004;339:217–226. [DOI] [PubMed] [Google Scholar]
- 42. Jungreis AM. The composition of larval‐pupal moulting fluid in the tobacco hornworm, Manduca sexta . J Insect Physiol. 1978;24:65–73. [Google Scholar]
- 43. Wyatt GR. The biochemistry of insect hemolymph. Annu Rev Entomol. 1961;6:75–102. [Google Scholar]
- 44. Phalaraksh C, Reynolds SE, Wilson ID, Lenz EM, Nicholson JK, Lindon JC. A metabonomic analysis of insect development: 1H‐NMR spectroscopic characterization of changes in the composition of the haemolymph of larvae and pupae of the tobacco hornworm, Manduca sexta . Science Asia. 2008;34:279. [Google Scholar]
- 45. Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anderson BF, Baker HM, Norris GE, Rice DW, Baker EN. Structure of human lactoferrin: crystallographic structure analysis and refinement at 2.8 A resolution. J Mol Biol. 1989;209:711–734. [DOI] [PubMed] [Google Scholar]
- 47. Kurokawa H, Mikami B, Hirose M. Crystal structure of diferric hen ovotransferrin at 2.4 A resolution. J Mol Biol. 1995;254:196–207. [DOI] [PubMed] [Google Scholar]
- 48. Noinaj N, Easley NC, Oke M, et al. Structural basis for iron piracy by pathogenic Neisseria. Nature. 2012;483:53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kabsch W. Automatic indexing of rotation diffraction patterns. J Appl Cryst. 1988;21:67–72. [Google Scholar]
- 50. Kabsch W. XDS. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vonrhein C, Flensburg C, Keller P, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Evans PR. An introduction to data reduction: Space‐group determination, scaling and intensity statistics. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Skubák P, Pannu NS. Automatic protein structure solution from weak X‐ray data. Nat Commun. 2013;4:2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sheldrick GM. Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. [DOI] [PubMed] [Google Scholar]
- 56. Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr, Sect D: Biol Crystallogr. 1996;52:30–42. [DOI] [PubMed] [Google Scholar]
- 57. Zhang KYJ, Cowtan K, Main P. Combining constraints for electron‐density modification Methods in Enzymol. Vol. 277. Macromolecular Crystallography Part B. Cambridge, Massachusetts: Academic Press, 1997; p. 53–64. [DOI] [PubMed] [Google Scholar]
- 58. Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:1002–1011. [DOI] [PubMed] [Google Scholar]
- 59. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen VB, Arendall WB, Headd JJ, et al. MolProbity: All‐atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Potterton L, McNicholas S, Krissinel E, et al. Developments in the CCP4 molecular‐graphics project. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2288–2294. [DOI] [PubMed] [Google Scholar]
- 64. Jurrus E, Engel D, Star K, et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018;27:112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinf. 2009;10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Chem. 2011;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sanner MF. Python: A programming language for software integration and development. J Mol Graph Model. 1999;17:57–61. [PubMed] [Google Scholar]
- 68. UniProt Consortium . UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Maderia F, Park YM, Lee J, et al. The EMBL‐EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Evans P. Scaling and assessment of data quality. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:72–82. [DOI] [PubMed] [Google Scholar]
- 71. Diederichs K, Karplus PA. Improved R ‐factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. [DOI] [PubMed] [Google Scholar]
- 72. Weiss MS. Global indicators of X‐ray data quality. J Appl Cryst. 2001;34:130–135. [Google Scholar]
- 73. Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Evans P. Resolving some old problems in protein crystallography. Science. 2012;336:986–987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Comparison of human serum transferrin in the holo‐ and apo‐form.
Figure S2. Topology diagram of MsTsf1.
Figure S3. Structural alignments of transferrin lobes and domains.
Figure S4. Electrostatic surface charge of MsTsf1.
Figure S5. Sequence alignment of MsTsf1, human lactoferrin and human serum transferrin.
Figure S6. Characteristics of Fe3+ and carbonate coordination in MsTsf1
Figure S7. Noncovalent interactions between the linker peptide and the N1, N2 and C1 domains.
Figure S8. The mediated contacts between the C1 and N2 domains.
Figure S9. Results from docking study of organic anions in the N‐lobe cleft of MsTsf1.
Table S1. Top 100 hits from the DALI search.
Table S2. Predicted carbonate binding residues of the N‐lobe of insect Tsf1 orthologs and other arthropod transferrins