

Abstract
How diversity is structured has been a central goal of microbial ecology. In freshwater ecosystems, selection has been found to be the main driver shaping bacterial communities. However, its relative importance compared with other processes (dispersal, drift, diversification) may depend on spatial heterogeneity and the dispersal rates within a metacommunity. Still, a decrease in the role of selection is expected with increasing dispersal homogenization. Here, we investigate the main ecological processes modulating bacterial assembly in contrasting scenarios of environmental heterogeneity. We carried out a spatiotemporal survey in the floodplain system of the Paraná River. The bacterioplankton metacommunity was studied using both statistical inferences based on phylogenetic and taxa turnover as well as co-occurrence networks. We found that selection was the main process determining community assembly even at both extremes of environmental heterogeneity and homogeneity, challenging the general view that the strength of selection is weakened due to dispersal homogenization. The ecological processes acting on the community also determined the connectedness of bacterial networks associations. Heterogeneous selection promoted more interconnected networks increasing β-diversity. Finally, spatiotemporal heterogeneity was an important factor determining the number and identity of the most highly connected taxa in the system. Integrating all these empirical evidences, we propose a new conceptual model that elucidates how the environmental heterogeneity determines the action of the ecological processes shaping the bacterial metacommunity.
Subject terms: Community ecology, Microbial ecology
Introduction
Determining the processes behind community assembly across Earth’s ecosystems is a major research topic in ecology [1–4]. The issue can be addressed using two alternative views. On one hand, the traditional niche-based theory states that deterministic processes, such as environmental filters and species interactions, govern community structure and determine species composition [5, 6]. On the other hand, the neutral theory considers that all species are ecologically equivalent and, therefore, the structure of each community is a consequence of stochastic processes such as birth, death, colonization, immigration, speciation, and probabilistic dispersal [1, 7, 8]. Although these perspectives are often considered contradictory, they are not mutually exclusive and a debate exists over each relative importance in shaping natural communities.
The metacommunity theory provides an adequate framework to identify these processes considering multiple spatial scales [9, 10]. Under this approach, communities are spatially connected in a network by dispersal, and the processes involved in their assembly are the result of the interplay between local factors and regional dynamics.
Recently, Vellend [3, 11] proposed a conceptual framework in which the structure of communities can be explained by the following four “high-level processes”: selection, dispersal, ecological drift, and diversification. These four processes are universally present across ecological communities, and frame the low-level processes involved in the community assembly (e.g., competition, predation, succession, colonization, local extinction). Selection consists in the deterministic factors that modify the community structure due to fitness differences among individuals. It can act under the so-called homogeneous or heterogeneous environmental conditions, leading to more similar or dissimilar structure among communities [12, 13]. When environmental conditions are homogeneous, selection leads to more similar communities (low community turnover), a process known as “homogeneous selection.” On the other hand, under heterogeneous conditions, selection promotes an increase in community turnover as different taxa are selected, and this is referred as “heterogeneous selection” [11, 14]. Dispersal results from both deterministic and stochastic factors that either favor or limit organism movement (active or passive) and the establishment of organisms in local communities. High dispersal rates have a homogenizing effect (homogeneous dispersal) leading to a low turnover within communities, whereas low dispersal rates (dispersal limitation), coupled with drift or weak selection, increase community turnover [12, 15]. Drift influences changes in communities due to demographic events of birth, death, and reproduction that occur at random, independently of species fitness [16]. Finally, diversification refers to the generation of new species by genetic variation mainly due to stochastic factors, and larger temporal scale with respect to the other processes is needed to observe its effects [17].
The ecological rules governing bacteria metacommunity assembly seem to be different in land than in water systems, mainly due to the differences in environmental heterogeneity, which is larger in land [18–22]. In addition, and within aquatic ecosystems, the complexity of freshwater bodies is relatively high in comparison with oceans [23] and, consequently, larger differences in bacterial community composition are observed in freshwater ecosystems [24–29]. In these ecosystems, deterministic processes (mainly selection) have been found to be the main factors shaping bacterial community structure [19, 27, 30–32]. However, it is not a general rule and the relative importance of selection compared with other processes may depend on the environmental heterogeneity and dispersion rates within the metacommunity [24, 28, 30, 33, 34]. For example, in systems with low environmental heterogeneity the stochastic processes seem to be more relevant, because environmental filters are not strong enough to act as selective forces exerting species sorting [24, 33]. Thus, bacterial communities in isolated systems are likely to be mainly structured by dispersal limitation, whereas as environmental connectivity increases, homogeneous dispersal as well as drift will become more important [21, 24, 31, 32, 35–37]. Most of these previous results have been generated from a snapshot of the metacommunity. However, in complex network systems subject to hydrological influence, the relative importance of the structuring ecological processes is likely to change over time according to the changes in the hydrological connectivity [29, 31, 37–42].
Another gap lies in understanding how the high-level ecological processes influence the interactions between taxa. Co-occurrence networks are an increasingly useful tool to infer microbial interactions [43–50]. Under this approach, interactive taxa are linked together either positively or negatively indicating mutualistic or antagonistic co-occurrence patterns. Even if taxa correlations do not necessarily reflect true interactions, network analyses allow to capture and summarize information of highly diverse communities [43–46].
Another useful feature of co-occurrence networks is that they allow to identify the highly connected taxa (commonly referred as hub taxa [44, 45, 51, 52]). These taxa play a significant role within the community and confer stability due to their high connectivity with other members. Therefore, a better understanding of the mechanisms that influence highly connected taxa composition and structure may provide an insight into the underlying response of the whole community [44, 52–55].
The Paraná River floodplain constitutes a hydrological network of linked environments, which can be considered a metacommunity [40], being an ideal system to address these issues. It is characterized by a wide range of temporal and spatial heterogeneity mediated by irregular hydrological fluctuations [56]. It comprises multiple shallow lakes, some of which are permanently connected to the main river or to secondary channels, whereas others remain isolated although high-water phases connect most of the environments [57]. The floods have a homogenization effect on most environmental features [58, 59], as observed for other large river systems [60]. In contrast, the environmental characteristics show a higher spatial heterogeneity during the low-water phases due to the intensified influence of local factors [59]. Here, we investigated bacterial community structure through 16S amplicon sequencing, in contrasting environmental heterogeneity scenarios of this complex floodplain system in order to (i) determine the relative importance of the high-level processes structuring the bacterial metacommunity, (ii) detect changes in bacterial co-occurrence networks, and (iii) identify highly connected taxa.
We hypothesize that the influence of different ecological processes shaping bacterioplankton metacommunities will depend mainly on the system’s environmental heterogeneity, in turn determined by the degree of landscape connectivity. To test this hypothesis, we first analyzed the processes shaping bacterioplankton community structure using the approach based on phylogenetic and taxa turnover proposed by Stegen et al. [12]. Then, we constructed co-occurrence networks for each hydrological scenario to explore how ecological processes affect putative interactions between taxa. We predicted that as the extent of hydrological connectivity increases and the system becomes more homogeneous, nonselective processes will have a larger relevance in structuring the bacterioplankton metacommunity, promoting less interconnected network associations.
Material and methods
Study site and sampling design
The Paraná River is the second largest river in South America and fifth larger in the world (Fig. 1) [57]. The middle stretch is composed by a main channel and a floodplain that encompasses a high number of temporary and permanent streams and lakes. The system is characterized by a complex spatiotemporal dynamic influenced by the hydro- and sedimentological regime. It has a fluctuating hydrological functioning with pulses of floods and drought that determine high- and low-water phases, which alternate in variable frequency with extreme hydrological phases [61]. During high-water phases, water drains the floodplain, connecting the environments in different degrees depending on the magnitude of the flood. The sediment pulse that mainly depends on suspended solids coming from the Bermejo Basin is not coupled with the hydrological pulses, affecting most abiotic characteristics of the environments connected with the main channel [62]. The dynamics of biological communities respond to a large extent to the hydrological fluctuations, since they have a direct influence not only on the environmental characteristics, but also on the dispersal processes, and habitat colonization [39–42, 63–65].
Fig. 1. The Paraná fluvial system.

a The river location in South America showing the study area and b the environments sampled. c Daily water level from 2013 to 2016 in the Paraná River. The sampled periods are indicated with arrows.
The temporal dynamics and the spatial heterogeneity of the system were captured by sampling different hydrological phases and environmental types, characteristic of the floodplain river (Fig. 1, Supplementary Fig. S1). Four sampling campaigns that lasted 10 days each were performed during a low-water phase (LW, November–December 2013), a low-water phase coupled with the sedimentological pulse (LWs, March–April 2014), a high-water phase (HW, September 2105), and an extraordinary high-water phase (eHW, March 2016). Four representative environmental types with different hydrological connectivity and morphological features were selected (Fig. 1): the main channel and large secondary channels (MC), minor secondary channels (SC), connected lakes (CL), and isolated lakes and swamps (IL). Lentic environments were classified as IL or CL according to their hydrological connectivity degree during LW phase. Overall, our dataset consists of 59 samples: 16 from LW, 17 from LWs, 13 from HW, and 13 from eHW.
Environmental data collection
Besides depth and Secchi depth, water temperature, pH, conductivity, and dissolved oxygen (DO) concentration were measured in situ with a HANNA checkers. Subsurface water samples (20 cm) were collected and transported within 5 h in polypropylene containers to the laboratory for turbidity, soluble reactive phosphorus (SRP), nitrate (NO3−), ammonium (NH4+), chromophoric dissolved organic matter (CDOM) concentration (A440), CDOM molecular weight (S275–295), and chlorophyll-a analyses. Technical details of the abiotic variable analyses are described in Supplementary Methods.
Bacterial samples collection and sequencing
Subsurface water samples for DNA (100–140 ml) were prefiltered with a 50 µm pore mesh, and then filtered through 0.22 µm pore-size cellulose filters (GSWP0-Millipore). The filters were frozen (−80 °C) until DNA extraction. Genomic DNA was extracted using a CTAB protocol [66] as described in Supplementary Methods.
Tagged amplicons of the 16S rRNA gene (V3–V4 region) were obtained with the primers 341F and 805R [67] and sequenced using Illumina MiSeq 2 × 250 paired-end reads approach [68] by Macrogen (Seoul, South Korea).
Sequence data analysis
Raw sequences were processed using a modified version of the pipeline proposed by Logares [69] (https://github.com/ramalok), described in Supplementary Methods. Operational taxonomic units (OTUs) were defined with no clustering (zero-radius OTUs [zOTUs]) using the UNOISE2 algorithm [70]. The zOTU table resulted in 10,253 zOTUs (2,560,053 reads). We constructed zOTU rarefaction curves to evaluate richness saturation (Supplementary Fig. S2).
A second zOTU table was generated discarding zOTUs with <10 reads as well those assigned to the Archaea domain or to chloroplasts. This table was normalized to an equal sampling depth to generate the subsampled zOTU table (rrarefy function, Vegan v.2.0.9 package [71] in the R environment [72]), which consisted of 8927 OTUs and average 18,747 reads per sample.
The sequence data obtained in this work were deposited at the European Nucleotide Archive public database with references ERS4228518–ERS4228576.
Environmental heterogeneity
Environmental heterogeneity was estimated by computing the average dissimilarity between sites () [73] based on nine abiotic variables: DO, turbidity, conductivity, pH, SRP, NO3−, NH4+, CDOM concentration, and CDOM molecular weight. For each hydrological phase (i.e., LW, LWs, HW, eHW), we computed a Euclidean distance matrix (Vegan package, R) and calculated the dissimilarity between sites (Ed) as follows:
where Euc is the Euclidean distance between two sites and Eucmax corresponds to the maximum Euclidean distance considering all the pairwise distances in the overall dataset. 0.001 was added to account for zero similarity between sites [73]. Then, we calculated the mean Ed () of each computed similarity matrix and used it as an index of environmental heterogeneity in each hydrological phase. In addition, we calculated the coefficient of variation (CV%) for each variable and hydrological phase, as the standard deviation divided by the mean of each variable.
To test whether the environmental conditions in the four hydrological phases differed significantly, we performed a one-way permutational multivariate analysis of variance (PERMANOVA) [74]. We first test for homogeneity of multivariate dispersion (PERMDISP, [75]) with betadisper function (Vegan package, R), which compares the within‐group spread among groups using the average value of the individual observation distances to the centroid of the own group. Since groups account for heterogeneous dispersion (Supplementary Table S2), we run a modified PERMANOVA pseudo-F statistics (F2) implemented by Anderson et al. [76] that solves permutation test sensitivity to differences in dispersion in unbalanced design. Differences in environmental conditions were also analyzed with a two-way PERMANOVA for testing the effects of hydrological phases, type of environments, and their interaction (adonis function in Vegan package, R).
PERMANOVAs were based on Euclidean distance matrix with 9999 permutations, and pairwise P value comparisons were tested with Bonferroni post-hoc test when significant differences were found.
Bacterial community structure and diversity
To describe a general view of bacterial community structures, we analyzed the taxonomic structure in each hydrological phase and constructed rank abundance curves (Supplementary Fig. S3). We characterized the “rare biosphere” and the “abundant fraction” (zOTUs relative abundances per sample <1% and >1%, respectively) [77].
The effect of hydrological conditions on bacterial metacommunity structures was assessed by calculating the community turnover in each hydrological phase. We computed a Bray–Curtis matrix on the basis of zOTU normalized abundances for each hydrological phase to calculated the dissimilarity (Xd) between the local communities as follows:
where Bray is the Bray–Curtis dissimilarity between two communities and Braymax corresponds to the maximum Bray–Curtis dissimilarity considering the overall dataset. Then, we calculated the mean Xd of each matrix computed, which was used as a value of community turnover.
To evaluate the significance of the effect of hydrological conditions on bacterial turnover, we performed a PERMANOVA with 9999 permutations on Bray–Curtis dissimilarity matrices and Bonferroni post-hoc pairwise comparisons. Since PERMDISP was significant (Supplementary Table S2), we run the modified PERMANOVA proposed by Anderson et al. [76]. We additionally calculated the Whittaker index to explore the β-diversity (Past software V3). To visualize the taxonomic similarity across local communities, a nonmetric multidimensional scaling (NMDS) was performed using the Bray–Curtis metric (Vegan package, R). zOTU richness and Shannon–Weaver diversity (H′) indices were calculated from the normalized bacteria zOTU table, and significant differences (P < 0.01) among hydrological phases were evaluated with Kruskal–Wallis analyses and Mann–Whitney U post-hoc tests (Past software V3).
Quantification of high-level process structuring the bacterial metacommunity
To quantify the relative importance of ecological processes in structuring the bacterial metacommunity, we used the approach proposed by Stegen et al. [12]. This approach analyzes the influence of environmental filtering on the community, independently of the considered environmental variables [12, 78], avoiding the problem of overestimating the effect of stochastic processes due to unmeasured environmental variables.
We first measured the influence of selection in each hydrological phase, comparing observed phylogenetic turnover to a random expectation using the βNTI Index (β-nearest taxon [79–81]). This metric is defined as the difference between the observed mean phylogenetic distance between each taxon and its closest relative in two communities (βMNTD metric) with the βMNTD obtained from the null distribution, divided by the standard deviation of the phylogenetic distances from the null data. Absolute βNTI values greater than 2 (|βNTI| > 2) indicate that coexisting taxa are more closely related than expected by chance, so selection strongly influenced community composition [12, 82]. Next, we estimated the percentage of homogeneous selection as the fraction of pairwise comparisons with a βNTI value of <−2 and heterogeneous selection as the fraction of pairwise comparisons with a βNTI value of >+2 [12].
As this approach considered that the habitat preferences of closely related taxa are more similar than the habitat preferences of distantly related taxa [12], we tested the phylogenetic signal performing a Mantel correlogram analysis between zOTU niche and zOTU phylogenetic distances (see Supplementary Methods) [12, 24, 83–85]. Phylogenetic signals were detected over short phylogenetic distances (Supplementary Fig. S4) consistent with previous studies [12, 14, 24, 85, 86].
The communities that were not structured by selection (i.e., |βNTI| < 2) were then analyzed in a second step, where the action of dispersal and drift was calculated based on the taxonomic (zOTU) turnover with the Raup–Crick metric [87] using Bray–Curtis dissimilarities (hereafter RCbray) [12]. RCbray compares the measured β-diversity from a metacommunity against the β-diversity obtained from a null model. RCbray values between –0.95 and +0.95 indicate significant departures from the degree of turnover, occurring when drift is acting alone [12]. In addition, RCbray values > +0.95 indicate that the communities are less similar than expected by chance as a result of dispersal limitation combined with drift, and RCbray values < −0.95 indicate that the communities are more similar than expected by chance as a result of homogeneous dispersion [12].
Phylogenetic and taxonomic turnovers were calculated in the R environment, we used the Picante package [88] for βNTI and the raup_crick_abundance function developed by Stegen et al. [12] for RCbray.
To identify the features that imposed selection or dispersal limitation during each hydrological phase, we performed a distance-based redundancy analysis (db-RDA) on the βNTI and RCbray matrices using spatial and environmental explanatory variables following Stegen et al. [12].
The spatial relationships between sampling sites were estimated using distance-based Moran’s eigenvector maps (dbMEMs, formerly called principal coordinates of neighbor matrices) [89]. A Euclidean distance matrix was generated from watercourse distances, a proxy of dispersal by water, measured as the minimal distance from each environment to the nearest major channel using satellite images (Google Earth Pro). dbMEM eigenvectors were computed from the truncated Euclidean matrix just at the distance at which IL become disconnected from the lotic influence (8.44 km for LW, and 6.98 km for LWs), or at the distance that keeps all the sites connected (for HW and eHW). The truncation value was calculated as the maximum distance of the minimum spanning tree [90].
The spatial eigenvectors and the nine measured abiotic variables were combined in a principal component analysis (PCA). The resulting PCA axes were used as independent variables in the db-RDA with either βNTI or RCbray as the dependent variables. Four separate sets of PCA axes and db-RDA analyses were run for each hydrological phase. Each βNTI and RCbray matrices were normalized adding the absolute magnitude of the minimum (negative) value to all values, and then dividing the resulting values by their maximum. Stepwise forward model selection was carried out on PCA axes evaluated using a P value significance level of 0.05 (as determined by 999 permutations) and the coefficient of determination R2 of the global model with all explanatory variables [91].
The significantly selected PCA axes in the db-RDA based on the βNTI characterized environmental variables that imposed selection. The extent to which PCA axes were related to abiotic variables or dbMEMs was evaluated by examining PCA axis loadings. Abiotic variables that loaded heavily on a significant PCA axis for βNTI were considered as measured environmental variables that imposed Selection. PCA axes that loaded on a dbMEMs were considered as an indicator of unmeasured spatially structured environmental variables (strong PCA loading with a dbMEM). The PCA axes were not related to the βNTI but were significantly selected in the db-RDA based on RCbray were considered as a feature of Dispersal Limitation. dbMEMs and db-RDA were performed with CANOCO software V5.0 [90].
Co-occurrence networks
A bacterial meta-network was constructed considering all the samples from the four hydrological phases. Furthermore, to infer whether bacterial associations were influenced by the different ecological processes structuring the metacommunity, one specific network association for each hydrological phase was constructed considering zOTUs with >50 reads and present in at least 50% of the samples. As previous works have shown that the number of taxa and samples have a strong impact on the network properties [92], the networks were constructed from the same number of taxa and samples randomly selected. Thus for each network, the initial zOTU matrix consisted in 13 samples and 328 zOTUs.
The networks were constructed using the CoNet software V1.1.1.beta [93] implemented in Cytoscape V3.7.1 [94]. Four measures were calculated: Bray–Curtis and Kullback–Leibler nonparametric dissimilarity indices, and Pearson and Spearman rank correlations. The combination of their results allows the appropriateness of scoring measures to determine the statistical significance of correlations, as stated by the authors [93]. The initial edge selection was set to include the 2000 positive and 2000 negative edges consistent across all four correlation measures. The significance of the edges was calculated using the ReBoot method [95] based on 1000 permutations with renormalization and 1000 bootstrap iterations. Only edge supported by at least three methods was considered. Then, edge-specific P values were merged using Brown’s method [96], followed by Benjamini–Hochberg for false discovery rate correction, edges with merged P values below 0.05 were kept [93].
In the microbial network, two taxa could be related because of a true ecological association or alternatively, because they are correlated to an abiotic or biotic environmental factor [46, 93]. To explore indirect associations driven by environmental factors, in the network construction, we included environmental information in an additional matrix containing the following variables: DO, turbidity, conductivity, pH, SRP, NO3−, NH4+, CDOM concentration, and CDOM molecular weight. No environmentally driven indirect edges (i.e., environmental node—taxa node) were detected.
To validate the nonrandom co-occurrence patterns, we evaluated networks against their randomized versions using the Barabasi–Albert model available in the Randomnetworks plugin in Cytoscape. NetworkAnalyzer tool [97] was used to calculate four network topology properties: number of nodes (bacterial zOTUs), number of edges (associations between bacteria), network density, and clustering coefficient. These structural attributes were used to infer the connectedness in bacterial networks [46, 92, 98, 99]. The network density indicates the average connectivity of a node in the network with values varying between 0 and 1 [92, 100]. Values close to 1 are expected when the connectivity of the network increases [100]. The clustering coefficient indicates how nodes are embedded in their neighborhood and, thus, the degree to which they tend to cluster together [92, 100, 101]. As a consequence, low values of this metric are interpreted as less interconnected association networks. Mathematically, it is the average clustering coefficient of all nodes in the graph. The clustering coefficient of a node is the ratio between the numbers of connections in its neighborhood to the number of possible connections. In graph theory, the neighborhood of a node is the set of nodes that are connected to it. Thus, for example, if the node has three neighbors, then it can have a maximum of three connections among them. If all three possible connections are realized, the clustering coefficient of the node is 1 (3/3). Contrarily, if none of the possible connections with the neighbors are realized, the local clustering coefficient value is 0 (0/3).
Highly connected bacterial taxa
To identify highly connected taxa we used three centrality metrics: node degree, closeness centrality, and betweenness centrality [47, 51–54]. The degree is the number of edges connecting each node to the rest of the network. Closeness centrality measures the length of the shortest path between two nodes, reflecting the importance of a node in disseminating information. Betweenness centrality quantifies how many steps away a particular node is from all the others in the web, denoting the role of a node as a bridge between the other components of a network [52, 54, 92].
For each network, we identified the highly connected taxa as those nodes with a high degree (>20), closeness centrality (>0.26), and low betweenness centrality (<0.06) (NetworkAnalyzer tool in Cytoscape) [53, 54, 98]. These metrics illustrate both the number of connections and how important those connections are to the overall network [53, 102, 103].
Redundancy analysis (RDA) was performed to evaluate the effect of abiotic variables on the abundance of the highly connected taxa (CANOCO software V5 [96]). The environmental variables tested as explanatory variables (DO, turbidity, conductivity, pH, SRP, NO3−, NH4+, CDOM concentration, CDOM molecular weight) were normalized as standard normal deviates. A forward selection procedure was run to find the subset of significant explanatory variables using Monte Carlo Permutations.
Results
Environmental heterogeneity
The average environmental dissimilarity between sites () as well as the CV% of the nine abiotic variables indicated a clear trend to higher spatial homogenization from LW (low hydrological connectivity) to HW (high hydrological connectivity) phases (Fig. 2). Thus, the four hydrological phases were significantly different according to their environmental characteristics (PERMANOVA F2 = 7.6799, P = 0.001, Supplementary Tables S1 and S2, Fig. S5). The two-way PERMANOVA showed a significant effect of environmental type (F1 = 2.06, P < 0.01) mainly due to IL, besides the effect of hydrological phases (F1 = 2.5, P < 0.01) (Supplementary Table S3).
Fig. 2. Environmental heterogeneity in the Paraná fluvial system in different hydrological phases.
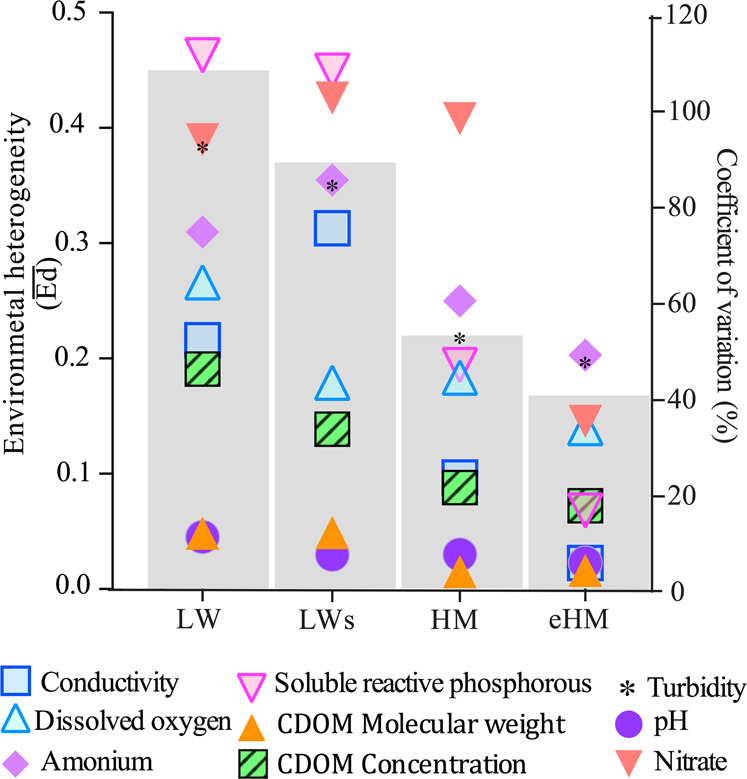
The environmental heterogeneity is expressed as average environmental dissimilarity between sites (, gray bars) and the variability (coefficient of variation, CV%) of nine selected abiotic variables among environments. LW low water, LWs low water with sedimentological pulse, HW high water, eHW extraordinary high water.
Bacterial community structure and diversity
In the 59 samples analyzed, a total of 8927 zOTUs within the bacterial domain were defined. The metacommunity was mainly dominated by Proteobacteria (33%), Actinobacteria (23%), Bacteroidetes (18%), and Verrucomicrobia (11%). The community structure according to zOTUs abundance varied with hydrological conditions with significant differences among phases (PERMANOVA, F2 = 6.9265, P = 0.001, Supplementary Fig. S6, Table S3), except between the two LW phases (LW and LWs). In the NMDS, the samples were distributed following a clear gradient of homogenization, from LW to HW phases (Fig. 3). In the eHW phase, local communities were more similar grouped in a distinct cluster. In contrast, those from LW phases were more spread, mainly due to differences in the community composition of IL (Fig. 3). The community turnover varied markedly among hydrological phases, being significantly lower (PERMANOVA, P < 0.005) in HW phases (: HW = 0.58; eHW = 0.29) than in LW phases (: LW = 0.76; LWs = 0.75). In line with this observation, the highest values of ß-diversity were obtained in both LW phases (Whittaker: LW = 3.50; LWs = 3.27; HW = 1.97; eHW = 1.95).
Fig. 3. Bacterial community structure and diversity in the Paraná fluvial system in different hydrological phases.

a Sample ordination in a nonmetric multidimensional scaling (NMDS) according to the similarity in bacterial communities structure (zOTU relative abundance) among the four hydrological phases (indicated with color) at the different type of environments (indicated with symbols) of the Paraná fluvial system. Stress value: 0.112. b, c Richness and Shannon–Weaver diversity index H′ of local bacterial communities in the four hydrological phases. LW low water, LWs low water with sedimentological pulse, HW high water, eHW extraordinary high water, IL isolated lake, CL connected lake, SC secondary channel, MC main channel.
Species richness and Shannon diversity (H′) presented similar average values in the different hydrological phases (Kruskal–Wallis, P = 0.366, Fig. 3). However, differences among local communities were pronounced in LW phases, when IL had a higher variability showing lower values than the mean (Fig. 3).
High-level process structuring the bacterial metacommunity
Having demonstrated that bacterial community structure was clearly linked to changes in environmental heterogeneity, we quantified the relative importance of selection, dispersal, and drift processes in structuring the metacommunity.
The phylogenetic turnover analysis (βNTI) revealed that selection was the most important structuring process regardless of the hydrological phases. However, its relative importance as well as the type of selection, changed according to the hydrological conditions and hence, to environmental heterogeneity. During both LW phases, heterogeneous selection had the major role in structuring the metacommunity (76.66 and 84.55% of the community turnover for LW and LWs, respectively), whereas the role of homogeneous selection and the nonselective processes was less significant. In the HW phase, heterogeneous selection was also the most important process (39.74% of the overall community turnover), but the relative importance of homogenous selection and nonselective processes were twice as high as in the LW phases (Fig. 4). In contrast, homogenous selection was the most important process in eHW (88.46% of the overall community turnover) (Fig. 4). Regarding nonselective processes (RCbray), dispersal limitation combined with drift had a prevalent role in all the hydrological phases, except in eHW when homogeneous dispersion became more important (Fig. 4).
Fig. 4. Relative importance of high-level processes structuring the bacterial metacommunity of the Paraná fluvial system in different hydrological phases.
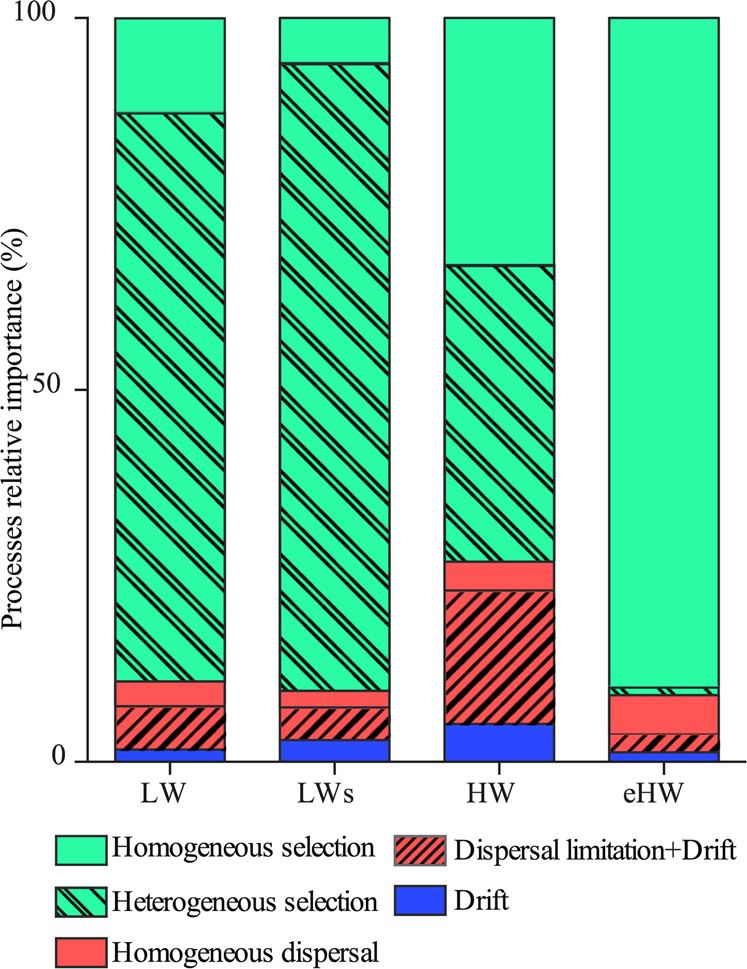
Values indicate percentage of community turnover associated to each process: homogeneous selection (βNTI < −2), heterogeneous selection (βNTI > +2), homogeneous dispersal (RCbray < −0.95), dispersal limitation combined with drift (RCbray > +0.95), and drift (|RCbray| < 0.95). LW low water, LWs low water with sedimentological pulse, HW high water, eHW extraordinary high water.
In each of the four hydrological phases, selection was imposed by a different set of environmental factors. In LW, three PCA axes were significant for the βNTI model (PCA1, PCA3, and PCA4; Table 1 and Supplementary Table S4). Within PCA1 the major loading values were observed for DO, pH, and CDOM molecular weight. Weak loading values of the measured abiotic variables were observed in the PCA3 and PCA4, indicating that these axes represent an unmeasured, spatially structured environmental variable that impose some degree of selection (Table 1 and Supplementary Table S4). In the LWs phase, model selection identified PCA1, PCA3, and PCA11, as significant axes. The stronger loadings on these axes were due to environmental features related with nutrient concentration (SRP and NO3−), CDOM molecular weight, pH, and an unmeasured, spatially structured environmental variable (Table 1 and Supplementary Table S4). In the HW phases, the βNTI model selection identified one significant PCA axis (PCA1) in HW, and three in eHW (PCA1, PCA5, and PCA6; Table 1 and Supplementary Table S4). In the HW phase, conductivity and CDOM molecular weight had the strongest loading values on PCA1, whereas in the eHW phase the environmental variables related were CDOM and SRP.
Table 1.
Features that imposed selection and dispersal limitation in the Paraná fluvial system during different hydrological phases based on the model selection for βNTI and RCbray analysis.
| Hydrological phase | Features imposing selection | Features imposing dispersal limitation |
|---|---|---|
| Low water (LW) | PCA1: DO, pH, CDOM molecular weight | NA |
| PCA3: unmeasured abiotic variable | ||
| PCA4: unmeasured abiotic variable | ||
| Low water + sedimentological pulse (LWs) | PCA1: SPR, nitrate | NA |
| PCA3: pH + unmeasured abiotic variable | ||
| PCA11: pH, CDOM molecular weight | ||
| High water (HW) | PCA1: conductivity, DO, CDOM molecular weight | PCA2 |
| PCA3 | ||
| Extraordinary high water (eHW) | PCA1: CDOM molecular weight, CDOM concentration | NA |
| PCA5: SPR | ||
| PCA6: SPR |
The PCA axes retained during model selection for βNTI were considered as imposing selection and those significant for RCbray as imposing dispersal limitation. Within each PCA significant for βNTI, strong loading was considered as a measure or unmeasured variable imposing selection. Loading values for each PCA axes are presented in Supplementary Table S4.
DO dissolved oxygen, SPR soluble reactive phosphorous, CDOM chromophoric dissolved organic matter, NA indicated that nonsignificant PCA axes were retained in RCbray model selection.
Regarding the features imposing dispersal limitations, only in HW phase, the RCbray model selection identified significant PCA axes (PCA2 and PCA3) that were not retained in the βNTI model selection (Table 1 and Supplementary Table S4).
Bacterial community associations
The bacterial meta-network consisted in 470 nodes (bacterial zOTUs) and 936 edges (associations between bacteria). It was mainly represented by taxa from Bacteroidetes, Betaproteobacteria, and Verrucomicrobia (which together account for the 45% of the total nodes). The network density was 0.008 and the coefficient of clustering was 0.364. The majority of OTUs presented positive associations (Supplementary Fig. S7).
The co-occurrence networks at each hydrological phases (Fig. 5) varied in the number of edges, being the highest in both LW phases, whereas the number of nodes did not vary considerable. The connectedness of the networks appeared to be higher during the dominance of heterogeneous selection, as the network density was considerably higher in both LW phases in contrast to HW phases, especially in the HW (Fig. 5). In agreement, the same behavior was shown by the clustering coefficient, indicating higher network connectivity when heterogeneous selection had the major role in structuring the metacommunity. Finally, the networks from HW phases presented a higher percentage of positive edges compared to those from LW phases (Fig. 5).
Fig. 5. Bacterial co-occurrence networks for each hydrological phase of the Paraná fluvial system, arranged according to the bacterial taxonomy (classes or orders).

The size of each node is proportional to the number of connections (degree). Gray dashed and red solids lines represent positive and negative correlation, respectively. The number of nodes, number of edges, positive edges percentage, network density, and clustering coefficient of the overall network is indicated below each network.
Metacommunity highly connected taxa
The number as well as the taxonomic composition of the highly connected taxa (i.e., those taxa that play a significant role within the community) were notably different in the four hydrological phases (Supplementary Table S5). In LW and LWs, 54 and 44, respectively, zOTUs were defined as highly connected taxa, while these were reduced to 10 in the HW phases, and to 3 in the eHW. Most of these taxa were exclusive of each hydrological phase and none was present in all the four phases. The highly connected taxa were recruited differentially from each phylum in each hydrological phase, being Actinobacteria and Bacteroidetes the more represented phyla (Supplementary Fig. S8).
The arrangement of highly connected taxa in the RDA analysis was mainly related to the hydrological phase in which they were defined (Supplementary Fig. S9). The first two axes accounted for 28% of the variance (axis 1: 16.7%; axis 2: 11.3%) being CDOM concentration (A440), CDOM molecular weight (S275–295), DO, and nutrients (SRP and NO3−) the main explanatory variables (P < 0.01). The first axis was mostly defined by the NO3− concentration and CDOM molecular weight (intraset correlation coefficients: −0.59 and 0.57, respectively). The second axis was principally associated with CDOM concentration and molecular weight (intraset correlation coefficients: 0.58 and −0.45, respectively). Taxa from the LW phases grouped together by the left of the graph and were positively related to NO3− and inversely related to CDOM molecular weight. The taxa from HW phases were mainly positioned toward the upper part of the graph (Supplementary Fig. S9), more associated with high CDOM molecular weight, SRP concentration, and high CDOM concentration (A440).
Discussion
This study provides the first empirical evidence that selection is the most important process structuring the bacterial metacommunity at both extremes of environmental heterogeneity and homogeneity. This finding challenges the general view that selection is weakened by dispersal homogenization [21, 36, 37]. In addition, we show that at intermediary environmental heterogeneity stochastic processes become more important.
Based on the underpinning of Vellend’s approach [3], we predicted that the influence of nonselective processes would increase as the system becomes more homogeneous. Contrarily, we found that selection was the main process shaping the bacterial metacommunity at both high and low environmental heterogeneity.
Several studies have found evidence of the pivotal role of selection in determining bacterial community structure in different environments, and a decrease in its relative importance as dispersal rates increase and the systems become more homogeneous in their environmental conditions [14, 31, 32, 104]. Particularly, Wang et al. [24] in a comparative survey of a broad range of ecosystems (i.e., soil, stream biofilm, and lake), observed a clearly dominant role of deterministic processes controlling the assembly of bacterial communities, whereas stochastic processes became more relevant as environmental heterogeneity decreased and dispersal rates increased. The authors proposed a theoretical threshold of selective strength above which communities should be mainly structured by deterministic processes as a consequence of high environmental heterogeneity and low dispersal rates.
However, these previous attempts considered only variability in the spatial scale. To address the full scope of the question, we extended these observations considering the spatial and the temporal variability in a complex ecosystem. We summarize our findings in three possible scenarios: (i) With high environmental heterogeneity characterized by low connectivity, the structure of the bacterial assemblage was mainly driven by heterogeneous selection (Fig. 6a). In this scenario, species with different fitness may strongly be filtered by different selective forces in each local community, leading to a high β-diversity, whereas the influence of drift and dispersion became irrelevant. (ii) At intermediate environmental heterogeneity, both heterogeneous and homogeneous selections had similar weight and the relative influence of stochastic processes increased (Fig. 6a). Here, the magnitude of ordinary floods seems to be insufficient to thoroughly mix water among all the floodplain, and some environments begin to homogenize their environmental filters while others retain their identity [105]. Selection may act with similar strength both homogenizing and segregating local communities, leading to an overall reduction of community turnover and β-diversity. Simultaneously, the metacommunity should be relatively random due to intermediate dispersal rates. (iii) At extreme environmental homogeneity, homogeneous selection was the main ecological rule structuring the metacommunity (Fig. 6a). Despite stochasticity is expected to increase due to the high dispersal rates, the bacterial species should strongly be filtered by common environmental factors and the strength of homogeneous selection may dampen the influence of drift and dispersion. For instance, low community turnover and β-diversity are expected. These evidences support the early assumptions made by Stegen et al. [85] and Dini-Andreote et al. [14] and provide a new view of bacterial community assembly in which the action of selective and nonselective processes both play a role.
Fig. 6. Conceptual model that synthetizes how environmental heterogeneity determines the action of the ecological processes assembling the bacterial metacommunity.

Empirical evidence (a, b) that support the proposed conceptual model (c). At high environmental heterogeneity, community structure is mainly determined by heterogeneous selection. The heterogeneous selection promotes greater β-diversity increasing divergence in local communities, and higher interconnected bacterial co-occurrence network. At intermediate values of environmental heterogeneity both heterogeneous and homogeneous selection act with similar strength, and stochastic processes reach more importance. In this scenario, the selection acting in opposite ways does not allow homogenization or differentiation of local community structure, leading to an overall reduction of turnover and β-diversity. Simultaneously, the metacommunity will have a relatively high randomness due to intermediate dispersal rates. As a consequence, the association network would tend to be loosely interconnected. In extremely homogeneous environmental conditions, the low diversity of niche habitats leads to an increase of homogeneous selection, and local communities are strongly filtered by common environmental factors. Homogeneous selection then leads to a low turnover and β-diversity, promoting less interconnected associations. ND network density.
The factors that impose selection changed in the different hydrological phases. They were mainly related with inorganic nutrient concentration, the quality and quantity of CDOM, as well as conductivity and DO. This could be explained by marked lateral gradient that characterize our system, which changes according to the landscape connectivity [59]. During the LW phases, and from the MC to the IL, we observed a clear decreasing trend in pH, DO, and SRP concentration, together with an increasing trend in conductivity, NO3−, and low molecular weight CDOM concentration [59, 106]. During eHW phase, the effect of the floods led to a reduction of the differences among environments, and the variables became more similar to those in the MC. In addition, features that impose dispersal limitation were only detected in the HW phase.
At this point, it is appropriate to mention that Stegen’s approach is based on the assumption that phylogenetic relatedness is indicative of shared environmental response traits, setting aside the ecological traits that do not have a strong phylogenetic signal. This may lead to an overestimation of the relative importance of dispersal limitation as it could mask the effects of phylogenetically nonconserved selection processes [12, 107]. In our study, the results of the estimated quantitative processes were in concordance with those from the identification of features imposing selection and dispersal limitation, giving robust support to our conclusions. More explicitly, even though in our system the bacterial communities were heavily governed by selection, significant variables imposing dispersal limitation were identified when this process showed the major influence (i.e., in HW phase).
The understanding of the ecological processes acting on the river floodplain metacommunity was extended studying the bacterial co-occurrence patterns. Network analyses-based approaches have the potential to infer intertaxa correlations and can be applied to investigate structure complexity [95, 108], and the ecological rules guiding community assembly [29, 109]. Particularly, the influence of ecological processes on bacterial associations is a poorly explored field. Emerging studies in soil communities have revealed that systems under organic farming harbor more interconnected networks than conventional farming, mainly associated to higher habitat heterogeneity [98]. In addition, it has been demonstrated that greater microbial diversity ensures greater association complexity [29, 109].
The networks’ topology was different according to the weight of the ecological processes acting on the bacterial community assemblage. At heterogeneous selection dominance (LW and LWs phases), the network associations seem to be more interconnected than HW phases (Fig. 6b). We assign this finding to the fact that heterogeneous selection promotes a greater taxonomic β-diversity [29, 109]. Contrarily, homogeneous selection may be associated to fewer interconnected networks as decreasing β-diversity. In addition, under multiple ecological processes acting with similar strength, the network tends to be less interconnected (Fig. 6b).
The dominance of different ecological processes structuring the community appeared to be especially important for shaping the mutualistic and antagonistic co-occurrence patterns. For instance, there were considerably more negative correlations when systems became more homogeneous, compared with periods of low water. Several ecological interpretations had been made about the extent to which we can interpret negative and positive correlations according to how the networks have been constructed (e.g., whether the network represents one or more trophic levels, or if it results in spatial or temporal associations) [44, 45, 48, 110]. At the metacommunity level negative correlations may reflect competition that it is expected to be more common under a homogeneous scenario, as homogeneous selection favors taxa with similar ecological requirements that will compete for similar sources. Contrarily, the dominance of positive correlations in our survey under the dominance of heterogeneous selection may reflect the coexistence of multiple taxa in the metacommunity occupying multiple different niches [44, 45, 48, 110]. Our analysis represents one of the first studies that have empirically linked bacterioplankton networks to community structuring processes. Future studies comparing networks for communities of different systems, and also structured by different ecological processes, will allow for a deepen exploration and generation of new ecological hypotheses.
A useful feature of network analysis is that it allows to identify the strongly connected taxa that have a core effect on the community assemblage [53, 111] and are adequate predictors of overall community changes [55]. Each hydrological phase presented a particular set of highly connected taxa that were determined by different environmental factors. Since conditions significantly varied among the hydrological phases this was not surprising (Fig. 1). Perhaps the most interesting fact of our findings was the huge number of highly connected taxa found under the highest environmental heterogeneity, which could be attribute to a higher niche segregation over this condition. As it was mentioned above, a marked lateral hydrological gradient (i.e., from the MC to IL) is observed in many variables during LW phases. Remarkably, all the significant variables in the RDA are related to quality and quantity of essential resources for bacteria (i.e., N, P, and CDOM). Even though caution is needed as these estimated taxa derive from a co-occurrence approach [43–45], we found a concordance between the factors imposing selection to the whole community, and those more related with the highly connected taxa. These results spotted the spatiotemporal heterogeneity as an important factor determining the number and identity of the highly connected taxa (hubs) and support their importance as target organisms for a better understanding of the whole community. A challenge for future research will be to elucidate ecological functions of hubs in the system.
Taking together, our results demonstrated that Stegen’s framework, strengthened with co-occurrence network analyses, can provide relevant insights about the ecological processes structuring microbial communities, although a good understanding on the method’s limitations must be exerted. Finally, we would like to highlight the importance of incorporating temporal variability in order to understand metacommunity assembly.
Concluding remarks
Integrating all the empirical evidence obtained here, we propose a conceptual model that synthetizes how environmental heterogeneity determines the action of the ecological processes assembling the bacterial metacommunity (Fig. 6c). In systems with high environmental heterogeneity, heterogeneous selection plays the major role in structuring the community, promoting a greater β-diversity, and more interconnected networks associations. At intermediate values of environmental heterogeneity, the action of stochastic processes reaches more importance and both, heterogeneous and homogeneous selection, have similar contribution. This leads to a decrease in β-diversity and to loosely interconnected networks associations. In systems with low environmental heterogeneity, the strength of homogeneous selection dampens the influence of drift and dispersion, and local communities become more similar and the association networks less interconnected. While this model was devised based on the empirical evidence of a complex fluvial system, it certainly would be needed to evaluate its robustness and limitation in other types of environments.
A particular strength of this model is that it was conceived relying on different metacommunity features (e.g., taxonomic and phylogenetic turnover, network associations) but taking into account the spatial and temporal variability scales. Furthermore, we used a step-forward analysis combining different statistical inferences, which allowed us to take advantage of the strengths of each method alone and in synergy with the others. Therefore, the proposed model represents a significant improvement on our knowledge of bacterial community’s assembly across freshwater ecosystems and provides a new framework to be tested in future studies in other communities.
Supplementary information
Acknowledgements
This study was supported by Agencia Nacional de Promoción Científica y Tecnológica (PICT 2016-0465 PI, MD), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET). HS’s work was supported by Conselho Nacional de Desenvolvimento e Pesquisa Tecnológica (CNPq process 309514/2017-7). We thank C. Debonis, E. Creus, and M. Piacenza for their field assistance, and A. Dabin for bioinformatic support. Bioinformatics analyses were performed at the PIRAYU cluster (https://cimec.org.ar/c3/pirayu/index.php) via grants obtained from the Agencia Santafesina de Ciencia, Tecnología e Innovación (ASACTEI; Res N° 117/14). We thank the members of Metacommunity Project particularly M. Marchese, P. Scarabotti, D. Borzone, and M. Licursi, for the long discussions that largely improved the manuscript. Finally, we thank J.M. Gasol for critical revision of the manuscript, R. Logares for his helpful feedback on network analyses, and three anonymous reviewers for valuable comments on the manuscript.
Author contributions
PH and MD conceived and designed the study; MD conducted sampling with the help of GM; PH performed the molecular analysis; GM performed the chemical analyses; PH and SM ran statistical analyses; PH and MD wrote the manuscript with contributions from FU and HS; and all authors edited the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41396-020-0723-2) contains supplementary material, which is available to authorized users.
References
- 1.Hubbell SP. The unified neutral theory of biodiversity and biogeography. Princeton: Princeton University Press; 2001. [DOI] [PubMed] [Google Scholar]
- 2.Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL, et al. Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol. 2006;4:102–12. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- 3.Vellend M. The theory of ecological communities. 1st ed. Woodstock, UK: Princeton University Press; 2016. [Google Scholar]
- 4.McGill BJ. Towards a unification of unified theories of biodiversity. Ecol Lett. 2010;13:627–42. doi: 10.1111/j.1461-0248.2010.01449.x. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert B. Joint consequences of dispersal and niche overlap on local diversity and resource use. J Ecol. 2012;100:287–96. [Google Scholar]
- 6.Chase JM, Leibold MA. Ecological niches: linking classical and contemporary approaches. Biodivers Conserv. 2004;13:1791–3. [Google Scholar]
- 7.Rosindell J, Hubbell SP, Etienne RS. The unified neutral theory of biodiversity and biogeography at age ten. Trends Ecol Evol. 2011;26:340–8. doi: 10.1016/j.tree.2011.03.024. [DOI] [PubMed] [Google Scholar]
- 8.Bell G. Neutral macroecology. Science. 2001;293:2413–8. doi: 10.1126/science.293.5539.2413. [DOI] [PubMed] [Google Scholar]
- 9.Leibold MA, Holyoak M, Mouquet N, Amarasekare P, Chase JM, Hoopes MF, et al. The metacommunity concept: a framework for multi-scale community ecology. Ecol Lett. 2004;7:601–13. [Google Scholar]
- 10.Wilson DS. Complex interactions in metacommunities, with implications for biodiversity and higher levels of selection. Ecology. 1992;73:1984–2000. [Google Scholar]
- 11.Vellend M. Conceptual synthesis in community ecology. Q Rev Biol. 2010;85:183–206. doi: 10.1086/652373. [DOI] [PubMed] [Google Scholar]
- 12.Stegen JC, Lin X, Fredrickson JK, Chen X, Kennedy DW, Murray CJ, et al. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013;7:2069–79. doi: 10.1038/ismej.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacArthur RH, Wilson EO. The theory of island biogeography. Woodstock, UK: Princeton University Press; 2001. [Google Scholar]
- 14.Dini-Andreote F, Stegen JC, Van Elsas JD, Salles JF. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci USA. 2015;112:E1326–E1332. doi: 10.1073/pnas.1414261112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowe WH, McPeek MA. Is dispersal neutral? Trends Ecol Evol. 2014;29:444–50. doi: 10.1016/j.tree.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Vellend M, Srivastava DS, Anderson KM, Brown CD, Jankowski JE, Kleynhans EJ, et al. Assessing the relative importance of neutral stochasticity in ecological communities. Oikos. 2014;123:1420–30. [Google Scholar]
- 17.Rundle HD, Nosil P. Ecological speciation. Ecol Lett. 2005;8:336–52. [Google Scholar]
- 18.Graham EB, Knelman JE, Schindlbacher A, Siciliano S, Breulmann M, Yannarell A, et al. Microbes as engines of ecosystem function: when does community structure enhance predictions of ecosystem processes? Front Microbiol. 2016;7:214. doi: 10.3389/fmicb.2016.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindström ES, Langenheder S. Local and regional factors influencing bacterial community assembly. Environ Microbiol Rep. 2012;4:1–9. doi: 10.1111/j.1758-2229.2011.00257.x. [DOI] [PubMed] [Google Scholar]
- 20.Isabwe A, Ren K, Wang Y, Peng F, Chen H. Community assembly mechanisms underlying the core and random bacterioplankton and microeukaryotes in a river–reservoir system. Water. 2019;11:1–17. [Google Scholar]
- 21.Fodelianakis S, Lorz A, Valenzuela-Cuevas A, Barozzi A, Booth JM, Daffonchio D. Dispersal homogenizes communities via immigration even at low rates in a simplified synthetic bacterial metacommunity. Nat Commun. 2019;10:1. doi: 10.1038/s41467-019-09306-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiao S, Yang Y, Xu Y, Zhang J, Lu Y. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 2020;14:202–16. doi: 10.1038/s41396-019-0522-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grosberg RK, Vermeij GJ, Wainwright PC. Biodiversity in water and on land. Curr Biol. 2012;22:R900–R903. doi: 10.1016/j.cub.2012.09.050. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Shen J, Wu Y, Tu C, Soininen J, Stegen JC, et al. Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J. 2013;7:1310–21. doi: 10.1038/ismej.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zinger L, Amaral-Zettler LA, Fuhrman JA, Horner-Devine MC, Huse SM, Welch DBM, et al. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS ONE. 2011;6:e24570. doi: 10.1371/journal.pone.0024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barberán A, Casamayor EO. Global phylogenetic community structure and β-diversity patterns in surface bacterioplankton metacommunities. Aquat Micro Ecol. 2010;59:1–10. [Google Scholar]
- 27.Nemergut DR, Schmidt SK, Fukami T, O’Neill SP, Bilinski TM, Stanish LF, et al. Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev. 2013;77:342–56. doi: 10.1128/MMBR.00051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruiz-González C, Niño-García JP, del Giorgio PA. Terrestrial origin of bacterial communities in complex boreal freshwater networks. Ecol Lett. 2015;18:1198–206. doi: 10.1111/ele.12499. [DOI] [PubMed] [Google Scholar]
- 29.Widder S, Besemer K, Singer GA, Ceola S, Bertuzzo E, Quince C, et al. Fluvial network organization imprints on microbial co-occurrence networks. Proc Natl Acad Sci USA. 2014;111:12799–804. doi: 10.1073/pnas.1411723111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comte J, Lovejoy C, Crevecoeur S, Vincent WF. Co-occurrence patterns in aquatic bacterial communities across changing permafrost landscapes. Biogeosciences. 2016;13:175–90. [Google Scholar]
- 31.Heino J, Melo AS, Siqueira T, Soininen J, Valanko S, Bini LM. Metacommunity organisation, spatial extent and dispersal in aquatic systems: Patterns, processes and prospects. Freshw Biol. 2015;60:845–69. [Google Scholar]
- 32.Logares R, Deutschmann IM, Giner CR, Krabberød AK. Different processes shape prokaryotic and picoeukaryotic assemblages in the sunlit ocean microbiome. Environ Microbiol. 2018;20:37–49. [Google Scholar]
- 33.Östman Ö, Drakare S, Kritzberg ES, Langenheder S, Logue JB, Lindström ES. Importance of space and the local environment for linking local and regional abundances of microbes. Aquat Micro Ecol. 2012;67:35–45. [Google Scholar]
- 34.Niño-García JP, Ruiz-González C, del Giorgio PA. Interactions between hydrology and water chemistry shape bacterioplankton biogeography across boreal freshwater networks. ISME J. 2016;1:1–12. doi: 10.1038/ismej.2015.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ofiţeru ID, Lunn M, Curtis TP, Wells GF, Criddle CS, Francis CA, et al. Combined niche and neutral effects in a microbial wastewater treatment community. Proc Natl Acad Sci USA. 2010;107:15345–50. doi: 10.1073/pnas.1000604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans S, Martiny JBH, Allison SD. Effects of dispersal and selection on stochastic assembly in microbial communities. ISME J. 2017;11:176–85. doi: 10.1038/ismej.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Declerck SAJ, Winter C, Shurin JB, Suttle CA, Matthews B. Effects of patch connectivity and heterogeneity on metacommunity structure of planktonic bacteria and viruses. ISME J. 2013;7:533–42. doi: 10.1038/ismej.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandes IM, Henriques-Silva R, Penha J, Zuanon J, Peres-Neto PR. Spatiotemporal dynamics in a seasonal metacommunity structure is predictable: the case of floodplain-fish communities. Ecography. 2014;37:464–75. [Google Scholar]
- 39.José de Paggi SB, Paggi JC. Hydrological connectivity as a shaping force in the zooplankton community of two lakes in the Paraná River floodplain. Int Rev Hydrobiol. 2008;93:659–78. [Google Scholar]
- 40.Devercelli M, Scarabotti P, Mayora G, Schneider B, Giri F. Unravelling the role of determinism and stochasticity in structuring the phytoplanktonic metacommunity of the Paraná River floodplain. Hydrobiologia. 2016;764:139–56. [Google Scholar]
- 41.Devercelli M. Changes in phytoplankton morpho-functional groups induced by extreme hydroclimatic events in the Middle Paraná river (Argentina) Hydrobiologia. 2010;639:5–19. [Google Scholar]
- 42.Scarabotti PA, López JA, Pouilly M. Flood pulse and the dynamics of fish assemblage structure from neotropical floodplain lakes. Ecol Freshw Fish. 2011;20:605–18. [Google Scholar]
- 43.Weiss S, Van Treuren W, Lozupone C, Faust K, Friedman J, Deng Y, et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016;10:1669–81. doi: 10.1038/ismej.2015.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuhrman JA, Cram JA, Needham DM. Marine microbial community dynamics and their ecological interpretation. Nat Rev Microbiol. 2015;13:133–46. doi: 10.1038/nrmicro3417. [DOI] [PubMed] [Google Scholar]
- 45.Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10:538–50. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 46.Röttjers L, Faust K. From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol Rev. 2018;42:761–80. doi: 10.1093/femsre/fuy030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duran-Pinedo AE, Paster B, Teles R, Frias-Lopez J. Correlation network analysis applied to complex biofilm communities. PLoS ONE. 2011;6:e28438. doi: 10.1371/journal.pone.0028438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Freilich MA, Wieters E, Broitman BR, Marquet PA, Navarrete SA. Species co-occurrence networks: can they reveal trophic and non-trophic interactions in ecological communities? Ecology. 2018;99:690–9. doi: 10.1002/ecy.2142. [DOI] [PubMed] [Google Scholar]
- 49.Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Structure and function of the global ocean microbiome. Science. 2015;348:1261359–1261359. doi: 10.1126/science.1261359. [DOI] [PubMed] [Google Scholar]
- 50.Liu J, Zhu S, Liu X, Yao P, Ge T, Zhang XH. Spatiotemporal dynamics of the archaeal community in coastal sediments: assembly process and co-occurrence relationship. ISME J. 2020;14:1463–78. doi: 10.1038/s41396-020-0621-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Layeghifard M, Hwang DM, Guttman DS. Disentangling interactions in the microbiome: a network perspective. Trends Microbiol. 2017;25:217–28. doi: 10.1016/j.tim.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agler MT, Ruhe J, Kroll S, Morhenn C, Kim ST, Weigel D, et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016;14:1–31. doi: 10.1371/journal.pbio.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banerjee S, Schlaeppi K, van der Heijden MGA. Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol. 2018;16:567–76. doi: 10.1038/s41579-018-0024-1. [DOI] [PubMed] [Google Scholar]
- 54.Berry D, Widder S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol. 2014;5:1–14. doi: 10.3389/fmicb.2014.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herren CM, McMahon KD. Keystone taxa predict compositional change in microbial communities. Environ Microbiol. 2018;20:2207–17. doi: 10.1111/1462-2920.14257. [DOI] [PubMed] [Google Scholar]
- 56.Neiff JJ. Large rivers of South America: toward the new approach. Verh Intern Ver Limnol. 1996;26:167–80. [Google Scholar]
- 57.Drago EC. The middle Paraná River: limnology of a subtropical wetland. Berlin, Heidelberg: Springer-Verlag; 2007. The physical dynamics of the river-lake floodplain system; pp. 83–122. [Google Scholar]
- 58.Mayora G, Devercelli M, Giri F. Spatial variability of chlorophyll-a and abiotic variables in a river-floodplain system during different hydrological phases. Hydrobiologia. 2013;717:51–63. [Google Scholar]
- 59.Mayora G, Scarabotti P, Schneider B, Alvarenga P, Marchese M. Multiscale environmental heterogeneity in a large river-floodplain system. J South Am Earth Sci. 2020;100:102546. [Google Scholar]
- 60.Thomaz SM, Bini LM, Bozelli RL. Floods increase similarity among aquatic habitats in river-floodplain systems. Hydrobiologia. 2007;579:1–13. [Google Scholar]
- 61.Camilloni IA, Barros VR. Extreme discharge events in the Paraná River and their climate forcing. J Hydrol. 2003;278:94–106. [Google Scholar]
- 62.Depetris PJ, Probst JL, Pasquini AI, Gaiero DM. The geochemical characteristics of the Paraná River suspended sediment load: an initial assessment. Hydrol Process. 2003;17:1267–77. [Google Scholar]
- 63.Marchese M, de Drago IE. Benthos of the lotic environments in the middle Paraná River system: transverse zonation. Hydrobiologia. 1992;237:1–13. [Google Scholar]
- 64.Zilli FL, Paggi AC. Ecological responses to different degrees of hydrologic connectivity: assessing patterns in the bionomy of benthic chironomids in a large river-floodplain system. Wetlands. 2013;33:837–45. [Google Scholar]
- 65.José de Paggi SB, Devercelli M, Molina FR. Zooplankton and their driving factors in a large subtropical river during low water periods. Fundam Appl Limnol/Arch für Hydrobiol. 2014;184:125–39. [Google Scholar]
- 66.Fernandez Zenoff V, Sineriz F, Farias ME. Diverse responses to UV-B radiation and repair mechanisms of bacteria isolated from high-altitude aquatic environments. Appl Environ Microbiol. 2006;72:7857–63. doi: 10.1128/AEM.01333-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Herlemann DPR, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011;5:1571–9. doi: 10.1038/ismej.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Logares R. Workflow for analysing MiSeq amplicons based on Uparse v1.5.: https://github.com/ramalok/amplicon_processing. 2017. 10.5281/zenodo.259579.
- 70.Edgar RC. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. 2016;081257. 10.1101/081257.
- 71.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, et al. Vegan: community ecology package. R package version 2.0-9. 2013. http://CRAN.R-project.org/package=vegan.
- 72.R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. https://www.R-project.org/.
- 73.Ranjard L, Dequiedt S, Chemidlin Prévost-Bouré N, Thioulouse J, Saby NPA, Lelievre M, et al. Turnover of soil bacterial diversity driven by wide-scale environmental heterogeneity. Nat Commun. 2013;4:1–10. doi: 10.1038/ncomms2431. [DOI] [PubMed] [Google Scholar]
- 74.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- 75.Anderson MJ. Distance-based tests for homogeneity of multivariate dispersions. Biometrics. 2006;62:245–53. doi: 10.1111/j.1541-0420.2005.00440.x. [DOI] [PubMed] [Google Scholar]
- 76.Anderson MJ, Walsh DCI, Robert Clarke K, Gorley RN, Guerra-Castro E. Some solutions to the multivariate Behrens–Fisher problem for dissimilarity-based analyses. Aust N Zeal J Stat. 2017;59:57–79. [Google Scholar]
- 77.Pedrós-Alió C. The rare bacterial biosphere. Ann Rev Mar Sci. 2012;4:449–66. doi: 10.1146/annurev-marine-120710-100948. [DOI] [PubMed] [Google Scholar]
- 78.Zhou J, Ning D. Stochastic community assembly: does it matter in microbial ecology? Microbiol Mol Biol Rev. 2017;81:1–32. doi: 10.1128/MMBR.00002-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fine PVA, Kembel SW. Phylogenetic community structure and phylogenetic turnover across space and edaphic gradients in western Amazonian tree communities. Ecography. 2011;34:552–65. [Google Scholar]
- 80.Webb CO, Ackerly DD, McPeek MA, Donoghue MJ. Phylogenies and community ecology. Annu Rev Ecol Syst. 2002;33:475–505. [Google Scholar]
- 81.Webb CO, Ackerly DD, Kembel SW. Phylocom: software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics. 2008;24:2098–2100. doi: 10.1093/bioinformatics/btn358. [DOI] [PubMed] [Google Scholar]
- 82.Kembel SW. Disentangling niche and neutral influences on community assembly: assessing the performance of community phylogenetic structure tests. Ecol Lett. 2009;12:949–60. doi: 10.1111/j.1461-0248.2009.01354.x. [DOI] [PubMed] [Google Scholar]
- 83.Oden NL, Sokal RR. Directional autocorrelation: an extension of spatial correlograms to two dimensions. Syst Zool. 1986;35:608–17. [Google Scholar]
- 84.Diniz-Filho JAF, Terribile LC, Da Cruz MJR, Vieira LCG. Hidden patterns of phylogenetic non-stationarity overwhelm comparative analyses of niche conservatism and divergence. Glob Ecol Biogeogr. 2010;19:916–26. [Google Scholar]
- 85.Stegen JC, Lin X, Konopka AE, Fredrickson JK. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012;6:1653–64. doi: 10.1038/ismej.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andersson AF, Riemann L, Bertilsson S. Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J. 2010;4:171–81. doi: 10.1038/ismej.2009.108. [DOI] [PubMed] [Google Scholar]
- 87.Chase JM, Myers JA. Disentangling the importance of ecological niches from stochastic processes across scales. Philos Trans R Soc B Biol Sci. 2011;366:2351–63. doi: 10.1098/rstb.2011.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chase JM. Picante: R tools for integrating phylogenies and ecology. Oecologia. 2003;136:489–98. [Google Scholar]
- 89.Dray S, Legendre P, Peres-Neto PR. Spatial modelling: a comprehensive framework for principal coordinate analysis of neighbour matrices (PCNM) Ecol Modell. 2006;196:483–93. [Google Scholar]
- 90.ter Braak CJF, Šmilauer P. CANOCO reference manual and CanoDraw for Windows user’s guide: software for canonical community ordination (Version 5) Ithaca, NY: Section on Permutation Methods Microcomputer Power; 2012. [Google Scholar]
- 91.Legendre P, Legendre L. Numerical ecology. Oxford, U.K.: Elsevier Scientific; 1998.
- 92.Faust K, Lima-Mendez G, Lerat JS, Sathirapongsasuti JF, Knight R, Huttenhower C, et al. Cross-biome comparison of microbial association networks. Front Microbiol. 2015;6:1–13. doi: 10.3389/fmicb.2015.01200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Faust K, Raes J. CoNet app: inference of biological association networks using Cytoscape. F1000Research. 2016;5:1–16. doi: 10.12688/f1000research.9050.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, et al. Microbial co-occurrence relationships in the Human Microbiome. PLoS Comput Biol. 2012;8. 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed]
- 96.Volterra V. Variazioni flultuazioni del numero d’invididui in specie convirenti. Men Acad Lincei. 1926;2:31–113. [Google Scholar]
- 97.Assenov Y, Ramírez F, Schelhorn SESE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24:282–4. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 98.Banerjee S, Walder F, Büchi L, Meyer M, Held AY, Gattinger A, et al. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019;13:1722–36. doi: 10.1038/s41396-019-0383-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cao X, Zhao D, Xu H, Huang R, Zeng J, Yu Z. Heterogeneity of interactions of microbial communities in regions of Taihu Lake with different nutrient loadings: a network analysis. Sci Rep. 2018;8:1–11. doi: 10.1038/s41598-018-27172-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Newman MEJ. The structure and function of complex networks. Soc Ind Appl Math Rev. 2003;45:167–256. [Google Scholar]
- 101.Barabási AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–13. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 102.Newman MEJ. A measure of betweenness centrality based on random walks. Soc Netw. 2005;27:39–54. [Google Scholar]
- 103.Brandes U. A faster algorithm for betweenness centrality. J Math Sociol. 2001;25:163–77. [Google Scholar]
- 104.Powell JR, Karunaratne S, Campbell CD, Yao H, Robinson L, Singh BK. Deterministic processes vary during community assembly for ecologically dissimilar taxa. Nat Commun. 2015;6:1–10. doi: 10.1038/ncomms9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weilhoefer CL, Pan Y, Eppard S. The effects of river floodwaters on floodplain wetland water quality and diatom assemblages. Wetlands. 2008;28:473–86. [Google Scholar]
- 106.Unrein F. Changes in phytoplankton community along a transversal section of the lower Paraná floodplain, Argentina. Hydrobiologia. 2002;468:123–34. [Google Scholar]
- 107.Vass M, Székely AJ, Lindström ES, Langenheder S. Using null models to compare bacterial and microeukaryotic metacommunity assembly under shifting environmental conditions. Sci Rep. 2020;10:1–13. doi: 10.1038/s41598-020-59182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wagg C, Schlaeppi K, Banerjee S, Kuramae EE, van der Heijden MGA. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat Commun. 2019;10:4841. doi: 10.1038/s41467-019-12798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barberán A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6:343–51. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fuhrman JA. Microbial community structure and its functional implications. Nature. 2009;459:193–9. doi: 10.1038/nature08058. [DOI] [PubMed] [Google Scholar]
- 111.Newman MEJ. The structure and function of networks. Comput Phys Commun. 2002;147:40–47. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.