

Abstract
The respiratory complex I is a gigantic (1 MDa) redox-driven proton pump that reduces the ubiquinone pool and generates proton motive force to power ATP synthesis in mitochondria. Despite resolved molecular structures and biochemical characterization of the enzyme from multiple organisms, its long-range (∼300 Å) proton-coupled electron transfer (PCET) mechanism remains unsolved. We employ here microsecond molecular dynamics simulations to probe the dynamics of the mammalian complex I in combination with hybrid quantum/classical (QM/MM) free energy calculations to explore how proton pumping reactions are triggered within its 200 Å wide membrane domain. Our simulations predict extensive hydration dynamics of the antiporter-like subunits in complex I that enable lateral proton transfer reactions on a microsecond time scale. We further show how the coupling between conserved ion pairs and charged residues modulate the proton transfer dynamics, and how transmembrane helices and gating residues control the hydration process. Our findings suggest that the mammalian complex I pumps protons by tightly linked conformational and electrostatic coupling principles.
Introduction
NADH:ubiquinone oxidoreductase or complex I is an L-shaped ca. 1 MDa membrane-bound redox enzyme that initiates cellular respiration in mitochondrial inner membranes.1−4 Fourteen of its 45 subunits comprise the catalytic core that is responsible for coupled electron transfer and proton pumping across distances up to ∼300 Å (Figure 1). The core domains are surrounded by up to 31 supernumerary subunits in the mammalian enzyme (Figure 1), with possible relevance for activity and assembly.2,5 The long-range proton-coupled electron transfer (PCET) process reduces the ubiquinone pool (Q10) that shuttles electrons to respiratory chain complexes III and IV, whereas the proton motive force (pmf) generated by proton pumping across the membrane powers ATP synthesis and active transport of solutes against their concentration gradients.6,7 The eight iron–sulfur (FeS) centers of the hydrophilic domain shuttle electrons from nicotinamide adenine dinucleotide (NADH) to ubiquinone (UQ10),8−10 triggering proton transfer reactions in the antiporter-like subunits ND2, ND4, and ND5, and possibly in ND1/ND3/ND4L/ND6 (Figure 1), by mechanistic principles that are still not fully understood.1−5 In recent years, the structures of complex I from various species were resolved,11−15 which together with biophysical and biochemical experiments,16−20 and molecular simulations,1,20−30 have advanced our understanding of the energy transduction principles in complex I.
Figure 1.
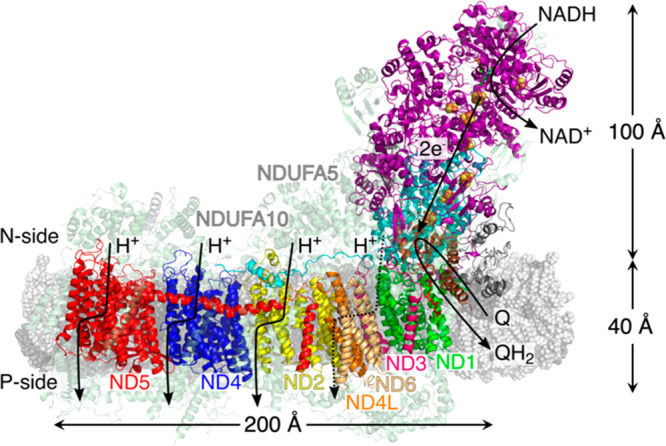
Structure and function of the mouse heart complex I. The mouse heart complex I embedded in a lipid membrane model (in gray). Electron transfer from NADH to Q in the hydrophilic domain triggers proton pumping in the membrane domain. The core subunits are depicted in solid colors, whereas the supernumerary subunits are shown in transparent colors.
The proton translocation process is catalyzed by the antiporter-like subunits in the membrane domain that form proton channels around buried charged residues at symmetry-related locations.1,20−23,28 In addition to protonatable residues that extend along a central axis of the membrane domain, water molecules are likely to provide essential elements that enable Grotthuss-type proton transfer reactions, with similarities to other energy-transducing enzymes.1,31 On the basis of computational studies of the bacterial complex I, it was suggested that the conformational state of conserved buried ion pairs modulate the thermodynamics and kinetics of the lateral proton transfer reaction within the antiporter-like subunits.20,21 These ion pairs could provide key coupling elements between the individual pumping modules and establish an electrostatic wave that propagates in both forward and reverse directions along the membrane domain.1 The complex interplay between electrostatic and conformational coupling is further highlighted by a recently proposed second membrane-bound quinone site at the interface of the ND1/NDUFS2/NDUFS7 subunits26 (cf. also refs (13, 29, 30, and 32)) and by conformational changes in surrounding loop structures.1,11,12,16,24
Despite these recent advances in the mechanistic understanding, the principles of proton pumping in the highly intricate mammalian complex I, which shares a 30–40% sequence identity with the bacterial isoform for the core subunits (Table S6), still remain unexplored. Although we expect overall similar mechanistic principles for all complex I variants, the dynamics of the mammalian enzyme is modulated by its 31 additional supernumerary subunits. In order to provide mechanistic insight into this highly complex enzyme, we study here the dynamics and proton transfer energetics of the mammalian respiratory complex I from mouse heart mitochondria by large-scale molecular simulations and hybrid quantum/classical (QM/MM) calculations.
Results
Proton Pathways Revealed from Microsecond Molecular Dynamics Simulations
To probe the dynamics underlying proton transfer in the mammalian complex I, we performed atomistic molecular dynamics (MD) simulations on the complete 45 subunit mouse heart enzyme32 that we embedded in a cardiolipin/POPC/POPE (1:2:2) lipid membrane, mimicking the composition of the inner mitochondrial membrane that forms tight interactions with complex I (Figure S12). During the microsecond simulations of the system with around 1 million modeled atoms (see Materials and Methods), the initially dry complex I structure undergoes significant hydration changes, in which ca. 350 water molecules diffuse from the bulk solvent into the protein interior (Figure 2, Figure S1A,B). Approximately 250 of these water molecules associate with the membrane domain and form along S-shaped pathways in ND2, ND4, and ND5, as well as a proton pathway that enters from ND1 and propagates via ND4L/ND6/ND3 to the interface of ND2 (Figure 2A, Figure S1A,B). Around 70 water molecules associate with buried parts of the electron transfer module (Figure S1A,B) and establish water arrays between the bulk and the Q-binding site (Figure 2A, Figure S1D). Water molecules also associate around the FeS centers, particularly around N3 and N2 at the top and lower parts of the hydrophilic arm and around a gap region between N5 and N6a (Figure S1C,G). Similar hydration dynamics is observed in our four independent simulations (simulations S1–S4, Figure S1A–D), which supports that the overall behavior is robust. However, in contrast to the flooded picture suggested by the ensemble-averaged MD analysis (Figure 2), the individual snapshots of the simulation trajectory suggest that the hydration dynamics is gated by bulky residues (Figure 3, Figure S3B–D).
Figure 2.

Overview of hydration dynamics during microsecond MD simulations. (A) Accumulated hydration dynamics (red spheres) during 3.5 μs MD simulation (simulations S1–S4, Table S1). (B) Water counts for different modules (left) and subunits (right) in complex I. (C) Average channel hydration (orange surface) and (D) dynamic flexibility of the broken helix elements TM7a/b (solid color in foreground) after MD simulations in ND5 (red), ND4 (blue), and ND2 (yellow) (0 ns white, 1000 ns colored, see also Figure S2). (E) Conformational changes and water molecules during hydration dynamics in ND1 (0 ns white, 1000 ns green, left) and average channel hydration of the E-channel in ND1 (orange surface, right).
Figure 3.

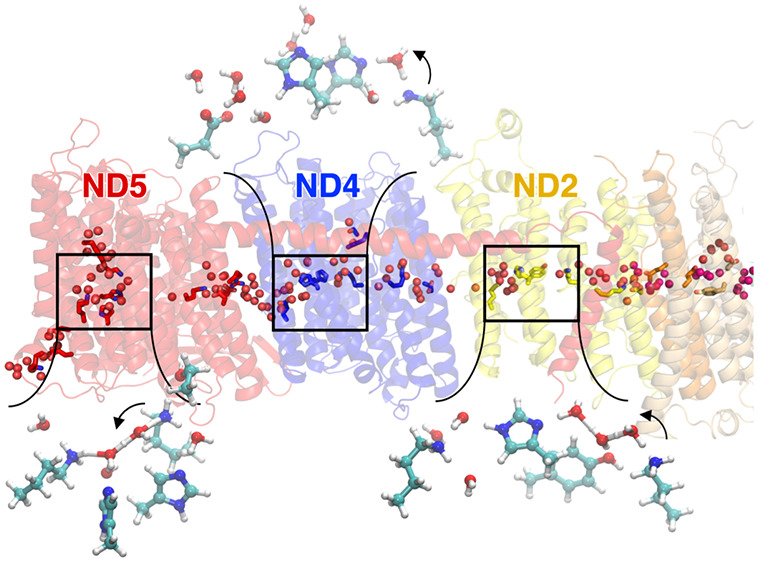
Proton wires in complex I. (A) Snapshot of the hydration structure in proton channels for an individual structure extracted after 1 μs MD simulation (simulation S1, Table S1) surrounded by close-up views with key hydration sites. Hydration dynamics for ND4L/ND6 is also shown from simulation with shifted protonation states (pink spheres, simulation S9, see Table S1). (B, C) Ion pair dynamics in ND2, ND4, and ND5 during MD simulations (see also Figure S4).
The water molecules establish proton conduction wires along several carboxylates in ND1 (E-channel) near the Q10 cavity and continue via ND4L/ND3/ND6 toward the ND2 interface (Figures 2E and 3). In the ensemble-averaged picture, the protonation sites seem to extend directly across the ND1 subunit to the P-side, similar to what was recently suggested for the bacterial complex I,33 but by analyzing individual structures, we do not observe wiring across the membrane, suggesting that the region could function as a dielectric well rather than as a proton channel (Figures 2E and 3). This could buffer the charge separation process, similar to the putative H-channel function in cytochrome c oxidase.34
The proton wires enter the ND2, ND4, and ND5 subunits at TM7a through a subtle tilting of the broken helices (Figures 2D and 3, Figure S2) and establish lateral proton wires in the three antiporter-like subunits. These wires connect the middle lysine residue (Lys135ND2, Lys237ND4, Lys336ND5) via three to four water molecules to one or two histidine residues (His186ND2, His319ND4/His293ND4, His332ND5/His328ND5). The wires continue further along a bridging water molecule to a terminal glutamate (Glu378ND2) in ND4 or a terminal lysine residue (Lys263ND2, Lys392ND5) in ND2 and ND5 (Figure 3, Figure S3G). We also observe conformational changes in broken helices TM12a/b of ND2, ND4, and ND5, suggesting that the terminal helices have the dynamic flexibility to support wetting/dewetting transitions (Figure S2). We note that the hydration dynamics and conformational changes in the TM helices are highly correlated and equilibrate on a ca. 500 ns time scale (Figure S2E).
In all MD simulations, ND5 becomes more extensively hydrated as compared to ND4 and ND2 (Figures 2 and 3, Figure S1). A nonpolar region blocks the continuous hydrogen-bonded connectivity from the N-side in ND5, but the P-side channel around TM12a/b is well hydrated and connected to the bulk solvent via an ionic gate at Asp393ND5/Glu397ND5/Lys487ND5 that wires the terminal Lys392ND5 and a nonpolar gap region via water molecules to the bulk solvent (see below). Similar P-side output channels are not clearly visible in ND2 or ND4 in the modeled states, although water molecules diffuse to the bulk solvent during the simulation trajectories at similar locations as in ND5 (Figure 2). In ND4, we observe an overall well-wired connection that effectively extends from the N-side bulk to the terminal Glu378ND4, although the hydrogen-bonded connectivity is partially broken by a nonpolar gating region formed by Leu216ND4/Leu236ND4/Val221ND4/Val291ND4/Phe327ND4 that blocks the connection from the N-side of the membrane in the simulation (Figure S3). The N-side proton channel in ND2 is partially blocked by the supernumerary subunit NDUFA10 that could shield the channel opening in ND2 (Figures S3A and S15).
The lysine/glutamate ion pairs in ND2 (Lys105ND2/Glu34ND2) and ND4 (Lys206ND4/Glu123ND4), and the double ion pair in ND5 (Lys223ND5/Asp179ND5; Arg176ND5/Glu145ND5) are bridged by three to four water molecules and undergo partial dissociation during the simulations (Figure 3B,C, Figure S4). However, we do not observe a complete shifting of the carboxylates toward the surrounding subunits in the modeled charged states during the simulations (cf. refs (20 and 21)). The ion pairs also remain disconnected from the lateral proton wires within their subunits, but we note that the terminal residues in ND4L/ND6, ND2, and ND4 form hydrogen-bonded water arrays that connect with the ion pairs in the respective neighboring subunits ND2, ND4, and ND5 (Figure S3E,F), with possible importance in modulation of the ion pair conformation (see below).
Lateral Proton Transfer Energetics
To probe the energetic feasibility of the proton transfer reaction along the observed water arrays, we next performed quantum chemical calculations, using both hybrid quantum/classical (QM/MM) free energy calculations and density functional theory (DFT) based cluster models (see Materials and Methods). The calculations were initiated from a structural snapshot extracted after a 1 μs classical MD simulation, followed by relaxation of the proton at the intermediate residues. We probed the dynamical effect by using QM/MM umbrella sampling simulations and by testing the effect of using different starting structures along the 1 μs simulation trajectory (see below). The modeled states are thus likely to represent the system under possible turnover conditions.
The quantum chemical calculations suggest that the water-mediated proton transfer reactions have modest reaction barriers in all antiporter-like subunits, with free energy barriers of 6–12 kcal mol–1 that are expected to result in water-mediated proton transfer reactions on a <50 μs time scale based on transition state theory (Figure 4A–C, Figure S5). The predicted barriers are thus compatible with the millisecond turnover time scale of complex I. Although it is challenging to model how larger-scale conformational changes affect the proton transfer barriers, we estimate that the side chain dynamics modulates the energetics by ca. 2–3 kcal mol–1 (Figure S8). Free energy effects, modeled here using QM/MM umbrella sampling simulations as well as vibrational analysis, are modest (ca. 3 kcal mol–1, Figure S5, Table S4). Moreover, electron correlation effects estimated using ab initio random-phase approximation (RPA) are also rather small (ca. 2 kcal mol–1 for transition states, Table S4), suggesting that hybrid density functionals can capture the energetics rather well. Although the statistical errors in the QM/MM free energy profiles are rather small (Figure S6), systematic errors can easily propagate when modeling extended multistep proton transfer reactions. We note, however, that the energetics predicted using the DFT-based cluster approach are similar to our QM/MM free energy calculations, suggesting that overall results are robust.
Figure 4.

Proton transfer energetics and dynamics. Free energy profiles for proton transfer from quantum chemical (DFT, top) and QM/MM umbrella sampling calculations (QM/MM bottom) of (A) ND2, (B) ND4, and (C) ND5. The modeled effect of ion pair (ip) opening (closed ip in subunit color/open ip in orange) on the proton transfer reactions are shown in the free energy profiles (see Table S4, Figures S5–S7). (D–F) Structural models used for the quantum chemical calculations shown in (A)–(C). (G–I) Proton transfer from the middle residue (in blue) toward the terminal edge (in red) or ion pair (in green) induces conformational changes in the terminal residues and ion pairs shown for (G) ND2, (H) ND4, and (I) ND5 (see also Figure S9). (J, K) Proton transfer between Lys237ND4 and the terminal Glu378ND4 induces dewetting of the N-side proton channel (transparent sphere) during classical MD simulations (see also Figure S8).
Importantly, the simulations indicate that the bridging water molecules provide an important prerequisite for a kinetically accessible proton transfer, without which the individual proton donor/acceptor distances have gaps of up to 8 Å and thus are expected to be blocked. The proton transfer reactions involve Zundel-like (H5O2+) and hydronium-like (H3O+) transition states, where the proton charge is delocalized between two water molecules (Figure 4D–F, Figure S7), and the bridging histidine residues provide intermediate protonation sites for transfer toward the terminal edge in ND2 and ND4. In ND5, we note that the water-mediated proton transfer between Lys336ND5 and Lys392ND5 could take place in a process where His332ND5 and His328ND5 stabilize the hydrogen-bonded network (Figure 3A, Figure S5C), but a histidine-mediated pathway could also be kinetically accessible during the conformational dynamics. Overall, our quantum chemical calculations support that the observed hydration dynamics is a crucial prerequisite that enables lateral proton transfer reactions along the antiporter-like subunits.
Protonation-Driven Conformational Dynamics
Next we tested the effect of shifting the protons within each antiporter-like subunit (ND2, ND4, ND5) to the terminal Lys or Glu residues in the classical MD simulations, mimicking a state that could occur after the lateral proton transfer reactions, as suggested by our quantum chemical calculations. We find that deprotonation of the middle lysine triggers partial dehydration of the well-connected lateral water wires by breaking the hydrogen-bonded contacts to the N-terminal channel and its nearby histidine residue (Figure 4J,K, Figure S4). This dehydration is accompanied by flipping of the bridging histidine residues that could stabilize the protonated terminal residue and possibly prevent proton backflow toward the N-side (Figure S9). In ND2, the Lys105ND2/Glu34ND2 ion pair undergoes a ca. 3–5 Å shift toward a more extended state, and the terminal Lys263ND2 moves toward the intersubunit space facing ND4 (Figure S4). Similar conformational and hydration changes are also observed in ND4, with a pronounced ion pair opening and dehydration around the middle Lys237ND4 that leads to the flipping of Glu378ND4 toward the ND5 interface (Figure 4I, Figure S9C) and rearrangement of the double ion pair (Asp179ND5/Arg176ND5; Glu145ND5/Lys223ND5) in ND5 (Figure 4F,I, Figure S9D). Protonation of the terminal Asp393ND5 triggers conformational changes in an ionic gate (Asp393ND5/Glu397ND5/Lys487ND5) at the terminal edge of ND5, opening up a connection to the P-side bulk (Figure S9). Interestingly, a sodium ion associates with the terminal carboxylates in ND5 during the MD simulations but is released during opening of this ionic gate, suggesting possible events that could also take place during proton release (Figure S9H). These findings, should not, however, be taken as a support for a previously suggested sodium pumping function of ND5.35,36
The water-wired terminal residues (Lys263ND2, Glu378ND4, Lys392ND5) and the protonation-dependent conformational shift within the ion pair region suggest that the subunit interfaces could be involved in the propagation of the protonation signal and/or in proton release to the P-side of the membrane. We therefore tested the effect of further shifting the proton to the carboxylate of the interface ion pair, a state that could possibly arise with a high (15–20 kcal mol–1) but kinetically accessible reaction barrier (Figure S8A). In this fully shifted state, we observe rapid dissociation of the ion pair in ND2, ND4, and ND5 that further dehydrates the region around the middle lysine (Figure 4J,K, Figure S9G). Arg176ND5 undergoes a pronounced flip toward the ND4–ND5 interface, and a similar dissociation is also observed for the ND4 ion pair (Lys206ND4/Glu123ND4) that could modulate the protonation dynamics by electrostatic effects. Our quantum chemical calculations (Figure 4A–C) suggest that the ion pair dissociation, induced here by a shift in protonation and/or conformation, could both thermodynamically and kinetically favor the proton transfer reactions toward the terminal edge of each subunit (Figure 4, Figures S5 and S8). We note that the conformational changes in the ion pair require exhaustive sampling that is not accessible on the current simulation time scale. Overall our current results further support that the ion pair conformation could modulate the lateral proton transfer energetics, and vice versa (cf. also refs (1, 20, 21, and 28)).
Discussion
Here we have shown that the mammalian respiratory complex I employs proton pathways formed around broken transmembrane-helix elements within its antiporter-like subunits, with evolutionary conserved topologies observed in the simpler bacterial and photosynthetic complex I variants (Figure S10).14,20,21,23,37 These proton pathways resemble the position of water molecules resolved in the simpler yeast (Yarrowia lipolytica) complex38 as well as in ovine complex I,43 which were released during finalization of this work, further supporting that the water-binding positions in the protein are evolutionary and functionally conserved (see Figure S11) and compare well with our predicted pathways. Interestingly, ND2 of the Yarrowia complex I is more hydrated than the other antiporter-like subunits in the yeast complex that contrast with our findings of the more hydrated mammalian ND4 and ND5 subunits (Figures S10 and S11). This difference could arise from the lack of the 42 kDa subunit (NDUFA10) in the Yarrowia enzyme that interacts with the ND2 subunit (Figure S3A) and undergoes conformational changes during the active–deactive transition of complex I.11,39
The overall pumping cycle of complex I takes place on a time scale of a few milliseconds, and although such time scales cannot currently be reached in our simulation systems, we have here partially circumvented longer dwelling steps by modeling transient steps along the pumping cycle and employed free energy calculations to explore the chemical reaction barriers possibly associated with longer time scales. We note that the MD simulations explore global twisting and bending motions (Figure S13A) that resemble those previously observed in cryoEM studies12,40 and network models,39 and our predicted proton pathways are also in good agreement with previous site-directed mutagenesis data (Figure S14, Table S7) and structural data (Figure S11). On the basis of the simulation data, we identify several potentially interesting sites that could be further probed experimentally (Figure S14).
We could further show that the hydration dynamics provides a prerequisite for the lateral proton transfer reactions along the hydrophilic arm that take place between the middle lysine residues (Lys135ND2/Lys237ND4/Lys336ND5) and terminal lysine/glutamate (Lys263ND2/Glu378ND4/Lys392ND5). The proton transfer reaction barriers are modulated by the conformation of the conserved ion pairs (Glu34/Lys105ND2; Glu123/Lys206ND4; Glu145/Lys223ND5 and Asp179/Arg176ND5) at the interface of each antiporter-like subunit. We observe proton input and output channels around TM7a/b and TM12a/b that open up via hydration changes, which are in turn modulated by the protonation state of the initial/terminal proton acceptor/donor residues (Figure 4J,K). The N- and P-side proton channels are not continuously in an open conducting state during the simulations, but are gated by nonpolar bulky residues (Figure S3B–D) that might have the same function in the bacterial homologues. The additional supernumerary subunits in the mammalian complex I undergo subtle conformational changes upon water entry, and they are dynamically more flexible in the simulations as compared to the core subunits (Figure S13C). Such gating regions could provide possible mechanisms for preventing proton leaks under high proton motive force and/or strongly changing respiratory conditions.
The terminal lysine/glutamate residues of each antiporter-like subunit and the conserved ion pairs of the neighboring subunit are tightly coupled in both their conformation and protonation states, with the ion pairs found to undergo conformational changes upon proton transfer to the terminal edges of the antiporter-like subunits (Figure 4, Figure S9). Interestingly, we also observed water arrays between these terminal proton loading sites and the ion pairs that could be involved in proton exchange, e.g., during ejection of the proton across the membrane or during the charge signal propagation between subunits. However, based on the estimated high barriers (>15 kcal mol–1) of such putative intersubunit proton transfer reactions, the processes are likely to kinetically compete with the ion pair opening dynamics with free energy barriers in the 5–10 kcal mol–1 range, depending on the state.20,21 We note that both protonation changes and ion pair opening electrostatically favor flipping of ion pair lysine (Lys105ND2/Lys206ND4/Lys223ND5) toward the middle proton donating lysine residue (Lys135ND2/Lys237ND4/Lys336ND5) that, in turn, favors the lateral proton transfer energetics toward the terminal edges of the antiporter-like subunit.
The proton transfer reactions were modeled here based on protonation states determined from electrostatic calculations (Table S2), and besides the residues that participate in the predicted proton transfer reactions themselves, we find only a few titratable residues that could affect the reaction energetics. Of these, the charged state of Asp393ND5, located at the P-side output channel along ND5 and His220ND4 at the N-side input channel of ND4, could influence the proton transfer energetics (Figure S6D), as also supported by our MD simulations (Figure S9G).
The free energy powering the proton pumping process originates from reduction and protonation of the ubiquinone (Q) to ubiquinol (QH2) by Tyr108/His59NDUFS2 and the nearby N2 FeS center24,25 that could trigger coupled conformational and electrostatic changes,16,24 and lead to diffusion of the QH2 to its second membrane-bound recognition region around ND1.26 This process is likely to release part of the redox energy, between Em ∼ −320 mV estimated in the primary binding site1,25,41 and Em ∼ +90 mV for Q in membranes that could shift the protons from the membrane-bound second Q-binding site toward the ND2 interface via the ND4L/ND6/ND3 proton pathway. Protonation of the ND2 interface triggers conformational changes in the ion pair and subsequent proton transfer reactions in ND2, ND4, and further in ND5.
ND5 is well-wired to the P-side of the membrane in our MD simulations, and therefore once the protonation signal reaches the terminal cluster of Lys/Glu residues, the proton ejection across the membrane is expected to initiate opening of the ion gate at the terminal edge of the subunit, resembling the function of putative gates also in other ion pumps.42 The proton release then follows reprotonation of the middle lysine that rearranges the ion pair network in ND5 and triggers proton release from the ND4/ND5 interface. A similar proton uptake in ND5 is expected to trigger ion pair shifting in ND4 and proton release at the ND4/ND2 interfaces. This could follow a similar conformational change in the ion pair coupled to proton uptake and release in ND2 and at the ND4L/ND2 interface, whereas subsequent proton uptake and quinol release around the ND1/Q-channel could initiate a new reaction cycle.
During revisions of this work, a pumping model was proposed43 in which two forward waves pump protons via ND5 only. This suggestion is based on three experimentally resolved water molecules at the P-side exit site of ND5, but not at the respective other antiporter-like subunits. Although we note that the directionality of the electrostatic waves cannot be inferred from the structural data, the proposed model might also conflict with prior experimental studies, where terminal subunits have been removed but pumping is still observed.44 The coupling between ion pairs, hydration dynamics, and proton transfer dynamics within and between the subunits explored here by molecular simulations of the mammalian complex I is consistent with our previously suggested forward/backward electrostatic wave-propagation model.1 The backward wave arises from principles of microscopic reversibility, where the back-propagation is similarly, as in a Newton’s cradle device, initiated by a pulse from the terminal unit. Such behavior is indeed also expected to arise if the last subunit is perturbed, leading to back-propagation from the penultimate subunit and resulting in reduced pumping as observed for dissected complex I variants.44
Conclusions
We have studied here the hydration dynamics and proton transfer energetics of the mammalian mitochondrial complex I by combination of atomistic molecular dynamics simulations and hybrid quantum/classical calculations. Our simulations suggest that the mammalian complex I undergoes extensive hydration dynamics that establishes hydrogen-bonded water arrays across the membrane domain and enables proton transfer reactions on a microsecond time scale. The hydration dynamics is modulated by the protonation reactions themselves, whereas the conformations of the buried ion pairs tune the proton transfer barriers along the hydrophilic axis. Our predicted protonation and conformational changes could in principle be observed with NMR or IR spectroscopic studies in combination with mutagenesis experiments. Water molecules also associate around the hydrophilic domain of complex I during the atomistic simulations and could provide protons to the primary ubiquinone oxidoreduction site,27 but possibly also tune electron transfer rates along the 100 Å long FeS wire by modulating electronic couplings, reorganization energies, and/or redox potentials of the redox cofactors.45,46 The molecular insight gained from the simulations provides important input for structural and biochemical experiments (see Table S7), and for understanding the coupling between conformational and electrostatic effects during the long-range proton pumping process.
Materials and Methods
Classical Molecular Dynamics Simulations
The 45 subunit cryoEM structure of complex I from mouse heart (PDB ID 6ZTQ)32 was embedded in a 1:2:2 cardiolipin:POPC:POPE membrane and solvated with TIP3P water molecules and 150 mM NaCl. Titratable residues were modeled based on protonation states suggested by Poisson–Boltzmann electrostatics calculations coupled to Monte Carlo sampling47,48 (Table S2). Ubiquinone (Q10) was modeled in the active site, and the N2 iron–sulfur center was modeled in a reduced state. The atomistic simulation system comprised ca. 991 000 atoms and was treated by use of the CHARMM36 force field49 and in-house DFT-based parametrization of the redox cofactors.20−25 MD simulations were performed by using a 2 fs time step at T = 310 K and p = 1 atm and treating the long-range electrostatics with particle mesh Ewald approach (PME) with a PME grid size of 1 Å. All classical simulations were performed with NAMD ver. 2.12.50 Visual molecular dynamics (VMD),51 CAVER,52 WATCLUST,53 and PyMol54 were used for visualization and/or analysis. Principal component analysis (PCA) was performed on the MD trajectories S1–S4 to probe large-scale motion during the MD simulations. The PCA was conducted on the backbone Cα atoms of the protein structure by using ProDy55 with highly flexible terminal loops removed from the analysis. See Table S5 for water selections and Tables S1 and S2 for a summary of classical MD simulations.
QM/MM and DFT Calculations
Hybrid quantum/classical (QM/MM) and quantum chemical cluster models were employed to probe the proton transfer energetics in the antiporter-like subunits based on a structure extracted after the 1 μs classical MD simulation or by using extracted structures along the MD trajectory (Table S3, Figures S6 and S8D). Quantum chemical cluster models with around 80 atoms, comprising amino acid side chains, terminated at the Cα–Cβ bond, and water molecules along the proton wires, were constructed for the ND2, ND4, and ND5 regions (see Table S3). The models with the protons on each intermediate Lys/His/Glu residue and transition states were optimized at the B3LYP-D3/def2-SVP level56−59 and by modeling the surroundings as a polarizable low dielectric medium with ε = 4. Transition states were validated by computation of the molecular Hessian. Using the same methodology, we also optimized the ion pairs of ND2, ND4, and ND5 in both closed and open conformations, stabilized by protonating the carboxylate within the ion pair (Table S3). Single point energy calculations were performed at the B3LYP-D3/def2-TZVP/ε=4 level, with correlated electronic energy corrections obtained from random phase approximation (RPA) calculations,60 and entropic and zero point energy corrections estimated at the B3LYP-D3/def2-SVP level. For the RPA/complete basis set (CBS) limit, single point energies were extrapolated from def2-TZVPPD and def2-QZVPPD61 based on molecular orbitals obtained at the TPSSh level.62 The energetics is in good agreement with results obtained by using other density functionals that have been suggested to perform well in modeling proton transfer reactions (see Figure S8E–H).63 The quantum chemical optimizations and single point energy calculations were performed by using TURBOMOLE ver. 6.6, ver. 7.3, and ver. 7.464 and thermodynamic corrections were obtained by using ORCA ver. 4.2.65
For the hybrid QM/MM calculations, the lateral proton transfer reactions were modeled at the B3LYP-D3/def2-SVP level, and the remaining MM system, cut 25 Å surrounding the QM part, was treated at the CHARMM36 level. Link atoms were introduced at the Cα–Cβ position, and proton transfer reaction pathways were optimized between the initial proton donating lysine residue and terminal proton acceptor (lysine/glutamate) by restrained reaction pathway optimization in both the forward and reverse directions, until convergence was reached (Figure S5). Heavy atoms of the MM region were kept fixed during the reaction pathway optimizations. A linear combination of all bond formation (ri) and bond-breaking (rj) distances within the proton wire was used as a reaction coordinate, R = ∑i>j ri – rj, spanning a range reported in Table S3 and Figure S6. Single QM/MM point energy calculations were performed at the B3LYP-D3/def2-TZVP/CHARMM36 level (Figure S5).
QM/MM free energies were computed at the B3LYP-D3/def2-SVP level by umbrella sampling (US)/weighted histogram analysis method (WHAM) across R divided into 27 (ND5)/48 (ND4)/39 (ND2) windows and restrained with a harmonic potential of 100 kcal mol–1 Å–2 with sampling of 2 ps per window at T = 310 K, with a total sampling of 228 ps (see Table S3, Figure S6). QM/MM free energy profiles were also computed for the open ion pair conformations (Figure 4) with 27 (ND5)/47 (ND4)/39 (ND2) windows and 2 ps per window umbrella sampling with the same setup as for the closed ion pair models, resulting in a total of 226 ps of sampling for the open state. QM/MM energy decomposition analysis was performed to probe the effect of individual residues on the proton transfer energetics. To this end, residue side chains within 5 Å of proton transfer wires were individually deleted and the difference in energy barrier and reaction energies were estimated (see Figure S6D). All QM/MM calculations were performed by using an in-house version of the CHARMM/TURBOMOLE interface,66 CHARMM ver. 38,67 and TURBOMOLE ver. 7.2–ver. 7.4.64
Free energy profiles for the proton transfer reaction in ND2, ND4, and ND5 were computed based on both the QM and QM/MM models in combination with thermodynamic, dynamic sampling, and correlation corrections according to
| 1 |
where ΔE[DFT/TZVP] are single point energies at the B3LYP-D3/def2-TZVP/ε=4 level; TΔS[DFT/SVP] and ΔZPE[DFT/SVP] are entropic and zero point energy corrections to the electronic energy, estimated at the B3LYP-D3/def2-SVP level; ΔE[DFT/TZVP → RPA/CBS] are electron correlation corrections estimated by using random-phase approximation based on TPSSh molecular orbitals, extrapolated to the complete basis set limit (see above); ΔEint[closed → open] are energy corrections for the ion pair opening; ΔEsurr[DFT→ QM/MM] are the surrounding protein effects estimated from the energy difference between the QM part of the QM/MM model and the QM/MM energy at the B3LYP-D3/def2-TZVP level. See Table S4 for summary of the energy analysis.
Acknowledgments
This work received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program/grant agreement 715311. The project was also supported by the Knut and Alice Wallenberg (KAW) Foundation. The authors are thankful for the computing time provided by PRACE for awarding us access to MareNostrum hosted by the Barcelona Supercomputing Center. This work was also supported by the Swedish National Infrastructure for Computing (SNIC 2020/1-38) at PDC Center, partially funded by the Swedish Research Council through grant agreement 2016-07213, and by SuperMuc (project pn34he) at the Leibniz Supercomputing Center (LRZ).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c09209.
Hydration, proton transfer and conformational dynamics, DFT and QM/MM energetics, convergence of free energy calculations, summary of QM/MM and MD simulations (PDF)
Author Contributions
§ M.R. and P.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Kaila V. R. I. Long-range proton-coupled electron transfer in biological energy conversion: Towards mechanistic understanding of respiratory complex I. J. R. Soc., Interface 2018, 15, 20170916. 10.1098/rsif.2017.0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J. Mitochondrial Complex I. Annu. Rev. Biochem. 2013, 82, 551–575. 10.1146/annurev-biochem-070511-103700. [DOI] [PubMed] [Google Scholar]
- Brandt U. Energy Converting NADH: Quinone Oxidoreductase (Complex I). Annu. Rev. Biochem. 2006, 75, 69–92. 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
- Sazanov L. A. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. 10.1038/nrm3997. [DOI] [PubMed] [Google Scholar]
- Hirst J.; Carroll J.; Fearnley I. M.; Shannon R. J.; Walker J. E. The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim. Biophys. Acta, Bioenerg. 2003, 1604, 135–150. 10.1016/S0005-2728(03)00059-8. [DOI] [PubMed] [Google Scholar]
- Mitchell P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–148. 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Muneyuki E.; Hisabori T. ATP Synthase - A marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2001, 2, 669–677. 10.1038/35089509. [DOI] [PubMed] [Google Scholar]
- Verkhovskaya M. L.; Belevich N.; Euro L.; Wikström M.; Verkhovsky M. I. Real-time electron transfer in respiratory complex I. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3763–3767. 10.1073/pnas.0711249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries S.; Dörner K.; Strampraad M. J.; Friedrich T. Electron tunneling rates in respiratory complex I are tuned for efficient energy conversion. Angew. Chem., Int. Ed. 2015, 54, 2844–2848. 10.1002/anie.201410967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov L. A.; Hinchcliffe P. Structure of the Hydrophilic Domain of Respiratory Complex I from Thermus thermophilus. Science 2006, 311, 1430–1436. 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- Agip A. N. A.; Blaza J. N.; Bridges H. R.; Viscomi C.; Rawson S.; Muench S. P.; Hirst J. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 2018, 25, 548–556. 10.1038/s41594-018-0073-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedorczuk K.; Letts J. A.; Degliesposti G.; Kaszuba K.; Skehel M.; Sazanov L. A. Atomic structure of the entire mammalian mitochondrial complex I. Nature 2016, 538, 406–410. 10.1038/nature19794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parey K.; Haapanen O.; Sharma V.; Köfeler H.; Züllig T.; Prinz S.; Siegmund K.; Wittig I.; Mills D. J.; Vonck J.; Kühlbrandt W.; Zickermann V. High-resolution cryo-EM structures of respiratory complex I: Mechanism, assembly, and disease. Sci. Adv. 2019, 5, eaax9484. 10.1126/sciadv.aax9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller J. M.; Saura P.; Thiemann J.; Schuller S. K.; Gamiz-Hernandez A. P.; Kurisu G.; Nowaczyk M. M.; Kaila V. R. I. Redox-coupled proton pumping drives carbon concentration in the photosynthetic complex I. Nat. Commun. 2020, 11, 494. 10.1038/s41467-020-14347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin T. G.; Bayne A. N.; Trempe J.-F.; Savage D. F.; Davies K. M. Structure of the complex I-like molecule NDH of oxygenic photosynthesis. Nature 2019, 566, 411–41. 10.1038/s41586-019-0921-0. [DOI] [PubMed] [Google Scholar]
- Cabrera-Orefice A.; Yoga E. G.; Wirth C.; Siegmund K.; Zwicker K.; Guerrero-Castillo S.; Zickermann V.; Hunte C.; Brandt U. Locking loop movement in the ubiquinone pocket of complex I disengages the proton pumps. Nat. Commun. 2018, 9, 4500. 10.1038/s41467-018-06955-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euro L.; Belevich G.; Verkhovsky M. I.; Wikström M.; Verkhovskaya M. Conserved lysine residues of the membrane subunit NuoM are involved in energy conversion by the proton-pumping NADH:ubiquinone oxidoreductase (Complex I). Biochim. Biophys. Acta, Bioenerg. 2008, 1777, 1166–1172. 10.1016/j.bbabio.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Kervinen M.; Pätsi J.; Finel M.; Hassinen I. E. A Pair of Membrane-Embedded Acidic Residues in the NuoK Subunit of Escherichia coli NDH-1, a Counterpart of the ND4L Subunit of the Mitochondrial Complex I, Are Required for High Ubiquinone Reductase Activity. Biochemistry 2004, 43, 773–781. 10.1021/bi0355903. [DOI] [PubMed] [Google Scholar]
- Michel J.; DeLeon-Rangel J.; Zhu S.; van Ree K.; Vik S. B. Mutagenesis of the L, M, and N subunits of complex I from Escherichia coli indicates a common role in function. PLoS One 2011, 6, e17420. 10.1371/journal.pone.0017420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlbauer M. E.; Saura P.; Nuber F.; Di Luca A.; Friedrich T.; Kaila V. R. I. Water-gated proton transfer dynamics in respiratory complex I. J. Am. Chem. Soc. 2020, 142, 13718–13828. 10.1021/jacs.0c02789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Luca A.; Gamiz-Hernandez A. P.; Kaila V. R. I. Symmetry-related proton transfer pathways in respiratory complex I. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6314–E6321. 10.1073/pnas.1706278114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila V. R. I.; Wikström M.; Hummer G. Electrostatics, hydration, and proton transfer dynamics in the membrane domain of respiratory complex I. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 6988–6993. 10.1073/pnas.1319156111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapanen O.; Sharma V. Role of water and protein dynamics in proton pumping by respiratory complex I. Sci. Rep. 2017, 7, 7747. 10.1038/s41598-017-07930-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Belevich G.; Gamiz-Hernandez A. P.; Róg T.; Vattulainen I.; Verkhovskaya M. L.; Wikström M.; Hummer G.; Kaila V. R. I. Redox-induced activation of the proton pump in the respiratory complex I. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 11571–11576. 10.1073/pnas.1503761112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamiz-Hernandez A. P.; Jussupow A.; Johansson M. P.; Kaila V. R. I. Terminal Electron-Proton Transfer Dynamics in the Quinone Reduction of Respiratory Complex I. J. Am. Chem. Soc. 2017, 139, 16282–16288. 10.1021/jacs.7b08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnau J.; Sharma V.; Gamiz-Hernandez A. P.; Di Luca A.; Haapanen O.; Vattulainen I.; Wikström M.; Hummer G.; Kaila V. R. I. Redox-Coupled Quinone Dynamics in the Respiratory Complex I. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E8413–E8420. 10.1073/pnas.1805468115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jussupow A.; Di Luca A.; Kaila V. R. I. How cardiolipin modulates the activity of complex I. Sci. Adv. 2019, 5, eaav1850. 10.1126/sciadv.aav1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Luca A.; Mühlbauer M. E.; Saura P.; Kaila V. R. I. How inter-subunit contacts in the membrane domain of complex I affect proton transfer energetics. Biochim. Biophys. Acta, Bioenerg. 2018, 1859, 734–741. 10.1016/j.bbabio.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoias Teixeira M.; Menegon Arantes G. Balanced internal hydration discriminates substrate binding to respiratory complex I. Biochim. Biophys. Acta, Bioenerg. 2019, 1860, 541–548. 10.1016/j.bbabio.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Gupta C.; Khaniya U.; Chan C. K.; Dehez F.; Shekhar M.; Gunner M. R.; Sazanov L.; Chipot C.; Singharoy A. Charge Transfer and Chemo-Mechanical Coupling in Respiratory Complex I. J. Am. Chem. Soc. 2020, 142, 9220–9230. 10.1021/jacs.9b13450. [DOI] [PubMed] [Google Scholar]
- Kaila V. R. I.; Verkhovsky M. I.; Wikström M. Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem. Rev. 2010, 110 (12), 7062–7081. 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- Bridges H. R.; Fedor J. G.; Blaza J. N.; Di Luca A.; Jussupow A.; Jarman O. D.; Wright J. J.; Agip A.-N. A.; Gamiz-Hernandez A. P.; Roessler M. M.; Kaila V. R. I.; Hirst J. Structure of Inhibitor-Bound Mammalian Complex I. Nat. Commun. 2020, 11 (1), 5261. 10.1038/s41467-020-18950-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaniya U.; Gupta C.; Cai X.; Mao J.; Kaur D.; Zhang Y.; Singharoy A.; Gunner M. R. Hydrogen bond network analysis reveals the pathway for the proton transfer in the E-channel of T. thermophilus Complex I. Biochim. Biophys. Acta, Bioenerg. 2020, 1861, 148240. 10.1016/j.bbabio.2020.148240. [DOI] [PubMed] [Google Scholar]
- Rich P. R.; Maréchal A. Functions of the hydrophilic channels in protonmotive cytochrome c oxidase. J. R. Soc., Interface 2013, 10, 20130183. 10.1098/rsif.2013.0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marreiros B. C.; Batista A. P.; Pereira M. M. Respiratory complex I from Escherichia coli does not transport Na+ in the absence of its NuoL subunit. FEBS Lett. 2014, 588, 4520–4525. 10.1016/j.febslet.2014.10.030. [DOI] [PubMed] [Google Scholar]
- Roberts P. G.; Hirst J. The deactive form of respiratory complex I from mammalian mitochondria is a Na+/H+ antiporter. J. Biol. Chem. 2012, 287, 34743–345. 10.1074/jbc.M112.384560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura P.; Kaila V. R. I. Molecular dynamics and structural models of the cyanobacterial NDH-1 complex. Biochim. Biophys. Acta, Bioenerg. 2019, 1860, 201–208. 10.1016/j.bbabio.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grba D. N.; Hirst J. Mitochondrial complex I structure reveals ordered water molecules for catalysis and proton translocation. Nat. Struct. Mol. Biol. 2020, 27, 892–900. 10.1038/s41594-020-0473-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Luca A.; Kaila V. R. I. Global collective motions in the mammalian and bacterial respiratory complex I. Biochim. Biophys. Acta, Bioenerg. 2018, 1859, 326–332. 10.1016/j.bbabio.2018.02.001. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Vinothkumar K. R.; Hirst J. Structure of Mammalian Respiratory Complex I. Nature 2016, 536, 354–358. 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovskaya M.; Wikström M. Oxidoreduction properties of bound ubiquinone in Complex I from Escherichia coli. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 246–250. 10.1016/j.bbabio.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Suomivuori C. M.; Gamiz-Hernandez A. P.; Sundholm D.; Kaila V. R. I. Energetics and dynamics of a light-driven sodium-pumping rhodopsin. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 7043–7048. 10.1073/pnas.1703625114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampjut D.; Sazanov L. A. The Coupling Mechanism of Mammalian Respiratory Complex I. Science 2020, 370 (6516), eabc4209. 10.1126/science.abc4209. [DOI] [PubMed] [Google Scholar]
- Dröse S.; Krack S.; Sokolova L.; Zwicker K.; Barth H. D.; Morgner N.; Heide H.; Steger M.; Nübel E.; Zickermann V.; Kerscher S.; Brutschy B.; Radermacher M.; Brandt U. Functional Dissection of the Proton Pumping Modules of Mitochondrial Complex I. PLoS Biol. 2011, 9, e1001128–e1001128. 10.1371/journal.pbio.1001128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Stuchebrukhov A. A. Electron tunneling in Respiratory Complex I. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19157–19162. 10.1073/pnas.1009181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila V. R. I.Multi-scale Molecular Simulations on Respiratory Complex I. In Mechanisms of Primary Energy Transduction in Biology; Wikström M., Ed.; Chemical Biology 5; The Royal Society of Chemistry: 2017; p 81. 10.1039/9781788010405-00081. [DOI] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 10037–10041. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabenstein B.; Knapp E.-W. Calculated pH-Dependent Population and Protonation of Carbon-Monoxy-Myoglobin Conformers. Biophys. J. 2001, 80, 1141–1150. 10.1016/S0006-3495(01)76091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E. M.; Mittal J.; Feig M.; MacKerell A. D. Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD - Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Chovancova E.; Pavelka A.; Benes P.; Strnad O.; Brezovsky J.; Kozlikova B.; Gora A.; Sustr V.; Klvana M.; Medek P.; Biedermannova L.; Sochor J.; Damborsky J. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012, 8, e1002708. 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López E. D.; Arcon J. P.; Gauto D. F.; Petruk A. A.; Modenutti C. P.; Dumas V. G.; Marti M. A.; Turjanski A. G. WATCLUST: a tool for improving the design of drugs based on protein-water interactions. Bioinformatics 2015, 31, 3697–3699. 10.1093/bioinformatics/btv411. [DOI] [PubMed] [Google Scholar]
- Delano W. L.PyMOL Molecular Graphics System. Schrödinger LLC, 2002. https://sourceforge.net/projects/pymol/.
- Bakan A.; Meireles L. M.; Bahar I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27 (11), 1575–1577. 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Furche F. Developing the random phase approximation into a practical post-Kohn–Sham correlation model. J. Chem. Phys. 2008, 129, 114105. 10.1063/1.2977789. [DOI] [PubMed] [Google Scholar]
- Halkier A.; Klopper W.; Helgaker T.; Jørgensen P.; Taylor P. R. Basis set convergence of the interaction energy of hydrogen-bonded complexes. J. Chem. Phys. 1999, 111, 9157. 10.1063/1.479830. [DOI] [Google Scholar]
- Tao J.; Perdew J. P.; Staroverov V.; Scuseria G. E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- Mangiatordi G. F.; Brémond E.; Adamo C. DFT and Proton Transfer Reactions: A Benchmark Study on Structure and Kinetics. J. Chem. Theory Comput. 2012, 8, 3082–3088. 10.1021/ct300338y. [DOI] [PubMed] [Google Scholar]
- Ahlrichs R.; Bär M.; Häser M.; Horn H.; Kölmel C. Electronic structure calculations on workstation computers: The program system Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Neese F. The ORCA Program System. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Riahi S.; Rowley C. N. The CHARMM-TURBOMOLE interface for efficient and accurate QM/MM molecular dynamics, free energies, and excited state properties. J. Comput. Chem. 2014, 35, 2076–2086. 10.1002/jcc.23716. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. 10.1002/jcc.540040211. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.