

Abstract
Coevolution between transposable elements (TEs) and their hosts can be antagonistic, where TEs evolve to avoid silencing and the host responds by reestablishing TE suppression, or mutualistic, where TEs are co-opted to benefit their host. The TART-A TE functions as an important component of Drosophila telomeres but has also reportedly inserted into the Drosophila melanogaster nuclear export factor gene nxf2. We find that, rather than inserting into nxf2, TART-A has actually captured a portion of nxf2 sequence. We show that TART-A produces abundant Piwi-interacting small RNAs (piRNAs), some of which are antisense to the nxf2 transcript, and that the TART-like region of nxf2 is evolving rapidly. Furthermore, in D. melanogaster, TART-A is present at higher copy numbers, and nxf2 shows reduced expression, compared to the closely related species Drosophila simulans. We propose that capturing nxf2 sequence allowed TART-A to target the nxf2 gene for piRNA-mediated repression and that these 2 elements are engaged in antagonistic coevolution despite the fact that TART-A is serving a critical role for its host genome.
Co-evolution between transposable elements (TEs) and their hosts can be antagonistic, where TEs evolve to avoid silencing and the host responds by re-establishing TE suppression, or mutualistic, where TEs are co-opted to benefit their host. This study shows that a specialized Drosophila retrotransposon that functions as a telomere has captured a portion of a host piRNA gene which may allow it to evade silencing.
Introduction
Transposable elements (TEs) must replicate faster than their host to avoid extinction. The vast majority of new TE insertions derived from this replicative activity are deleterious to their host: They can disrupt and/or silence protein-coding genes and lead to chromosome rearrangements [1–3]. In response to the mutational burden imposed by TEs, TE hosts have evolved elaborate genome surveillance mechanisms to identify and target TEs for suppression.
One of the most well-known genome defense pathways in metazoan species involves the production of Piwi-interacting small RNAs, also known as piRNAs [4]. PiRNA precursors are produced from piRNA clusters, which are located in heterochromatin and contain fragments of many families of TEs, whose insertions have accumulated in these regions. These precursors are processed into phased piRNAs, which use sequence homology to guide Piwi proteins to complementary transcripts produced by active TEs [4,5]. Piwi proteins induce posttranscriptional silencing through cleavage of the TE transcript. The sense-strand cleavage product of the TE transcript can then aid in processing piRNA precursors though a process known as the ping-pong cycle, which amplifies the silencing signal [4,5]. Alternatively, the cleaved transcript can be processed by the endonuclease Zucchini into additional phased piRNAs starting from the cleavage site and proceeding in the 3′ direction.
Several recent studies have identified a novel protein complex in Drosophila that connects the piRNA-mediated targeting of mRNAs by Piwi with the establishment of cellular heterochromatin [6–9]. This complex contains a heterodimer consisting of Nxf2 and Nxt1 and Panoramix [6–9]. Nxf2 belongs to a family of nuclear export proteins, yet has lost the ability to export RNA and instead plays a specialized role in the piRNA pathway [6–9]. Nxf2 interacts with Piwi-targeted transcripts, while Panoramix likely recruits Lsd1 to demethylate H3K4me2 and SetDB1 to establish H3K9me3 [6–9] Interestingly, 2 paralogs of Nxf2, Nxf1, and Nxf3 play an important role in piRNA precursor export: Nxf1 exports flamenco piRNA precursors [10,11], and Nxf3 exports germline piRNA precursors [12,13], which raises the possibility that the nuclear export factor gene family may have diversified in response to TE–host conflict [6].
The ubiquity of active TEs suggests that host silencing mechanisms are not completely effective, which may be due to the fact that selection for complete TE repression is relatively weak [14–16] or because the TE and its host genome are involved in an evolutionary “arms race” where TEs are continuously evolving novel means to avoid host silencing and the host genome is constantly reestablishing TE suppression [17]. On the host side, many TE silencing components have been shown to be evolving rapidly under positive selection [18–26], in agreement with ongoing host–TE conflict.
On the transposon side, a TE can mount a counter-defense by silencing or blocking host factors [27–29] or simply evade host silencing by replicating in permissive cells [30] or cloaking themselves in virus-like particles [31]. However, there are surprisingly few examples of any of these strategies [32]. In fact, there is some evidence that, rather than an evolutionary arms race, the rapid evolution of host silencing genes is related to avoiding gene silencing due to off-target effects (i.e., piRNA autoimmunity [33,34]) and/or coevolution with viruses (reviewed in [32]).
While there are currently only a few examples of TE counter-defense strategies, there are many examples of TEs being co-opted by their host genome for its own advantage (see reviews [32,35–38]). TEs can disperse regulatory sequences across the genome [39–46] and have been co-opted as a source of host genes and noncoding RNAs [37,38,47–49]. TEs can also act as structural components of the genome. There is evidence that TEs may play a role in centromere specification in a variety of species [50–52], and in Drosophila, which lacks telomerase, specific TEs serve as telomeres by replicating to chromosome ends [53,54].
In Drosophila melanogaster, 3 related non-long terminal repeat (non-LTR) retrotransposons occupy the telomeres: HeT-A, TAHRE, and TART, which are often abbreviated as HTT elements [53,55–57]. These elements belong to the Jockey clade of Long Interspersed Nuclear Elements (LINEs), which contain open reading frames for gag (ORF1) and an endonuclease/reverse transcriptase protein (ORF2, lost in HeT-A) [58,59]. These elements form head-to-tail arrays at the chromosome ends, and their replication solves the chromosome “end-shortening” problem without the need for telomerase [60].
These telomeric elements represent a unique case of TE domestication. They serve a critical role for their host genome, yet they are still active elements, capable of causing mutational damage if their activity is left unchecked [61–63]. All 3 elements have been shown to produce abundant piRNAs and RNA interference (RNAi) knockdown and/or mutation of piRNA pathway components, including nxf2, leads to their up-regulation [6–9,62,64,65], consistent with the host genome acting to constrain their activity and raising the possibility that, despite being domesticated, these elements are still in conflict with their host [66]. There are multiple lines of evidence that this is indeed the case: The protein components of Drosophila telomeres are rapidly evolving under positive selection, potentially due to a role in preventing the HTT elements from overproliferation [66]. There is a high rate of gain and loss of HTT lineages within the melanogaster species group [67], and there is dramatic variation in telomere length among strains from the Drosophila Genetic Reference Panel (DGRP) [68]. These observations are more consistent with evolution under conflict rather than a stable symbiosis [67]. Furthermore, the nucleotide sequence of the HTT elements evolves extremely rapidly, especially in their unusually long 3′ UTRs [69,70]. Within D. melanogaster, 3 TART subfamilies have been identified which contain completely different 3′ UTRs, and which are known as TART-A, TART-B, and TART-C [57].
In this study, we have characterized the presence of sequence within the coding region of the D. melanogaster nxf2 gene that was previously annotated as an insertion of the TART-A transposon [71]. We find that the shared homology between TART-A and nxf2 is actually the result of TART-A acquiring a portion of the nxf2 gene, rather than the nxf2 gene gaining a TART-A insertion. Our findings support a model where TART-A produces antisense piRNAs that target nxf2 for suppression as a counter-defense strategy in response to host silencing. We identified nxf2 cleavage products from degradome sequencing (degradome-seq) data that are consistent with Aub-directed cleavage of nxf2 transcripts and we find that, across the DGRP, TART-A copy number is negatively correlated with nxf2 expression, and nxf2 piRNA production is positively correlated with TART-A piRNA production. Furthermore, the D. melanogaster nxf2 sequence is evolving rapidly in the region of shared homology with TART-A, and TART-A insertions are more abundant in D. melanogaster compared to Drosophila simulans. Our findings suggest that TEs can selfishly manipulate host silencing pathways in order to increase their own copy number and that a single TE family can benefit, as well as antagonize, its host genome.
Results
The TART-like region of nxf2 is conserved across the melanogaster group
It was previously reported that the homology between nxf2 and TART-A is due to an insertion of the TART-A TE in the nxf2 gene that became fixed in the ancestor of D. melanogaster and D. simulans [71] (Fig 1A). To investigate the homology between these elements in more detail, we used BLAST [72] to search the nxf2 transcript against the TART-A RepBase sequence, which was derived from a full-length TART-A element cloned from the iso1 D. melanogaster reference strain [73]. There are 4 regions of homology between nxf2 and the 3′ UTR of TART-A that lie within a 700-bp segment of nxf2. These regions are between 63 bp and 228 bp in length and 93% to 96% sequence identity (Fig 1B). The 5′ UTR of TART-A is copied from a portion of the 3′ UTR during reverse transcription [74], which means that the nxf2-like region in the 3′ UTR is therefore mirrored in the 5′ UTR as well (Fig 1B).
Fig 1. Shared homology between the D. melanogaster nxf2 gene and the TART-A TE.

(A) Gene, transcript, and TE annotations from FlyBase showing the nxf2 gene models along with the annotated TART-A TE insertion. Note that the TART-A annotation overlaps the 3′ CDS of nxf2. (B) BLAST hits between the RepBase TART-A sequence and the nxf2 transcript. Each colored box represents a single BLAST alignment. The 5′ UTR of TART-A is copied from a portion of the 3′ UTR during replication. The homology between nxf2 and the TART-A 3′ UTR is therefore mirrored in the 5′ UTR. The red arrow shows the region of the 3′ UTR that the 5′ UTR is copied from. (C). A zoomed-out multiple sequence alignment of nxf2 orthologs for 6 species from the melanogaster species group shows that the TART-like region of nxf2 is present in all 6 species. The actual alignment can be found in S1 Data. CDS, coding sequence; ORF, open reading frame; TE, transposable element.
To investigate the evolutionary origin of the homology between nxf2 and TART-A, we identified nxf2 orthologs in D. simulans, Drosophila yakuba, Drosophila erecta, Drosophila biarmipes, and Drosophila elegans. The TART-like region of nxf2 is clearly present in all 6 of these species. Therefore, if this portion of the nxf2 gene was derived from an insertion of a TART-A element, the most recent time point at which the insertion could have occurred is in the common ancestor of the melanogaster group, approximately 15 million years ago [75] (Fig 1C, S1 Data). At the nucleotide level, there is only weak homology between nxf2 coding sequence (CDS) and transcripts from more distantly related Drosophila species, such as Drosophila pseudoobscura. However, at the peptide level, the carboxyl-terminal region of Nxf2, which was thought to be derived from TART-A, is actually conserved across Drosophila, from D. melanogaster to Drosophila virilis (S1 Fig), suggesting that, if a TART-A element did insert into the nxf2 gene, it was not a recent event.
A portion of nxf2 was captured by the D. melanogaster TART-A element
If an ancestral TART-A element was inserted into the nxf2 gene in the common ancestor of the melanogaster group, the shared homology between nxf2 and TART-A should be present in most, if not all, extant species in the group. To test this prediction, we obtained the sequences for previously identified TART-A homologs from D. yakuba and Drosophila sechellia [59,69]. We aligned these sequences to D. melanogaster TART-A and found that the TART-A region that shares homology with the nxf2 gene is only present in the D. melanogaster TART-A sequence (Fig 2A, S2 and S3 Figs, S2 Data).
Fig 2. The TART-A/nxf2 homology is unique to D. melanogaster.

(A) Dotplot comparing D. melanogaster TART-A to its homologs in D. yakuba and D. sechellia. The diagonal lines denote regions of homology, while the light gray boxes show the location of the nxf2-like sequence in the D. melanogaster TART-A. Neither the D. yakuba nor the D. sechellia TART-A sequences contain nxf2-like sequence. However, the regions directly flanking the nxf2-like sequence in D. melanogaster are also present in D. yakuba (see S2 Fig for magnified view). Underlying data can be found in S2 Data. (B) Gene tree showing relative age of shared homology. We aligned the nxf2-like sequences from 9 copies of TART-A in the D. melanogaster reference genome to the nxf2 transcripts from 6 Drosophila species and inferred a maximum likelihood phylogeny using RAxML. D. melanogaster nxf2 is most closely related to the nxf2-like sequences present in the D. melanogaster TART-A copies, suggesting the shared homology occurred after the divergence between D. melanogaster and D. simulans.
We identified 9 TART-A elements (5 full length and 4 fragments) in the D. melanogaster reference genome assembly that contain the nxf2-like sequence. We added these 9 sequences to the multiple sequence alignment in Fig 1C and inferred a maximum likelihood phylogeny in order to better understand the evolutionary history of the nxf2/TART shared homology (Fig 2B, S3 Data). The youngest node in the phylogeny represents the split between the D. melanogaster nxf2 and TART-A elements, suggesting that the event leading to the shared homology between these sequences occurred relatively recently, which is consistent with the high degree of sequence similarity between the D. melanogaster TART-A and nxf2 subsequences. Based on these results, we conclude that the nxf2/TART-A shared homology is much more likely to have arisen via the recent acquisition of nxf2 sequence by TART-A after the split of D. melanogaster from D. simulans/sechellia, rather than an insertion of TART-A into the nxf2 gene. The mechanism by which TART-A could have acquired a portion of nxf2 is not clear; however, one possibility is via transduction, a process where genomic regions flanking a TE insertion can be incorporated into the TE itself due to aberrant retrotransposition [76,77].
The nxf2-captured region is likely to be fixed in D. melanogaster
We next sought to determine whether the nxf2-like TART-A variant is present in all dispersed copies of TART-A in D. melanogaster. We compared the RepBase (strain iso1) TART-A sequence to other TART-A sequences present in GenBank that included the 3′ UTR. We found 2 additional sequences: 1 cloned from the strain A4-4 and another cloned from the strain Oregon-R, both of which contained the nxf2-like region. We then searched the RepBase TART-A sequence against the D. melanogaster reference genome assembly plus long-read assemblies of 16 other D. melanogaster strains [78,79] (see Methods). We identified a total of 71 TART-A sequences that included at least a portion of the 3′ UTR. All 71 sequences also included the nxf2-like region (S4 Fig).
In order to survey additional strains, we next used a coverage-based approach along with Illumina (San Diego, California, United States of America) data from the DGRP [80,81]. We aligned Illumina reads to the TART-A RepBase sequence and compared sequencing coverage for the nxf2-like region to both the upstream and downstream flanking regions. We divided read coverage for these regions by each strain’s median coverage of TART-A ORF1 and ORF2 to control for copy number differences between strains. If the nxf2-like region is only present in some TART-A elements and missing from others, we would expect that coverage of this region should be lower than the 2 flanking regions. Across 151 DGRP strains, we found that the coverage of the nxf2-like region was lower than the coverage of the upstream region but similar to the coverage of the downstream region (S5 Fig). There are only 6 individuals where the coverage of the nxf2-like region is lower than both flanking regions. In these cases, the difference between the coverage of the nxf2-like region and the downstream region is very small (mean reduction of 6.3%). This pattern is not consistent with polymorphism, but rather, truncation of the 5′ UTR, which has previously been described for TART [74]. Because the nxf2-like sequence is present in both UTRs, truncation of the 5′ UTR, which is fairly common, should reduce coverage of the nxf2-like region by as much as 50% compared to the upstream region, which is not present in the 5′ UTR (Fig 1B). We observed a reduction in coverage of approximately 30%, consistent with a mixture of TART-A copies, some with truncated 5′ UTRs and some without.
The nxf2 gene plays a role in suppressing the activity of D. melanogaster telomeric elements
Nxf2 is part of an evolutionarily conserved gene family with functions related to export of RNA from the nucleus [82]. In Drosophila, there are 4 nuclear export factor paralogs: nxf1 is involved in the export of mRNAs and flamenco piRNA precursors from the nucleus [10,11], while nxf3 plays a more specialized role in germline piRNA precursor export [12,13]. Nxf4 shows testes specific expression; however, its exact function remains unknown. The nxf2 gene was identified as a member of the germline piRNA pathway via an RNAi screen [10,11], and more recently, several studies have independently shown that Nxf2 is involved in the co-transcriptional silencing of transposons as part of a complex with Nxt1 and Panoramix [6–9]. Batki and colleagues reported TE derepression in a nxf2 null mutant, including an approximately 80-fold increase in TART-A expression. To confirm the involvement of nxf2 in the suppression of TART-A, we used a short hairpin RNA (shRNA) from the Drosophila Transgenic RNAi Project (TRiP) with a nos-GAL4 driver to target and knockdown expression of nxf2 in the ovaries. The nanos promoter drives Gal4 expression in germline stem cells and starting in stage 5 of oogenesis, with weak expression in young egg chambers [83]. We sequenced total RNA from the nxf2 knockdown and a control knockdown of the white gene. We observed a strong increase in expression for a variety of TE families upon knockdown of nxf2 (S6 Fig). The 3 telomeric elements, HeT-A, TAHRE, and TART-A, are among the top 10 most highly up-regulated TEs, with HeT-A showing approximately 300-fold increase in expression in the nxf2 knockdown (TAHRE: approximately 110-fold increase, TART-A: approximately 30-fold increase) (Fig 3). We repeated the experiment using a shRNA that targeted a different region of nxf2 and observed a similar pattern and strong correlation between TE expression profiles of both knockdowns (Spearman’s rho = 0.94, S7 Fig). The nxf paralogs (i.e., nxf1-4) are highly diverged (<25% amino acid identity); therefore, it is unlikely that there would be RNAi off-target effects among paralogs. These results support previous findings that nxf2 is a component of the germline piRNA pathway and show that this gene is particularly important for the suppression of the telomeric TEs HeT-A, TAHRE, and TART-A.
Fig 3. RNAi knockdown of nxf2 leads to strong up-regulation of HTT elements.
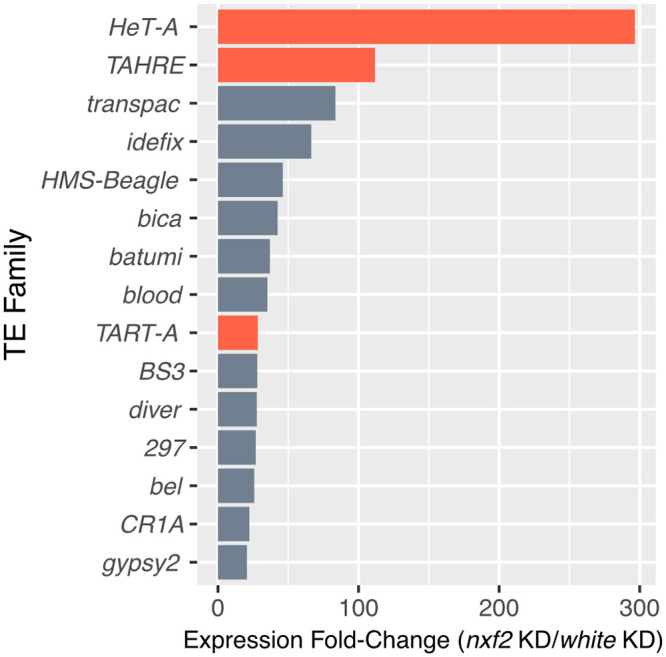
We examined TE expression profiles using RNA-seq of total RNA from ovaries in an nxf2 knockdown versus a control knockdown of the white gene. We found that a variety of TEs show increased expression in the nxf2 knockdown (see S6 Fig for all TEs); however, the 3 telomeric HTT elements (red bars) are among the top 10 most highly up-regulated TEs. Underlying data can be found in S2 Data. HTT, HeT-A, TAHRE, and TART; RNAi, RNA interference; RNA-seq, RNA sequencing; TE, transposable element.
TART-A piRNAs may target nxf2 for silencing
Previous studies have reported abundant piRNAs derived from the telomeric TEs, HeT-A, TAHRE, and TART-A [62,65,84]. We sought to determine whether piRNAs arising from the nxf2-like region of TART-A could be targeting the nxf2 gene for down-regulation via the piRNA pathway. We used previously published piRNA data from 16 wild-derived strains from the DGRP [85]. Among the 16 strains, we found wide variation in TART-A piRNA production ranging from 60 to 12,300 reads per million (RPM). From the pool of 16 strains, we identified approximately 1.3 million reads that aligned to TART-A, 98% of which map uniquely (see Methods) (Fig 4A). TART-A piRNAs have previously been shown to exhibit the 10-bp overlap signature of ping-pong cycle amplification [86], and we identified both sense and antisense piRNAs arising from TART-A (Fig 4B) as well as an enrichment of alignments where the 5′ end of 1 piRNA is found directly after the 3′ end of the previous piRNA (i.e., 3′ to 5′ distance of 1), consistent with piRNA phasing (Fig 4C). We identified approximately 95,000 piRNAs arising from the TART-A region that shares homology with nxf2. Of these reads, 59% are antisense to TART-A, and 41% are sense.
Fig 4. piRNAs are produced from both TART-A and nxf2.
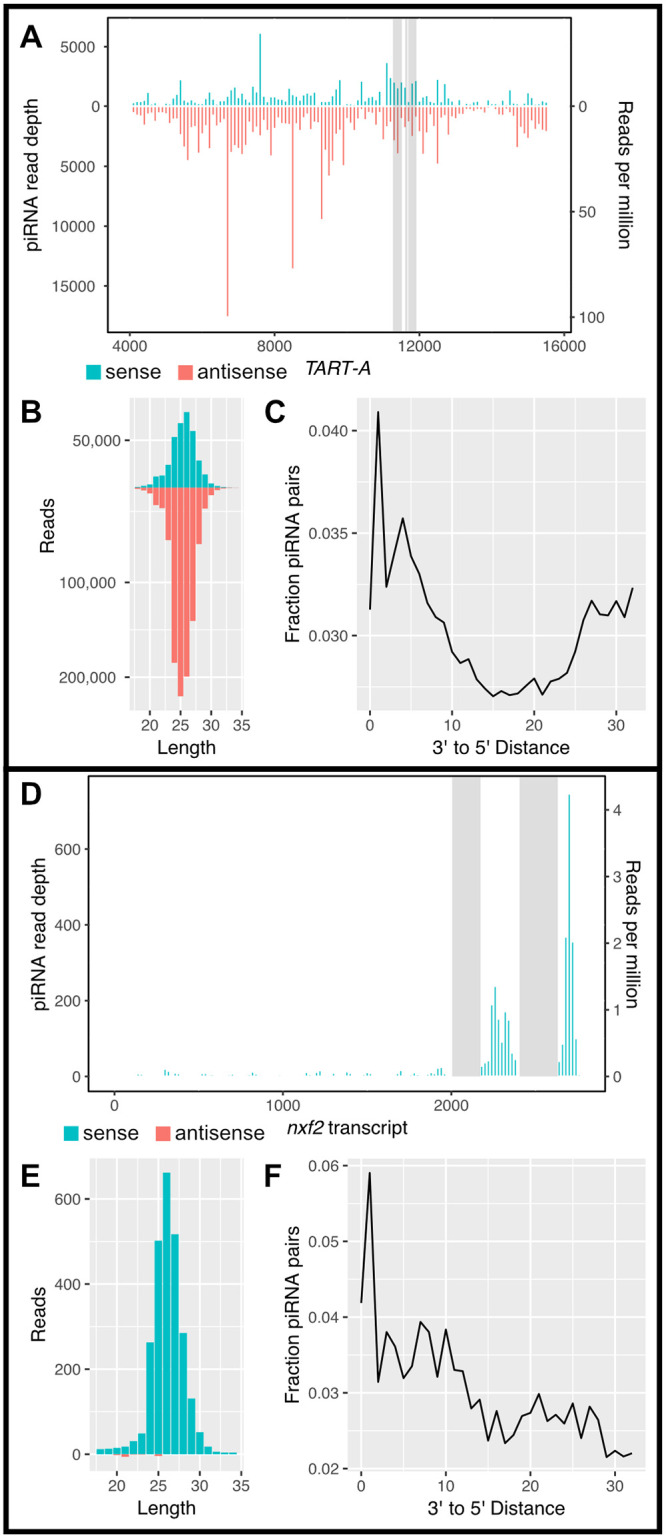
(A) We aligned previously published piRNA data from the D. melanogaster DGRP [85] to TART-A and examined read coverage across the element. We find abundant sense (blue bars) and antisense (red bars) piRNA production across most of the element, including the regions containing the nxf2-like sequence (gray boxes). Note that the 5′ UTR of TART-A is copied from the 3′ UTR during replication and is therefore identical in sequence. We masked the 5′ UTR (positions 1–4,000) for this analysis. (B) The length of aligned reads are consistent with that expected for piRNAs and the TART-A derived piRNAs are biased toward the minus strand. (C) TART-A piRNAs show an enrichment of alignments where the 5′ end of 1 piRNA is found directly after the 3′ end of the previous piRNA (i.e., distance of 1), consistent with piRNA phasing. (D) Unlike TART-A, nxf2 produces piRNAs primarily in the regions directly downstream from its TART-like sequence (gray boxes). The vast majority of these piRNAs are only from the sense strand of nxf2 (E) and also show the signature of phasing (F). Note that the TART-like sequence of nxf2 was masked for this analysis to avoid cross-mapping of TART-derived piRNAs to the nxf2 transcript. Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA; TE, transposable element.
We used an allele-specific approach to confirm that these antisense piRNAs are derived from TART-A rather than nxf2. We identified 17 positions within the region of shared homology where TART-A and nxf2 have different variants. A total of 9,301 bases from TART-derived antisense piRNAs aligned to one of these positions, 9,244 (99.4%) of which matched the TART-A variant and 27 (0.29%) of which matched the nxf2 variant. Another 30 bases were different from either TART-A or nxf2 (S1 Table).
We next focused on piRNA production from nxf2. We reasoned that, if nxf2 expression is subject to piRNA-mediated regulation, we should see piRNAs derived from the nxf2 transcript, outside of the region that shares homology with TART-A. We masked the nxf2/TART-A region of shared homology and aligned the piRNA sequence data to the nxf2 transcript. We found low but consistent production of piRNAs from nxf2 across all 16 DGRP strains (between 1.5 and 41 RPM), with 99.7% of nxf2-aligned reads mapping uniquely. To increase sequencing depth, we pooled the data from all 16 strains (2,624 nxf2 reads total) and examined piRNA abundance along the nxf2 transcript (Fig 4D). We found that the most abundant production of piRNAs from nxf2 occurs at the 3′ end of the transcript, downstream from the region of shared homology with TART-A (Fig 4D). Overall, 99.4% of reads from nxf2 are derived from the sense strand of the transcript (Fig 4E), and the nxf2 piRNAs also show evidence of phasing (Fig 4F). We quantified sense-strand piRNA production from all D. melanogaster protein-coding genes and found that nxf2 falls within the top 5% of genes in terms of the abundance of sense-strand piRNAs. The enrichment of nxf2-derived piRNAs downstream from the region of shared homology with TART-A, along with our observation that almost all nxf2 piRNAs are derived from the sense strand, suggests that these piRNAs are not amplified via the ping-pong cycle, but are instead produced by the Zucchini-mediated phasing process. Furthermore, the lack of antisense piRNAs suggests that nxf2 is not converted into a dual-strand piRNA cluster [87].
These results are consistent with a model where antisense piRNAs from the nxf2-like region of TART-A are bound by Aubergine and targeted to sense transcripts from the nxf2 gene. Aub cleaves target transcripts between the bases paired to the 10th and 11th nucleotides of its guide piRNA, resulting in a cleavage product with a 5′ monophosphate that shares a 10-bp sense:antisense overlap with the guide piRNA that triggered the cleavage. These cleavage products can be enriched and sequenced using an approach known as degradome-seq [88]. We analyzed published degradome-seq and Aub-immunoprecipitated piRNA data from wild-type D. melanogaster ovaries [89] to determine whether we could detect nxf2 cleavage products resulting from targeting by antisense TART-A piRNAs. We first aligned the antisense TART-A piRNAs to the nxf2 transcript, which resulted in 3,601 aligned reads (676 alignments with 0 mismatches, 2,145 with 1 mismatch, and 780 with 2 mismatches). We found 11 locations within the TART-like region of nxf2 where we observe degradome cleavage products that share the characteristic 10-bp sense:antisense overlap with TART-A antisense piRNAs (S8 Fig). We performed a permutation test to assess the statistical significance of these overlaps by shuffling the piRNA and degradome-seq read alignments. We found that the degradome-seq alignment locations are associated with more abundant piRNA alignments than expected by chance (P = 0.002), and overall, we observe more 10-bp sense:antisense overlaps than expected by chance (P = 0.001). These results can be explained under the following model: TART-A antisense piRNAs are produced by the ping-pong cycle and bound to Aubergine. A subset of these piRNAs (those from the nxf2-like region of TART-A) guide Aub to nxf2 transcripts which are then cleaved. Aub cleavage products can be further processed by Zucchini in the 5′ to 3′ direction, thereby producing phased piRNAs from nxf2 transcripts downstream from the nxf2/TART-A regions of shared homology (Fig 5).
Fig 5. Model describing generation of phased piRNAs from nxf2.
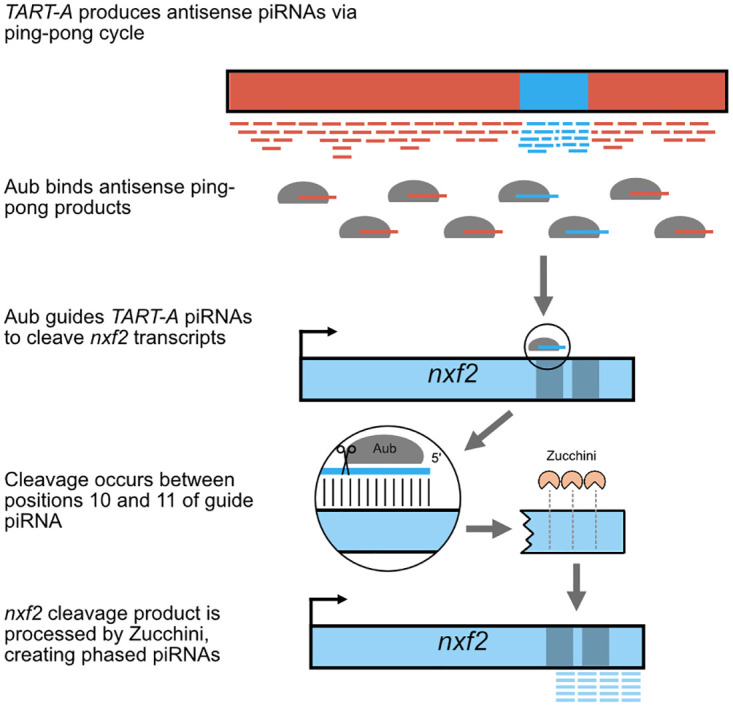
TART-A produces abundant antisense piRNAs derived from ping-pong amplification, including from the TART-A/nxf2 region of shared homology (blue box on red background). The PIWI protein Aubergine binds antisense ping-pong piRNAs, a subset of which share homology with nxf2. These piRNAs guide Aub to nxf2 and result in cleavage of the transcript between the 10th and 11th nucleotide of the guide piRNA. Transcript cleavage creates an nxf2 cleavage product that shares a 10-bp sense:antisense overlap with the guide piRNA (see S8 Fig). The nxf2 cleavage product can by subsequently processed by the Zucchini endonuclease, creating phased piRNAs starting from the site of Aub cleavage and proceeding to the 3′ end of the nxf2 transcript. piRNA, Piwi-interacting small RNA.
If piRNAs from TART-A are targeting nxf2 and down-regulating its expression, knockdown of piRNA pathway components that either decrease piRNA production from TART-A (ping-pong and/or phased piRNA pathway components) or disrupt silencing of nxf2 (phased piRNA components) should result in an increase in expression of nxf2. We analyzed published RNA sequencing (RNA-seq) data from nos-GAL4 driven knockdowns of 16 genes that were identified as components of the piRNA pathway and that were specifically shown to be involved in repression of HeT-A and TAHRE [11]. We compared the expression of nxf2 in each piRNA component knockdown to its expression in the control knockdown of the white gene and found that nxf2 shows increased expression in the majority of knockdowns; however, the observed increase in nxf2 expression is relatively mild (Fig 6A).
Fig 6. Knockdown of piRNA pathway components is associated with up-regulation of nxf2.
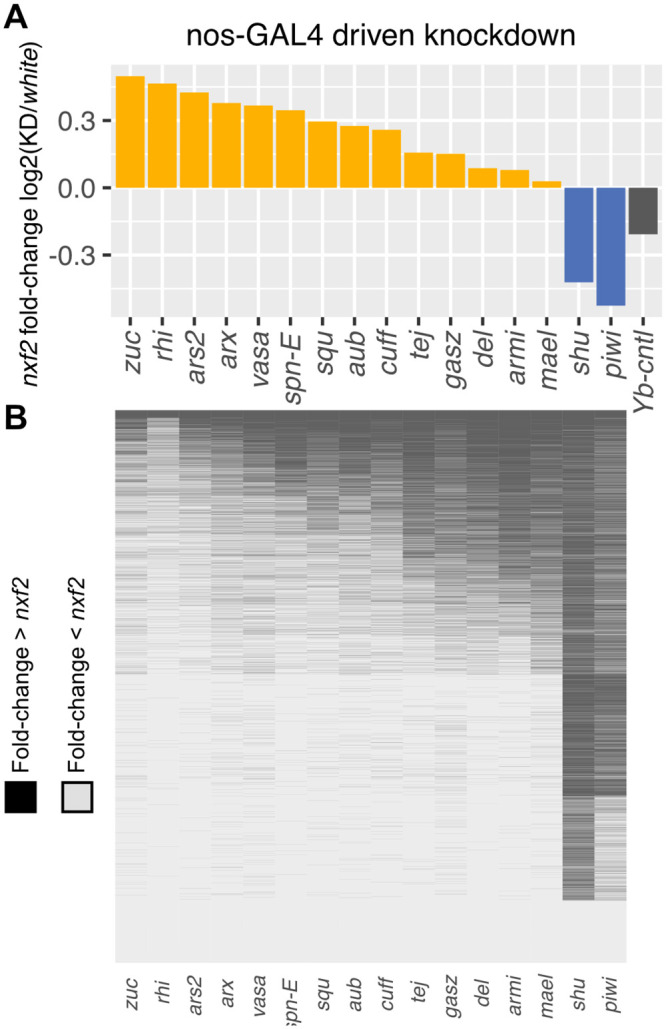
If TART-derived piRNAs are targeting nxf2 for suppression, disruption of the piRNA pathway should relieve this suppression. We examined previously published RNA-seq data from 16 piRNA component knockdowns, as well as a control (Yb) [11]. (A) Nxf2 expression increased in the majority of the 16 knockdowns (B) We compared the fold change in nxf2 expression across the 16 knockdowns to all expressed genes. Each row in the heatmap corresponds to a gene and each cell is colored based on whether the fold change in expression for that gene is larger (black) or smaller (gray) than what we observed for nxf2. Only 168 genes show larger fold change in expression across all 16 knockdowns, which places nxf2 in the top 1.4% of expressed genes in terms of its pattern of up-regulation. Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA; RNA-seq, RNA sequencing.
The small change in expression is likely due to the fact that the nxf2 expression data are from bulk ovaries containing both somatic and germ cells, whereas the knockdown is germline specific. Nxf2 is expressed at much higher levels in somatic follicle cells compared to germ cells [6]. The somatic expression, which should be unchanged between knockdown and control, will therefore mask a larger fold change that is specific to the germline. However, nxf2 should still show an increased fold change in expression relative to other genes in the genome, even though the true magnitude of expression fold change may be obscured by the mix of somatic and germ cells. We therefore compared the fold change in nxf2 expression to the genome-wide pattern of fold changes to assess the significance of nxf2 up-regulation.
For each gene with nonzero expression in the control knockdown, we determined whether its fold change in expression was greater than or equal to that of nxf2, for each of the 16 piRNA component knockdowns. We found only 168 genes with fold changes equal to or larger than nxf2 for each of the 16 knockdowns, which places nxf2 among the top approximately 1.4% of expressed genes in terms of its pattern of up-regulation (Fig 6B). Interestingly, in wild-type DGRP strains, these 168 genes have significantly more piRNAs aligning to them, compared to the remainder of expressed genes, suggesting their expression may also be regulated by piRNAs (Wilcoxon test P = 4.1e-06) (S9 Fig).
For the same 16 knockdown experiments, we also assessed the pattern of nxf2 up-regulation relative to other known piRNA pathway genes. We obtained a list of 41 germline-specific and germline/soma piRNA pathway genes from [90]. None of these 41 genes are among the 168 genes with larger fold changes than nxf2. To further confirm that there is not a general up-regulation of piRNA pathway genes upon disruption of the piRNA pathway, we examined the expression of these 41 genes for each of the 16 piRNA component knockdowns, excluding the targeted gene from analysis for each knockdown. We found no evidence of a uniform up-regulation of piRNA pathway genes across each knockdown. Instead, the median fold change of piRNA pathway genes is near 1 for each experiment (S10 Fig).
Natural variation in TART-A copy number is correlated with nxf2 expression levels
Previous work has shown that there is a large variation in HTT element copy number at the telomeres of wild Drosophila strains [68,91]. Our results predict that, if TART-A piRNAs are targeting nxf2 for suppression, then strains with more copies of TART-A should have lower expression of nxf2 and vice versa: Isofemale lines with low nxf2 expression should accumulate more copies of TART-A. To test this prediction, we used previously published Illumina genomic sequencing data and microarray gene expression profiles from the DGRP [80,81]. We used the Illumina data to infer TART-A copy number for 151 DGRP strains (see Methods) and obtained nxf2 microarray gene expression levels from whole adult females for these same strains. Nxf2 is predominantly expressed in the ovary [6]; therefore, the expression of nxf2 in whole females mainly reflects the expression of nxf2 in the ovary. We found that, as predicted, there is a strong negative correlation between TART-A copy number and nxf2 gene expression levels among the DGRP (Fig 7A) (Spearman’s rho = −0.48, P = 4.6e-10). We obtained a similar result using a replicate microarray dataset (S11 Fig). Furthermore, this pattern is unique to nxf2: correlation coefficients comparing TART-A copy number to gene expression of other piRNA pathway components are at least 2-fold smaller in magnitude (S12 and S13 Figs).
Fig 7. TART-A copy number is negatively correlated with nxf2 expression across the DGRP.

(A) We inferred TART-A copy number for 151 DGRP strains using published Illumina sequencing data [80,81] and retrieved expression values for nxf2 from microarray data from whole adult females [125]. We found that TART-A copy number is significantly negatively correlated with nxf2 expression levels, as expected if TART-A piRNAs are targeting nxf2 for suppression (Spearman’s rho = −0.48, P = 4.6e-10). (B) We also compared piRNA production from TART-A and nxf2 across 16 DGRP individuals using data from [85]. There is a strong positive correlation between TART-A piRNA production and nxf2 piRNA production (Spearman’s rho = 0.89, P < 2.2e-16) across the 16 DGRP strains. (C) We also compared nxf2 expression to the amount of TART-derived piRNAs that align to nxf2 across these same strains. In this case, we observe a negative correlation, slightly larger in magnitude than what we see for our comparison of nxf2 expression and TART-A copy number (Spearman’s rho = −0.51, P = 0.046). Underlying data can be found in S2 Data. DGRP, Drosophila Genetic Reference Panel; piRNA, Piwi-interacting small RNA; RPM, reads per million.
We predict that TART-A piRNAs are targeting nxf2 for suppression, leading to the production of additional nxf2-derived piRNAs downstream from the region of shared homology. Therefore, DGRP strains with larger production of TART-A piRNAs should also have larger amounts of nxf2-derived piRNAs. Consistent with this expectation, we find a strong positive correlation between TART-A piRNA production and nxf2 piRNA production (Spearman’s rho = 0.89, P < 2.2e-16) across 16 DGRP strains [85] (Fig 7B, S14 Fig). We find a similar correlation when we compare the TART-derived piRNAs that align to nxf2 versus the nxf2 piRNAs downstream from the region of shared homology (Spearman’s rho = 0.88, P < 2.2e-16) (S15 Fig) We also compared nxf2 expression to the amount of TART-derived piRNAs that align to nxf2 across these same strains. In this case, we observe a negative correlation, slightly larger in magnitude than what we see for our comparison of nxf2 expression and TART-A copy number (Spearman’s rho = −0.51, P = 0.046) (Fig 7C).
Evidence for genetic conflict between nxf2 and TART-A
If nxf2 is being repressed by TART-A specifically in D. melanogaster, nxf2 expression should be reduced in D. melanogaster relative to other closely related species. We performed mRNA sequencing (mRNA-seq) of ovaries for D. simulans strain w501 as well as 5 DGRP strains whose median nxf2 expression level is similar to that of the population as a whole based on the DGRP microarray data (S16 Fig). Nxf2 showed reduced expression in all 5 DGRP strains relative to D. simulans (average melanogaster/simulans fold change: 0.76). We then reanalyzed previously published mRNA-seq data from the ovaries of 4 other D. simulans strains [92], corrected for batch effects, and compared nxf2 expression between the 2 species. We found that nxf2 expression in D. melanogaster is significantly reduced compared to D. simulans, consistent with D. melanogaster–specific repression (P = 0.0039) (Fig 8A). If reduced nxf2 expression in D. melanogaster relative to D. simulans increases TART-A activity, then TART-A should show higher copy numbers in D. melanogaster. We inferred TART-A copy number using genomic sequencing data from 90 individuals from a North American population of D. simulans [93]. We found that the D. melanogaster DGRP population has a significantly larger number of TART-A copies per individual (1 to 13 copies per strain, median = 4) compared to the D. simulans population (1 to 4 copies per strain, median = 2) (Wilcoxon test P < 2.2e-16) (Fig 8B).
Fig 8. Evidence for genetic conflict between nxf2 and TART-A.

(A) We used mRNA-seq from ovaries to compare nxf2 expression between D. melanogaster and D. simulans (5 isofemale lines per species). We found that nxf2 expression in D. melanogaster is significantly reduced compared to D. simulans, consistent with D. melanogaster–specific repression (Wilcoxon test P = 0.031). Bars show mean expression level and whiskers show standard deviation. (B) We inferred TART-A copy number using genomic sequencing data from 90 D. simulans individuals [93]. We found that the D. melanogaster DGRP population has a significantly larger number of TART-A copies per individual (1–13 copies per strain, median = 4) compared to the D. simulans population (1–4 copies per strain, median = 2) (Wilcoxon test P < 2.2e-16). (C) We identified lineage-specific mutations in nxf2 CDS for both D. melanogaster and D. simulans using D. yakuba as an outgroup and used Tajima relative rate test to compare the evolutionary rate of nxf2 since it diverged from the common ancestor of D. melanogaster and D. simulans. For the TART-like region of nxf2, the evolutionary rate is significantly accelerated along the D. melanogaster branch, relative to D. simulans (13 D. melanogaster–specific substitutions, 3 D. simulans substitutions, P = 0.012), whereas there is no difference in evolutionary rate for the remainder of the gene (37 D. melanogaster–specific substitutions, 40 D. simulans–specific substitutions, P = 0.73). Underlying data can be found in S2 Data. CDS, coding sequence; mRNA-seq, mRNA sequencing.
If nxf2 is expressed at suboptimal levels due to repression by TART-derived piRNAs, selection should favor mutations within the TART-like region of nxf2 that reduce the amount of shared homology. To test for this pattern, we identified lineage-specific mutations in nxf2 CDS for both D. melanogaster and D. simulans using D. yakuba as an outgroup. If nxf2 is evolving to escape TART-mediated suppression, we would expect an accelerated rate of evolution of D. melanogaster nxf2 specifically within the region of shared homology, since its common ancestor with D. simulans. We used the Tajima relative rate test [94] to compare the evolutionary rate of nxf2 since it diverged from the common ancestor of D. melanogaster and D. simulans. For the TART-like region of nxf2, we found that the evolutionary rate is significantly accelerated along the D. melanogaster branch, relative to D. simulans (13 D. melanogaster–specific substitutions, 3 D. simulans substitutions, χ2 test statistic = 6.25, P = 0.012, 1 degree of freedom), whereas there is no difference in evolutionary rate for the remainder of the gene (37 D. melanogaster–specific substitutions, 40 D. simulans–specific substitutions, χ2 test statistic = 0.12, P = 0.73, 1 degree of freedom) (Fig 8C). We also found that the majority of the D. melanogaster–specific substitutions in the TART-like region of nxf2 occurred after the capture of nxf2 by TART-A: Of the 13 D. melanogaster–specific substitutions, TART-A is identical to D. simulans at 10 of the sites, and identical to D. melanogaster at only 3 sites (S1 Data), consistent with D. melanogaster nxf2 evolving away from TART-A.
Discussion
If the CDS of a gene shares sequence homology with a known TE, the most likely explanation for this shared homology is that a portion of the gene was derived from a TE insertion. This is, understandably, what was previously reported by Sackton and colleagues for the nxf2 gene and the TART-A TE [71]; however, our analyses are not consistent with such a scenario. Specifically, based on sequence similarity and phylogenetic clustering, the event that created the shared homology between nxf2 and TART-A must have occurred relatively recently, after D. melanogaster diverged from D. simulans, yet the putative insertion of TART-A in the nxf2 gene is shared across Drosophila.
A scenario that is more consistent with these observations is one where, rather than the nxf2 gene gaining sequence from TART-A, the TART-A element captured a portion of the nxf2 gene, likely via aberrant transcription that extended past the internal TART-A poly-A signal to another poly-A signal in the flanking genomic region. This process has been observed for other TEs and is known as exon shuffling or transduction [76,77]. Notably, the nxf2-like sequence of TART-A is located in its 3′ UTR, which would be expected if it were acquired via transduction (Fig 1). Interestingly, TART is part of the LINE family of non-LTR retrotransposons, and Human LINE-L1 elements are known to undergo transduction fairly frequently [76,77,95]. However, transduction would require that an active TART-A element was inserted somewhere upstream of the 3′ region of nxf2 at some point in the D. melanogaster lineage but has since been lost from the population. Is this possible given that TART-A should only replicate to chromosome ends? The TIDAL-fly database of polymorphic TEs in D. melanogaster reports several polymorphic TART-A insertions far from the chromosome ends, which suggests that this element is occasionally capable of inserting into locations outside of the telomeres [96]. The aberrant TART-A copy that acquired a portion of the nxf2 gene likely arose as a single polymorphic insertion in an ancestral D. melanogaster population yet has now probably replaced the ancestral TART-A element: Our results show that the nxf2-like region of TART-A is now present in most, if not all, full-length TART-A elements in D. melanogaster.
Nxf2 suppresses TART-A activity via its role in the piRNA pathway, whereas the acquisition of nxf2 sequence appears to allow TART-A to suppress nxf2. Our results are consistent with a scenario where TART-derived piRNAs guide Aub proteins to the nxf2 transcript. The TART-A piRNAs may then act as “trigger” piRNAs that catalyze cleavage of nxf2 transcripts while also resulting in the production of phased piRNAs starting in the region of shared homology and proceeding in the 3′ direction to the end of the nxf2 transcript (Fig 5). The piRNA-mediated cleavage of nxf2 transcripts, which is supported by degradome-seq data (see S8 Fig), should result in a reduction in nxf2 expression levels.
The fact that Nxf2 is known to interact with Piwi-targeted transcripts makes it very likely that it is directly interacting with TART-A. Telomeric piRNAs are bound by Piwi [63,97,98], and Piwi has previously been described as playing an important role in maintenance of telomeric chromatin: In piwi mutants, the telomeres become depleted of H3K9me3 and move from the nuclear periphery to the interior [99]. Interestingly, Zhao and colleagues observed a similar translocation of telomeres to the nuclear interior in their nxf2 mutant [9].
Given that nxf2 plays a role in suppressing TART-A activity, reduced nxf2 levels should relieve TART-A suppression, which would presumably increase TART-A fitness by allowing it to make more copies of itself. Indeed, in the DGRP, we find that individuals with lower nxf2 expression levels tend to have higher numbers of TART-A copies and vice versa (Fig 7). If additional copies of TART-A act to further suppress nxf2 expression, which then further derepresses TART-A, why is there not run-away accumulation of telomere length in D. melanogaster? Previous work has shown that long telomeres in D. melanogaster are associated with both reduced fertility and fecundity [91], so it is possible that a run-away trend toward increasing telomere length is balanced by a fitness cost.
Our results provide several lines of evidence that nxf2 and TART-A are evolving in conflict. In D. melanogaster, nxf2 is expressed at lower levels and TART-A has proliferated to higher copy numbers compared to D. simulans, consistent with TART-A benefitting from nxf2 suppression (Fig 8B and 8C). If nxf2 expression level is suboptimal in D. melanogaster due to TART-A suppression, there should be selection to disrupt the shared homology between these 2 elements, which is supported by our finding that the TART-like region of nxf2 is experiencing accelerated evolution in the D. melanogaster lineage (Fig 8A).
Targeting of host transcripts by transposon-derived piRNAs has been previously observed in Drosophila. Most notably, piRNAs from the LTR retrotransposons roo and 412 play a critical role in embryonic development by targeting complementary sequence in the 3′ UTR of the gene nos, leading to its repression in the soma [100]. More recent results suggest hundreds of maternal transcripts could be regulated in a similar fashion [101]. However, these represent cases where TE piRNAs have been co-opted to regulate host transcripts, whereas our results suggest that the piRNA targeting of nxf2 is a counter-defense strategy by TART-A. This type of strategy has only been previously observed in plants [32]. In rice, a CACTA DNA transposon produces a micro-RNA that targets a host methyltransferase gene known to be involved in TE suppression [28], while in Arabidopsis, siRNAs from Athila6 retrotransposons target the stress granule protein UBP1b, which is involved in suppressing Athila6 GAG protein production [29].
Given that viruses and other pathogens have evolved a variety of methods to block or disrupt host defense mechanisms, it is surprising that there is much less evidence for TEs adopting similar strategies [32]. However, unlike viruses, TEs depend heavily on vertical transmission from parent to offspring. Any counter-defense strategy that impacts host fitness would therefore decrease the fitness of the TE as well. Our finding that the nxf2-like region of TART-A appears to be fixed in D. melanogaster is therefore unexpected: TART-A variants lacking nxf2 homology should benefit from the presence of the nxf2-like TART-A element without incurring the fitness cost. The nxf2-like TART-A variant should therefore be under frequency dependent selection and would be expected to remain polymorphic. One possibility that would explain the fixation of the novel TART-A variant is if the nxf2-like sequence also enhances TART-A retrotransposition. Unusually long UTRs are a hallmark of all HTT elements that have been identified across multiple Drosophila species; however, their specific functional role in retrotransposition remains unknown.
Another reason that TE counter-defense may be rare is that disruption of host silencing is likely to lead to up-regulation of other TEs, making it more likely that there will be a severe decrease in host fitness, similar to what is observed in hybrid dysgenesis. TART-A may be targeting nxf2 for its own advantage, but our knockdown experiment shows that nxf2 suppression causes up-regulation of many other TEs besides TART-A (Fig 3, S6 Fig), and other studies have shown that nxf2 mutants are sterile [6, 7]. Why then, does TART-A appear to be targeting nxf2 in spite of these potentially deleterious consequences? One possibility is that the suppression of nxf2 expression caused by TART-A is relatively mild (i.e., much less than the level of down-regulation caused by the RNAi knockdown), which is enough to provide a slight benefit to TART-A without causing widespread TE activation. It is also possible that the suppression effect was initially much larger but has since been counterbalanced by cis-acting variants that increase nxf2 expression and/or reduction in shared homology caused by substitutions in the TART-like region of nxf2. Future work examining TE activation under varying levels of nxf2 expression may help to determine whether there is a tipping point where nxf2 suppression becomes catastrophic.
In summary, our results show that so-called domesticated TEs, if active, can still be in conflict with their host and raise the possibility that TE counter-defense strategies may be more common than previously recognized, despite the potentially deleterious consequences for the host.
Methods
TART-A sequence analysis
We used the TART-A sequence from RepBase [102], which is derived from the sequence reported in [73] (GenBank accession AJ566116). This sequence represents a single full-length TART-A element cloned from the D. melanogaster iso1 reference strain. The nxf2-like portion of this sequence is 100% identical to another TART-A element cloned and sequenced from D. melanogaster strain A4-4 (GenBank DMU02279) [53] as well as the TART-A sequence from the FlyBase canonical set of transposon sequences (version 9.42) [103] (cloned from D. melanogaster strain Oregon-R: GenBank AY561850) [104].
We used BLAST [72] to compare the TART-A sequence to the D. melanogaster nxf2 transcript and visualized BLAST alignments with Kablammo [105]. To compare TART-A among Drosophila species, we used the D. yakuba TART-A sequence reported in [69](GenBank AF468026), which includes the 3′ UTR. We also used the D. sechellia TART-A ORF2 reported by [59] (GenBank AM040251) to search the D. sechellia FlyBase r1.3 genome assembly for a TART-A copy that included the 3′ UTR, which we found on scaffold_330:4944–14419. We attempted a similar approach for D. simulans, but were unable to find a TART-A copy in the D. simulans FlyBase r2.02 assembly that included the 3′ UTR. We aligned the D. melanogaster, D. yakuba, and D. sechellia TART-A sequences to each other, and to the D. melanogaster nxf2 transcript (FlyBase FBtr0089479), using nucmer [106]. We then used mummerplot [106] to create a dotplot to visualize the alignments.
To determine whether the TART-A nxf2-like sequence was fixed or polymorphic in D. melanogaster, we used BLAST (e-value cutoff 1e-50) to search the TART-A 3′ UTR against genome assemblies from the following D. melanogaster strains: the iso1 release 6 reference genome [107], nanopore assemblies for DGRP379 and DGRP732 [79], and PacBio (Pacific Biosciences, Menlo Park, California, USA) assemblies for 14 D. melanogaster strains [78]. We extended the coordinates of each BLAST hit to match the length of the full UTR and extracted the sequences from their assemblies. We then further filtered them by requiring each sequence to have at least 1,000 aligned bases between it and the TART-A RepBase sequence, and we required the best BLAST hit of each sequence to be TART-A, when searched against the RepBase TE database. We then created a multiple sequence alignment of all sequences using MAFFT [108].
nxf2 sequence analysis
We downloaded nxf2 transcripts from the NCBI RefSeq database for D. simulans (XM_016169386.1), yakuba (XM_002095083.2), erecta (XM_001973010.3), biarmipes (XM_017111057.1), and elegans (XM_017273027.1) and created a codon-aware multiple sequence alignment using PRANK [109], which we visualized with JalView [110]. To compare Nxf2 peptide sequences, we used the web version of NCBI BLAST to search the D. melanogaster Nxf2 peptide sequence against all Drosophila peptide sequences present in the RefSeq database. We then used the NCBI COBALT [111] multiple-sequence alignment tool to align the sequences shown in S1 Fig. We used MEGA [112] to conduct Tajima relative rates test for nxf2.
TART-A/nxf2 gene tree
We extracted the nxf2-like sequences from all TART-A copies present in the D. melanogaster reference genome and aligned them to the TART-like nxf2 sequences from 7 Drosophila species using PRANK. We then inferred a maximum likelihood phylogeny with 100 bootstrap replicates using RAxML [113].
nxf2 knockdown
We used 2 different strains from the Drosophila TRiP that express dsRNA for RNAi of nxf2 (Bloomington #34957 and #33985), as well as a control strain for RNAi of the white gene (Bloomington #33613). These 3 RNAi strains were all generated from the same attP2 progenitor strain. Seven males of each of these strains were crossed to seven, 3- to 5-day-old, virgin females carrying the nos-GAL4 driver (Bloomington #25751). After 6 days of mating, we discarded the parental flies and then transferred F1 offspring to fresh food for 2.5 days before collecting ovaries from 6 females for each cross. We performed 2 biological replicates for each of the 3 crosses, dissected the ovaries in 1× PBS and immediately transferred them to RNAlater. We extracted RNA using Trizol/Phenol-Chloroform and used the AATI Fragment Analyzer to assess RNA integrity. We then prepared stranded, total RNA-seq libraries by first depleting rRNA with ribo-zero and then using the NEBnext ULTRA II library prep kit (New England Biolabs, Ipswich, Massachusetts, USA) to prepare the sequencing libraries. The libraries were sequenced on the Illumina NextSeq machine with 150-bp paired-end reads.
nxf2 knockdown RNA-seq analysis
The average insert sizes of the total RNA-seq libraries were less than 300 bp, which resulted in overlapping mate pairs for the majority of sequenced fragments. Instead of analyzing these data as paired-end reads, we instead merged the overlapping mates to generate single-end reads using BBmerge [114]. We removed rRNA and tRNA contamination from the merged reads by aligning them to all annotated rRNA and tRNA sequences in the D. melanogaster reference genome using Hisat2 [115] and retained all unaligned reads. In order to quantify expression from genes as well as TEs, we combined all D. melanogaster transcript sequences (FlyBase version 6.26) with D. melanogaster RepBase TE consensus sequences. We accounted for multi-mapping reads by using bowtie2 [116] to align each read to all possible alignment locations (using—all and—very-sensitive-local) and then using eXpress [117] to estimate FPKM values, accounting for the multi-mapped alignments. We averaged FPKM values between biological replicates and assessed the reproducibility of both TE and gene expression profiles in the nxf2 knockdown by comparing the results from the 2 different dsRNA hairpins.
piRNA analysis
We analyzed previously published piRNA data from 16 strains from the DGRP [85]. We used cutadapt [118] to trim adapter sequences from each library and then removed rRNA and tRNA sequences by using bowtie [119] to align the reads to all annotated rDNA and tRNA genes in the D. melanogaster reference genome, retaining the reads that did not align. We then created a reference database composed of the following sequence sets: a hard-masked version of the D. melanogaster reference genome assembly (release 6) where all TE sequences and the nxf2 gene were replaced by N’s using RepeatMasker, the full set of D. melanogaster RepBase TE consensus sequences, and the nxf2 transcript, with its TART-like region replaced by N’s. Because the 5′ UTR is copied from the 3′ UTR, we also masked the 5′ UTR of TART-A. We used the unique-weighting mode in ShortStack [120,121] to align the piRNA reads to this reference database. With this mode, ShortStack probabilistically aligns multi-mapping reads based on the abundance of uniquely mapping reads in the flanking region. We then used the ShortStack alignments and Bedtools [122] to calculate coverage for sense and antisense alignments to TART-A as well as nxf2. To test for evidence of piRNA phasing, we used the formula described in [123].
piRNA component knockdowns
We used the raw read counts reported in GEO accession GSE117217 from 16 RNAi knockdowns of piRNA pathway components as well as a control knockdown of the Yb gene [11]. We used the DESeq2 median of ratios approach to normalize raw counts [124].
Degradome-seq analysis
We used degradome-seq and Aub-immunoprecipitated small RNA data from wild-type D. melanogaster strain w1 [89]. We used bowtie2 to align the degradome-seq data to the same reference sequence used in the piRNA analysis except we unmasked the nxf2 transcript. The degradome-seq data are 100-bp paired-end reads which are long enough to distinguish between the TART-like region of nxf2 and the nxf2-like region of TART-A. We analyzed the small RNA data as described under “piRNA analysis,” except we allowed 2 mismatches for alignments between TART-derived piRNAs and nxf2. We then used Bedtools to extract degradome read alignments whose 5′ end was located in the TART-like region of nxf2 and antisense small RNA alignments whose 5′ end was located in the nxf2-like region of TART-A and whose length was consistent with piRNAs (23 to 30 bp). We then used bowtie to align the minus strand piRNAs to the nxf2 transcript and used bedtools to identify piRNAs whose 5′ end overlapped the 5′ of degradome reads by 10 bp.
For the permutation test, we determined the number of antisense piRNAs whose 5′ ends aligned at each position within the TART-like region of nxf2. From the degradome-seq alignments, we determined there were 18 unique locations within the TART-like region of nxf2 where at least 1 degradome-seq read aligned. Eleven of these locations also showed the 10-bp sense:antisense overlap with at least 1 piRNA. To test whether the 10-bp sense:antisense overlaps that we observed between degradome and piRNA reads were associated with higher abundance piRNAs, we randomly sampled 11 piRNA alignment locations from the TART-like region of nxf2 and calculated the number of piRNAs that aligned at each location using Bedtools. We repeated this process 1,000 times and counted the number of times where the random sample had the same or more piRNAs at each location compared to the true alignments. To test whether we observed more 10-bp sense:antisense overlaps than expected by chance, we randomly sampled 18 positions within the TART-like region of nxf2 and counted how many of the 18 positions also had a 10-bp sense:antisense overlap with at least 1 piRNA. We repeated this process 1,000 times and determined the number of times where the random sample had the same or more 10-bp sense:antisense overlaps compared to the true sample.
TART-A copy number variation and nxf2 expression
We obtained nxf2 expression values from previously published microarray gene expression profiles from whole adult females for all DGRP strains [125] and used Illumina genomic sequencing data from the DGRP [80,81] to estimate TART-A copy number. Across strains, the DGRP Illumina data differs in terms of coverage, read length, and paired versus single-end data. To attempt to control for these differences, we trimmed all reads to 75 bp and treated all data as single-end. We also downsampled all libraries to approximately 13 million reads. We first trimmed each strain’s complete dataset (unix command: zcat file.fastq.gz | cut -c 75) and then aligned the trimmed reads to the D. melanogaster release 6 genome assembly using bowtie2 with the—very-sensitive option. We then corrected the resulting bam file for GC bias using DeepTools [126] and counted the number of aligned reads in the corrected bam file using samtools [127]. We removed all strains with less than 13 million aligned reads and, for each remaining strain, we calculated the fraction of reads to keep by dividing the smallest number of aligned reads across all remaining individuals (13,594,737) by the total number of aligned reads for that strain. We then used this fraction to randomly downsample the GC corrected bam file using the subsample option from samtools view [127]. We converted each bam file to a fastq file with samtools fastq and aligned the fastq file to the D. melanogaster RepBase TE sequences with bowtie2 using the—very-sensitive,—local, and—all options. With—all, bowtie2 reports every possible alignment for each multi-mapping sequence. We then used eXpress to retain a single alignment for each multi-mapping sequence based on the abundance of neighboring unique alignments. We used the eXpress bam files to calculate the median per-base coverage (excluding positions with coverage of 0) for the TART-A CDS (i.e., ORF1 and ORF2), for each individual. To estimate TART-A copy number, we divided the median TART-A coverage of each strain by that strain’s median per-base coverage of all uniquely mappable positions in the D. melanogaster reference genome (calculated from the GC corrected, downsampled bam file). Uniquely mappable positions were identified using mirth (https://github.com/EvolBioInf/mirth).
We used the same pipeline to infer TART-A copy numbers for D. simulans. We used Illumina genomic sequencing data from [93] and, rather than aligning to RepBase TE consensus sequences, we identified TEs de novo using RepeatModeler (http://www.repeatmasker.org) with the long-read D. simulans genome assembly from [128]. The D. simulans TART-A sequence fragment is provided in (S4 Data).
Nxf2 expression in D. simulans versus D. melanogaster ovaries
For D. simulans strain w501 and DGRP strains 313, 362, 379, 391, and 732, we used 5 to 20 pairs of ovaries from mated females. The ovaries were dissected in 1× PBS and then immediately transferred to 200-μL RNAlater Solution. Total RNA was extracted using 200-μL Trizol and DNase treated using the Ambion TURBO DNA-free Kit (Invitrogen, Carlsbad, California, USA). The mRNA-seq libraries were prepared using Bioo Scientific NEXTflex Poly(A) Beads and NEXTflex Rapid Directional RNA-Seq Kit (PerkinElmer, Austin, Texas, USA). We obtained additional D. simulans ovary expression data from [92]. We aligned the RNA-seq data to their respective reference genome assembly using hisat2 [115] and then used htseq-count [129] to obtain raw read counts for each gene. We only counted reads overlapping CDS features in case the UTR annotations differed between species. We corrected the raw counts for batch effects using ComBat-seq [130] and used DESeq2 to normalize the batch-corrected counts and test for differential expression. We only considered genes identified as 1-to-1 orthologs between D. melanogaster and D. simulans by FlyBase and excluded orthologs whose CDS length differed by more than 10 bp.
Supporting information
We used NCBI web BLAST to search the D. melanogaster Nxf2 peptide sequence against the RefSeq peptide database and identified homologs in 22 Drosophila species. The carboxyl-terminal region of Nxf2 derives from CDS which shares homology with the TART-A TE (gray box). At the peptide level, this region is conserved out to D. virilis, which suggests that, if it was acquired from an insertion of the TART-A TE, the insertion would have occurred in the common ancestor of the entire genus. CDS, coding sequence; TE, transposable element.
(TIFF)
The pink boxes show the 2 segments of shared homology between D. melanogaster TART-A and D. melanogaster nxf2. D. yakuba TART-A aligns to D. melanogaster TART-A at regions directly adjacent to, but not including, the TART-A/nxf2 shared homology. Underlying data can be found in S2 Data.
(TIFF)
We compared nxf2 transcript sequences from D. melanogaster (A), D. yakuba (B), and D. sechellia (C) to TART-A sequences from the same species using mummer [106]. There is sequence homology present between D. melanogaster nxf2 and TART-A but not for D. yakuba nxf2/TART-A nor for D. sechellia nxf2/TART-A. Underlying data can be found in S2 Data.
(TIFF)
We identified 71 TART-A elements with 3′ UTRs from 17 long-read D. melanogaster genome assemblies. All 71 elements contain the nxf2-like sequence (gray box) suggesting that this region is present in most, if not all, TART-A elements in D. melanogaster. Note that a portion of the nxf2-like region appears to have been deleted in one of the TART-A elements.
(TIFF)
We compared genomic sequencing coverage for the nxf2-like region of TART-A (blue shading) to its upstream and downstream flanking regions (yellow shading). For each DGRP strain, we divided read coverage by the median coverage of that strain’s TART-A ORF1 and ORF2 to control for copy number differences between strains. We calculated coverage for each strain in 10-bp windows across the region. Each box in the figure summarizes the per-strain coverage values for a single 10-bp segment. Within each box, the internal line represents the median coverage and the hinges correspond to the 25th and 75th percentiles. The whiskers extend to 1.5× the interquartile range. The coverage of the nxf2-like region is similar to the coverage of the downstream region, both of which are reduced relative to the upstream region. This pattern is consistent with truncation of the UTR, which has previously been described for TART [74]. Because the nxf2-like sequence is present in both UTRs, truncation of the 5′ UTR, which is fairly common, should reduce coverage of both the nxf2-like region and downstream flanking region by approximately 50% compared to the upstream region, which is not present in the 5′ UTR (Fig 1B). We observed a reduction in coverage of approximately 30%, consistent with a mixture of TART-A copies, some with truncated 5′ UTRs and some without. The median coverage across all boxes within a region is shown by the colored horizontal bars. Underlying data can be found in S2 Data. ORF, open reading frame.
(PDF)
Each RepBase repeat for which we observed expression in total RNA-seq data from female ovaries is shown on the y-axis, and the fold change in expression in the nxf2 RNAi knockdown versus a control knockdown of the white gene is shown on the x-axis with a log2 scale. Expression values are the mean of 2 biological replicates for both knockdown and control. For LTR retrotransposons, LTRs are shown separately from the rest of the TE. Underlying data can be found in S2 Data. LTR, long terminal repeat; TE, transposable element.
(PDF)
We used 2 shRNAs that target different regions of the nxf2 transcript and calculated expression values for genes as well as TEs for each knockdown. We found that the expression values are highly correlated between the 2 experiments (Spearman’s rho = 0.92 [Genes] and 0.94 [TEs]). Underlying data can be found in S2 Data. shRNA, short hairpin RNA; TE, transposable element.
(PDF)
We analyzed published degradome-seq and Aub-immunoprecipitated small RNA data to determine whether there were nxf2 degradome-seq reads showing the 10-bp sense:antisense overlap with TART-A piRNAs, consistent with cleavage by a Piwi protein. We identified 11 locations (A–K) within the TART-like region of nxf2 where degradome-seq cleavage products (red) overlap with antisense piRNAs (blue) by 10 bp at their 5′ ends. The nxf2 transcript is shown in black. degradome-seq, degradome sequencing; piRNA, Piwi-interacting small RNA.
(TIFF)
We identified 168 genes whose fold change in expression was greater than or equal to nxf2 across RNAi knockdowns of 16 piRNA pathway components. These genes have a significantly larger abundance of aligned piRNAs compared to the remainder of expressed genes, suggesting their expression may be regulated by piRNAs (Wilcoxon test P = 4.1e-06). Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA; RNAi, RNA interference.
(PDF)
We examined the fold change in expression of 41 known piRNA pathway genes across RNAi knockdowns of 16 piRNA pathway components, excluding the targeted gene from analysis for each experiment. PiRNA pathway genes show a median fold change near 1 (horizontal red line) for most experiments. Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA; RNAi, RNA interference.
(PDF)
We repeated the analysis shown in Fig 7A using a replicate microarray dataset from [125] and found a similar correlation (Spearman’s rho = −0.49), which suggests that the microarray expression measurements are highly reproducible. Underlying data can be found in S2 Data.
(PDF)
We were able to obtain expression values for 39 other piRNA pathway genes from the same microarray dataset that we used for nxf2 expression. For each of these genes, we calculated Spearman correlation coefficient for its expression compared to TART-A copy number. All correlation coefficients are at least 2-fold smaller in magnitude than what we observed for nxf2. Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA.
(TIFF)
The histogram summarizes the Spearman correlation coefficients between 39 piRNA pathway genes and TART-A copy number (shown in S12 Fig). The red line shows the correlation coefficient for nxf2. Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA.
(PDF)
We plotted piRNA read depth (normalized as RPM mapped) along the nxf2 transcript for each of the 16 DGRP strains shown in Fig 7. For each strain, the abundance of TART piRNAs is listed in the plot title. We masked the locations of the TART/nxf2 shared homology (gray boxes) before alignment to avoid cross-mapping of TART-derived piRNAs. Underlying data can be found in S2 Data. DGRP, Drosophila Genetic Reference Panel; piRNA, Piwi-interacting small RNA; RPM, reads per million.
(PDF)
There is a strong positive correlation between TART-derived piRNAs that align to nxf2 versus the nxf2 piRNAs downstream from the region of shared homology, across 16 DGRP strains (Spearman’s rho = 0.88, P < 2.2e-16). Underlying data can be found in S2 Data. DGRP, Drosophila Genetic Reference Panel; piRNA, Piwi-interacting small RNA.
(PDF)
We used the microarray dataset from [125] to select 5 DGRP strains whose median nxf2 expression level is similar to that of the full DGRP population. Underlying data can be found in S2 Data. DGRP, Drosophila Genetic Reference Panel; RNA-seq, RNA sequencing.
(TIFF)
(DOCX)
(XLSX)
(XLSX)
(TXT)
(TXT)
Acknowledgments
The authors acknowledge the Office of Advanced Research Computing (OARC) at Rutgers, The State University of New Jersey for providing access to the Amarel cluster and associated research computing resources that have contributed to the results reported here. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study.
Abbreviations
- CDS
coding sequence
- degradome-seq
degradome sequencing
- DGRP
Drosophila Genetic Reference Panel
- HTT
HeT-A, TAHRE, and TART
- LINE
Long Interspersed Nuclear Element
- LTR
long terminal repeat
- mRNA-seq
mRNA sequencing
- ORF
open reading frame
- piRNA
Piwi-interacting small RNA
- RNAi
RNA interference
- RNA-seq
RNA sequencing
- RPM
reads per million
- shRNA
short hairpin RNA
- TE
transposable element
- TRiP
Transgenic RNAi Project
Data Availability
The RNA-seq data generated for this study are available from the NCBI Short Read Archive (BioProject number PRJNA606690).
Funding Statement
The authors received no specific funding for this work.
References
- 1.Lee YC. The Role of piRNA-Mediated Epigenetic Silencing in the Population Dynamics of Transposable Elements in Drosophila melanogaster. PLoS Genet. 2015;11(6):e1005269 Epub 2015/06/05. 10.1371/journal.pgen.1005269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee YCG, Karpen GH. Pervasive epigenetic effects of Drosophila euchromatic transposable elements impact their evolution. Elife. 2017;6 Epub 2017/07/12. 10.7554/eLife.25762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrov DA, Fiston-Lavier AS, Lipatov M, Lenkov K, Gonzalez J. Population genomics of transposable elements in Drosophila melanogaster. Mol Biol Evol. 2011;28(5):1633–44. Epub 2010/12/22. 10.1093/molbev/msq337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128(6):1089–103. Epub 2007/03/10. 10.1016/j.cell.2007.01.043 . [DOI] [PubMed] [Google Scholar]
- 5.Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, et al. A slicer-mediated mechanism for repeat-associated siRNA 5’ end formation in Drosophila. Science. 2007;315(5818):1587–90. Epub 2007/02/27. 10.1126/science.1140494 . [DOI] [PubMed] [Google Scholar]
- 6.Batki J, Schnabl J, Wang J, Handler D, Andreev VI, Stieger CE, et al. The nascent RNA binding complex SFiNX licenses piRNA-guided heterochromatin formation. Nat Struct Mol Biol. 2019;26(8):720–31. Epub 2019/08/07. 10.1038/s41594-019-0270-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fabry MH, Ciabrelli F, Munafo M, Eastwood EL, Kneuss E, Falciatori I, et al. piRNA-guided co-transcriptional silencing coopts nuclear export factors. Elife. 2019;8 Epub 2019/06/21. 10.7554/eLife.47999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murano K, Iwasaki YW, Ishizu H, Mashiko A, Shibuya A, Kondo S, et al. Nuclear RNA export factor variant initiates piRNA-guided co-transcriptional silencing. EMBO J. 2019;38(17):e102870 Epub 2019/08/02. 10.15252/embj.2019102870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao K, Cheng S, Miao N, Xu P, Lu X, Zhang Y, et al. A Pandas complex adapted for piRNA-guided transcriptional silencing and heterochromatin formation. Nature Cell Biology. 2019;21:1261–72. 10.1038/s41556-019-0396-0 [DOI] [PubMed] [Google Scholar]
- 10.Dennis C, Brasset E, Sarkar A, Vaury C. Export of piRNA precursors by EJC triggers assembly of cytoplasmic Yb-body in Drosophila. Nat Commun. 2016;7:13739 Epub 2016/12/09. 10.1038/ncomms13739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Czech B, Preall JB, McGinn J, Hannon GJ. A transcriptome-wide RNAi screen in the Drosophila ovary reveals factors of the germline piRNA pathway. Mol Cell. 2013;50(5):749–61. Epub 2013/05/15. 10.1016/j.molcel.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ElMaghraby MF, Andersen PR, Puhringer F, Hohmann U, Meixner K, Lendl T, et al. A Heterochromatin-Specific RNA Export Pathway Facilitates piRNA Production. Cell. 2019;178(4):964–79 e20. Epub 2019/08/10. 10.1016/j.cell.2019.07.007 . [DOI] [PubMed] [Google Scholar]
- 13.Kneuss E, Munafo M, Eastwood EL, Deumer US, Preall JB, Hannon GJ, et al. Specialization of the Drosophila nuclear export family protein Nxf3 for piRNA precursor export. Genes Dev. 2019;33(17–18):1208–20. Epub 2019/08/17. 10.1101/gad.328690.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charlesworth B, Langley CH. The evolution of self-regulated transposition of transposable elements. Genetics. 1986;112(2):359–83. Epub 1986/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee YC, Langley CH. Transposable elements in natural populations of Drosophila melanogaster. Philos Trans R Soc Lond B Biol Sci. 2010;365(1544):1219–28. Epub 2010/03/24. 10.1098/rstb.2009.0318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelleher ES, Barbash DA. Analysis of piRNA-mediated silencing of active TEs in Drosophila melanogaster suggests limits on the evolution of host genome defense. Mol Biol Evol. 2013;30(8):1816–29. Epub 2013/04/30. 10.1093/molbev/mst081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parhad SS, Theurkauf WE. Rapid evolution and conserved function of the piRNA pathway. Open Biol. 2019;9(1):180181 Epub 2019/04/09. 10.1098/rsob.180181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levine MT, Vander Wende HM, Hsieh E, Baker EP, Malik HS. Recurrent Gene Duplication Diversifies Genome Defense Repertoire in Drosophila. Mol Biol Evol. 2016;33(7):1641–53. Epub 2016/03/17. 10.1093/molbev/msw053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelleher ES, Edelman NB, Barbash DA. Drosophila interspecific hybrids phenocopy piRNA-pathway mutants. PLoS Biol. 2012;10(11):e1001428 Epub 2012/11/29. 10.1371/journal.pbio.1001428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolaczkowski B, Hupalo DN, Kern AD. Recurrent adaptation in RNA interference genes across the Drosophila phylogeny. Mol Biol Evol. 2011;28(2):1033–42. Epub 2010/10/26. 10.1093/molbev/msq284 . [DOI] [PubMed] [Google Scholar]
- 21.Obbard DJ, Jiggins FM, Bradshaw NJ, Little TJ. Recent and recurrent selective sweeps of the antiviral RNAi gene Argonaute-2 in three species of Drosophila. Mol Biol Evol. 2011;28(2):1043–56. Epub 2010/10/28. 10.1093/molbev/msq280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Obbard DJ, Jiggins FM, Halligan DL, Little TJ. Natural selection drives extremely rapid evolution in antiviral RNAi genes. Curr Biol. 2006;16(6):580–5. Epub 2006/03/21. 10.1016/j.cub.2006.01.065 . [DOI] [PubMed] [Google Scholar]
- 23.Simkin A, Wong A, Poh YP, Theurkauf WE, Jensen JD. Recurrent and recent selective sweeps in the piRNA pathway. Evolution. 2013;67(4):1081–90. Epub 2013/04/05. 10.1111/evo.12011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helleu Q, Levine MT. Recurrent Amplification of the Heterochromatin Protein 1 (HP1) Gene Family across Diptera. Mol Biol Evol. 2018;35(10):2375–89. Epub 2018/06/21. 10.1093/molbev/msy128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crysnanto D, Obbard DJ. Widespread gene duplication and adaptive evolution in the RNA interference pathways of the Drosophila obscura group. BMC Evol Biol. 2019;19(1):99 Epub 2019/05/10. 10.1186/s12862-019-1425-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs FM, Greenberg D, Nguyen N, Haeussler M, Ewing AD, Katzman S, et al. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature. 2014;516(7530):242–5. Epub 2014/10/03. 10.1038/nature13760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu Y, Kawabe A, Etcheverry M, Ito T, Toyoda A, Fujiyama A, et al. Mobilization of a plant transposon by expression of the transposon-encoded anti-silencing factor. EMBO J. 2013;32(17):2407–17. Epub 2013/08/01. 10.1038/emboj.2013.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nosaka M, Itoh J, Nagato Y, Ono A, Ishiwata A, Sato Y. Role of transposon-derived small RNAs in the interplay between genomes and parasitic DNA in rice. PLoS Genet. 2012;8(9):e1002953 Epub 2012/10/03. 10.1371/journal.pgen.1002953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCue AD, Nuthikattu S, Slotkin RK. Genome-wide identification of genes regulated in trans by transposable element small interfering RNAs. RNA Biol. 2013;10(8):1379–95. Epub 2013/07/19. 10.4161/rna.25555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Dou K, Moon S, Tan FJ, Zhang ZZ. Hijacking Oogenesis Enables Massive Propagation of LINE and Retroviral Transposons. Cell. 2018;174(5):1082–94 e12. Epub 2018/07/31. 10.1016/j.cell.2018.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mari-Ordonez A, Marchais A, Etcheverry M, Martin A, Colot V, Voinnet O. Reconstructing de novo silencing of an active plant retrotransposon. Nat Genet. 2013;45(9):1029–39. Epub 2013/07/16. 10.1038/ng.2703 . [DOI] [PubMed] [Google Scholar]
- 32.Cosby RL, Chang NC, Feschotte C. Host-transposon interactions: conflict, cooperation, and cooption. Genes Dev. 2019;33(17–18):1098–116. Epub 2019/09/05. 10.1101/gad.327312.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blumenstiel JP, Erwin AA, Hemmer LW. What Drives Positive Selection in the Drosophila piRNA Machinery? The Genomic Autoimmunity Hypothesis. Yale J Biol Med. 2016;89(4):499–512. Epub 2016/12/27. [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Barbash DA, Kelleher ES. Divergence of piRNA pathway proteins affects piRNA biogenesis and off-target effects, but not TE transcripts, revealing a hidden robustness to piRNA silencing. bioRxiv. 2019. [Google Scholar]
- 35.Chuong EB, Elde NC, Feschotte C. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 2017;18(2):71–86. Epub 2016/11/22. 10.1038/nrg.2016.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9(5):397–405. Epub 2008/03/28. 10.1038/nrg2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volff JN. Turning junk into gold: domestication of transposable elements and the creation of new genes in eukaryotes. Bioessays. 2006;28(9):913–22. Epub 2006/08/29. 10.1002/bies.20452 . [DOI] [PubMed] [Google Scholar]
- 38.Bohne A, Brunet F, Galiana-Arnoux D, Schultheis C, Volff JN. Transposable elements as drivers of genomic and biological diversity in vertebrates. Chromosome Res. 2008;16(1):203–15. Epub 2008/02/23. 10.1007/s10577-007-1202-6 . [DOI] [PubMed] [Google Scholar]
- 39.Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016;351(6277):1083–7. Epub 2016/03/05. 10.1126/science.aad5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lynch VJ, Nnamani MC, Kapusta A, Brayer K, Plaza SL, Mazur EC, et al. Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep. 2015;10(4):551–61. Epub 2015/02/03. 10.1016/j.celrep.2014.12.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunn-Fletcher CE, Muglia LM, Pavlicev M, Wolf G, Sun MA, Hu YC, et al. Anthropoid primate-specific retroviral element THE1B controls expression of CRH in placenta and alters gestation length. PLoS Biol. 2018;16(9):e2006337 Epub 2018/09/20. 10.1371/journal.pbio.2006337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pontis J, Planet E, Offner S, Turelli P, Duc J, Coudray A, et al. Hominoid-Specific Transposable Elements and KZFPs Facilitate Human Embryonic Genome Activation and Control Transcription in Naive Human ESCs. Cell Stem Cell. 2019;24(5):724–35 e5. Epub 2019/04/23. 10.1016/j.stem.2019.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuentes DR, Swigut T, Wysocka J. Systematic perturbation of retroviral LTRs reveals widespread long-range effects on human gene regulation. Elife. 2018;7 Epub 2018/08/03. 10.7554/eLife.35989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellison C, Bachtrog D. Contingency in the convergent evolution of a regulatory network: Dosage compensation in Drosophila. PLoS Biol. 2019;17(2):e3000094 Epub 2019/02/12. 10.1371/journal.pbio.3000094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellison CE, Bachtrog D. Dosage compensation via transposable element mediated rewiring of a regulatory network. Science. 2013;342(6160):846–50. Epub 2013/11/16. 10.1126/science.1239552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Notwell JH, Chung T, Heavner W, Bejerano G. A family of transposable elements co-opted into developmental enhancers in the mouse neocortex. Nat Commun. 2015;6:6644 Epub 2015/03/26. 10.1038/ncomms7644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joly-Lopez Z, Bureau TE. Exaptation of transposable element coding sequences. Curr Opin Genet Dev. 2018;49:34–42. Epub 2018/03/12. 10.1016/j.gde.2018.02.011 . [DOI] [PubMed] [Google Scholar]
- 48.Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013;9(4):e1003470 Epub 2013/05/03. 10.1371/journal.pgen.1003470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joly-Lopez Z, Hoen DR, Blanchette M, Bureau TE. Phylogenetic and Genomic Analyses Resolve the Origin of Important Plant Genes Derived from Transposable Elements. Mol Biol Evol. 2016;33(8):1937–56. Epub 2016/05/18. 10.1093/molbev/msw067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chueh AC, Northrop EL, Brettingham-Moore KH, Choo KH, Wong LH. LINE retrotransposon RNA is an essential structural and functional epigenetic component of a core neocentromeric chromatin. PLoS Genet. 2009;5(1):e1000354 Epub 2009/01/31. 10.1371/journal.pgen.1000354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klein SJ, O’Neill RJ. Transposable elements: genome innovation, chromosome diversity, and centromere conflict. Chromosome Res. 2018;26(1–2):5–23. Epub 2018/01/15. 10.1007/s10577-017-9569-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang CH, Chavan A, Palladino J, Wei X, Martins NMC, Santinello B, et al. Islands of retroelements are major components of Drosophila centromeres. PLoS Biol. 2019;17(5):e3000241 Epub 2019/05/16. 10.1371/journal.pbio.3000241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levis RW, Ganesan R, Houtchens K, Tolar LA, Sheen FM. Transposons in place of telomeric repeats at a Drosophila telomere. Cell. 1993;75(6):1083–93. Epub 1993/12/17. 10.1016/0092-8674(93)90318-k . [DOI] [PubMed] [Google Scholar]
- 54.Traverse KL, Pardue ML. A spontaneously opened ring chromosome of Drosophila melanogaster has acquired He-T DNA sequences at both new telomeres. Proc Natl Acad Sci U S A. 1988;85(21):8116–20. Epub 1988/11/01. 10.1073/pnas.85.21.8116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biessmann H, Valgeirsdottir K, Lofsky A, Chin C, Ginther B, Levis RW, et al. HeT-A, a transposable element specifically involved in "healing" broken chromosome ends in Drosophila melanogaster. Mol Cell Biol. 1992;12(9):3910–8. Epub 1992/09/01. 10.1128/mcb.12.9.3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abad JP, De Pablos B, Osoegawa K, De Jong PJ, Martin-Gallardo A, Villasante A. TAHRE, a novel telomeric retrotransposon from Drosophila melanogaster, reveals the origin of Drosophila telomeres. Mol Biol Evol. 2004;21(9):1620–4. Epub 2004/06/04. 10.1093/molbev/msh180 . [DOI] [PubMed] [Google Scholar]
- 57.Sheen FM, Levis RW. Transposition of the LINE-like retrotransposon TART to Drosophila chromosome termini. Proc Natl Acad Sci U S A. 1994;91(26):12510–4. Epub 1994/12/20. 10.1073/pnas.91.26.12510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malik HS, Burke WD, Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol. 1999;16(6):793–805. Epub 1999/06/16. 10.1093/oxfordjournals.molbev.a026164 . [DOI] [PubMed] [Google Scholar]
- 59.Villasante A, Abad JP, Planello R, Mendez-Lago M, Celniker SE, de Pablos B. Drosophila telomeric retrotransposons derived from an ancestral element that was recruited to replace telomerase. Genome Res. 2007;17(12):1909–18. Epub 2007/11/09. 10.1101/gr.6365107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Biessmann H, Mason JM. Telomere maintenance without telomerase. Chromosoma. 1997;106(2):63–9. Epub 1997/07/01. 10.1007/s004120050225 . [DOI] [PubMed] [Google Scholar]
- 61.Savitsky M, Kravchuk O, Melnikova L, Georgiev P. Heterochromatin protein 1 is involved in control of telomere elongation in Drosophila melanogaster. Mol Cell Biol. 2002;22(9):3204–18. Epub 2002/04/10. 10.1128/mcb.22.9.3204-3218.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Savitsky M, Kwon D, Georgiev P, Kalmykova A, Gvozdev V. Telomere elongation is under the control of the RNAi-based mechanism in the Drosophila germline. Genes Dev. 2006;20(3):345–54. Epub 2006/02/03. 10.1101/gad.370206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khurana JS, Xu J, Weng Z, Theurkauf WE. Distinct functions for the Drosophila piRNA pathway in genome maintenance and telomere protection. PLoS Genet. 2010;6(12):e1001246 Epub 2010/12/24. 10.1371/journal.pgen.1001246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shpiz S, Kalmykova A. Role of piRNAs in the Drosophila telomere homeostasis. Mob Genet Elements. 2011;1(4):274–8. Epub 2012/05/01. 10.4161/mge.18301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shpiz S, Olovnikov I, Sergeeva A, Lavrov S, Abramov Y, Savitsky M, et al. Mechanism of the piRNA-mediated silencing of Drosophila telomeric retrotransposons. Nucleic Acids Res. 2011;39(20):8703–11. Epub 2011/07/19. 10.1093/nar/gkr552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee YC, Leek C, Levine MT. Recurrent Innovation at Genes Required for Telomere Integrity in Drosophila. Mol Biol Evol. 2017;34(2):467–82. Epub 2016/11/12. 10.1093/molbev/msw248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saint-Leandre B, Nguyen SC, Levine MT. Diversification and collapse of a telomere elongation mechanism. Genome Res. 2019;29(6):920–31. Epub 2019/05/30. 10.1101/gr.245001.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wei KH, Reddy HM, Rathnam C, Lee J, Lin D, Ji S, et al. A Pooled Sequencing Approach Identifies a Candidate Meiotic Driver in Drosophila. Genetics. 2017;206(1):451–65. Epub 2017/03/05. 10.1534/genetics.116.197335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Casacuberta E, Pardue ML. Coevolution of the telomeric retrotransposons across Drosophila species. Genetics. 2002;161(3):1113–24. Epub 2002/07/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Danilevskaya ON, Tan C, Wong J, Alibhai M, Pardue ML. Unusual features of the Drosophila melanogaster telomere transposable element HeT-A are conserved in Drosophila yakuba telomere elements. Proc Natl Acad Sci U S A. 1998;95(7):3770–5. Epub 1998/05/09. 10.1073/pnas.95.7.3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sackton TB, Kulathinal RJ, Bergman CM, Quinlan AR, Dopman EB, Carneiro M, et al. Population genomic inferences from sparse high-throughput sequencing of two populations of Drosophila melanogaster. Genome Biol Evol. 2009;1:449–65. Epub 2009/01/01. 10.1093/gbe/evp048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. Epub 1990/10/05. 10.1016/S0022-2836(05)80360-2 . [DOI] [PubMed] [Google Scholar]
- 73.Abad JP, De Pablos B, Osoegawa K, De Jong PJ, Martin-Gallardo A, Villasante A. Genomic analysis of Drosophila melanogaster telomeres: full-length copies of HeT-A and TART elements at telomeres. Mol Biol Evol. 2004;21(9):1613–9. Epub 2004/05/28. 10.1093/molbev/msh174 . [DOI] [PubMed] [Google Scholar]
- 74.George JA, Traverse KL, DeBaryshe PG, Kelley KJ, Pardue ML. Evolution of diverse mechanisms for protecting chromosome ends by Drosophila TART telomere retrotransposons. Proc Natl Acad Sci U S A. 2010;107(49):21052–7. Epub 2010/11/20. 10.1073/pnas.1015926107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Obbard DJ, Maclennan J, Kim KW, Rambaut A, O’Grady PM, Jiggins FM. Estimating divergence dates and substitution rates in the Drosophila phylogeny. Mol Biol Evol. 2012;29(11):3459–73. Epub 2012/06/12. 10.1093/molbev/mss150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moran JV, DeBerardinis RJ, Kazazian HH Jr. Exon shuffling by L1 retrotransposition. Science. 1999;283(5407):1530–4. Epub 1999/03/05. 10.1126/science.283.5407.1530 . [DOI] [PubMed] [Google Scholar]
- 77.Pickeral OK, Makalowski W, Boguski MS, Boeke JD. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 2000;10(4):411–5. Epub 2000/04/26. 10.1101/gr.10.4.411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chakraborty M, Emerson JJ, Macdonald SJ, Long AD. Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits. Nat Commun. 2019;10(1):4872 Epub 2019/10/28. 10.1038/s41467-019-12884-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ellison CE, Cao W. Nanopore sequencing and Hi-C scaffolding provide insight into the evolutionary dynamics of transposable elements and piRNA production in wild strains of Drosophila melanogaster. Nucleic Acids Res. 2020;48(1):290–303. Epub 2019/11/23. 10.1093/nar/gkz1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang W, Massouras A, Inoue Y, Peiffer J, Ramia M, Tarone AM, et al. Natural variation in genome architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res. 2014;24(7):1193–208. Epub 2014/04/10. 10.1101/gr.171546.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mackay TF, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, et al. The Drosophila melanogaster Genetic Reference Panel. Nature. 2012;482(7384):173–8. Epub 2012/02/10. 10.1038/nature10811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Herold A, Suyama M, Rodrigues JP, Braun IC, Kutay U, Carmo-Fonseca M, et al. TAP (NXF1) belongs to a multigene family of putative RNA export factors with a conserved modular architecture. Mol Cell Biol. 2000;20(23):8996–9008. Epub 2000/11/14. 10.1128/mcb.20.23.8996-9008.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Van Doren M, Williamson AL, Lehmann R. Regulation of zygotic gene expression in Drosophila primordial germ cells. Curr Biol. 1998;8(4):243–6. Epub 1998/03/21. 10.1016/s0960-9822(98)70091-0 . [DOI] [PubMed] [Google Scholar]
- 84.Shpiz S, Kwon D, Uneva A, Kim M, Klenov M, Rozovsky Y, et al. Characterization of Drosophila telomeric retroelement TAHRE: transcription, transpositions, and RNAi-based regulation of expression. Mol Biol Evol. 2007;24(11):2535–45. Epub 2007/09/25. 10.1093/molbev/msm205 . [DOI] [PubMed] [Google Scholar]
- 85.Song J, Liu J, Schnakenberg SL, Ha H, Xing J, Chen KC. Variation in piRNA and transposable element content in strains of Drosophila melanogaster. Genome Biol Evol. 2014;6(10):2786–98. Epub 2014/10/01. 10.1093/gbe/evu217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hur JK, Luo Y, Moon S, Ninova M, Marinov GK, Chung YD, et al. Splicing-independent loading of TREX on nascent RNA is required for efficient expression of dual-strand piRNA clusters in Drosophila. Genes Dev. 2016;30(7):840–55. Epub 2016/04/03. 10.1101/gad.276030.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mohn F, Sienski G, Handler D, Brennecke J. The rhino-deadlock-cutoff complex licenses noncanonical transcription of dual-strand piRNA clusters in Drosophila. Cell. 2014;157(6):1364–79. Epub 2014/06/07. 10.1016/j.cell.2014.04.031 . [DOI] [PubMed] [Google Scholar]
- 88.Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol. 2008;18(10):758–62. Epub 2008/05/13. 10.1016/j.cub.2008.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang W, Yoshikawa M, Han BW, Izumi N, Tomari Y, Weng Z, et al. The initial uridine of primary piRNAs does not create the tenth adenine that Is the hallmark of secondary piRNAs. Mol Cell. 2014;56(5):708–16. Epub 2014/12/03. 10.1016/j.molcel.2014.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Handler D, Meixner K, Pizka M, Lauss K, Schmied C, Gruber FS, et al. The genetic makeup of the Drosophila piRNA pathway. Mol Cell. 2013;50(5):762–77. Epub 2013/05/15. 10.1016/j.molcel.2013.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walter MF, Biessmann MR, Benitez C, Torok T, Mason JM, Biessmann H. Effects of telomere length in Drosophila melanogaster on life span, fecundity, and fertility. Chromosoma. 2007;116(1):41–51. Epub 2006/11/08. 10.1007/s00412-006-0081-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lerat E, Fablet M, Modolo L, Lopez-Maestre H, Vieira C. TEtools facilitates big data expression analysis of transposable elements and reveals an antagonism between their activity and that of piRNA genes. Nucleic Acids Res. 2017;45(4):e17 Epub 2017/02/17. 10.1093/nar/gkw953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Signor SA, New FN, Nuzhdin S. A Large Panel of Drosophila simulans Reveals an Abundance of Common Variants. Genome Biol Evol. 2018;10(1):189–206. Epub 2017/12/12. 10.1093/gbe/evx262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tajima F. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics. 1993;135(2):599–607. Epub 1993/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goodier JL, Ostertag EM, Kazazian HH Jr. Transduction of 3’-flanking sequences is common in L1 retrotransposition. Hum Mol Genet. 2000;9(4):653–7. Epub 2000/03/04. 10.1093/hmg/9.4.653 . [DOI] [PubMed] [Google Scholar]
- 96.Rahman R, Chirn GW, Kanodia A, Sytnikova YA, Brembs B, Bergman CM, et al. Unique transposon landscapes are pervasive across Drosophila melanogaster genomes. Nucleic Acids Res. 2015;43(22):10655–72. Epub 2015/11/19. 10.1093/nar/gkv1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H, et al. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell. 2009;137(3):509–21. Epub 2009/04/28. 10.1016/j.cell.2009.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, et al. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell. 2009;137(3):522–35. Epub 2009/04/28. 10.1016/j.cell.2009.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Radion E, Morgunova V, Ryazansky S, Akulenko N, Lavrov S, Abramov Y, et al. Key role of piRNAs in telomeric chromatin maintenance and telomere nuclear positioning in Drosophila germline. Epigenetics Chromatin. 2018;11(1):40 Epub 2018/07/13. 10.1186/s13072-018-0210-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rouget C, Papin C, Boureux A, Meunier AC, Franco B, Robine N, et al. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature. 2010;467(7319):1128–32. Epub 2010/10/19. 10.1038/nature09465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barckmann B, Pierson S, Dufourt J, Papin C, Armenise C, Port F, et al. Aubergine iCLIP Reveals piRNA-Dependent Decay of mRNAs Involved in Germ Cell Development in the Early Embryo. Cell Rep. 2015;12(7):1205–16. Epub 2015/08/11. 10.1016/j.celrep.2015.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16(9):418–20. Epub 2000/09/06. 10.1016/s0168-9525(00)02093-x . [DOI] [PubMed] [Google Scholar]
- 103.Thurmond J, Goodman JL, Strelets VB, Attrill H, Gramates LS, Marygold SJ, et al. FlyBase 2.0: the next generation. Nucleic Acids Res. 2019;47(D1):D759–D65. Epub 2018/10/27. 10.1093/nar/gky1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Berloco M, Fanti L, Sheen F, Levis RW, Pimpinelli S. Heterochromatic distribution of HeT-A- and TART-like sequences in several Drosophila species. Cytogenet Genome Res. 2005;110(1–4):124–33. Epub 2005/08/12. 10.1159/000084944 . [DOI] [PubMed] [Google Scholar]
- 105.Wintersinger JA, Wasmuth JD. Kablammo: an interactive, web-based BLAST results visualizer. Bioinformatics. 2015;31(8):1305–6. Epub 2014/12/07. 10.1093/bioinformatics/btu808 . [DOI] [PubMed] [Google Scholar]
- 106.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12 Epub 2004/02/05. 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hoskins RA, Carlson JW, Wan KH, Park S, Mendez I, Galle SE, et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 2015;25(3):445–58. Epub 2015/01/16. 10.1101/gr.185579.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. Epub 2013/01/19. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Loytynoja A. Phylogeny-aware alignment with PRANK. Methods Mol Biol. 2014;1079:155–70. Epub 2013/10/31. 10.1007/978-1-62703-646-7_10 . [DOI] [PubMed] [Google Scholar]
- 110.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–91. Epub 2009/01/20. 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Papadopoulos JS, Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007;23(9):1073–9. Epub 2007/03/03. 10.1093/bioinformatics/btm076 . [DOI] [PubMed] [Google Scholar]
- 112.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018;35(6):1547–9. Epub 2018/05/04. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. Epub 2014/01/24. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bushnell B, Rood J, Singer E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE. 2017;12(10):e0185056 Epub 2017/10/27. 10.1371/journal.pone.0185056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60. Epub 2015/03/10. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. Epub 2012/03/06. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10(1):71–3. Epub 2012/11/20. 10.1038/nmeth.2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011;17(1):3 Epub 2011-08-02. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 119.Langmead B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. 2010;Chapter 11:Unit 11 7 Epub 2010/12/15. 10.1002/0471250953.bi1107s32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Axtell MJ. ShortStack: comprehensive annotation and quantification of small RNA genes. RNA. 2013;19(6):740–51. Epub 2013/04/24. 10.1261/rna.035279.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Johnson NR, Yeoh JM, Coruh C, Axtell MJ. Improved Placement of Multi-mapping Small RNAs. G3 (Bethesda). 2016;6(7):2103–11. Epub 2016/05/14. 10.1534/g3.116.030452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. Epub 2010/01/30. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Han BW, Wang W, Li C, Weng Z, Zamore PD. Noncoding RNA. piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science. 2015;348(6236):817–21. Epub 2015/05/16. 10.1126/science.aaa1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550 Epub 2014/12/18. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huang W, Carbone MA, Magwire MM, Peiffer JA, Lyman RF, Stone EA, et al. Genetic basis of transcriptome diversity in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2015;112(44):E6010–9. Epub 2015/10/21. 10.1073/pnas.1519159112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42(Web Server issue):W187–91. Epub 2014/05/07. 10.1093/nar/gku365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. Epub 2009/06/10. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Miller DE, Staber C, Zeitlinger J, Hawley RS. Highly Contiguous Genome Assemblies of 15 Drosophila Species Generated Using Nanopore Sequencing. G3 (Bethesda). 2018;8(10):3131–41. Epub 2018/08/09. 10.1534/g3.118.200160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang Y, Parmigiani G, Johnson WE. ComBat-Seq: batch effect adjustment for RNA-Seq count data. bioRxiv. 2020:2020.01.13.904730. 10.1093/nargab/lqaa078 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
We used NCBI web BLAST to search the D. melanogaster Nxf2 peptide sequence against the RefSeq peptide database and identified homologs in 22 Drosophila species. The carboxyl-terminal region of Nxf2 derives from CDS which shares homology with the TART-A TE (gray box). At the peptide level, this region is conserved out to D. virilis, which suggests that, if it was acquired from an insertion of the TART-A TE, the insertion would have occurred in the common ancestor of the entire genus. CDS, coding sequence; TE, transposable element.
(TIFF)
The pink boxes show the 2 segments of shared homology between D. melanogaster TART-A and D. melanogaster nxf2. D. yakuba TART-A aligns to D. melanogaster TART-A at regions directly adjacent to, but not including, the TART-A/nxf2 shared homology. Underlying data can be found in S2 Data.
(TIFF)
We compared nxf2 transcript sequences from D. melanogaster (A), D. yakuba (B), and D. sechellia (C) to TART-A sequences from the same species using mummer [106]. There is sequence homology present between D. melanogaster nxf2 and TART-A but not for D. yakuba nxf2/TART-A nor for D. sechellia nxf2/TART-A. Underlying data can be found in S2 Data.
(TIFF)
We identified 71 TART-A elements with 3′ UTRs from 17 long-read D. melanogaster genome assemblies. All 71 elements contain the nxf2-like sequence (gray box) suggesting that this region is present in most, if not all, TART-A elements in D. melanogaster. Note that a portion of the nxf2-like region appears to have been deleted in one of the TART-A elements.
(TIFF)
We compared genomic sequencing coverage for the nxf2-like region of TART-A (blue shading) to its upstream and downstream flanking regions (yellow shading). For each DGRP strain, we divided read coverage by the median coverage of that strain’s TART-A ORF1 and ORF2 to control for copy number differences between strains. We calculated coverage for each strain in 10-bp windows across the region. Each box in the figure summarizes the per-strain coverage values for a single 10-bp segment. Within each box, the internal line represents the median coverage and the hinges correspond to the 25th and 75th percentiles. The whiskers extend to 1.5× the interquartile range. The coverage of the nxf2-like region is similar to the coverage of the downstream region, both of which are reduced relative to the upstream region. This pattern is consistent with truncation of the UTR, which has previously been described for TART [74]. Because the nxf2-like sequence is present in both UTRs, truncation of the 5′ UTR, which is fairly common, should reduce coverage of both the nxf2-like region and downstream flanking region by approximately 50% compared to the upstream region, which is not present in the 5′ UTR (Fig 1B). We observed a reduction in coverage of approximately 30%, consistent with a mixture of TART-A copies, some with truncated 5′ UTRs and some without. The median coverage across all boxes within a region is shown by the colored horizontal bars. Underlying data can be found in S2 Data. ORF, open reading frame.
(PDF)
Each RepBase repeat for which we observed expression in total RNA-seq data from female ovaries is shown on the y-axis, and the fold change in expression in the nxf2 RNAi knockdown versus a control knockdown of the white gene is shown on the x-axis with a log2 scale. Expression values are the mean of 2 biological replicates for both knockdown and control. For LTR retrotransposons, LTRs are shown separately from the rest of the TE. Underlying data can be found in S2 Data. LTR, long terminal repeat; TE, transposable element.
(PDF)
We used 2 shRNAs that target different regions of the nxf2 transcript and calculated expression values for genes as well as TEs for each knockdown. We found that the expression values are highly correlated between the 2 experiments (Spearman’s rho = 0.92 [Genes] and 0.94 [TEs]). Underlying data can be found in S2 Data. shRNA, short hairpin RNA; TE, transposable element.
(PDF)
We analyzed published degradome-seq and Aub-immunoprecipitated small RNA data to determine whether there were nxf2 degradome-seq reads showing the 10-bp sense:antisense overlap with TART-A piRNAs, consistent with cleavage by a Piwi protein. We identified 11 locations (A–K) within the TART-like region of nxf2 where degradome-seq cleavage products (red) overlap with antisense piRNAs (blue) by 10 bp at their 5′ ends. The nxf2 transcript is shown in black. degradome-seq, degradome sequencing; piRNA, Piwi-interacting small RNA.
(TIFF)
We identified 168 genes whose fold change in expression was greater than or equal to nxf2 across RNAi knockdowns of 16 piRNA pathway components. These genes have a significantly larger abundance of aligned piRNAs compared to the remainder of expressed genes, suggesting their expression may be regulated by piRNAs (Wilcoxon test P = 4.1e-06). Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA; RNAi, RNA interference.
(PDF)
We examined the fold change in expression of 41 known piRNA pathway genes across RNAi knockdowns of 16 piRNA pathway components, excluding the targeted gene from analysis for each experiment. PiRNA pathway genes show a median fold change near 1 (horizontal red line) for most experiments. Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA; RNAi, RNA interference.
(PDF)
We repeated the analysis shown in Fig 7A using a replicate microarray dataset from [125] and found a similar correlation (Spearman’s rho = −0.49), which suggests that the microarray expression measurements are highly reproducible. Underlying data can be found in S2 Data.
(PDF)
We were able to obtain expression values for 39 other piRNA pathway genes from the same microarray dataset that we used for nxf2 expression. For each of these genes, we calculated Spearman correlation coefficient for its expression compared to TART-A copy number. All correlation coefficients are at least 2-fold smaller in magnitude than what we observed for nxf2. Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA.
(TIFF)
The histogram summarizes the Spearman correlation coefficients between 39 piRNA pathway genes and TART-A copy number (shown in S12 Fig). The red line shows the correlation coefficient for nxf2. Underlying data can be found in S2 Data. piRNA, Piwi-interacting small RNA.
(PDF)
We plotted piRNA read depth (normalized as RPM mapped) along the nxf2 transcript for each of the 16 DGRP strains shown in Fig 7. For each strain, the abundance of TART piRNAs is listed in the plot title. We masked the locations of the TART/nxf2 shared homology (gray boxes) before alignment to avoid cross-mapping of TART-derived piRNAs. Underlying data can be found in S2 Data. DGRP, Drosophila Genetic Reference Panel; piRNA, Piwi-interacting small RNA; RPM, reads per million.
(PDF)
There is a strong positive correlation between TART-derived piRNAs that align to nxf2 versus the nxf2 piRNAs downstream from the region of shared homology, across 16 DGRP strains (Spearman’s rho = 0.88, P < 2.2e-16). Underlying data can be found in S2 Data. DGRP, Drosophila Genetic Reference Panel; piRNA, Piwi-interacting small RNA.
(PDF)
We used the microarray dataset from [125] to select 5 DGRP strains whose median nxf2 expression level is similar to that of the full DGRP population. Underlying data can be found in S2 Data. DGRP, Drosophila Genetic Reference Panel; RNA-seq, RNA sequencing.
(TIFF)
(DOCX)
(XLSX)
(XLSX)
(TXT)
(TXT)
Data Availability Statement
The RNA-seq data generated for this study are available from the NCBI Short Read Archive (BioProject number PRJNA606690).