

Abstract
As technology advances, whole genome sequencing (WGS) is likely to supersede other genotyping technologies. The rate of this change depends on its relative cost and utility. Variants identified uniquely through WGS may reveal novel biological pathways underlying complex disorders and provide high-resolution insight into when, where, and in which cell type these pathways are affected. Alternatively, cheaper and less computationally intensive approaches may yield equivalent insights. Understanding the role of rare variants in the noncoding gene-regulating genome, through pilot WGS projects, will be critical to determine which of these two extremes best represents reality. With large cohorts, well-defined risk loci, and a compelling need to understand the underlying biology, psychiatric disorders have a role to play in this preliminary WGS assessment. The WGSPD consortium will integrate data for 18,000 individuals with psychiatric disorders, beginning with autism spectrum disorder, schizophrenia, bipolar disorder, and major depressive disorder, along with over 150,000 controls.
Genetic variation is a major contributor to neuropsychiatric disorders. The variants responsible likely include the complete range of sizes, from single nucleotides to large structural variants, and the full spectrum of population frequency, from common variants to rare variants unique to a family or individual. For severe, early onset neuropsychiatric disorders, such as autism spectrum disorder (ASD) and schizophrenia, natural selection limits the population frequency of variants so that variants with larger effect sizes are extremely rare1,2. Over the past decade, genomic technologies have advanced our understanding of neuropsychiatric disorders, yet remaining limitations in technology and cohort sizes have limited progress in identifying inherited rare variants.
Genome-wide association studies (GWAS) using genotyping arrays have detected over 100 regions (loci) at which common genetic variants (population frequency ≥2%), are associated with a psychiatric diagnosis (Table 1). Individually, these variants exert small effects and thus require very large sample sizes for detection (Table 1). Common risk variants can provide a window into the molecular architecture of these disorders. For example, common variants suggest a previously unrecognized role for the complement cascade in schizophrenia3.
Table 1.
The largest genomic studies to date in autism spectrum disorder, schizophrenia, bipolar disorder, and major depression.
| Study design |
Platform | Variant detected |
Disorder | Patients | Controls | Genome- wide hits |
Reference |
|---|---|---|---|---|---|---|---|
| Case-control | Genotyping microarray | SNP (GWAS) | ASD | 16,539 | 157,234 | 1 | Anney et al, Mol Autism, 201764 |
| SCZ | 36,989 | 113,075 | 108 | Ripke et al, Nature 20147 | |||
| BPD | 11,974 | 51,792 | 2 | Sklar et al, Nature Genetics 201139 | |||
| MDD | 121,380 | 338,101 | 15 | Hyde et al, Nature Genetics 201665 | |||
| CNV | SCZ | 21,094 | 20,227 | 8 | Marshall et al, Nature Genetics 201766 | ||
| BPD | 9,129 | 81,802 | 1 | Green et al, Mol Psychiatry 201567 | |||
| MDD | 2,591 | 8,842 | 0 | Rucker et al, Biol Psychiatry 201568 | |||
| Exome sequencing | Rare PTV mutation | ASD | 5,563 | 1,881 | 0 | Sanders et al, Neuron 20156 | |
| SCZ | 2,536 | 2,543 | 0 | Purcell et al, Nature 201469 | |||
| Ultra rare PTV mutation | SCZ | 4,877 | 6,203 | 0 | Genovese et al Nature Neuroscience 201670 | ||
| Family-based | Genotyping microarray | CNV | ASD | 4,687 | 2,100 | 8 | Sanders et al, Neuron 20156 |
| Exome sequencing | De novo PTV mutation | ASD | 5,563 | 1,881 | 65 | Sanders et al, Neuron 20156 | |
| SCZ | 617 | 731 | 0 | Fromer et al, Nature 201471 | |||
| Meta-analysis | Exome sequencing | Rare and de novo PTV mutations | SCZ | 7,776 | 13,028 | 1 | Singh et al, Nature Neuroscience 201672 |
SNP, single nucleotide polymorphism; CNV, copy number variant; PTV, protein-truncating variant. ASD, autism spectrum disorder; SCZ, schizophrenia; BPD, bipolar disorder; MDD, major depressive disorder.
Exome sequencing, which identifies genetic variants in the ~1% of the genome that encodes proteins, has identified over 50 genes in ASD (Table 1). The majority of this discovery was through de novo protein truncating variants (PTVs) observed in a patient but not in either unaffected parent. Such mutations are very rare, e.g. population frequency ≤0.000002%, but they can have large effect sizes, up to a ~50-fold increase in risk. As with common variation, these very rare variants have advanced our understanding of the etiology of these disorders, for example by implicating chromatin remodeling in ASD4,5.
Although much remains to be discovered, these results have yielded critical starting points for studies of pathogenesis,6,7 and indicate the feasibility and importance of discovering sufficient additional variation to fully delineate the key biological pathways underlying these disorders.
Insights from whole genome sequencing
By assaying most of the genome at single nucleotide resolution, WGS holds the potential to extend rare variant discovery to the ~99% of the genome that is noncoding (Box 1). While GWAS identifies common noncoding variants, the rare noncoding variants assayed by WGS might have substantially higher effect sizes1, increasing tractability for biological experimentation. WGS also enables detection of most structural variation including translocations, inversions, and copy number variants (CNVs)8,9. Furthermore, WGS can improve detection of common variants in existing GWAS by statistically inferring SNPs not directly genotyped (imputation) and identifying the specific risk variants within a risk region (fine mapping). Similarly, WGS data may allow detection of common structural variants, including CNVs, that can be missed by current SNP-based approaches10, facilitating common CNV association studies.
Box 1: Types of genetic variation reliably detected by genomic technologies.
Karyotype (≤1% common; ≤1% rare): Chromosomal aneuploidies, massive structural variation (e.g. translocations, inversions, CNVs of millions of nucleotides), some fragile sites with special protocols.
Microarray (~90% common; ~1% rare): Protein coding and noncoding common SNVs, large rare CNVs (over ~20,000 nucleotides).
Exome sequencing (~1% common; ~1% rare): Protein coding common SNVs and indels, protein coding rare SNVs and indels, some CNVs.
Low coverage WGS (~95% common; ~85% rare): Protein coding and noncoding common SNVs, most protein coding and noncoding rare SNVs.
Deep coverage WGS (~99% common; ~99% rare): Protein coding and noncoding common SNVs and indels, protein coding and noncoding rare SNVs and indels, rare and common CNVs (over ~1,000 nucleotides), multi-allelic CNVs (e.g. over 3 copies), mobile element insertions, other structural variation (e.g. translocations, inversions)
Long-read (>10,000bp), deep coverage WGS (100% common; 100% rare): As for deep coverage WGS plus: small CNVs (50–1,000 nucleotides), complex structural variation, variants in repetitive DNA, direct assessment of phasing (whether two variants are on the same allele)
SNV: Single nucleotide variant
Indel: Insertion/Deletion (gain or loss of ≤50bp)
CNV: Copy number variant (gain or loss of >50bp)
The role of noncoding variation
There is considerable evidence that noncoding variation influences brain function and neuropsychiatric disorders. Over 90% of disease-associated GWAS loci discovered by assaying common variants map to noncoding regions11,12. In humans, at least 4% of the noncoding genome has been under strong purifying selection13. Additionally, epigenomic studies have identified many functional noncoding elements involved in regulation of gene expression underlying neurogenesis, cell differentiation, and neurodevelopment14.
Noncoding variation influences which exons are expressed within a gene, in which cells, and under what circumstances. While such insights can be gained from gene association15, noncoding variation studies should increase the resolution of such analyses by identifying regulatory regions of genes restricted to fewer cell types, developmental periods, or brain regions. Given the multiple biological roles (pleiotropy) of genes implicated in psychiatric disorders, such WGS-derived hypotheses may be critical for biological follow-up.
The role of rare noncoding variation
While common noncoding variation clearly plays a role in neuropsychiatric disorders, the role of rare noncoding variation is less clear. A pessimist could note that in Mendelian disorders few linkage peaks were resolved to noncoding causal variants and that systematic deletion of noncoding regions proximate to the HPRT1 gene (Lesch–Nyhan syndrome) had little impact on protein activity16. In contrast, an optimist could argue that Fragile X, the first psychiatric linkage peak resolved to a gene, is a triplet repeat expansion in the 5` untranslated region (UTR) of the FMRP gene, and that there are several clear examples of Mendelian traits (e.g. OCA2 enhancer in eye color) and disorders (e.g. TBX5 enhancer in congenital heart disease) with penetrant noncoding variants17.
The role and utility of rare variation in the noncoding genome is likely to be a function of the number of noncoding regions that, when mutated, disrupt gene expression or function to a high degree. While this can be estimated in model systems, there will be experimental confounds (e.g. species, cell type, developmental stage) that limit interpretation. Direct analysis of WGS offers a complementary and irreplaceable approach to identify and characterize the role of rare noncoding variants in human disease.
WGS technology is sufficiently novel that we cannot accurately evaluate its potential in neuropsychiatric disorders without generating pilot data in human cohorts. It may implicate novel biological pathways missed by previous genomic efforts and identify disease-associated regulatory elements specific to certain cell types, developmental stages, or brain regions. Alternatively, WGS may prove less efficient than cheaper methods in identifying experimentally actionable disease-associated variation. Optimal allocation of future resources rests on efforts, such as the WGSPD, that seriously test the utility of WGS.
Estimating our ability to find rare noncoding variants
Finding disease-associated loci or variants by WGS will prove more challenging than with GWAS or WES. With WGS there are two orders of magnitude more sites to consider (~3 billion) compared to potential loci in GWAS (~20 million) or variants in WES (~30 million). Furthermore, we cannot predict functional changes, e.g., to transcriptional rate, in the straightforward way we can predict changes to amino acids from coding variation.
To evaluate our power to detect noncoding variants in WGS data, we estimated the power to detect de novo protein truncating variants that contribute to risk in ASD4,18,19 if they were in the noncoding genome. Without any additional information to help us distinguish signal from noise, for every one risk-mediating variant in the WGS data there would be about 25,000 non-risk variants (a ratio of 1:25000, Table S2). By only considering variants with some evidence of functional effect (e.g. conservation) or proximity to a gene with genome-wide significant association to ASD, we would expect to reduce the noise of non-risk variants, making the risk-mediating variant signal easier to detect. We considered a range of annotation scenarios, from an optimistic 1:5 to a pessimistic 1:500 (see Table S2). Moreover, we do not know what penetrance to expect for these noncoding variants so we considered a wide range, shown as relative risk. For context, the highest relative risks for common variants and de novo mutations in psychiatric disorders are about 1.3 and 50, respectively.
We first considered our ability to detect an overall excess of noncoding variants between cases and controls (a burden analysis). Such an analysis could identify a class of variants that mediate risk in psychiatric disorders, for example promoters in proximity to ASD-associated genes, providing insight into regions of the noncoding genome most likely to yield specific risk variants for neuropsychiatric disorders. Since there is no clear category of noncoding variation equivalent to de novo protein truncating variants, we adjusted for testing 1,000 annotation categories. The results for de novo and case-control analyses are shown in Figure 1a and 1b respectively (see Supplemental Methods).
Figure 1. Statistical power in the noncoding genome.
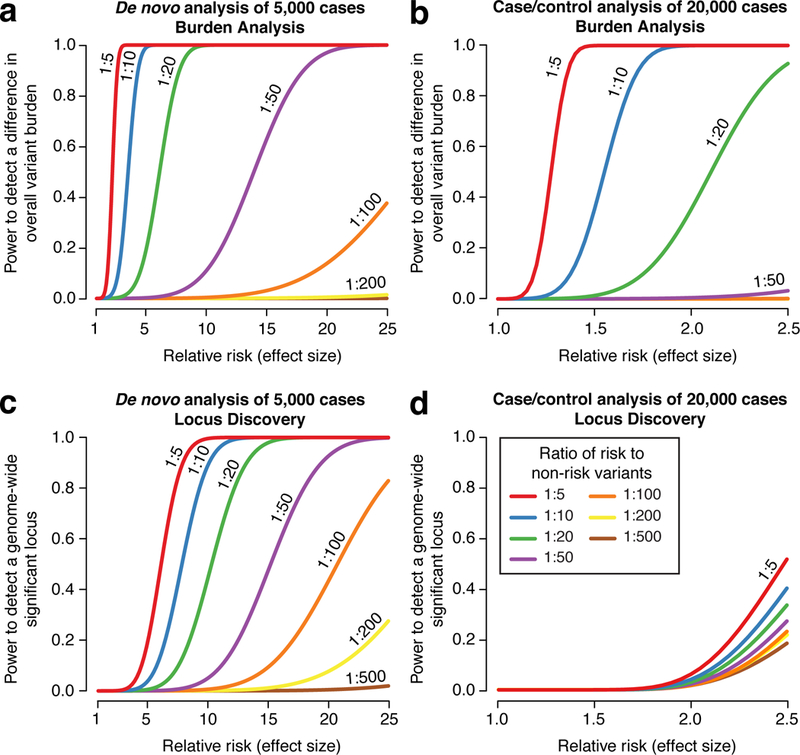
We estimated the power at a significance threshold (alpha) of 5 × 10−5, selected to account for 1,000 categories of noncoding variants, to detect an excess of noncoding variants at 122,500 risk loci in cases vs. controls as we varied the relative risk and risk:non-risk ratio, which represents annotation quality (Table S2). In a) we assessed the power for detecting an excess of de novo mutations in 5,000 cases vs. 5,000 controls as the relative risk increases. With a risk:non-risk ratio of 1:20, approximately equivalent to assessing protein truncating variants in the coding genome, we achieve >80% power with a relative risk of 5. In b) the power to detect an excess burden of rare variants (allele frequency ≤0.1%) is assessed in 20,000 cases vs. 20,000 controls. In c) we assessed the power to identify an excess of de novo mutations at a specific genomic locus, e.g. the noncoding region regulating a single gene. Consequently, we set the significance threshold (alpha) at 2.5×10−6. In d) we assessed the power to identify an excess of rare variants (allele frequency ≤0.1%) at a specific nucleotide (alpha = 1.7×10−11), since this yielded better power than testing for burden at a locus (alpha = 2.5×10−6).
We next considered our ability to identify a specific genetic variant, functional element, or group of functional elements (e.g. enhancers that regulate one gene) associated with risk that could be assessed in larger patient cohorts. The results for de novo and case-control analyses are shown in Figure 1c and 1d respectively (see Supplemental Methods).
From these analyses, it is clear that we will need: 1) large cohorts, and 2) methods to decrease background noise (to obtain a high risk to non-risk ratio), e.g. through predicting functional effects or regulation of known risk loci.
Why perform WGS in psychiatric disorders?
Given current uncertainty over the utility of WGS, we could wait until WGS for non-psychiatric phenotypes provide sufficient insight to enable better power analyses. However, even large case-control cohorts may not be informative of the utility of WGS in ASD, for which de novo mutations have provided a more efficient approach to identifying specific genes and genetic loci6,20 (Figure 1). Additionally, there is a pressing need to identify specific cell types, tissues, and developmental stages involved in brain-based disorders due to the complexity of the nervous system, limited understanding of how molecular changes lead to disorder, and difficulty in interpreting model systems. In short, the potential benefits of WGS in psychiatric disorders may be greater than in other phenotypes and the availability of family-based cohorts may offer insights otherwise unobtainable.
Implications for neuroscientists
Interpreting the biology downstream of variants identified by existing WES and GWAS analyses remains a challenge; this is especially true in neuroscience due to the inaccessibility and complexity of neural tissue.
The interface of human genetics and neuroscience has typically focused on rare, highly penetrant variants that permit generation of transgenic animals with a robust phenotype5,21–24. Neuroscientists now face the challenge of obtaining biological insights through investigation of the multiple weakly penetrant variants, identified through modern genomics, that act through unknown neurological mechanisms, in a manner highly dependent on genetic background25. Noncoding variants will pose yet harder challenges. Their effect sizes are likely to be small, and the relevant biology likely to be restricted to specific cell types, developmental stages, or cell states. Analysis of 3D chromatin structure must often be performed to identify the genes that a noncoding variant regulates. Finally, a proportion of noncoding variants may have human-specific functions absent in model organisms. For example, human accelerated regions (HARs), which are conserved across multiple species but differ within humans, are enriched for homozygous variants in consanguineous ASD cases26.
Notwithstanding such challenges, many variants identified by genomic technologies have strong evidence of association with the disorders, creating a foundation for investigating pathogenesis. Furthermore, the presence of numerous variants allows systems analyses that identify biological convergences5, thus generating mechanistic hypotheses.
Strategies to improve locus discovery in WGS
Sample selection:
As with other genomic technologies, large sample sizes will be key (see Figure 1 and S2); the simplest way to achieve large cohorts will be through case-control studies, see Table 2.
Table 2.
Individuals with WGS data generated by, or accessible to, the WGSPD.
| Data being generated by the WGSPD | ||||
|---|---|---|---|---|
| Project | Disorder | Cases | Controls | Details |
| 1 | Schizophrenia | 3,333 | 1,667 | Case-control analysis; African American ancestry |
| 1 | Bipolar Disorder | 3,333 | 1,667 | Case-control analysis; African American ancestry |
| 2 | ASD | 378 | 1,512 | Simplex families with two parents, affected child, unaffected child |
| 2 | Schizophrenia | 281 | 843 | Families with two parents and one or more affected children |
| 3 | Schizophrenia | 1,000 | 1,400 | Case-control analysis of individuals from Finland |
| 3 | Bipolar Disorder | 1,000 | 500 | Case-control analysis of individuals from Finland |
| 3 | Schizophrenia | 650 | 325 | Case-control analysis of individuals from Netherlands |
| 3 | Bipolar Disorder | 650 | 325 | Case-control analysis of individuals from Netherlands |
| 3 | Bipolar Disorder | 62 | 138 | Multiplex families with affected and unaffected from Colombia |
| 3 | Bipolar Disorder | 83 | 170 | Multiplex families with affected and unaffected from Costa Rica |
| 4 | Schizophrenia | 271 | 280 | Multiplex families with affected and unaffected |
| 4 | Bipolar Disorder | 299 | 309 | Multiplex families with affected and unaffected |
| 4 | Major depression | 476 | 492 | Multiplex families with affected and unaffected |
| Data being generated by other funding mechanisms with consistent analysis pipelines | ||||
| Disorder | Cases | Controls | Details | |
| ASD* | 5,302 | 15,856 | Families with two parents, affected child, +/− unaffected child | |
| ASD* | 150 | 150 | Multiplex families with affected and unaffected | |
| Schizophrenia | 118 | 198 | Multiplex families with affected and unaffected | |
| Bipolar Disorder | 118 | 198 | Multiplex families with affected and unaffected | |
| Major depression | 478 | 804 | Multiplex families with affected and unaffected | |
| TOPMed† | 0 | 68,950 | Heart, lung, blood and sleep disorders | |
| CCDG† | 0 | 63,950 | Heart, vascular, lung, bowel, neurological, and endocrine disorders | |
| Totals | 17,957 | 165,834 | ||
ASD samples are being generated by several groups: Centers for Common Disease Genomics (CCDG) of the National Human Genome Research Institute (NHGRI), Simons Foundation Autism Research Initiative (SFARI)73, Autism Sequencing Consortium (ASC)74.
6,100 samples are shared between Trans-Omics for Precision Medicine (TOPMed) of the National Heart, Lung, and Blood Institute (NHLBI) and CCDG, therefore the total number of samples was reduced by 3,050 for each cohort. These cohorts are composed of individuals ascertained for non-psychiatric disorders and for whom their psychiatric disorder status is generally unknown.
Several recent studies have shown an excess of deleterious variants in isolated populations that have expanded rapidly following recent bottlenecks27–30, including deletions of the TOP3B gene, associated with schizophrenia and intellectual disability29, in ~3% of individuals in Northern Finland compared to 0.05% in other European populations. Large multiplex pedigrees with multiple affected individuals may be enriched for rare, inherited variants with high effect sizes31,32. Simplex pedigrees, with only one affected individual, are enriched for de novo mutations with very high effect sizes given the lack of exposure to natural selection. This strategy has succeeded in severe early-onset disorders, including ID and ASD4,6,19,33. Finally, consanguineous pedigrees may be enriched for homozygous variants that, like de novo mutations, are extremely rare with very high relative risks26,34,35. Homozygous variants may also play a role in non-consanguineous cases (Table S4) and have been found to contribute to risk in some outbred ASD families36,37. Determining which of these sample selection strategies will be most successful will require WGS pilot projects under each strategy.
Integrating phenotypic data:
Broadly, two contrasting approaches have been employed in integrating phenotypes in genomic studies, both with the aim of improving statistical power: 1) Combining clinically- or genetically-related diagnoses to increase sample size; and 2) Subdividing cohorts by shared phenotypes to decrease heterogeneity of the underlying genetics (subtyping or endophenotypes). GWAS data demonstrate substantial common variant sharing across current conventional diagnostic categories, e.g., bipolar disorder and schizophrenia38. Similarly, genes identified by de novo mutations are frequently shared between ASD, intellectual disability, and developmental delay4,19. Thus, combining data from related diagnoses, can increase sample size, hastening variant discovery39.
The alternative approach, dividing by shared phenotypes, was critical for discovery of Mendelian disorders by linkage methods, in which mis-classifying one individual could prevent discovery. However, such an approach is risky for common, non-Mendelian psychiatric disorders given: 1) current lack of insight into relevant subtypes; and 2) reduced sample size. A GWAS based on ~2,500 cases in the Simons Simplex Collection ASD cohort showed no improvement in the proportion of genetic heritability explained by the top SNPs accounting for changes in sample size for over 10 phenotypic characteristics40. In contrast, a GWAS of a nonpsychiatric phenotype, bone mineral density, showed benefits of subgrouping, leading to the identification of 16 new loci41.
Phenotypic subtyping also poses practical challenges. Genetic analysis is comparatively cheap, while deep phenotyping is cumbersome and costly, effectively diminishing sample size. The relative ease of using pre-existing cohorts and registries to inexpensively boost sample size has favored “phenotype-light” sample collection. This balance could be shifted by the adoption of consistent phenotyping schema42,43, identification of reliable neuropsychiatric biomarkers, or utilization of electronic medical records. Several large-scale initiatives are already working in this direction, for example deCODE44, UK biobank45, Geisinger46, and the All of Us Research Program (formerly the Precision Medicine Initiative).
Identifying functional variants:
Our assessment of statistical power (Figure 1) shows that distinguishing variants that are likely to be functional and risk-mediating (i.e. high risk to non-risk ratio) will maximize discovery of specific noncoding variants. Several strategies might help.
Annotating the noncoding genome:
Annotations may predict functional variants, including: 1) Conservation of DNA sequence across species; 2) Regions of open chromatin, where DNA is exposed allowing proteins to bind (detected by DNase-Seq or ATAC-Seq); 3) Regions of active chromatin, where epigenetic marks suggest transcription of a nearby gene (detected by ChIP-Seq); 4) Transcription factor binding sites (detected by ChIP-Seq); and 5) Predicting the regulatory gene target using proximity to the variant (<40% accurate47) or physical interactions with target loci (e.g. ChIA-Pet) or genome-wide (e.g. Hi-C, 5C)47. Of note, many of these annotations may be tissue and developmental stage specific48–51.
Large-scale endeavors such as ENCODE52 and the Roadmap Epigenome Consortium (REC)53 have created a reference for human epigenome annotation. Parallel efforts focused on brain tissue, such as the PsychENCODE Consortium54, will help extend these resources55.
Cataloguing human variation:
Building a database of human variation has proven invaluable in interpreting the coding genome56 and the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org) extends this approach to WGS. Such data can be used to estimate regions of constraint, (with less variation than expected), suggesting functionality57–59.
Regions associated with psychiatric disorders:
GWAS and WES have defined specific regions of the genome that contribute to psychiatric disorders, particularly in ASD4,6,19 and schizophrenia7. It is plausible that noncoding variation in proximity to these regions will be enriched for risk-mediating variants.
Large variants:
On average, large variants, especially deletions, have greater potential to mediate risk than small variants6. However, while large indels and small CNVs may have a greater impact on noncoding function, there are considerably fewer such variants compared to SNVs8. The utility of this strategy will depend on the balance between these two opposing effects.
Functional validation:
Methods have been developed to assess the functional effects of large numbers of potential regulatory regions. These Massively Parallel Reporter Assays (MPRA)60, including Self-Transcribing Active Regulatory Region Sequencing (STARR-Seq)61, assess the function of a regulatory region by its potential to transcribe itself, or a specific sequence of DNA (barcode). Of note, this ability to functionally validate noncoding variants en masse is a major benefit over interpreting coding missense variants, for which protein-specific functional assays are usually required.
The Whole Genome Sequencing Consortium for Psychiatric Disorders (WGSPD)
The potential for WGS to help understand neuropsychiatric disorders, and the absence of insight into the role of rare noncoding variants, prompted the United States National Institute of Mental Health (NIMH) to fund four pilot projects aimed at generating WGS data in neuropsychiatric disorders to provide a more complete understanding of genomic architecture.
Big questions in biology are akin to solving problems of similar complexity in other disciplines such as particle physics or astronomy and require a ‘Team Science’ approach62. Recognizing the need for large samples sizes to make progress (Table 1, Figure 1), the NIMH, the Stanley Center for Psychiatric Research, and researchers at 11 academic institutions across the USA that were funded in the four selected projects, have formed a public-private partnership: the Whole Genome Sequencing Consortium for Psychiatric Disorders (WGSPD). This consortium aims to establish a repository of WGS data, processed in a consistent manner, to facilitate large-scale analyses within and across four psychiatric disorders (Figure 2). This approach can make more efficient use of funding and resources, for example, by using a central data repository, consistent analysis pipelines, and collaborative methods development to help all researchers access and use the data.
Figure 2.
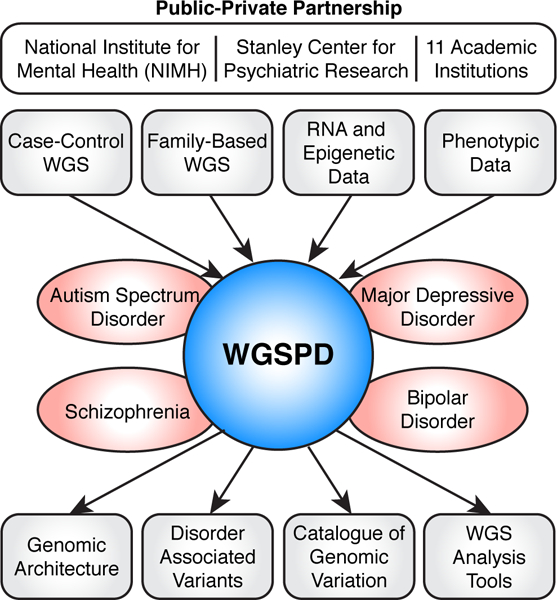
Overview of the WGSPD.
The WGSPD will need to expand, both beyond the founding members and these four disorders. Investigators with relevant WGS data will be invited to join the WGSPD and participate in working groups focused on specific disorders or cross-disorder projects. Given the scale of WGS data, the cost of reprocessing the data in a consistent manner and storing the data will be substantial. Establishing a suitable funding strategy for such genomic integration is a key question that needs to be addressed urgently throughout the genomics community. In a first step to improve this, WGS analysis pipelines have been coordinated across several major sequencing centers and consortia (e.g. CCDG, TOPMed, WGSPD) to allow direct comparison of results. To obtain the sample sizes necessary (Figure 1), a similar consensus will need to be established internationally.
Cloud-based analysis
The sheer scale of WGS datasets necessitates new models for data analysis, since data storage and computation is likely to be beyond the resources at any single institution. Fortunately, the development of cloud-based computing has coincided with the generation of WGS data. Under this model, a single cloud-based data repository can be accessed by teams at each collaborating site, and cloud-based analysis eliminates the need for cumbersome and costly downloads. This approach has the further advantage of facilitating the sharing of preinstalled algorithms and pipelines, encouraging consistent consortium-wide analysis.
The scale of WGS data can make simple analytical tasks overwhelming. Therefore, the WGSPD is committed to developing Application Program Interfaces (APIs) and software solutions for the wider community to simplify cloud-based data access (e.g. hail63). In doing so, computational biologists and analysts can focus on the development and application of methods for analysis, rather than on lower level data management and handling.
The analysis of deidentified genetic data on university-hosted remote servers is common practice, with contributing sites being responsible for securing non-genetic identifying information. So long as cloud environments meet equivalent security standards to existing remote servers, then existing informed consent will cover this use, except in rare instances where the consent specifically excludes this approach. Best practice guidelines for secure sharing of genomic data have been described by the NIH: https://www.ncbi.nlm.nih.gov/projects/gap/pdf/dbgap_2b_security_procedures.pdf. There is an urgent need for methods that allow such guidelines to be easily adopted and readily vetted across cloud providers and institutions.
The WGSPD projects and data
The four WGSPD projects, developed by independent sets of investigators, encompass the diverse strategies for improving locus discovery and therefore will provide some of the earliest opportunities to assess their relative utility in complex disorders. The four projects are:
-
1)
Case-control analysis of schizophrenia and bipolar disorder in individuals of African American ancestry.
-
2)
Family-based analysis of ASD in families with a single affected child, allowing the detection of de novo mutations.
-
3)
Case-control analysis of schizophrenia or bipolar disorder in isolated populations with recent population bottlenecks.
-
4)
Family-based analysis of schizophrenia, bipolar disorder, or major depression in families with multiple affected individuals.
Combining these WGS cohorts with consistently processed WGS data from other consortia will yield an initial dataset of 183,000 individuals, including 18,000 cases and 165,000 controls (Table 2). In addition to the genotype data, we are collating phenotype data that are comparable across projects, disorders, and ages to allow in-depth genotype-phenotype analysis.
Conclusion
The noncoding genome remains largely unexplored and major discoveries undoubtedly await intrepid explorers. Whole genome sequencing of neuropsychiatric cases and controls provides an important avenue in this exploration, potentially offering high resolution insight into the developmental stages, brain regions, cell types, and biological functions that underlie these disorders. If the cost of sequencing continues to fall, it is inevitable that WGS will ultimately replace both microarray and WES – the key question is at what price point this transition offers a good return for investment. Pooling preliminary WGS data between researchers and across disorders offers the most efficient mechanism to make this determination.
The creation of the WGSPD has allowed numerous researchers to pursue diverse scientific approaches on multiple psychiatric disorders, while simultaneously working towards a harmonized data set for integrated analysis. The pooling of expertise, methods, and data will accelerate progress towards understanding genetic contributions to brain development, function, and pathology and create a resource that will continue to yield scientific and clinical insights for years to come.
Supplementary Material
Acknowledgements
The authors would like to acknowledge and thank the study participants and their families. The WGSPD is a public-private partnership between the National Institute of Mental Health (NIMH), the Stanley Center for Psychiatric Research, and researchers at 11 academic institutions across the USA. This work was supported by grants from the National Institute of Mental Health (NIMH): U01 MH105653 (M.B.), U01 MH105641 (S.A.M.), U01 MH105573 (C.N.P.), U01 MH105670 (D.B.G.), U01 MH105575 (M.W.S., A.J.W.), U01 MH105669 (M.J.D., K.E.), U01 MH105575 (N.B.F., D.H.G., R.A.O.), U01 MH105666 (A.P.), U01 MH105630 (D.C.G.), U01 MH105632 (J.B.), U01 MH105634 (R.E.G), U01 MH100239-03S1 (M.W.S., S.J.S., A.J.W.), R01 MH095454 (N.B.F.), the Simons Foundation: (SFARI #385110, M.W.S., S.J.S., A.J.W., D.B.G., SFARI #401457 (D.H.G)), and a gift from the Stanley Foundation (S.E.H.).
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Owen MJ, Sawa A & Mortensen PB Schizophrenia. Lancet 86–97 (2016). doi: 10.1016/S0140-6736(15)01121-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Power RA et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA psychiatry 70, 22–30 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Sekar A et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Rubeis S et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders SJ First glimpses of the neurobiology of autism spectrum disorder. Curr. Opin. Genet. Dev. (2015). doi: 10.1016/j.gde.2015.10.002 [DOI] [PubMed] [Google Scholar]
- 6.Sanders SJ et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandler WM et al. Frequency and Complexity of De Novo Structural Mutation in Autism. Am. J. Hum. Genet. 1–13 (2016). doi: 10.1016/j.ajhg.2016.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins RL et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. 1–21 (2017). doi: 10.1186/s13059-017-1158-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang C et al. The impact of structural variation on human gene expression. Nat. Genet. 49, 55962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hindorff L a et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U. S. A. 106, 9362–7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maurano MT et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siepel A et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Visel A et al. A high-resolution enhancer atlas of the developing telencephalon. Cell 152, 895–908 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willsey AJ et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasperini M et al. CRISPR / Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am. J. Hum. Genet. 101, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scacheri CA & Scacheri PC Mutations in the non-coding genome. Curr Opin Pediatr 27, 659–664 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanders SJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McRae JF et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature (2017). doi: 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katz DM et al. Rett Syndrome: Crossing the Threshold to Clinical Translation. Trends Neurosci. 39, 100–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berrios J et al. Loss of UBE3A from TH-expressing neurons suppresses GABA co-release and enhances VTA-NAc optical self-stimulation. Nat. Commun. 7, 10702 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erickson CA et al. STX209 (Arbaclofen) for autism spectrum disorders: An 8-week open-label study. J. Autism Dev. Disord. 44, 958–964 (2014). [DOI] [PubMed] [Google Scholar]
- 24.de la Torre-Ubieta L, Won H, Stein JL & Geschwind DH Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 22, 345–61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sittig LJ et al. Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron 91, 1253–1259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doan RN et al. Mutations in Human Accelerated Regions Disrupt Cognition and Social Behavior. Cell 167, 341–354.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim ET et al. Distribution and Medical Impact of Loss-of-Function Variants in the Finnish Founder Population. PLoS Genet. 10, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Service SK et al. Re-sequencing Expands Our Understanding of the Phenotypic Impact of Variants at GWAS Loci. PLoS Genet. 10, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoll G et al. Deletion of TOP3β, a component of FMRP-containing mRNPs, contributes to neurodevelopmental disorders. Nat. Neurosci. 16, 1228–37 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gudbjartsson DF et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Cirulli ET & Goldstein DB Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 11, 415–425 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Leppa VM et al. Rare Inherited and De Novo CNVs Reveal Complex Contributions to ASD Risk in Multiplex Families. Am. J. Hum. Genet. 1–15 (2016). doi: 10.1016/j.ajhg.2016.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laumonnier F et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novarino G et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 338, 394–397 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gamsiz ED et al. Intellectual disability is associated with increased runs of homozygosity in simplex autism. Am. J. Hum. Genet. 93, 103–109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim ETT et al. Rare Complete Knockouts in Humans: Population Distribution and Significant Role in Autism Spectrum Disorders. Neuron 77, 235–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu TWW et al. Using Whole-Exome Sequencing to Identify Inherited Causes of Autism. Neuron 77, 259–273 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.PGC-Cross-disorder et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 45, 984–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Psychiatric GWAS Consortium Bipolar Disorder Working Group et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43, 977–983 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chaste P et al. A Genome-wide Association Study of Autism Using the Simons Simplex Collection: Does Reducing Phenotypic Heterogeneity in Autism Increase Genetic Homogeneity? Biol. Psychiatry 77, 775–784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saint-Pierre A et al. Bivariate association analysis in selected samples: application to a GWAS of two bone mineral density phenotypes in males with high or low BMD. Eur J Hum Genet 19, 710–716 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Köhler S et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res. 45, gkw1039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Insel T et al. Research Domain Criteria (RDoC): Toward a new classification framework for research on mental disorders. Am. J. Psychiatry 167, 748–751 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Stefansson H et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Kendall KM et al. Archival Report Cognitive Performance Among Carriers of Pathogenic Copy Number Variants: Analysis of 152,000 UK Biobank Subjects. Biol. Psychiatry 1–8 (2016). doi: 10.1016/j.biopsych.2016.08.014 [DOI] [PubMed] [Google Scholar]
- 46.Dewey FE et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 354, (2016). [DOI] [PubMed] [Google Scholar]
- 47.Sanyal A, Lajoie BR, Jain G & Dekker J The long-range interaction landscape of gene promoters. Nature 489, 109–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao SSP et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sahlén P et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biol. 16, 156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoenfelder S et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 25, 582–597 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Babaei S et al. Hi-C Chromatin Interaction Networks Predict Co-expression in the Mouse Cortex. PLOS Comput. Biol. 11, e1004221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.ENCODE et al. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306, 636–640 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Roadmap Epigenome Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akbarian S et al. The PsychENCODE project. Nat. Neurosci. 18, 1707–1712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Won H et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petrovski S, Wang Q, Heinzen EL, Allen AS & Goldstein DB Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9, e1003709 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kosmicki JA et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. (2017). doi: 10.1038/ng.3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Melnikov A, Zhang X, Rogov P, Wang L & Mikkelsen TS Massively parallel reporter assays in cultured mammalian cells. J. Vis. Exp. e51719 (2014). doi: 10.3791/51719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arnold CD et al. Genome-Wide Quantitative Enhancer Activity Maps Identified by STARR-seq. Science 339, 1074–1077 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Lehner T, Senthil G & Addington AM Convergence of advances in genomics, team science, and repositories as drivers of progress in psychiatric genomics. Biol. Psychiatry 77, 6–14 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Ganna A et al. Ultra-rare disruptive and damaging mutations influence educational attainment in the general population. Nat. Neurosci. 19, 1–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 8, 21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hyde CL et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 48, 1–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium et al. A contribution of novel CNVs to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Green EK et al. Copy number variation in bipolar disorder. Mol. Psychiatry 21, 89–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rucker JJH et al. Phenotypic association analyses with copy number variation in recurrent depressive disorder. Biol. Psychiatry 79, 329–336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Purcell SM et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–90 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Genovese G et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 19, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fromer M et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh T et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fischbach GD & Lord C The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68, 192–195 (2010). [DOI] [PubMed] [Google Scholar]
- 74.Buxbaum JD et al. The Autism Sequencing Consortium: Large-Scale, High-Throughput Sequencing in Autism Spectrum Disorders. Neuron 76, 1052–1056 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.