

Abstract
One of the novel interesting topics in the study of cardiovascular disease is the role of the oxidation system, since inflammation and oxidative stress are known to lead to cardiovascular diseases, their progression and complications. During decades of research, many complex interactions between agents of oxidative stress, oxidation, and antioxidant systems have been elucidated, and numerous important pathophysiological links to na number of disorders and diseases have been established. This review article will present the most relevant knowledge linking oxidative stress to vascular dysfunction and disease. The review will focus on the role of oxidative stress in endotheleial dysfunction, atherosclerosis, and other pathogenetic processes and mechanisms that contribute to the development of ischemic heart disease.
1. Introduction
Atherosclerosis is the most common form of large vessel pathology responsible for syndromes of vital organ ischemic damage like myocardial infarction [1].
The key pathophysiologic process of atherosclerosis is chronic inflammation, where oxidative stress plays an essential role in vascular homeostasis regulation including endothelial and smooth muscle cell growth, proliferation, and migration; angiogenesis; apoptosis; vascular tone; host defenses; and genomic stability. Imbalance in the oxidant/antioxidant mechanisms leads to oxidative stress and uncontrolled vascular injury [2–4].
The relation between heart failure and vascular disease is also marked by oxidative stress, caused by ischemia, left ventricular (LV) dysfunction, and neuroendocrinological activation. Reactive oxygen species (ROS) negatively affect myocardial calcium handling, cause arrhythmias, and contribute to cardiac remodeling by inducing hypertrophic signaling, apoptosis, and necrosis. Neurohumoral activation via the renin-angiotensin-aldosterone system (RAAS) and the sympathetic nervous system (SNS), combined with increased pre- and after-load, impose additional myocardial oxidative stress [5].
Ageing, traditional cardiovascular risk factors (arterial hypertension, dyslipidemia, diabetes mellitus and smoking), genetic predisposition, and environmental factors increase ROS generation and decrease endothelial nitric oxide (NO) production. Additional factors like mechanic vascular properties and geometry, hemodynamic forces, and endothelial gene regulation by biomechanical forces (atheroprone and atheroprotective phenotypes), disturbed flow in vascular regions like arches, branches, and bifurcations can promote vascular injury, ROS activity, coronary atherosclerosis, and ischemic heart failure development [6, 7]. The gut microbiota is involved in mediating metabolic processes associated with risk factors for coronary artery disease such as obesity, dyslipidemia, diabetes mellitus, and dyslipidemia. These comorbidities via its metabolites can induce development of atheroslerosis and aterosclerotic coronary artery disease. The main pathways for these processes are provided via oxidative stress, inflammation, cholesterol, and uric acid metabolism [8, 9].
The current therapeutic approach for atherosclerotic vascular plaque stabilization and disease includes RAAS inhibitors, statins, and acetylsalicylic acid, because of their pleiotropic antioxidative effects [10–12]. There is a need to elucidate oxidative stress physiology and pathophysiology, to identify novel therapeutic modalities for selective oxidative stress targeting in atherosclerosis [4].
2. Ischemic Heart Disease
Myocardial infarction (MI) is defined by clinical presentation, new ischemic electrocardiogram changes, and cardiac biomarkers elevation. The cause of MI is acute myocardial injury. Prolonged ischemia (a restriction in tissue blood supply, causing a deficiency of oxygen) can lead to myocardial necrosis and cell death. According to the Fourth Universal Definition of Myocardial Infarction 2018, MI can be divided into five categories (Figure 1).
Figure 1.
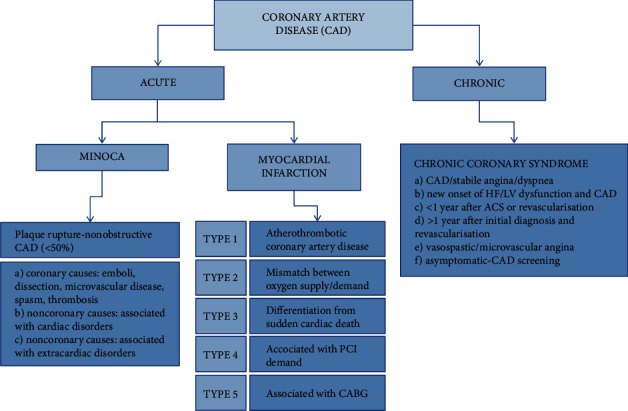
Acute and chronic coronary syndrome definitions and classification [14, 17].
MI type 1—caused by atherothrombotic coronary artery disease (CAD) and usually precipitated by atherosclerotic plaque disruption (rupture or erosion)
MI type 2—result of a mismatch between oxygen supply and demand: (a) reduced myocardial perfusion—coronary artery spasm, microvascular dysfunction, coronary embolism, coronary artery dissection, sustained bradyarrhythmia, hypotension or shock, respiratory failure, and severe anemia; (b) increased myocardial oxygen demand—sustained tachyarrhythmia and severe hypertension with or without LV hypertrophy
MI type 3—differentiation from sudden cardiac death
MI type 4—associated with percutaneous coronary intervention (PCI)
MI type 5—associated with coronary artery bypass grafting [7]
MINOCA (myocardial infarction with nonobstructive coronary arteries) can cause MI presenting with typical symptoms for acute coronary syndrome (ACS) and ST-segment elevation or equivalent. The underlying cause of disease may be nonobstructive (<50%) coronary artery disease stenosis, or mismatch between oxygen supply and demand, or secondary to myocardial disorders without involvement of the coronary arteries as myocarditis or Takotsubo syndrome [13].
Coronary artery disease (CAD) is a chronic, mostly progressive pathological process with predominant serious prognosis. This process can be modified by conservative and invasive treatment to achieve disease stabilization or regression [14]. Other cardiac conditions that are related to secondary myocardial injury are heart failure, myocarditis, any type of cardiomyopathy, Takotsubo syndrome, coronary revascularization procedure, cardiac procedure other than revascularization, catheter ablation, defibrillator shocks, cardiac contusion, systemic conditions, sepsis, infectious disease, chronic kidney disease, stroke, subarachnoid hemorrhage, pulmonary embolism, pulmonary hypertension, infiltrative diseases, amyloidosis, sarcoidosis, chemotherapeutic agents, critically ill patients, and strenuous exercise [15].
The leading symptom that initiates the diagnostic and therapeutic cascade in patients with suspected ACS is chest pain. Two groups of patients should be differentiated based on the electrocardiogram (ECG): those with persistent ST-segment elevation and those without persistent ST-segment elevation (transient ST-segment elevation, persistent or transient ST-segment depression, T wave inversion, flat T waves or pseudonormalization of T waves, or with normal ECG). The pathological finding at the myocardial level is cardiomyocyte necrosis or myocardial ischemia without cell loss [16].
Criteria for type 1 MI and type 2 MI detection are rise and/or fall of upper reference limit (URL) values with at least one value above the 99th percentile URL, with at least one of the following criteria: acute myocardial ischemia symptoms, new ischemic ECG changes, pathological Q wave development, and imaging evidence of new loss of viable myocardium or new regional wall motion abnormality in a pattern consistent with an ischemic etiology. For type 1 MI identification of a coronary thrombus by angiography including intracoronary imaging or by autopsy is needed as a one of the criteria, while evidence of an imbalance between myocardial oxygen supply and demand unrelated to acute coronary atherothrombosis for type 2 MI is a part of key definition [15].
The dynamic nature of the CAD results in various clinical presentations, which can be categorized as acute and chronic coronary syndromes. The diagnostic approach and management for patients with dyspnea and suspected ACS include assessment of symptoms and signs of disease, evaluation of the patient's general condition and quality of life, comorbidities evaluation, basic testing and assessment of LV function, risk assessment of obstructive CAD, diagnostic testing for CAD, and further event risk determination and treatment [14].
Management—including diagnosis and treatment—of acute ACS starts from the point of first medical contact. Out- and in-hospital treatment in acute setting is obligatory: relief of pain, breathlessness, and anxiety; arrhythmia management; reperfusion with PCI alone or/with fibrinolysis strategy, and periprocedural pharmacologic and nonpharmacologic therapy in a coronary unit [13].
Long-term management following acute treatment includes life style intervention, risk factor control, blood pressure and dyslipidemia treatment, glucose lowering therapy, antithrombotic therapy in acute and long-term settings, possible heart failure treatment, and arrhythmia management. Cardiac rehabilitation should be recommended [13].
CAD is a chronic, progressive disease with a predominantly serious prognosis. The outcome of MINOCA strongly depends on the underlying cause, and its overall prognosis is serious, with a 1-year mortality of about 3.5% [13, 14].
3. Endothelial Dysfunction and Oxidative Stress
Endothelial dysfunction caused by oxidative stress is an early event in the pathogenesis of many cardiovascular diseases including atherosclerosis, dyslipidemia, hypertension, diabetes, chronic kidney disease, heart failure, and ischaemia/reperfusion injury [18–23], and it is a hallmark of vascular diseases. An imbalance between NO bioavailability and ROS, also called oxidative stress, promotes endothelial dysfunction [24, 25] which is characterized by an altered modulation of vasomotor tone and vascular growth, impaired anti-inflammatory and antithrombotic endothelial characteristics, and disturbances of vascular remodeling [26].
The endothelium is a simple squamous layer of cells that forms an interface between the circulating blood and the vascular wall. A healthy endothelium provides endothelium-dependent vasorelaxation in response to vascular stress, controls vascular permeability, and prevents platelet aggregation [27]. It is very reactive to mechanical stimuli, chemical factors, and humoral agents by producing several mediators, such as NO, to maintain vasomotor tone and structural integrity. NO has a major role in endogenous antioxidant defense because of its potent vasodilatory, anti-inflammatory, and antithrombotic characteristics [28, 29]. Most of the vascular NO is produced by endothelial nitric oxide synthase (eNOS), a cytochrome p450 reductase-like enzyme which uses tetrahydrobiopterin to form NO from L-arginine [19]. The main causes of reduced NO bioavailability include increased NO degradation caused by ROS, decreased expression of eNOS, deficiency of substrates or cofactors for eNOS, and an inappropriate activation of eNOS caused by impaired cellular signaling [19, 30, 31]. Also, previous studies examined the phenomenon called eNOS uncoupling, causing reduced NO bioavailability by eNOS switching its enzymatic activity to generate superoxide (O2−) and H2O2 instead of NO [32, 33]. This occurs, for example, in the absence of NOS substrate L-arginine or the cofactor tetrahydrobiopterin in that process [34, 35]. Besides eNOS, which is mostly expressed in endothelial cells, there are two more isoforms of NO synthase with other functions—neuronal NOS (nNOS) and inducible NOS (iNOS) [36], which can also be a subject to uncoupling [33].
ROS are the products of the normal cellular aerobic metabolism generated during the reduction of oxygen [19]. ROS include unstable free radicals such as superoxide anion (O2−), hydroxyl radical or lipid radicals, and nonfree radicals such as hydrogen peroxide (H2O2), hypoclorous acid, or peroxynitrite which also have oxidizing effects that contribute to oxidative stress [19]. At moderate concentrations, ROS exert some physiological roles such as signaling [19, 37], but increased production of ROS which exceeds endogenous antioxidant defense mechanisms causes oxidizing of DNA, proteins, carbohydrates, lipids, and other biological macromolecules, leading to oxidative stress [6]. Enzymatic sources of ROS that are important in the cardiovascular system are NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidase, xanthine oxidase, and uncoupled eNOS with an addition of the mitochondrial electron transport chain, cyclooxygenase, and lipoxygenase as additional possible sources [6, 19, 32, 37]. Furthermore, production of ROS may be enhanced by free radical chain reactions. Several studies showed a very important role of NADPH oxidase, including Nox family oxidases Nox1, Nox2, Nox4, and Nox5, in endothelial dysfunction [19, 32]. NADPH oxidase is an enzyme located in the membrane of endothelial cells, smooth muscle cells, and fibroblasts, and it is the most powerful source of O2− production [38]. Angiotensin II, thrombin, platelet-derived growth factor, tumor growth factor-α, and lactosylceramide upregulate this enzyme and cause excessive ROS production [19]. Previous studies regarding angiotensin II-induced hypertension, diabetes mellitus, and hypercholesterolemia demonstrated the important impact of NADPH oxidase [23, 38, 39]. Xanthine oxidase is an enzyme that has a role in oxidation of hypoxanthine and xanthine in the metabolism of purines, leading to production of O2− and H2O2. The activity and expression of this enzyme are increased by interferon-γ [19]. The role of xanthine oxidase in ROS production in hypertension and hypercholesterolemia has been discovered in previous research [40–42]. As mentioned before, all three isoforms of NOS can be a source of ROS when uncoupling occurs, and NOS starts producing O2− and H2O2 instead of NO, but uncoupled eNOS products play a critical role in the pathogenetic processes of cardiovascular diseases [33]. This was shown in previous studies regarding hypertension, hypercholesterolemia, smoking, and diabetes mellitus [6, 43, 44]. Mitochondrial oxidative phosphorylation normally produces physiological levels of superoxide, which is converted to hydrogen peroxide and afterwards to water. Mitochondrial oxidative stress can be a consequence of excessive ROS production or insufficient ROS detoxification [39].
Excessive ROS production exceeding antioxidant defense systems leads to endothelial oxidative stress. The first step of endothelial dysfunction is called endothelial activation, which represents the expression of abnormal prothrombotic and proinflammatory characteristics of the endothelial cells, leading to other chronic changes [45]. Endothelial dysfunction includes impaired endothelium-mediated vasodilation; abnormal vascular reactivity and vasospasm; greater expression of chemotactic and adhesive molecules; increased platelet activation and thrombus formation; increased permeability of endothelium, leucocyte adhesion, and monocyte migration into the vascular wall; and impaired regeneration of endothelial cells with proliferation and migration of smooth muscle cells, leading to vascular damage [32, 46] (Figure 2).
Figure 2.
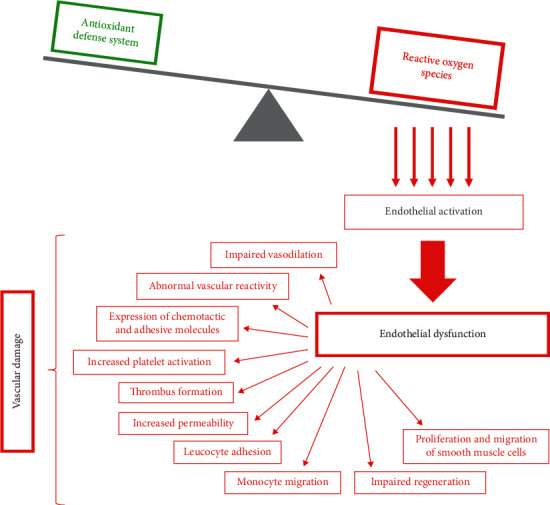
Endothelial dysfunction and development of vascular damage.
Many studies have demonstrated an important role of oxidative stress in endothelial dysfunction under the conditions of excessive oxidative stress. Cardiovascular risk factors cause imbalances between NO and ROS, so prevention of endothelial dysfunction by reducing oxidative stress and enhancement of endothelial NO production is seen as a reasonable therapeutic strategy in cardiovascular diseases [6, 46].
4. Oxidative Stress in Atherosclerosis
Atherosclerosis is a multisystemic, progressive, chronic inflammatory disease characterized by the interaction of immune and endothelial cells that is mediated by adhesion molecules on the surface of the vascular endothelium leading to the release of numerous proinflammatory mediators [47]. Specifically, it has been demonstrated that there is a close interaction between vascular endothelial inflammation and intense oxidative stress in triggering the atherosclerotic process [48].
The imbalance between the generation of excess reactive oxygen species (ROS) and the antioxidant mechanism leads to increased oxidative stress resulting in the formation of atherogenic oxidized low-density lipoprotein (Ox-LDL) which is a major determinant of atherogenesis [49].
Production of ROS from various sources (xanthine oxidase, lipoxygenase, nicotinamide adenine dinucleotide phosphate oxidase, eNOS, nNOs, and iNOS) leads to damage to mitochondrial capacity and to the development of mitochondrial dysfunction [50]. Free fatty acids in endothelial cells enter the tricarboxylic acid cycle during which oxidation results in the overformation of NADH, which is an important driver of ROS during oxidative phosphorylation [51]. Mitochondrial dysfunction leads to increased ROS formation and oxidative stress and thus plays a role in the initiation, formation, and progression of an atherosclerotic lesion [51].
Studies have shown that increasing ROS production in mitochondria is induced by age, obesity, smoking, hypertension, diabetes, and dyslipidemia [52]. Numerous studies have found that mitochondrial dysfunction significantly affects the regulation of inflammation, proliferation, and apoptosis in the onset and progression of atherosclerotic plaques [53–57].
During the atherosclerotic process, the accumulated neutrophils produced an additional amount of ROS [58]. ROS enhance the activation of poly (ADPribose) polymerase 1 (PARP1), which damages mtDNA and thus the mitochondrial transport chain, further enhancing ROS formation and further damaging endothelial cells [59]. The resulting ROS trigger the synthesis of inflammatory cytokines by different cellular pathways resulting in vascular inflammation and participating in the oxidation process of LDL [58]. Ox-LDL has a cytotoxic effect on vascular cells, and macrophage removal receptors can phagocytose them, forming foaming cells that deposit in the blood vessel wall forming an atherosclerotic plaque [60]. Ox-LDL exerts its various effects on cells such as endothelial cells, macrophages, platelets, fibroblasts, and smooth muscle cells through transmembrane glycoproteins such as SR-A, CD36, and LOX-1 [61]. The resulting Ox-LDL increases the NADPH oxidase activity, leading to an increase in ROS synthesis and to NO inactivation. It also causes eNOS dysfunction by displacing it from the alveolar membrane site and enhances the arginase II activity thereby reducing the amount of L-arginine cosubstrate for eNOS resulting in an additional decrease in NO synthesis [62]. Ox-LDL increases the synthesis of matrix metalloproteinases (MMP), namely MMP-1, MMP-3, and MMP-9, leading to a breakdown of the fibrotic cap and to a consequent rupture of the atherosclerotic plaque. LOX-1 is expressed on macrophages, vascular endothelial smooth muscle cells, cardiomyocytes, platelets, and fibroblasts. The binding of Ox-LDL to LOX-1 in macrophages and vascular smooth muscle cells results in the formation of foam cells [63]. The main inducers of the LOX-1 expression are tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-1), interferon-gamma (IFN-γ), CRP, and modified lipoproteins such as glyxidized LDL, lysophosphatidylcholine, and ROS, while the mediators and conditions regulating the gene expression are numerous: angiotensin II, cytokines, glycation end products, diabetes mellitus, hypertension, dyslipidemia, ischemia reperfusion injury (IRI), heart failure, psychological stress, and HIV infection [61]. TNF-α and NF-kB increase Ca2+ levels in the mitochondria and consequently increase ROS production. SR-A and CD36 take up 75% to 90% of LDL [64]. SR-A is expressed in the presence of oxidative stress and growth factors in endothelial and smooth muscle cells, while normally found only in myeloid cells [65]. CD36 is found on monocytes, macrophages, platelets, and adipocytes [63]. Human macrophages lacking CD36 have a 40% reduction in Ox-LDL binding and uptake [66]. Toll-like receptors (TLRs) constitute a major subset of pattern recognition receptors (PRRs) that are significantly expressed on different immune cells during atherogenesis in the coronary circulation [67]. TLR signaling cascades can be activated by a wide range of endogenous ligands associated with tissue damage, which plays a central role in the development of atherosclerotic plaques [67]. The main culprits involved in the immune response to oxLDL are TLR4 [67]. Ox-LDL has been shown to lead to increase the expression of TLR4 in macrophages, neutrophils, and dendritic cells with which it plays an important role in the development of atherosclerotic plaques in the coronary circulation by activating MAPK and NF-κB pathways [68, 69]. Activation of MAPK and NF-κB transcription factors results in enhanced activation of genes encoding proinflammatory cytokines and chemokines important for the progression of the atherosclerotic process, including TNF-α, IL-1, and Il-6 [67]. miR-590 has antiapoptotic effects on endothelial cells attacked by the atherosclerotic process by inactivating the TLR4/NF-κB pathway, which may be a potential therapeutic target [70]. TLR4 is required for Ox-LDL-induced differentiation of macrophages into foam cells in the early stages of atherosclerosis [71]. It plays a crucial role in plaque progression and rupture leading to occlusive thrombus formation in human coronary arteries [72]. Specific Ox-LDL derivatives act as TLR4 ligands by enhancing the MMP-9 expression [73]. Also, minimally modified low-density lipoproteins (mmLDL) via CD14 and TLR4 induce actin polymerization which together with MMP-9 leads to remodeling of the coronary artery wall, resulting in instability of atherosclerotic plaques and their rupture [74]. Cellular fibronectin (cFN) is an extracellular matrix protein (ECM) that is overexpressed only in chronically inflamed tissues and is synthesized by vascular smooth muscle cells and endothelial cells [75]. cFN activates macrophages and platelets via TLR4 resulting in platelet aggregation and arterial thrombosis within atherosclerotic lesions in the coronary arteries [75]. TLR2 activation stimulates VSMC migration from the intima in an IL-6-dependent manner, regulates inflammatory processes and ROS production after vascular injury, and contributes to coronary endothelial dysfunction after ischemic-reperfusion injury by activating neutrophils and creating ROS [67]. Ox-LDL can induce the expression of mRNAs of Wnt5a (Wnt family of glycoproteins) that are coexpressed with TLR2 and TLR4 and play a key role in the formation of foam cells, especially in advanced atherosclerotic plaques, which correlates with the severity of atherosclerotic lesions in human studies [76, 77]. TLR9 is expressed in the endoplasmic reticulum and not on cell surfaces such as TLR2 and TLR4 [67]. TLR9 is activated by CpG motifs in nucleic acids released during vascular necrosis and stimulates the transformation of macrophages into foam cells in a manner dependent on NF-κB and IRF7 (interferon regulatory factor 7) and stimulates the secretion of INF and increases cytotoxic activity CD4 + T cell versus coronary artery smooth muscle cells [78].
VSMCs are important components of atherosclerotic plaques that, under the influence of biostimulation or mechanical damage triggered by oxidative stress, change their phenotype and, through differentiation, become synthetic VSMCs that produce significantly less contractile proteins, increase proliferation and migration, and thus participate in the development of atherosclerosis [79]. Increased concentrations of Ox-LDL via LOX-1 cause smooth muscle cell apoptosis as they increase the expression of a proapoptotic protein such as the bcl-2-associated X protein (Bax) leading to instability and rupture of the atherosclerotic plaque. In addition, through inducers, CD147 can cause plaque instability by releasing extracellular MMP [52].
Oxidative stress caused by the production of ROS and RNS (nitric oxide (NO), peroxynitrite (ONOO−)) and S-nitrosothiol (RSNO)) can damage macromolecules because it reacts with specific amino acid residues and DNA and chromatin cause mutations or double-stranded breaks in a phenomenon overall known as “oxidative damage” [80]. The selenoprotein family is involved in the control of oxidative stress in the cardiovascular system by inhibiting oxidative stress, modulating inflammation, suppressing endothelial dysfunction, and protecting vascular cells from apoptosis and calcification [81]. Potent selenoproteins of particular importance to the cardiovascular system are glutathione peroxidase (GPX), thioredoxin reductase 1 (TXNRD), methionine sulfoxide reductase B1 (MSRB1), selenoprotein P (SELENOP), selenoprotein S (SELENOS), and selenoprotein T (SELENOT) [81]. Dysfunction of various selenoproteins can lead to congestive heart failure, coronary heart disease, and to damaged heart structure and function [80]. The main catalytic site of selenoprotein is called Sec [80]. GPXs are the major components of the antioxidant system that maintain oxidative homeostasis, using glutathione as a cofactor for catalyzing the reduction of hydrogen peroxide (H2O2) and/or phospholipid hydroperoxide [80]. GPX3 controls vascular tone and the thrombotic properties of vascular endothelium [80]. TXNRD, along with thioredoxin (Trx) and NADPH, represents the major disulfide reduction system in the cell [82]. MSRB1 acts synergistically with GPX and TXNRD primarily in the liver, kidneys, and heart [80]. Selenoprteins P, S, and T predominantly contribute to calcium ion (Ca2+) signaling, protein folding, and ER-related degradation [80]. SELENOS, SELENOK, SELENOM, SELENON, SELENOF, and SELENOT are involved in maintaining the homeostasis of oxidative stress in the ER of cardiac myocytes [80]. Studies have shown that decreased selenoprotein levels are associated with the increased Nrf2 expression which may represent an important compensatory response to the maintenance of homeostasis [83]. Selenoproteins play an important role in embryogenesis, since it was found that mice that had a genetic disorder of cytosolic TXNRD1, mitochondrial TXNRD2, and GPX4 experienced embryonic mortality [84].
Polyunsaturated fatty acids (PUFAs) exert anti-inflammatory, antiatherogenic, and antioxidant properties on the cardiovascular system [85–87]. These important effects are achieved by competing with arachidonic acid (AA) for enzymes involved in the biosynthesis of proinflammatory mediator molecules, by suppressing proinflammatory NF-κB by modulating TLR4 signaling, by activating PPAR-γ, and FFA4 receptors (before GPR120 d) in macrophages and metabolites such as esolvins, maresins, and protectins that have anti-inflammatory and antioxidant effects [88, 89]. The most studied molecular mechanisms are the activation of Nrf2 in the vascular tissue, leading to the production of antioxidant enzymes (HO-1, GPx) and the activation of FFA4 receptors, resulting in the preservation of κB inhibitors (IκB) and the prevention of NF-κB nuclear translocation [90–93]. F2-isoprostanes are prostaglandin-like molecules formed as a result of peroxidation of ROS-mediated esterified arachidonic acid [94]. n-3 PUFAs reduce 8-isoprostane levels in macrophages and reduce oxidative stress [88]. The peroxidation products of ω-3 PUFAs and ω-6 PUFAs can also have toxic effects in oxidative stress, and a diet rich in PUFAs can lead to tissue hypersensitivity to lipid peroxidation induced by oxidative stress [95, 96]. Therefore, in the future, it will be necessary to investigate individually each potential PUFA that has been shown to be an important protective factor in oxidative stress.
The most important antioxidants are glutathion peroxidase (Gpx), glutathion reductase, catalase, and superoxide dismutase [97]. Numerous studies have shown that efficient elimination of ROS from cells reduces the formation and progression of atherosclerotic plaques [98, 99]. The role of antioxidants on the progression of an atherosclerotic lesion needs to be further investigated as there are studies that confirm that certain antioxidants have no effect on the development of an atherosclerotic lesion [100]. A transcriptional coactivator that regulates a gene involved in energy metabolism in mitochondria called peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) which is an important mitochondrial protector that promotes the synthesis of NO enzymes, mitochondrial protein 2 (UCP-2), and the antioxidant defense of mitochondria (manganese SOD, catalase, and thioredoxin 2), and this way limits endothelial dysfunction [52, 101]. PGC-1α also reduces the activity of the inflammatory factors NF-κB and TNF-α and prevents the entry of Ox-LDL into cells [62, 102]. Twinkle mtDNA helicase plays a major role in stabilizing atheromatous plaques and reducing the development of atherosclerosis as it decreases apoptosis of VMSCs and macrophages [52]. Mitofusini 1 (Mfn1) is an important GTPase that regulates VMSC proliferation and apoptosis and acts as an important endogenous inhibitor of VSMC proliferation by inhibiting the Ras-Raf-ERK 1/2 pathway during the atherosclerotic process [103, 104]. Thus, prevention of vascular oxidative stress and improvement of NO production may be key future targets of new therapeutic strategies for the treatment of atherosclerosis [61]. Figure 3 summarizes the interactions of pathogenetic mechanisms linking oxidative stress to atherosclerosis, coronary artery disease, and consequently heart failure.
Figure 3.
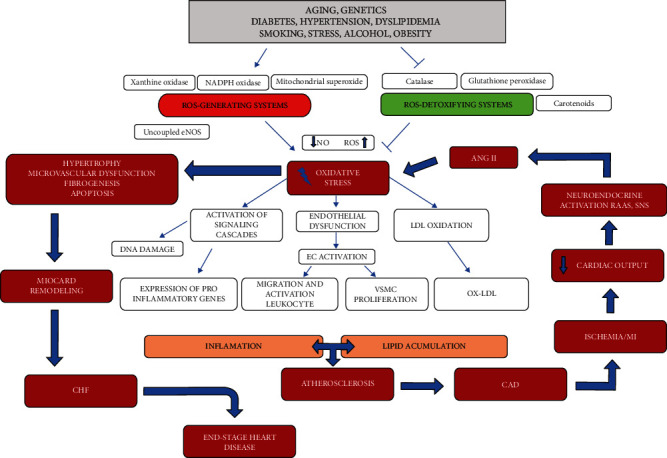
The role of oxidative stress in the onset of atherosclerosis and the pathophysiological implications of congestive heart failure. ANG II: angiotensin II; CAD: coronary artery disease; CHF: congestive heart failure; DNA: deoxyribonucleic acid; EC: endothelial cells; eNOS: endothelial nitric oxide synthase; LDL: low-density lipoprotein; MI: myocardial infarction; NO: nitric oxide; RAAS: renin–angiotensin–aldosterone system; ROS: reactive oxygen species; SNS: sympathetic nervous system; VSMC: vascular smooth muscle cells.
5. Oxidative Stress in Coronary Artery Disease
Dyslipidemia, as well as an imbalance between ROS production and enzymatic and nonenzymatic antioxidant protection systems, leads to endothelial dysfunction and atherosclerosis of the coronary arteries [1]. Numerous studies have shown impaired balance of prooxidants and antioxidants in patients with CAD [105–108]. Oxidative stress is today considered a new risk factor responsible for the development of CAD that affects the onset, prognosis, quality of life, and survival of patients [109].
In addition to being associated with atherosclerosis, oxidative stress can create oxidative modification or damage to lipid peroxidation at the level of deoxyribonucleic acids (DNA) and proteins with deleterious effects on the structure and function of the vascular system [110]. In addition to classical free oxygen radicals (superoxide radical (O2-), hydrogen peroxide (H2O2), hydroxyl (OH), peroxyl (RO), hydroperoxyl (HRO-)), reactive oxidative stress has also been shown to be involved in the oxidative stress process of nitrogen species (RNS), especially peroxynitrite (ONOO-) [111]. We know that ROS damages key molecules in signaling pathways involved in vascular inflammation, and it damages essential biomolecules in cells and participates in oxidative modification of lipids that make them atherogenic [112]. The most important sources of oxidative stress are the phagocytic isoform of NADPH oxidase (Nox2 and to a lesser extent Nox1) with its regulatory subunit p47phox, xanthine oxidase (XO), and dysregulated eNOS [113].
Common risk factors (hyperlipidemia, hyperglycemia, smoking, hypoxia, etc.) activate NADPH oxidase via different signaling pathways. It is now known that enhanced release of reactive oxygen species (ROS) by NADPH oxidases and mitochondrial enzymes results in cardiomyocyte hypertrophy, fibrosis, and an increase in metalloproteinase. The most studied mechanism of NADPH activation is mediated by one of the mechanisms of the PKCα/β2 signaling pathway in which protein kinase C plays a key role [114]. The p47phox subunit is the major Nox2 (gp91phox) regulatory subunit whose phosphorylation is required for Nox2 activation. The expression of p47phox was significantly increased in patients with CAD and overweight by about 60% and in obese patients with CAD by about 80%. So far, XO is known to be significantly elevated in CAD patients, and overweight is thought to be a potent driver of the enhanced XO expression [113].
In patients who have a BMI increase, suffer from CAD and will undergo CABG, increased ROS levels, increased expression of ROS-producing enzymes (P47phox, xanthine oxidase), decreased expression of antioxidant enzymes (mitochondrial aldehyde dehydrogenase, heme oxygenase-1, and eNOS), and increase in markers of inflammatory processes in serum and right atrial myocardial tissue (sVCAM-1 and CCL5/RANTES) have been demonstrated [115].
Endoplasmic reticulum stress (ERS) occurs in cardiac myocytes and cardiac tissue in response to various stressors, such as ischemia, hypoglycemia, hyperlipidemia, inflammation, and osmotic stress [116]. The resulting oxidative stress leads to changes in the redox status of the ER that interfere with the formation of protein disulfide and cause misfolding of the protein [116]. High cholesterol, fatty acids, and oxidative stress may induce ERS-induced apoptosis of macrophages and endothelial cells in atherosclerotic plaques [117]. ERS is associated with the development and progression of cardiac hypertrophy, ischemic heart disease, and heart failure [118]. The consequences of ERS are the accumulation of incorrectly posttranslationally modified secretory and transmembrane proteins that have important cellular functions [116]. During ERS, intracellular signaling pathways called unfolded protein response (UPR) are activated, restoring ER homeostasis, but if ERS persists chronically at high levels, terminal UPR activates cell apoptosis, which may be one of the important pathophysiological mechanisms for disease development [116]. Terminal UPR makes an important contribution to myocyte loss during myocardial infarction [119]. Also, it has been discovered that ER autophagy may be the last resort to restore ERS homeostasis [120]. There is also evidence that activation of UPR also activates Nrf2, which has been shown to be an important cardioprotective factor [121]. Improved understanding of the molecular mechanisms of regulated ERS in the future may lead to the discovery of new therapeutic targets [118].
ROS leads to the activation of the nuclear transcription factor kappa B (NF-κB), which regulates key genes for the encoding of proinflammatory cytokines, chemokines, and leukocyte adhesion molecules. Two important transcription factors—nuclear factor erythroid 2-related factor 2 (Nrf2) and peroxisome proliferator-activated receptor-β/δ (PPARβ/δ)—have been shown to protect coronary blood vessels from excessive exposure to oxidative stress. Oxidative stress and inflammation are thought to be major activators of these protective transcription factors [122]. Nrf2 stimulates genes for the synthesis of antioxidant and detoxifying enzymes and indirectly antagonizes the proinflammatory effects of NF-κB by removing ROS [123–125]. PPARβ/δ is predominantly located in the heart and has cardioprotective effects by suppressing the activity of several transcription factors, including NF-κB [126].
Bone marrow endothelial progenitor cells (EPCs) are responsible for neovascularization and reendothelialization after ischemia and/or tissue injury, and a decrease in EPC numbers and their function has been demonstrated in CAD patients [114, 127]. High levels of oxidative stress in CAD patients are thought to be closely related to the enhanced activation of NDPH oxidase mediated by the membrane component p47phox, which plays a major role in the regulation of the NADPH activity and thus reduced vascular capacity of EPCs in CAD patients [114]. Medications used today, such as AT blockers, ACE inhibitors, statins, and tazolidindiones, have a beneficial effect on the bioactivity of EPCs that maintain vascular homeostasis [2]. During oxidative stress, serum EPC levels drop significantly, suggesting that this may serve as a good biomarker of oxidative stress [128].
Cytotoxic products of the enzyme myeloperoxidase (MPO), such as hypochlorous acid, lead to oxidative damage to blood vessels. Human MPO is an important pathophysiological mediator and biomarker in CAD patients whose levels are significantly elevated, leading to the formation of dysfunctional lipoproteins with increased atherogenic potential, decreasing NO availability, weakening vasoreactivity, and leading to atherosclerotic plaque instability [129]. Malonyldialdehyde (MDA) is one of the last products of peroxidation of polyunsaturated fatty acids in cells whose levels increase significantly during oxidative stress. Therefore, the level of human MDA in blood plasma is a very important biomarker of ROS-induced lipid peroxidation [130]. In a study on 30 patients with angiographically defined CAD and 30 healthy control subjects, serum MDA levels were increased, although these values did not differ depending on the number of affected coronary vessels and were not correlated with the severity of vascular lesions [131]. The level of MDA and the percentage of MDA release were significantly elevated, while the level of glutathione (GSH), erythrocyte GPx activity, and total plasma antioxidant capacity (TAC) was significantly reduced in patients with acute coronary syndrome and with CAD, compared to healthy subjects (n = 30/group) [112]. The study thus found that in patients with CAD, there was a significant decrease in glutathione in erythrocytes and consequently elevated levels and increased release of MDA, confirming that the susceptibility of erythrocyte membranes to oxidative stress was significantly higher in patients with CAD than in healthy subjects. Also, the same study showed results in which the erythrocyte level and total antioxidant capacity (TAC) value were significantly lower compared to healthy controls [112].
Various study groups have reported significant decreases in the parameters of antioxidants in patients with CAD. It is important to emphasize that, according to current data, patients with multivessel coronary artery stenoses have significantly higher levels of MDA and significantly lower levels of GSH, TAC, and GPx activity than patients with double and single coronary artery disease, which clearly leads to the conclusion that the greater the number of coronary artery stenoses, the higher the level of oxidative stress [112].
Abolhasani et al. conducted a study showing that the serum concentrations of high-sensitivity C-reactive protein (hs-CRP), sialic acid (SA), vitronectin (VN), plasminogen activator inhibitor-1 (PAI-1), Ox-LDL, and MDA were significantly elevated in patients with CAD relative to the healthy control group [132]. ROS-mediated lipid peroxidation leads to the formation of unsaturated aldehydes, including acrolein and MDA, which have toxic effects [112]. A study conducted by Yilmaz et al. showed that serum MDA was significantly higher, and TAC was significantly lower in CAD patients [133]. A study by Ninic et al. showed that the major lipid peroxidation product thiobarbiturate acid-reactive substances (TBARS) was significantly higher in patients with CAD than in the control group, while the antioxidant effect of many serum antioxidants was significantly lower [1]. TBARS leads to further formation of ROS and acts on proteins and DNA that exert proatherogenic and mutagenic effects [134]. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a cytokine that acts as an apoptosis-inducing ligand, and research has shown that TRAIL levels are significantly reduced in animal CAD models, and that the unknown mechanism of TRAIL reduces oxidative stress and endothelial dysfunction [135].
Below, we present new insights into the numerous molecules, signaling pathways, and antioxidants involved in the highly complex development of oxidative stress in the coronary circulation. Iranian researchers have shown in a study that the increased expression of HSP27 mRNA in the peripheral blood mononuclear cell (PBMCs) is significantly associated with the severity of CAD and can serve as an important prognostic biomarker, indicating the degree of total oxidative stress [136]. A large study conducted by Khaper et al. showed that one week after acute myocardial infarction, the mRNA level for mitochondrial manganese superoxide dismutase (Mn SOD) decreased by 40% and after sixteen weeks by 73% compared with healthy subjects, an indicator of depleted antioxidant protection in patients with CAD [137].
In the animal model, the growth arrest-specific 5 (GAS5) overexpression in CAD rats has been shown to inhibit abnormal activation of the Wnt/β-catenin signaling pathway, leading to improvement of hyperlipidaemia, attenuation of myocardial injury, inhibition of cardiomyocyte apoptosis, and reduction of oxidative stress [138]. Decrease in leukocyte telomere length (TL) and mitochondrial DNA copy number (mtDNA-CN) are important indicators of the development of CAD, which are involved in the modulation of oxidative stress as independent risk factors, but this needs further investigation [139]. Polymorphisms in NRF2 and its target antioxidant genes: HMOX-1, NQO1, and MT significantly influence the level of oxidative stress in CAD formation [51, 140]. Inhibition of SAH hydrolase (SAHH) adenosine dialdehyde inhibitor in CAD patients leads to a significant increase in plasma S-adenosylhomocysteine (SAH) that promotes the production of free oxygen radicals and leads to endothelial dysfunction by epigenetic regulation of the oxidative stress pathway mediated by the p66shc gene promoter expression [141].
An antioxidant and an important component of the electron transport chain, coenzyme Q10 (CoQ10), has an effect on biomarkers of inflammation and oxidative stress, and the study found that CoQ10 significantly increased SOD and catalase (CAT) levels in CAD patients, significantly reduced MDA and dienes, and had significant effect on C-reactive protein (CRP), tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and GPx levels [142]. Supplementation with L-carnitine at a dose of 1000 mg/d after 12 weeks reduces oxidative stress (MDA level by 7%) and increases the activity of antioxidant enzymes (CAT by 16%, SOD by 47%, and GPx by 12%) in patients with CAD [143], while the administration of doses higher than 2000 mg/d showed a cardioprotective effect and reduced mortality rates in CAD patients [144].
Protein phosphatase and actin regulator 1 (PHACTR1), which regulates the reorganization of the actin cytoskeleton, is significantly expressed in atherosclerotic plaques of the coronary arteries. Inhibition of PHACTR1 synthesis led to a decrease in the Ox-LDL-induced expression of VCAM-1, ICAM-1, and VE-cadherin; attenuation of p47phox phosphorylation; and attenuation of the p65 and NF-κB activity without affecting IκBα and IKKα/β phosphorylation, all resulting in a decrease of intracellular oxidative stress [145].
Sirtuin 1 (SIRT1) is a protein that plays a role in mitochondrial biogenesis and deacetylation of proteins important for stimulating antioxidant defense. SIRT1 enhances the antioxidant enzyme activity and inhibits free radical-mediated oxidative injury by reducing NADPH oxidase activation, also reducing endothelial cell death caused by oxidative injury [146]. The main mechanism of its action is the inhibition of the LOX-1 expression by modulation of the LOX-1 promoter [147]. The SIRT1 expression level is suppressed, while the acetylated p53 expression levels are increased in monocytes of CAD patients. The mitochondrial function is significantly impaired in monocytes in patients with CAD, and it is thought that SIRT1 may increase the mitochondrial function. Also, a consequence of the decreased expression of SIRT1 is the increased adhesion of monocytes to endothelial cells [148].
We can conclude that oxidative stress plays a key role in the development and pathogenesis of CAD and the emergence of its complications [2]. By modulating very complex and numerous singular pathways and various biomolecules, oxidative stress can be reduced. In the future, there is a possibility and need to investigate more thoroughly all molecules involved in this highly complex biological process, which may open up new therapeutic targets and ultimately reduce the onset and complications of CAD.
6. Coronary Microvascular Dysfunction
Coronary microvascular dysfunction (CMD) is a disorder that leads to the development of myocardial ischemia, although there is no proven obstruction in coronary arteries on coronary angiography [149] (Figure 4). Risk factors that can trigger oxidative stress in coronary microvascular dysfunction are obesity, dyslipidemia, diabetes, and the metabolic syndrome. Some disorders such as hypertrophic cardiomyopathy, hypertensive heart disease, myocarditis, and vasculitis are examples where myocardial ischemia can develop without the presence of coronary artery obstruction. In addition to these conditions, structural and functional alterations in the coronary microcirculation may be responsible for the occurrence of myocardial ischemia, in up to 20% of patients with acute coronary syndromes (ACS) and up to 50% of patients with chronic coronary syndromes (CCS) [150].
Figure 4.
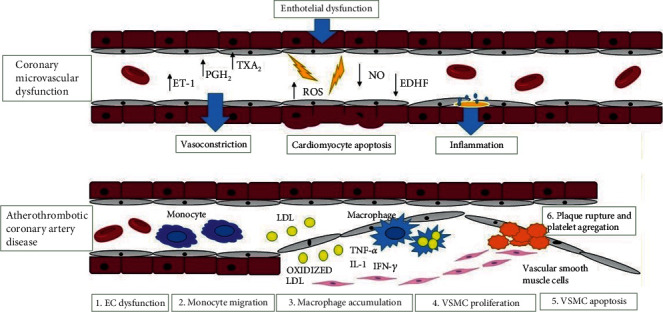
Difference between coronary microvascular dysfunction and atherothrombotic coronary artery disease. ET-1: endothelin -1; PGH2: prostaglandin H2; TXA2: thromboxane A2; ROS: reactive oxygen species; NO: nitric oxide; EDHF: endothelium-derived hyperpolarizing factor; LDL: low-density lipoprotein; TNF-α: tumor necrosis factor – α; IL-1: interleukin-1; IFN- γ:), interferon-gamma; VSMC: vascular smooth muscle cells; EC: endothelial cell.
6.1. CMD in Nonobstructive ACS
MINOCA is a term that refers to myocardial infarction with nonobstructive coronary arteries [13]. Today, the pathophysiology of MINOCA is not very well understood. Some studies show that MINOCA has two causes: epicardial causes which are represented by coronary plaque disease, coronary dissection, coronary artery spasm and micorvascular causes such as coronary microvascular spasms, Takotsubo syndrome, myocarditis, or coronary embolism [17, 151]. Conditions such as myocarditis and Takotsubo syndrome are considered nonobstructive ACS, but cardiac nonischemic aetiologies [152, 153].
6.2. CMD in Nonobstructive CCS
INOCA is a term denoting ischemia with non-obstructive coronary arteries, where endothelial dysfunction is a key mediator in the pathogenesis of CMD [154, 155]. Studies have shown that INOCA is present in approximately one-third of men and two-thirds of women undergoing angiography for suspected ischemic heart disease [155, 156]. Some studies show that factors originating from the blood and endothelium, as well as metabolic and neurohumoral influences, affect the regulation of the coronary microvascular tone. These include the influence of passive mechanical factors (extravascular contraction of contracting myocardium, distension by intravascular pressure) as well as active changes in the smooth muscle tone by myogenic responses (in response to changes in perfusion pressure) [157, 158]. An important role in the development of this disorder is played by the vascular endothelium where if there is dysfunction, inadequate release of NOS would result in coronary artery vasoconstriction [149]. More specifically, reduced endothelial NO synthesis or increased inactivation will result in endothelial dysfunction and vasoconstriction of blood vessels [159–161]. Endothelial dysfunction is also present as an imbalance between the release of vasorelaxant substances, such as prostacyclin (PGI2), endothelium-derived hyperpolarizing factors (EDHF), and vasoconstricting substances, such as endothelin-1, superoxide, hydrogen peroxide, and thromboxanes [162]. Endothelin-1 (ET-1), as a potent vasoconstrictor, plays a significant role in the pathogenesis of coronary microvascular dysfunction by acting through endothelin A receptors located on coronary vascular smooth muscle cells. Also, ET-1 participates in the regulation of vascular tone via endothelin B receptors located on coronary vascular smooth muscle cells and on endothelial cells where it has an effect on NO release and vasodilation [163]. Endothelium-derived NO is produced from L-arginine using NO synthase and released to the vascular smooth muscle layer, ultimately causing vasodilation. NO occurs in response to an increase in shear stress [164]. Endothelial NO has an effect on mitochondrial metabolism, reducing the production of ROS and thus inhibiting inflammation. In addition, it inhibits myocyte hypertrophy by activating cGMP-dependent protein kinase (PKG). It also prevents thrombosis and vascular inflammation by inhibiting platelet activation. Therefore, in conditions such as ischemia and metabolic diseases, there is an increased release of ET-1, thromboxane A2, and ROS, which ultimately results in increased cardiomyocyte apoptosis [165].
Corban et al. have combined numerous studies, pointing out that mutations of eNOS and ET-1 genes are crucial for the development of coronary microvascular dysfunction [162]. For example, an eNOS gene missense Glu298Asp variant is associated with reduced NO production and impaired endothelial cell response to physiological stimuli such as shear stress, then the T786 > C mutation in the eNOS gene compromises endothelial NO synthesis [166, 167].
Ford et al. conducted a multimodality investigation on patients with angina, investigating the role of ET-1 and the gene variant (rs9349379-G allele), chromosome 6 (PHACTR1/EDN1)] in the pathogenesis of CMD. Their goal was to investigate whether the G allele associates with noninvasive parameters of myocardial ischaemia. The second goal was to examine vascular mechanisms using isometric tension recordings in small peripheral resistance vessels isolated from patients according to genotype. In conclusion, peripheral small artery reactivity to endothelin-1 and ETA receptor antagonist affinity was conserved in the rs9349379-G allele group. Zibotentan tested at clinically relevant concentrations completely prevented the effect of endothelin-1. This study indicates that ETA receptor antagonism in this group of patients may have therapeutic benefits [168, 169].
Experimental studies conducted to date on large animal models such as swine, given that they show a remarkably similar cardiovascular anatomy as humans, have significantly helped in the understanding of the regulation of coronary microvascular function [170]. Experimental studies on animal models, with an emphasis on metabolic derangements as risk factors—in dogs, swine, rabbits, rats, and mice—today help to understand the pathophysiology of CMD. Metabolic derangements in animals are most commonly caused by a high-fat diet (HFD) and/or diabetes mellitus through an injection of alloxan or streptozotocin. There are also transgenic animal models in which metabolic derangements develop. All these animal models show disturbances in the function and structure of the coronary microvascular bed. Therefore, the application of these animal models will be useful in identifying novel therapeutic targets for the purpose of combating ischemic heart disease [171]. Experimental studies have shown that adipocytes, leptin, interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) are crucial in the development of oxidative stress [172, 173]. In patients with metabolic syndrome, the increased sympathetic activity produces exaggerated alpha-adrenergic coronary vasoconstriction and thus contributes to the development of coronary microvascular dysfunction [174]. Also, in patients with metabolic syndrome and prehypertension, the RAAS system is activated resulting in the formation of angiotensin II—causing vasoconstriction in the coronary circulation [175].
In spite of the conducted research efforts to date, there is still insufficient knowledge about the role of oxidative stress in the pathophysiology of coronary microvascular dysfunction, as a disorder leading to the development of myocardial ischemia despite a normal finding of coronary angiography.
7. The Impact of Environmental Factors on CAD
Research to date has shown that environmental factors may play an important role in the development of cardiovascular disease (CVD), but the mechanisms by which environmental factors affect CVD have not been fully explained [176]. Knowing how different environmental factors affect CVD risk would greatly improve the development of therapeutic and preventive strategies to combat CVD. In addition to the previously known fact that genetics, combined with environmental factors, is contributing to the development of CVD, the results of many studies have shown that environmental factors play a more dominant role, as many subjects have prevented CVD by maintaining a healthy lifestyle [177].
The study by Hill et al. investigated the influence of selected genetic and environmental factors on the clinical expression of heterozygous familial hypercholesterolemia. Men were shown to have a higher risk of developing CAD because they had lower high-density lipoprotein (HDL) cholesterol levels and were smokers. In women, CAD has been associated with elevated triglycerides and the presence of hypertension [178].
In order to understand how the environment affects CVD or how that risk is transmitted, we need to understand the complexity of the human environment. According to research, it has been shown that there is a mismatch between ancient human genes and the current human environment, and that the mismatch is the result of a rapid change in the human environment relative to genetic adaptation. First of all, the circadian rhythm is a fundamental feature of the natural environment and has an impact on the levels of neurohormones that regulate cardiovascular function, such as angiotensin II, renin, aldosterone, growth hormone, and atrial natriuretic peptide [179–181]. Therefore, an interesting link is that the frequency of adverse cardiovascular events varies with time. For example, myocardial infarction most commonly occurs between 6 a.m. and 12 p.m. and is more likely to occur early in the morning than at night [182, 183]. Also, a disturbed circadian rhythm increases the risk of diabetes mellitus, obesity, and hypertension [184–186].
The change of seasons has an impact on the development of CVD which is shown by research which found that in the northern and southern hemispheres, and the levels of blood pressure, HDL, LDL, and glucose are slightly higher in winter than in summer. More patients on statin therapy reach the target LDL level in summer than in winter [187–189]. Likewise, exposure to cold ambient temperature increases vascular resistance and blood pressure and can induce coronary vasospasm and lead to the development of myocardial infarction [190]. Also, heat waves, especially in the elderly who cannot adapt quickly to changes in temperature, can promote the development of CVD [191]. That high levels of sunlight early in life can delay CVD by 0.6 to 2.1 years has been shown by some studies [192, 193]. Vitamin D deficiency is associated with an increased risk of adverse cardiovascular events such as myocardial infarction, stroke, heart failure, and sudden cardiac death [194, 195].
Studies have shown that short-term irradiation of the whole body of healthy people with UVA has the effect of lowering blood pressure, on the principle of releasing NO, and increasing the level of S-nitrosoglutation, which reduces blood pressure [196–200]. Also, differences in solar exposure to UV radiation and synthesis of vitamin D increase at high altitudes [201]. Studies show that the proximity of vegetation is associated with lower levels of stress, diabetes mellitus, and CVD [202, 203]. Children who live in greener areas have lower levels of asthma, blood pressure, and insulin resistance [204, 205]. Socioeconomic conditions have an impact on CVD as evidenced by higher data on the incidence of the disease among the poor population. Which is also related to the supply of food and the availability of health care [206]. Exposure to synthetic chemicals and environmental pollutants can have an impact on the health of the population and is ubiquitous and unavoidable today [207]. There is evidence to suggest that chronic and persistent exposure to air pollution increases the progression of atherosclerotic lesions and has adverse effects on blood pressure regulation, peripheral thrombosis, endothelial function, and insulin sensitivity [201, 208–210]. Some studies have shown that constant exposure to noise induces stress and has an impact on cognitive function, autonomic homeostasis, and sleep, and that it increases the risk of CVD [211]. In animal models, chronic exposure to continuous noise (80-100 dB) has been shown to increase the heart rate and mean systemic arterial blood pressure, functional changes associated with increased plasma corticosterone, adrenaline, and endothelin-1 [212].
Smoking, as one of the environmental factors, has a great influence on the development of CVD. Data show that smoking reduces regional left ventricular function even in asymptomatic individuals and significantly (45% –80%) increases the risk of heart failure [213]. The reasons for the high vulnerability of cardiovascular tissue remain unclear, but may relate to poor xenobiotic metabolism in these tissues and their direct exposure to blood-borne toxins. Although the mechanisms by which smoking increases the risk of CVD are not fully known, they appear to affect CVD independently of other factors [214]. A meta-analysis of 54 different studies suggests that smoking increases LDL-C and decreases HDL, but lipid changes account for <10% of the excessive risk of CVD in smokers [215]. Similarly, although acute smoking affects blood pressure, smokers tend to maintain lower blood pressure. Smoking leads to coronary occlusion causes endothelial dysfunction and platelet adhesion to subintimal layers, thereby increasing lipid infiltration and platelet-derived growth factor- (PDGF-) mediated proliferation of smooth muscle cells [216].
Studies have shown that people with homocystinuria, which is one of the inherited recessive disorders in methionine metabolism, have a tendency to develop cardiovascular disease. Such persons have high levels of homocysteine in the circulation and urine, which has an impact on the development of atherosclerosis and in the coagulation system [217–219].
Also, patients with hyperuricemia have a tendency to develop CAD because serum uric acid levels are positively associated with arterial intima-media thickness, which is a precursor to atherosclerosis [220, 221]. In conclusion, we can greatly contribute to the prevention and severity of CVD by influencing environmental factors.
8. Pharmacological Therapeutic Possibilities
The therapeutic approach in patients with or without evidence of coronary atherosclerosis involves, first and foremost, lifestyle changes and the management of risk factors, including an effort to influence environmental factors. Beta-blockers are a class of medications that are used to protect the heart from a myocardial infarction because they may reduce myocardial oxygen consumption [222]. Potential therapeutic strategies are focused on the NO-cGMP (nitric oxide-cyclic guanosine monophosphate) pathway. Given that the NO-cGMP pathway has been implicated in the pathophysiology of heart failure, it is a promising target for therapy; although unfortunately, clinical data are not yet fully conclusive [222]. A beta-blocker such as nebivolol exerts its effect through beta-adrenoreceptors located on endothelial cells. In this way, it stimulates eNOS, which ultimately results in NO release and vasodilation. Data on the effect of nebivolol have been supported by studies such as the SENIORS (the Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors with Heart Failure) study conducted in elderly patients with heart failure [223–225].
Mihai et al. investigated the effect of vericiguat, a soluble guanylate cyclase (sGC) stimulator, on N-terminal prohormone of brain natriuretic peptide (NT-proBNP) levels in patients with chronic heart failure and reduced ejection fraction. The study concluded that among 351 patients with heart failure (HF) and reduced ejection fraction, compared with placebo, vericiguat did not have a statistically significant effect on NT-proBNP levels at 12 weeks. Therefore, the researchers suggested additional clinical trials of vericiguat based on the dose-response relationship to determine the potential role of this drug, and that phase III outcome trial is still ongoing [222]. Natriuretic peptides (NPs) via the natriuretic peptide receptor-A are known to increase intracellular cyclic guanosine monophosphate (cGMP) levels [226]. A drug such as sacubitril/valsartan that simultaneously inhibits neprilysin (neutral endopeptidase) via LBQ657 and the angiotensin II receptor has its effect in chronic heart failure with a reduced ejection fraction. The benefits of this drug are attributed to the increase in the amount of peptides that neprilysin breaks down, such as NPs, by LBQ657 and the simultaneous inhibition of the effects of angiotensin II by valsartan. NPs exert their effects by activating membrane-bound receptors paired with guanylate cyclase, which results in an increase in the second messenger cGMP and ultimately leads to vasodilation, natriuresis, diuresis, and decreased sympathetic activity. These insights are supported by the PARADIGM-HF administration trial [227]. Also, research such as PARAMOUNT, designed as a randomized, parallel-group, double-blind study in a phase II clinical trial of sacubitril/valsartan in the clinical syndrome of HF with preserved ejection fraction (HFpEF), suggested benefits in HFpEF at least in terms of NT-proBNP reduction [228].
Medication groups such as angiotensin-converting enzyme (ACE) inhibitors and statins are used in patients who have evidence of endothelial dysfunction and evidence of atherosclerosis. ACE inhibitors exert vasoprotective effects by inhibiting the renin-angiotensin axis. Statins, in addition to reducing cholesterol levels, also have an inhibitory effect on vascular inflammation, they upregulate eNOS, and enhance vascular NO bioavailability [229].
Studies to date have shown that antioxidants such as flavonoids and vitamins reduce the risk of stroke [230, 231]. Since ROS are known to occur during ischemia, reperfusion, and bleeding in the brain, several antioxidants of different chemical structures have been investigated as neuroprotective therapeutic agents for brain injuries. An example of this is the use of Vaccinium berries that have high antioxidant activity and that have been used in an animal model. They showed their neuroprotective effect due to the high total content of polyphenols [232–234]. It would be interesting to consider such antioxidants in ischemic heart disease, although conclusive evidence is lacking for now.
Resveratrol (chemical name: 3,5,4′-trihydroxy-trans-stilbene) is another polyphenol abundantly found in the skin and seeds of grapes [235, 236].
NXY-059 (chemical name: α(2,4-disulfophenyl)-N-tertbutylnitrone) is a novel nitrone free radical trapping (antioxidant) agent. This compound is a stable form of NO, capable of inhibiting the reaction of O2 - and NO to produce ONOO-. This chemical agent might thus be able to neutralize ROS [237, 238].
Therapeutic options for CMD are limited. Some studies show that inhibition of Rho-kinase might constitute one of the treatment options in patients with CMD and vasospastic angina, but this has not yet been proved [239]. Some studies show that targeting of perivascular adipose tissue to stimulate the production of vasoactive factors such as hydrogen sulphide [240] and adiponectin could be of benefit [241].
Studies show that the use of platelet inhibitors such as aspirin may have an effect on treatment in CAD but they have not been sufficiently implicated in the treatment of CMD [241]. Studied of Zhang et al. showed aspirin provides a new potential strategy for regulating cardiac microcirculation, preventing heat stress- (HS-) induced heart failure. In this study, they used a heat stress model of rat cardiac microvascular endothelial cell cultures in vitro and investigated the cell injuries and molecular resistance mechanisms of cardiac microvascular endothelial cells (CMVECs) caused by heat stress. In conclusion, aspirin treatment of CMVECs induced a significant expression of heat shock proteins (Hsp90), which promoted both Akt and M2 isoform of pyruvate kinase (PKM2) signals, which are beneficial for relieving HS damage and for maintaining the function of CMVECs [242].
Clinical research on the use of ticagrelor for microcirculation protection is still ongoing [243]. Nitrates are effective in inducing vasodilatation, and they relieve angina symptoms, but not in patients with nonobstructive CAD [244]. L-arginine is as precursor of NO, with attempted use in subjects with nonobstructive CAD [245, 246], but its use is controversial. Zibotentan and atrasentan are ETA receptor antagonists, and there are studies that have suggested them to be a potential therapeutic option in patients with microvascular dysfunction [247, 248].
Drugs or substances that modify TLR4 signaling can be very useful in treating the atherosclerotic process in the coronary arteries [249]. Some already known cardiovascular drugs may have pleiotropic anti-inflammatory and antiatherosclerotic effects achieved through TLR4 (). The well-known statin atorvastatin [249] and angiotensin-converting enzyme (ACE) inhibitors fosinopril [250] showed their antiatherosclerotic properties because they reduced the expression of the TLR4 protein in atherosclerotic lesions. Furthermore, combination treatment with atorvastatin and telmisartan (angiotensin II receptor blocker) or atorvastatin and enalapril (ACE inhibitors) in human PBMCs (peripheral blood mononuclear cells) resulted in decreased TLR4 receptor expression in patients with CAD [251]. Some studies have shown that thiazolidinediones (TZDs), such as rosiglitanose and pioglitazone, can exert their antiatherogenic effect by inhibiting the TLR4 singular pathways [252–254]. Carvedilol, a third-generation beta-blocker, decreased the TLR4 expression in AIM-induced rats [255]. Paclitaxel, an anticancer drug, has also been shown to inhibit TLR4 signaling [256]. The anesthetic propofol and ketamine have the ability to reduce ROS production and suppress the NF-κB expression and reduce IL-6 [256]. The exact mechanisms of action of these already known cardiological drugs remain to be explored in the future. Of course, there are a number of newly discovered potential TLR4 antagonists (eritoran, cyanobacterial product (CyP), EM-163, epigallocatechin-3-gallate, 6-shogaol, cinnamon extract, N-acetylcysteine, melatonin, molecular hydrogen, monoclonal antibody anti-hTLR4-IgG) which could be useful in preventing atherosclerosis in patients with CAD [257].
Also, epigenetic regulation (DNA methylation and histone acetylation) could become the most promising therapeutic target for the treatment of TLR4-mediated inflammatory disorders [258].
Tsai et al. conducted research in rat and in vitro models examining the role of IL-20 in the infarcted heart following ischemia/reperfusion injury, with the aim of discovering new therapeutic options in the treatment of ischemic heart disease. This study revealed that IL-20 and its receptors, IL-20R1 and IL-20R2, were increased in H2C2 cardiomyoblast cells and ventricular tissues subjected to prior hypoxia/reoxygenation (H/R) stimulation. The obtained results suggest that IL-20 causes an increase in Ca2+ and activation of the PKC/NADPH oxidase pathway, leading to an increase in oxidase stress and a decrease in AKT regulation. Also, IL-20 can mediate H/R-induced apoptosis via PKC/NADPH oxidase/AKT signaling. Therefore, regulation of IL-20 may contribute to cardiomyocyte apoptosis, and this might be helpful in future considerations of new therapeutic targets in the treatment of ischemic heart disease [259]. In their work, Samakova et al. combined insights into the phosphatidylinositol-3-kinase- (phosphoinositide-3-kinase-) protein kinase B (serine-threonine protein kinase) (PI3k/Akt) pathway and the association with oxidative stress, angiogenesis, and mesenchymal stem cell survival in pathophysiologic conditions in ischemia [260].
Cell therapy has long been known to be one of the options for treating ischemic heart disease when, in 1974, Friedenstein and his associates first isolated and characterized the use of mesenchymal stem cells (MSC) [261, 262]. Since then, numerous studies have been conducted to improve the use of mesenchymal stem cells in regenerative therapy. Also, the influences of biologically active molecules such as cytokines, growth factors, and chemokines were found to be important for any attempts at successful cell therapies. In addition, the PI3K/Akt pathway was determined to be one of the mechanisms of intracellular signaling that plays a role in regulating cell proliferation, differentiation, apoptosis, and migration. Therefore, the aforementioned contributors emphasized that preconditioning of MSCs is an important process for the improvement of the efficiency of signaling mechanisms [260].
Some studies show that fisetin protects against cardiac cell death through reduction of ROS production and caspase activity. In vitro studies of mammalian cardiac cell models have shown that fisetin increases the vitality of rat cardiomyocytes after hypoxia or starvation or reoxygenation. It also reduces ROS formation, activates caspases, protects from DNA damage, and ultimately inhibits apoptosis. Fisetin is a very promising drug for protection against ischemic damage after myocardial infarction and for counteracting ischemia reperfusion injury because it can, in addition, activate genes involved in cell proliferation [263].
Experimental studies in rat models have shown that cocoa flavonoids reduce inflammation, oxidative stress, and myocardial apoptosis after acute coronary ischemia-reperfusion. In these studies, cocoa extract treatment reversed membrane peroxidation and nitro-oxidative stress as well as lead to reduction of inflammatory marker levels such as IL-6 and NF-κB [264]. Verma et al. conducted an experimental study in rats that showed that morin, a bioflavonoid, has antioxidant and anti-inflammatory effects, and that it prevents apoptosis. It exerts its effects by regulating RISK/SAPK pathways. Extracellular regulated kinase (ERK), protein kinase A (Akt), and eNOS are involved in the RISK pathways. The p38 proteins and c-Jun N-terminal kinase (JNK) are involved in the SAPK pathway [265]. Syeda et al. in their study in mice investigated the cardioprotective potential of anthocyanidin against myocardial ischemia injury. In in vivo conditions, the left anterior descending coronary artery was ligated to induce myocardial ischemia in mice, whereas in in vitro conditions, neonatal mice cardiomyocytes were treated with H2O2 to induce oxidative stress. It was concluded that, in vivo and in vitro, anthocyanidin can induce a state of myocardial resistance against ischemic insult. Inhibition of the ROS/p-JNK/Bcl-2 pathway is the underlying mechanism of action of anthocyanidin [266]. Table 1 summarizes the discussed pharmacological therapeutic possibilities.
Table 1.
Discussed pharmacological therapeutic possibilities in ischemic heart disease.
| Reference | Study characteristic | Therapeutic options | Primary endpoint |
|---|---|---|---|
| Zhang X et al. [242] | Experimental study on animal models such as rat | Aspirin (platelet inhibitors) | Enhances the protection of Hsp90 from heat-stressed injury in cardiac microvascular endothelial cells through PI3K-Akt and PKM2 pathways |
| Rodius et al. [263] | In vitro studies of mammalian cardiac cell models | Fisetin (plant polyphenol from the flavonoid group) | Reduction of ROS production, protects from DNA damage |
| Verma et al. [265] | Experimental study on male albino Wistar rats | Morin (bioflavonoid) | Regulation of RISK/SAPK pathways |
| Syeda et al. [266] | Experimental study on mice | Anthocyanidins (plant pigments) | Inhibition of ROS/p-JNK/Bcl-2 pathway |
| Flather et al. Ambrosio et al. [223, 224] |
Randomized trial in elderly patients with heart failure | Nebivolol (beta-1-selective blocker), beta(3)-adrenoreceptor agonistic effect | Stimulates eNOS, NO release, vasodilatation |
| Mihai et al. [222] | Randomized trial in patients with heart failure and reduced ejection fraction | Vericiguat (a soluble guanylate cyclase stimulator) | Changes in the NT-proBNP level have not been achieved, but the phase III trial is ongoing |
| McMurray et al. [226] | Randomized, double-blind trial in patients with heart failure and reduced ejection fraction | Sacubitril/valsartan (NP degradation inhibitor/angiotensin II receptor inhibitor) vs. enalapril | Increase cGMP, vasodilatation |
| Solomon et al. [227] | Randomized, double-blind study in a phase II trials, in patients with heart failure and reduced ejection fraction | Sacubitril/valsartan (NP degradation inhibitor/angiotensin II receptor inhibitor) vs. valsartan | Changes in NT-proBNP |
| Carmine et al. [228] | Randomized, prospective, single-blind, placebo-controlled fashion in patients who have chest pain and angiographically normal epicardial vessels | Ramipril (ACE inhibitor) and atorvastatin (statins) | Reduced SOD activity, low superoxide anion level |
| Amir et al. [245] | Randomized study, double- blind in patients with patients without significant CAD on coronary angiography | L-arginine (substrate for NO synthase) | Improve endothelial function, increase NO and NO inhibits the production of endothelin via cGMP pathway |
| Martin et al. [247] | Single-center, double-blind, randomized controlled trial in patients with CMD | Atrasentan (ETA receptor antagonist) | Supports the role of the endogenous endothelin system |
eNOS: endothelial nitric oxide synthase; NO: nitric oxide; NT-proBNP: N-terminal prohormone of brain natriuretic peptide; cGMP: cyclic guanosine monophosphate; ACE inhibitor: angiotensin converting enzyme inhibitors; SOD: superoxide dismutase; Hsp90: heat shock proteins 90; PI3K/Akt: phosphoinositide-3-kinase–protein kinase; PKM2: M2 isoform of pyruvate kinase; ETA receptor: endothelin-A receptor; CMD: coronary microvascular dysfunction; ROS: reactive oxygen species; RISK/SAPK pathway: extracellular regulated kinase (ERK), protein kinase A (Akt) and eNOS/p38 proteins, and c-Jun N-terminal kinase (JNK); ROS/p-JNK/Bcl-2 pathway: reactive oxygen species/stress-activated c-Jun N-terminal kinase/B-cell lymphoma.
9. Biomarkers of Oxidative Stress in Ischemic Heart Disease
Many oxidative stress-related biomarkers have been recently proposed, reflecting different and independent pathways, including oxidative and antioxidant ones [267]. Some reliable and simple tests have been presented to estimate oxidative stress in vivo, and also a calculation of a global oxidative stress index (OSI) is described, which represents the ratio of total oxidant status to total antioxidant status [268], and it showed higher values in patients with CAD [269]. Some of the oxidative stress-related biomarkers seem promising for future clinical use in understanding the pathogenesis and predicting clinical outcomes of ischemic heart disease. Although there are a lot of common biological features between ACS and stable CAD, there are also many differences resulting in variation of levels of biomarkers included in different oxidative stress-related pathways [270].
Measurement of reactive oxygen metabolites (ROM) based on the conversion of hydroperoxides to alkoxyl and peroxyl radicals under acidic conditions in combination with estimation of total antioxidant capacity (OXY) can quantify oxidative stress levels [271, 272]. Previous studies evaluated levels of ROM and OXY in patients with cardiovascular disease in comparison with the general population and evaluated their prediction value in adverse CV events [268, 271, 273]. Lubrano et al. examined ROM and OXY levels during acute myocardial infarction (AMI) which showed a progressive increase and then decrease suggesting significant rise of oxidative stress level during AMI [270]. The level of ROM values was higher in stable CAD in comparison with ACS patients, indicating that this parameter reflects the chronic oxidative stress status [270]. OXY was progressively reduced in stable CAD and more in ACS compared with the control group, showing severe acute harm to the antioxidant system in ischemic disease, especially during myocardial reperfusion injury [270]. This fact is further enhanced by findings that different vitamins and antioxidant enzymes were also reduced during acute myocardial infarction [270, 274].
Low levels of NO are related to endothelial dysfunction and many CV events, but its direct quantification is difficult so it can be estimated by measurements of its stable metabolites—nitrite/nitrate (NOX) [275]. NOX are end-products of NO metabolism and a reliable index of NO production. In previous studies, there are controversial results regarding NOX levels in CV disease and CV risk. Some of them revealed higher levels of NOX in a group with CAD and AMI, which can be explained by the fact that increase in systemic NOX can be a consequence of activation of inducible NO synthase as a result of vascular injury, without restoration of endothelial NO release [276, 277]. Other studies showed reduced levels of NOX during acute myocardial infarction, pointing to deteriorated NO levels during an acute ischemic event. Further researches are needed to understand meaning of different levels of NOX [270].
Several studies suggested that Ox-LDL may play an important role in the pathogenesis of atherosclerosis, plaque rupture, and onset of ACS [278, 279]. Uptake of Ox-LDL by macrophages and activated smooth muscle cells probably transforms these cells into foam cells which are found in the atherosclerotic intima. Endothelial uptake of Ox-LDL depends on receptors expressed on the cell surface. LOX-1 is a major receptor for Ox-LDL, and its expression is induced by oxidative stress, hemodynamic stimuli, and inflammatory cytokines, and it is related to development of atherosclerosis and plaque instability. It is highly expressed in luminal endothelial cells in the early stage of atherogenesis as well as in intimal neovascular endothelial cells of advanced plaques and released during plaque rupture, promising to be a good marker of plaque instability [280]. Soluble LOX-1 (sLOX-1) is proposed as a potential marker for identification of ACS in the early stage [281, 282] with the peak time being even earlier than troponin T [281]. Some studies suggest that levels of sLOX-1 might begin to rise before onset of ACS but that could be a subject of further research [282].
In addition to these biomarkers, there is a large cohort study that found an association of urinary oxidized guanine/guanosine (OxGua) and 8-isoprostane levels with CVD mortality prediction and with myocardial infarction incidence in obese subjects [283]. Other studies also showed that elevated plasmatic levels of 8-isoprostane are associated with acute myocardial infarction and also with the severity and extent of CAD [284, 285].
10. Epigenetic (Dys)regulation MicroRNA in CAD
Small noncoding RNAs (microRNAs or miRs) of 19–25 nucleotides (nt) regulate the expression of more than 30% of human genes at the posttranscriptional level [286]. They are involved in intracellular and intercellular signaling and circulate in the blood in stable forms due to packaging into apoptotic bodies, microvesicles (MV), exosomes, and lipoproteins (Lp) [287]. Each miR from RNA is transcribed by RNA polymerase II and, less frequently, by RNA polymerase III [288, 289]. First, Pri-miRNA is formed, containing a canonical hairpin structure, a 50 cap, and a 30 poly-A tail, which is processed in the nucleus by Drosha-DGCR8 (Di George Syndrome Critical Region Gene 8), then pre-miRNA is formed to a hairpin form which is exported to the cytoplasm via Exportin-5 and then further truncated by the RNase III enzyme complex Dicer/TRBP (TAR RNA-binding protein), yielding a mature duplex of miRNA (miRNA-5p and miRNA3p) [290]. Mature miRs bind at a specific site of the messenger RNA (mRNA) in the Argonaute protein multiprotein complex, known as RNA-induced attenuation complex (RISC), providing sequence-specific attenuation by degrading messenger RNA (mRNA) and/or inhibiting its translation [286]. Each miR can target several mRNAs that act in several important cellular functions such as differentiation, proliferation, and apoptosis in the cardiovascular system [291]. MicroRNAs that can regulate cellular homeostasis of oxidative stress by modulating the expression of antioxidant genes and the expression of enzymes that generate ROS are called redox sensitive micorRNAs or redoximiR [292]. RedoximiR achieves its oxidative or antioxidant effects by directly regulating the posttranscriptional level of the redox-sensitive nuclear factor Nrf2. Of course, there are also miRs that achieve their effects independently of Nrf2 [293]. Nrf is considered a major regulator of cell survival in oxidative stress because it controls the basal and induced expression of a number of important antioxidant genes via a cis-acting element, designated the antioxidant-response element (ARE), in the promoter of target genes [293]. Some of these genes are the genes for heme oxygenase-1 (HO-1), gamma-glutamylcysteine synthetase, thioredoxin reductase, glutathione-S-transferase, and NAD (P) H: quinone oxidoreductase [293]. Protein kinase C (PKC), mitogen-activated protein kinase (MAPK), and phosphotidylinositol 3-kinase (PI3K) are involved in the regulation of Nrf2/ARE signaling [293]. Numerous microRNAs participate in the regulation of Nrf2 via certain coregulatory proteins such as Kelch-like ECH-associated protein 1 (Keap1), BTB and CNC homolog 1 (Bach1), Parkinson's protein 7 (PARK7/DJ-1), and small masculoaponeurotic fibrosarcoma (Maf) proteins. Most miRs act by the downregulation mechanism Nrf2 (miR-153, miR-27a, miR-142-5p, miR144 miR-28, and miR-34a), while some act by the upregulation mechanism Nrf2 (miR 200-a, miR-136-3p, miR-128). Also, miRs regulate the expression of key enzymes that generate ROS which can lead to modification of the biogenesis of miRs. Cellular oxidative stress can alter miR biogenesis during processing in the nucleus and cytoplasm, altering its stability, functionality, and binding affinity for target promoter sites [292] (Figure 5).
Figure 5.
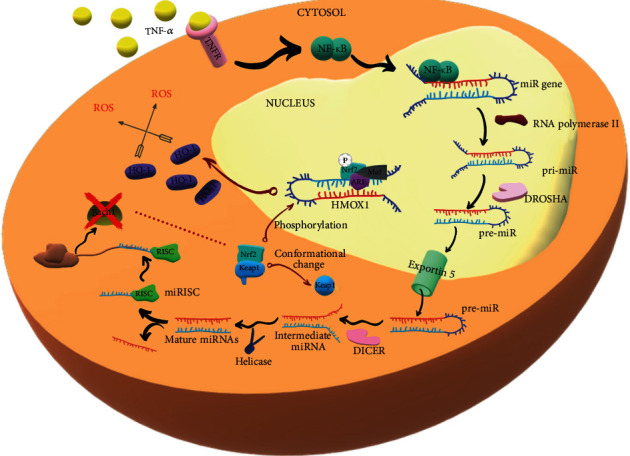
Biogenesis of miR-155. ARE: antioxidant responsive element; Bach 1: BTB domain and CNC homolog 1; DICER: ribonuclease DICER; DROSHA: ribonuclease DROSHA; HO-1: heme oxygenase-1; KEAP1: Kelch-like ECH-associated protein 1; MAF: musculoaponeurotic fibrosarcoma; miR: microRNA; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NRF2: nuclear factor erythroid 2-related factor 2; RISC: RNA-induced silencing complex; ROS: reactive oxygen species; TNF-α: tumor necrosis factor-α; TNFR: tumor necrosis factor receptor.
The primary pathological process that causes CAD is atherosclerosis triggered by oxidative stress [291]. miR is involved in almost all steps of atherosclerosis and CAD, such as endothelial damage and endothelial dysfunction, oxidative enzyme expression, inflammatory molecule expression, monocyte invasion and activation, LDL oxidation, platelet function, vascular smooth muscle response, and angiogenesis [294]. The miRs involved in oxidative stress and associated with CAD are miR-155, miR34a, and miR-136-3p.
miR-155 was expressed in mononuclear and endothelial cells [295]. Ox-LDL can induce the expression of miR-155 [295]. miR-155 may inhibit the inflammatory response thereby reducing enhanced lipid oxidation in macrophages during oxidative stress [295]. Inhibition of endogenous miR-155 in THP-1 macrophages resulted in increased lipid uptake and release of several cytokines, including interleukin (IL)-6, -8, and tumor necrosis factor-α (TNF-α) [296]. The overexpression of miR-155 may induce apoptosis of Ox-LDL attacked macrophages [295]. Levels of miR-155 were decreased in plasma and peripheral blood mononuclear cells (PBMCs) in patients with CAD. The study showed that miR-155 levels in peripheral blood mononuclear cells or plasma were inversely correlated with the severity of stenotic lesions in the coronary arteries [297]. miR-155 acts on the Nrf2 pathway whose activation of Nrf2 can reduce the degree of oxidative stress in mitochondria and can reduce oxidative stress and the inflammatory response of vascular endothelial cells [298]. Bach1 in association with the masculoaponeurotic fibrosarcoma (Maf) protein dominantly hides ARE sequences from Nrf2 binding of transcription factor [299, 300]. Oxidative stress and inflammation-induced tumor necrosis factor-α (TNF-α) activate TNFR (tumor necrosis factor receptor) [301]. NF-κB is in an inactive state bound to the inhibitor protein IκB, and activation of TNFR leads to activation of IκB kinases (IKKs) which by phosphorylation dislocates IκB with NF-κB [302]. Phosphorylated IB is degraded by the 26s proteasome [303]. Nuclear translocation of NF-κB stimulates the expression of miR-155 which inhibits the production of Bach1 protein, allowing the binding of the Nrf2 transcription factor to the ARE sequence [299]. Under normal physiological conditions, Nrf2 is bound to Kelch-like ECH-associated protein-1 (Keap1) within the cytoplasm [304].When the Bach1 expression is reduced and cells are attacked by oxidative stress, phosphorylation of Nrf2 via (MAPK), protein kinase C (PKC) and (phosphoinositide 3-kinase (PI3K) and its transport into the nucleus and binding to the ARE sequence together with Maf protein occurs [295]. Consequently, there is an increased synthesis of HO-1 (heme oxygenase-1) [299]. HO-1 is a microsomal enzyme induced in oxidative stress that metabolizes heme to biliverdin, carbon monoxide (CO), and iron, and CO has antiapoptotic and anti-inflammatory properties and may act as a vasodilator in atherogenesis when NO bioavailability is reduced due to ROS inactivation [305]. Numerous studies have shown a cardioprotective effect of HO-1 [306–308]. Thus, it is clear that suppression of the Bach1 protein expression alters cellular redox signaling and enhances the expression of antioxidant enzymes induced by Nrf2 [309].
miR-34a induced by oxidative stress via PI3K signaling in EPCs obtained from patients with CAD reduces the expression of the enzymes SIRT1 and SIRT6 involved in histone deacetylation and DNA repair [310, 311]. Silencing the entire miR-34 family may protect the heart from pathological myocardial remodeling [312]. miR-34a induces postacute MI, and inhibition of miR-34a improves recovery of cardiac contractile function after acute MI [313].
miR-136-3p can reduce oxidative stress and inflammatory response and consequent pathological damage to myocardial tissue by inhibiting the expression of the target EIF5A2 gene thereby blocking the Rho A/ROCK signaling pathway in the CAD rat myocardial tissue and models of cardiac microvascular endothelial cell (CMEC) injury [314].
Numerous studies have shown that miR can serve as an important biomarker for early detection of CAD, differentiation of patients with or without CAD, as well as patients with stable CAD or unstable CAD, and assessment of disease severity, as prognostic indicators and indicator of restenosis after stenting (in-stent restenosis, ISR) [291]. miRNAs are not specific because each miRNA can be elevated or decreased in different disease conditions, so this is a big challenge for researchers [291].
Increased levels of miR-31, miR-720, miR-181, and miR-208a may have potential roles for early CAD detection [315–317]. miR-208a is a highly selective cardiac RNA that is overexpressed 3 hours after myocardial infarction (MI) and correlates with increased cardiac troponin (cTn) I levels [318]. MiR-208a has been shown to have superiority in early diagnosis of MI over cTn [318]. Devaux et al. argue that miR-499-5p which is myosin gene-regulated has higher diagnostic accuracy in correlation with cTN than miR-208a [319].
To distinguish CAD from non-CAD patients, numerous miRNAs were detected that were significantly increased in patients with CAD (miR-149, miR-765, miR-424, miR-133a miR-206, miR-574-5p, miR-135a, miRNA-24, miRNA-33, miRNA-103a, miRNA-122). On the other hand, levels of miRNA-23a, miR-19a, miR-484, miR155, miR-222, miR-145, miR-29a, miR-378, miR-342, miR-181d, miR-150, and miR-30e-5p were found to be reduced in the blood of patients with CAD compared to healthy controls [320]. Faccin et al. state that the combination of three miRNAs (miRNA-155, -145, and flight-7c) has better classification power than just one miRNA [321]. Two studies have shown that increased plasma levels of miRNA-133a, miR-126, and miR-1 are useful for the diagnosis of unstable CAD [322, 323]. MiR-145 is significantly elevated in unstable angina compared to stable angina, but so far, no miR or cascade of miR has been detected in the blood of patients by which we will distinguish these two types of angina [287]. Another study showed that miR-134, miR-198, and miR-370 were increased in unstable versus stable angina pectoris [324]. Li suggested that six microRNAs (miR-1/134/186/208a and 208b/233/499-5p) have increased sensitivity and specificity in MI detection, although miR208 and miR499 were significantly higher in patients with pecotris angina compared to IM [325]. Ward et al. demonstrated that myocardial infarction miRNA-25-3p, miRNA-221-3p, and miRNA-374b-5p are highly present in the blood of patients with STEMI and miRNA 221-3p and 483-5 in patient with NSTEMI [326].
To assess the severity of coronary artery disease, miR-133a was presented as a potential biomarker showing the presence of coronary artery stenosis and is a better indicator of assessing the severity of CAD compared to cTn1 [327]. Other miRNA-208a, miRNA-155, and miRNA-223 strongly correlated with the CAD severity assessment [291]. Levels of miRNA-92a lipoprotein-2 (HDL-2) HDL-2 miRNA-92a, and HDL-3 miRNA-486 could be signals of severe CAD and threatened myocardial infarction [328]. Oxidative stress-induced microRNA-92a (miR-92a) leads to endothelial dysfunction caused by activation of sirtuin 1, Krüppel-like factor 2, and Krüppel-like factor 4, leading to NOD-like receptor family pyrin domain-containing 3 inflammasome activation and endothelial nitric oxide synthase inhibition [329]. The expression of miRNA-21 in the macrophages of uncalcified coronary artery lesions was significantly higher than in calcified lesions [330]. miR-100 can be released into the coronary circulation from sensitive coronary plaques and can therefore be useful as a biomarker of plaque vulnerability [331].
Some vascular miRs may have a prognostic potential for coronary artery disease. The increased expression of miRNA-126 and miRNA-199a in circulating microvesicles is associated with a lower cardiovascular mortality rate [332]. Also, elevated levels of miRNA-197, miRNA-223, miRNA-133a, and miRNA-208b were significantly associated with higher mortality rates in patients with CAD [333, 334]. In obese patients, miR-181a levels within polymorphonuclear cells are increased, which is associated with an increased risk of developing CAD [316].
As new noninvasive potential biomarkers for assessing the occurrence of ISR, levels of circulating miRNA-143, miRNA-145, and miRNA-181b and increased levels of miRNA-185 and miRNA-155 were reduced compared to non-ISR [335, 336].
During the development and progression of atherosclerotic plaques, miR-92a, miR-100, miR-126, miR-127, and miR-145 are mostly released as a result of vascular damage. The miRNAs released from myeloid cells involved in the formation of atherosclerotic lesions are miR-155 and miR-223. During myocardial injury in patients with CAD, miR-133a, miR-208a, and miR-499 are mostly released into the coronary circulation [337].
RedoximiR is an important regulator of the cellular redox status and new valuable biomarkers that constitute a key step in the pathogenesis of CAD. Due to their cell-type specificity, abundance, and stability in most solid and liquid clinical specimens, they provide the opportunity for further study to expand our understanding of CAD pathogenesis and open up new innovative diagnostic and therapeutic approaches. For now, the following therapeutic strategies are being studied: inhibition of premicroRNA export from the nucleus, inhibition of premicroRNA transcription into mature microRNAs, or competitive inhibition via complementary binding to specific microRNAs [287]. Whether we can block or prevent the progression of atherosclerosis and CAD development in the future remains to be patiently awaited.
11. Final Remarks
This review article discussed evidence associating oxidative stress with the pathogenesis and occurrence of ischemic heart disease. Since oxidative stress is an important group of processes in a number of disorders connected with vascular structure and function, it is not surprising that there are many indications that some of the factors implicated in oxidative stress play roles in vascular disease mechanisms. From this review, however, it is likewise clear that the interactions of the many factors of oxidative stress that contribute to vascular disease mechanisms in ischemic heart disease are very complex and not yet clearly nor completely understood. At the same time, it would be desirable and interesting to therapeutically target oxidative stress, in hopes of developing better therapeutic strategies for ischemic heart disease, which after many years of various treatment approaches and strategy changes is still not managed optimally and with satisfactory results in a large number of affected patients. The main prerequisite for the development of such therapeutic strategies targeting oxidative stress is, however, a much better understanding of all the specific roles of ROS in specific pathophysiological mechanisms, as well as the interactions of ROS with other signaling systems. Only then can a targeted therapeutic approach be successful, effective, and with a limited spectrum of adverse effects. To achieve such an understanding of the roles of oxidative stress in ischemic heart disease, more research in this area is warranted.
Conflicts of Interest
The authors have no conflict of interest to declare.
References
- 1.Gimbrone M. A., Jr., García-Cardeña G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovascular Pathology. 2013;22(1):9–15. doi: 10.1016/j.carpath.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ninić A., Bogavac-Stanojević N., Sopić M., et al. Superoxide dismutase isoenzymes gene expression in peripheral blood mononuclear cells in patients with coronary artery disease. Journal of Medical Biochemistry. 2019;38(3):284–291. doi: 10.2478/jomb-2018-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin C. P., Lin F. Y., Huang P. H., et al. Endothelial progenitor cell dysfunction in cardiovascular diseases: role of reactive oxygen species and inflammation. BioMed Research International. 2013;2013:10. doi: 10.1155/2013/845037.845037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kattoor A. J., Pothineni N. V. K., Palagiri D., Mehta J. L. Oxidative stress in atherosclerosis. Current Atherosclerosis Reports. 2017;19(11) doi: 10.1007/s11883-017-0678-6. [DOI] [PubMed] [Google Scholar]
- 5.Münzel T., Camici G. G., Maack C., Bonetti N. R., Fuster V., Kovacic J. C. Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. Journal of the American College of Cardiology. 2017;70(2):212–229. doi: 10.1016/j.jacc.2017.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Förstermann U., Xia N., Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circulation Research. 2017;120(4):713–735. doi: 10.1161/CIRCRESAHA.116.309326. [DOI] [PubMed] [Google Scholar]
- 7.Thygesen K., Alpert J. S., Jaffe A. S., et al. Fourth universal definition of myocardial infarction (2018) Circulation. 2018;138(20):e618–e651. doi: 10.1161/CIR.0000000000000617. [DOI] [PubMed] [Google Scholar]
- 8.Liu H., Zhuang J., Tang P., Li J., Xiong X., Deng H. The role of the gut microbiota in coronary heart disease. Current Atherosclerosis Reports. 2020;22(12) doi: 10.1007/s11883-020-00892-2. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez-Rodriguez E., Egea-Zorrilla A., Plaza-Díaz J., et al. The gut microbiota and its implication in the development of atherosclerosis and related cardiovascular diseases. Nutrients. 2020;12(3) doi: 10.3390/nu12030605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amin A. R., Attur M. G., Pillinger M., Abramson S. B. The pleiotropic functions of aspirin: mechanisms of action. Cellular and Molecular Life Sciences. 1999;56(3-4):305–312. doi: 10.1007/s000180050432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chrysant S. G., Chrysant G. S. The pleiotropic effects of angiotensin receptor blockers. The Journal of Clinical Hypertension. 2006;8(4):261–268. doi: 10.1111/j.1524-6175.2005.05264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Profumo E., Buttari B., Saso L., Rigano R. Pleiotropic effects of statins in atherosclerotic disease: focus on the antioxidant activity of atorvastatin. Current Topics in Medicinal Chemistry. 2014;14(22):2542–2551. doi: 10.2174/1568026614666141203130324. [DOI] [PubMed] [Google Scholar]
- 13.Ibanez B., James S., Agewall S., et al. 2017 ESC Guidelines for the management of acute myocrdial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC) European Heart Journal. 2018;39(2):119–177. doi: 10.1093/eurheartj/ehx393. [DOI] [PubMed] [Google Scholar]
- 14.Knuuti J., Wijns W., Saraste A., et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. European Heart Journal. 2020;41(3):407–477. doi: 10.1093/eurheartj/ehz425. [DOI] [PubMed] [Google Scholar]
- 15.Thygesen K., Alpert J. S., Jaffe A. S., et al. Fourth universal definition of myocardial infarction (2018) European Heart Journal. 2019;40(3):237–269. doi: 10.1093/eurheartj/ehy462. [DOI] [PubMed] [Google Scholar]
- 16.Roffi M., Patrono C., Collet J. P., et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC) European Heart Journal. 2016;37(3):267–315. doi: 10.1093/eurheartj/ehv320. [DOI] [PubMed] [Google Scholar]
- 17.Agewall S., Beltrame J. F., Reynolds H. R., et al. ESC working group position paper on myocardial infarction with non-obstructive coronary arteries. European Heart Journal. 2017;38(3):143–153. doi: 10.1093/eurheartj/ehw149. [DOI] [PubMed] [Google Scholar]
- 18.Urso C., Caimi G. Oxidative stress and endothelial dysfunction. Minerva Medica. 2011;102(1):59–77. [PubMed] [Google Scholar]
- 19.Cai H., Harrison D. G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circulation Research. 2000;87(10):840–844. doi: 10.1161/01.RES.87.10.840. [DOI] [PubMed] [Google Scholar]
- 20.Channon K. M., Qian H. S., George S. E. Nitric oxide synthase in atherosclerosis and vascular Injury. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(8):1873–1881. doi: 10.1161/01.ATV.20.8.1873. [DOI] [PubMed] [Google Scholar]
- 21.Panza J. A., García C. E., Kilcoyne C. M., Quyyumi A. A., Cannon R. O., III Impaired endothelium-dependent vasodilation in patients with essential hypertension: evidence that nitric oxide abnormality is not localized to a single signal transduction pathway. Circulation. 1995;91(6):1732–1738. doi: 10.1161/01.CIR.91.6.1732. [DOI] [PubMed] [Google Scholar]
- 22.Schiffrin E. L., Lipman M. L., Mann J. F. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116(1):85–97. doi: 10.1161/CIRCULATIONAHA.106.678342. [DOI] [PubMed] [Google Scholar]
- 23.Hink U., Li H., Mollnau H., et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circulation Research. 2001;88(2):E14–E22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- 24.Higashi Y., Maruhashi T., Noma K., Kihara Y. Oxidative stress and endothelial dysfunction: clinical evidence and therapeutic implications. Trends in Cardiovascular Medicine. 2014;24(4):165–169. doi: 10.1016/j.tcm.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Gimbrone M. A., Jr. Vascular endothelium: an integrator of pathophysiologic stimuli in atherosclerosis. The American Journal of Cardiology. 1995;75(6):67B–70B. doi: 10.1016/0002-9149(95)80016-L. [DOI] [PubMed] [Google Scholar]
- 26.Drexler H. Endothelial dysfunction: clinical implications. Progress in Cardiovascular Diseases. 1997;39(4):287–324. doi: 10.1016/S0033-0620(97)80030-8. [DOI] [PubMed] [Google Scholar]
- 27.Mensah G. A. Healthy endothelium: the scientific basis for cardiovascular health promotion and chronic disease prevention. Vascular Pharmacology. 2007;46(5):310–314. doi: 10.1016/j.vph.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 28.Zhang D. X., Gutterman D. D. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. American Journal of Physiology. Heart and Circulatory Physiology. 2007;292(5):H2023–H2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]
- 29.Ignarro L. J., Buga G. M., Wood K. S., Byrns R. E., Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proceedings of the National Academy of Sciences. 1987;84(24):9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilcox J. N., Subramanian R. R., Sundell C. L., et al. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(11):2479–2488. doi: 10.1161/01.ATV.17.11.2479. [DOI] [PubMed] [Google Scholar]
- 31.Harrison D. G. Endothelial function and oxidant stress. Clinical Cardiology. 1997;20(S2):II-11–II-17. doi: 10.1002/j.1932-8737.1997.tb00007.x. [DOI] [PubMed] [Google Scholar]
- 32.Montezano A. C., Touyz R. M. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic & Clinical Pharmacology & Toxicology. 2012;110(1):87–94. doi: 10.1111/j.1742-7843.2011.00785.x. [DOI] [PubMed] [Google Scholar]
- 33.Luo S., Lei H., Qin H., Xia Y. Molecular mechanisms of endothelial NO synthase uncoupling. Current Pharmaceutical Design. 2014;20(22):3548–3553. doi: 10.2174/13816128113196660746. [DOI] [PubMed] [Google Scholar]
- 34.Munzel T., Daiber A., Ullrich V., Mulsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(8):1551–1557. doi: 10.1161/01.ATV.0000168896.64927.bb. [DOI] [PubMed] [Google Scholar]
- 35.Forstermann U., Munzel T. Endothelial nitric oxide synthase in vascular disease. Circulation. 2006;113(13):1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 36.Förstermann U., Sessa W. C. Nitric oxide synthases: regulation and function. European Heart Journal. 2012;33(7):829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanders S. A., Eisenthal R., Harrison R. NADH oxidase activity of human xanthine oxidoreductase–generation of superoxide anion. European Journal of Biochemistry. 1997;245(3):541–548. doi: 10.1111/j.1432-1033.1997.00541.x. [DOI] [PubMed] [Google Scholar]
- 38.Guzik T. J., West N. E., Black E., et al. Vascular superoxide production by NAD (P) H oxidase: association with endothelial dysfunction and clinical risk factors. Circulation Research. 2000;86(9):E85–E90. doi: 10.1161/01.res.86.9.e85. [DOI] [PubMed] [Google Scholar]
- 39.Laursen J. B., Rajagopalan S., Galis Z., Tarpey M., Freeman B. A., Harrison D. G. Role of superoxide in angiotensin II–induced but not catecholamine-induced hypertension. Circulation. 1997;95(3):588–593. doi: 10.1161/01.CIR.95.3.588. [DOI] [PubMed] [Google Scholar]
- 40.Nakazono K., Watanabe N., Matsuno K., Sasaki J., Sato T., Inoue M. Does superoxide underlie the pathogenesis of hypertension? Proceedings of the National Academy of Sciences. 1991;88(22):10045–10048. doi: 10.1073/pnas.88.22.10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki H., Swei A., Zweifach B. W., Schmid-Schonbein G. W. In vivo evidence for microvascular oxidative stress in spontaneously hypertensive rats: hydroethidine microfluorography. Hypertension. 1995;25(5):1083–1089. doi: 10.1161/01.HYP.25.5.1083. [DOI] [PubMed] [Google Scholar]
- 42.Ohara Y., Peterson T. E., Harrison D. G. Hypercholesterolemia increases endothelial superoxide anion production. The Journal of Clinical Investigation. 1993;91(6):2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kerr S., Brosnan M. J., McIntyre M., Reid J. L., Dominiczak A. F., Hamilton C. A. Superoxide anion production is increased in a model of genetic hypertension: role of the endothelium. Hypertension. 1999;33(6):1353–1358. doi: 10.1161/01.HYP.33.6.1353. [DOI] [PubMed] [Google Scholar]
- 44.Heitzer T., Brockhoff C., Mayer B., et al. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circulation Research. 2000;86(2):E36–E41. doi: 10.1161/01.res.86.2.e36. [DOI] [PubMed] [Google Scholar]
- 45.Incalza M. A., D'Oria R., Natalicchio A., Perrini S., Laviola L., Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascular Pharmacology. 2018;100:1–19. doi: 10.1016/j.vph.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 46.Förstermann U. Nitric oxide and oxidative stress in vascular disease. Pflügers Archiv - European Journal of Physiology. 2010;459(6):923–939. doi: 10.1007/s00424-010-0808-2. [DOI] [PubMed] [Google Scholar]
- 47.Sima P., Vannucci L., Vetvicka V. Atherosclerosis as autoimmune disease. Annals of Translational Medicine. 2018;6(7) doi: 10.21037/atm.2018.02.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malekmohammad K., Sewell R. D. E., Rafieian-Kopaei M. Antioxidants and atherosclerosis: mechanistic aspects. Biomolecules. 2019;9(8) doi: 10.3390/biom9080301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kattoor A. J., Goel A., Mehta J. L. LOX-1: regulation, signaling and its role in atherosclerosis. Antioxidants. 2019;8(7) doi: 10.3390/antiox8070218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yeh H. L., Kuo L. T., Sung F. C., Yeh C. C. Association between polymorphisms of antioxidant gene (MnSOD, CAT, and GPx1) and risk of coronary artery disease. BioMed Research International. 2018;2018:8. doi: 10.1155/2018/5086869.5086869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tibaut M., Petrovi| D. Oxidative stress genes, antioxidants and coronary artery disease in type 2 diabetes mellitus. Cardiovascular & Hematological Agents in Medicinal Chemistry. 2016;14(1):23–38. doi: 10.2174/1871525714666160407143416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peng W., Cai G., Xia Y., et al. Mitochondrial dysfunction in atherosclerosis. DNA and Cell Biology. 2019;38(7):597–606. doi: 10.1089/dna.2018.4552. [DOI] [PubMed] [Google Scholar]
- 53.Yu E., Calvert P. A., Mercer J. R., et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128(7):702–712. doi: 10.1161/CIRCULATIONAHA.113.002271. [DOI] [PubMed] [Google Scholar]
- 54.Yu E. P. K., Reinhold J., Yu H., et al. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37(12):2322–2332. doi: 10.1161/ATVBAHA.117.310042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahn S. Y., Choi Y. S., Koo H. J., et al. Mitochondrial dysfunction enhances the migration of vascular smooth muscles cells via suppression of Akt phosphorylation. Biochimica et Biophysica Acta. 2010;1800(3):275–281. doi: 10.1016/j.bbagen.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Z., Meng P., Han Y., et al. Mitochondrial DNA-LL-37 complex promotes atherosclerosis by escaping from autophagic recognition. Immunity. 2015;43(6):1137–1147. doi: 10.1016/j.immuni.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 57.Chistiakov D. A., Shkurat T. P., Melnichenko A. A., Grechko A. V., Orekhov A. N. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Annals of Medicine. 2018;50(2):121–127. doi: 10.1080/07853890.2017.1417631. [DOI] [PubMed] [Google Scholar]
- 58.Ruan C. Endothelial progenitor cells and atherosclerosis. Frontiers in Bioscience. 2013;18(4):1194–1201. doi: 10.2741/4172. [DOI] [PubMed] [Google Scholar]
- 59.Virag L. Structure and function of poly (ADP-ribose) polymerase-1: role in oxidative stress-related pathologies. Current Vascular Pharmacology. 2005;3(3):209–214. doi: 10.2174/1570161054368625. [DOI] [PubMed] [Google Scholar]
- 60.Le Guezennec X., Brichkina A., Huang Y. F., Kostromina E., Han W., Bulavin D. V. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metabolism. 2012;16(1):68–80. doi: 10.1016/j.cmet.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 61.Perrotta I., Aquila S. The role of oxidative stress and autophagy in atherosclerosis. Oxidative Medicine and Cellular Longevity. 2015;2015:10. doi: 10.1155/2015/130315.130315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kluge M. A., Fetterman J. L., Vita J. A. Mitochondria and endothelial function. Circulation Research. 2013;112(8):1171–1188. doi: 10.1161/CIRCRESAHA.111.300233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plüddemann A., Neyen C., Gordon S. Macrophage scavenger receptors and host-derived ligands. Methods. 2007;43(3):207–217. doi: 10.1016/j.ymeth.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 64.Kunjathoor V. V., Febbraio M., Podrez E. A., et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. The Journal of Biological Chemistry. 2002;277(51):49982–49988. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- 65.Linton M. F., Fazio S. Class A scavenger receptors, macrophages, and atherosclerosis. Current Opinion in Lipidology. 2001;12(5):489–495. doi: 10.1097/00041433-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 66.Nozaki S., Kashiwagi H., Yamashita S., et al. Reduced uptake of oxidized low density lipoproteins in monocyte-derived macrophages from CD36-deficient subjects. Journal of Clinical Investigation. 1995;96(4):1859–1865. doi: 10.1172/JCI118231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roshan M. H., Tambo A., Pace N. P. The role of TLR2, TLR4, and TLR9 in the pathogenesis of atherosclerosis. International Journal of Inflammation. 2016;2016:11. doi: 10.1155/2016/1532832.1532832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cole J. E., Georgiou E., Monaco C. The expression and functions of toll-like receptors in atherosclerosis. Mediators of Inflammation. 2010;2010:18. doi: 10.1155/2010/393946.393946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghosh S., May M. J., Kopp E. B. NF-κB and rel proteins: evolutionarily conserved mediators of immune responses. Annual Review of Immunology. 1998;16(1):225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 70.Yang L., Gao C. MiR-590 Inhibits endothelial cell apoptosis by inactivating the TLR4/NF-κB pathway in atherosclerosis. Yonsei Medical Journal. 2019;60(3):298–307. doi: 10.3349/ymj.2019.60.3.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Howell K. W., Meng X., Fullerton D. A., Jin C., Reece T. B., Cleveland J. C. Toll-like receptor 4 mediates oxidized LDL-induced macrophage differentiation to foam cells. Journal of Surgical Research. 2011;171(1):e27–e31. doi: 10.1016/j.jss.2011.06.033. [DOI] [PubMed] [Google Scholar]
- 72.Ishikawa Y., Satoh M., Itoh T., Minami Y., Takahashi Y., Akamura M. Local expression of Toll-like receptor 4 at the site of ruptured plaques in patients with acute myocardial infarction. Clinical Science. 2008;115(4):133–140. doi: 10.1042/CS20070379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gargiulo S., Gamba P., Testa G., et al. Relation between TLR4/NF-κB signaling pathway activation by 27-hydroxycholesterol and 4-hydroxynonenal, and atherosclerotic plaque instability. Aging Cell. 2015;14(4):569–581. doi: 10.1111/acel.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miller Y. I., Viriyakosol S., Binder C. J., Feramisco J. R., Kirkland T. N., Witztum J. L. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. The Journal of Biological Chemistry. 2003;278(3):1561–1568. doi: 10.1074/jbc.M209634200. [DOI] [PubMed] [Google Scholar]
- 75.Prakash P., Kulkarni P., Lentz S. R., Chauhan A. K. Cellular fibronectin containing extra domain A promotes arterial thrombosis in mice through platelet Toll-like receptor 4. Blood. 2015;125(20):3164–3172. doi: 10.1182/blood-2014-10-608653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.M Bhatt P. Increased Wnt5a mRNA expression in advanced atherosclerotic lesions, and oxidized LDL treated human monocyte-derived macrophages. The Open Circulation and Vascular Journal. 2012;5(1):1–7. doi: 10.2174/1877382601205010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ackers I., Szymanski C., Duckett K. J., McCall K., Malgor R. Wnt 5a signaling in atherosclerosis, its effect on OxLDL uptake and foam cell differentiation. The FASEB Journal. 2015;29(supplement 1) [Google Scholar]
- 78.Sorrentino R., Morello S., Bonavita E., Pinto A. The activation of liver X receptors inhibits toll-like receptor-9-induced foam cell formation. Journal of Cellular Physiology. 2009;223(1) doi: 10.1002/jcp.22022. [DOI] [PubMed] [Google Scholar]
- 79.Durham A. L., Speer M. Y., Scatena M., Giachelli C. M., Shanahan C. M. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovascular Research. 2018;114(4):590–600. doi: 10.1093/cvr/cvy010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rocca C., Pasqua T., Boukhzar L., Anouar Y., Angelone T. Progress in the emerging role of selenoproteins in cardiovascular disease: focus on endoplasmic reticulum-resident selenoproteins. Cellular and Molecular Life Sciences. 2019;76(20):3969–3985. doi: 10.1007/s00018-019-03195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu H., Xu H., Huang K. Selenium in the prevention of atherosclerosis and its underlying mechanisms. Metallomics. 2017;9(1):21–37. doi: 10.1039/C6MT00195E. [DOI] [PubMed] [Google Scholar]
- 82.Ursini F., Maiorino M., Brigelius-Flohé R., et al. [5] Diversity of glutathione peroxidases. Methods in Enzymology. 1995;252:38–53. doi: 10.1016/0076-6879(95)52007-4. [DOI] [PubMed] [Google Scholar]
- 83.Suzuki T., Kelly V. P., Motohashi H., et al. Deletion of the selenocysteine tRNA gene in macrophages and liver results in compensatory gene induction of cytoprotective enzymes by Nrf2. The Journal of Biological Chemistry. 2008;283(4):2021–2030. doi: 10.1074/jbc.M708352200. [DOI] [PubMed] [Google Scholar]
- 84.Conrad M. Transgenic mouse models for the vital selenoenzymes cytosolic thioredoxin reductase, mitochondrial thioredoxin reductase and glutathione peroxidase 4. Biochimica et Biophysica Acta. 2009;1790(11):1575–1585. doi: 10.1016/j.bbagen.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 85.Casós K., Zaragozá M. C., Zarkovic N., et al. A fish oil-rich diet reduces vascular oxidative stress in apoE–/–mice. Free Radical Research. 2010;44(7):821–829. doi: 10.3109/10715762.2010.485992. [DOI] [PubMed] [Google Scholar]
- 86.Penumetcha M., Song M., Merchant N., Parthasarathy S. Pretreatment with n-6 PUFA protects against subsequent high fat diet induced atherosclerosis--potential role of oxidative stress-induced antioxidant defense. Atherosclerosis. 2012;220(1):53–58. doi: 10.1016/j.atherosclerosis.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Siddiqui R. A., Harvey K. A., Ruzmetov N., Miller S. J., Zaloga G. P. n-3 fatty acids prevent whereastrans-fatty acids induce vascular inflammation and sudden cardiac death. The British Journal of Nutrition. 2009;102(12):1811–1819. doi: 10.1017/s0007114509992030. [DOI] [PubMed] [Google Scholar]
- 88.Meital L. T., Windsor M. T., Perissiou M., et al. Omega-3 fatty acids decrease oxidative stress and inflammation in macrophages from patients with small abdominal aortic aneurysm. Scientific Reports. 2019;9(1, article 12978) doi: 10.1038/s41598-019-49362-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ishikado A., Morino K., Nishio Y., et al. 4-Hydroxy hexenal derived from docosahexaenoic acid protects endothelial cells via Nrf2 activation. PLoS One. 2013;8(7, article e69415) doi: 10.1371/journal.pone.0069415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anderson E. J., Thayne K., Harris M., Carraway K., Shaikh S. R. Aldehyde stress and up-regulation of Nrf2-mediated antioxidant systems accompany functional adaptations in cardiac mitochondria from mice fed n−3 polyunsaturated fatty acids. The Biochemical Journal. 2012;441(1):359–366. doi: 10.1042/bj20110626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang Y. C., Lii C. K., Wei Y. L., et al. Docosahexaenoic acid inhibition of inflammation is partially via cross-talk between Nrf2/heme oxygenase 1 and IKK/NF-κB pathways. The Journal of Nutritional Biochemistry. 2013;24(1):204–212. doi: 10.1016/j.jnutbio.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 92.Oh D. Y., Talukdar S., Bae E. J., et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142(5):687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Farías J. G., Carrasco-Pozo C., Carrasco Loza R., et al. Polyunsaturated fatty acid induces cardioprotection against ischemia-reperfusion through the inhibition of NF-kappaB and induction of Nrf2. Experimental Biology and Medicine. 2016;242(10):1104–1114. doi: 10.1177/1535370216649263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morrow J. D., Awad J. A., Boss H. J., Blair I. A., Roberts L. J. Non-cyclooxygenase-derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10721–10725. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Di Nunzio M., Valli V., Bordoni A. PUFA and oxidative stress. Differential modulation of the cell response by DHA. International Journal of Food Sciences and Nutrition. 2016;67(7):834–843. doi: 10.1080/09637486.2016.1201790. [DOI] [PubMed] [Google Scholar]
- 96.O'Farrell S., Jackson M. J. Dietary polyunsaturated fatty acids, vitamin E and hypoxia/reoxygenation- induced damage to cardiac tissue. Clinica Chimica Acta. 1997;267(2):197–211. doi: 10.1016/s0009-8981(97)00147-2. [DOI] [PubMed] [Google Scholar]
- 97.Naik E., Dixit V. M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. The Journal of Experimental Medicine. 2011;208(3):417–420. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mercer J. R., Yu E., Figg N., et al. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE−/− mice. Free Radical Biology and Medicine. 2012;52(5):841–849. doi: 10.1016/j.freeradbiomed.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 99.Wang Y., Li L., Zhao W., et al. Targeted therapy of atherosclerosis by a broad-spectrum reactive oxygen species scavenging nanoparticle with intrinsic anti-inflammatory activity. ACS Nano. 2018;12(9):8943–8960. doi: 10.1021/acsnano.8b02037. [DOI] [PubMed] [Google Scholar]
- 100.Williams K. J., Fisher E. A. Oxidation, lipoproteins, and atherosclerosis: which is wrong, the antioxidants or the theory? Current Opinion in Clinical Nutrition and Metabolic Care. 2005;8(2):139–146. doi: 10.1097/00075197-200503000-00006. [DOI] [PubMed] [Google Scholar]
- 101.Kadlec A. O., Chabowski D. S., Ait-Aissa K., Gutterman D. D. Role of PGC-1α in vascular regulation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016;36(8):1467–1474. doi: 10.1161/atvbaha.116.307123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stein S., Lohmann C., Handschin C., et al. ApoE−/− PGC-1α−/− mice display reduced IL-18 levels and do not develop enhanced atherosclerosis. PLoS One. 2010;5, article e13539(10) doi: 10.1371/journal.pone.0013539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen K. H., Guo X., Ma D., et al. Dysregulation of HSG triggers vascular proliferative disorders. Nature Cell Biology. 2004;6(9):872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 104.Guo X., Chen K.-H., Guo Y., Liao H., Tang J., Xiao R.-P. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circulation Research. 2007;101(11):1113–1122. doi: 10.1161/circresaha.107.157644. [DOI] [PubMed] [Google Scholar]
- 105.Kunsch C., Medford R. M. Oxidative stress as a regulator of gene expression in the vasculature. Circulation Research. 1999;85(8):753–766. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- 106.Madamanchi N. R., Moon S.-K., Hakim Z. S., et al. Differential activation of mitogenic signaling pathways in aortic smooth muscle cells deficient in superoxide dismutase isoforms. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(5):950–956. doi: 10.1161/01.atv.0000161050.77646.68. [DOI] [PubMed] [Google Scholar]
- 107.Shrivastava A. K., Singh H. V., Raizada A., Singh S. K. C-reactive protein, inflammation and coronary heart disease. The Egyptian Heart Journal. 2015;67(2):89–97. doi: 10.1016/j.ehj.2014.11.005. [DOI] [Google Scholar]
- 108.Wronska-Nofer T., Nofer J.-R., Stetkiewicz J., et al. Evidence for oxidative stress at elevated plasma thiol levels in chronic exposure to carbon disulfide (CS2) and coronary heart disease. Nutrition, Metabolism and Cardiovascular Diseases. 2007;17(7):546–553. doi: 10.1016/j.numecd.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 109.Yang X., He T., Han S., et al. The role of traditional chinese medicine in the regulation of oxidative stress in treating coronary heart disease. Oxidative Medicine and Cellular Longevity. 2019;2019:13. doi: 10.1155/2019/3231424.3231424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Leopold J. A., Loscalzo J. Oxidative risk for atherothrombotic cardiovascular disease. Free Radical Biology and Medicine. 2009;47(12):1673–1706. doi: 10.1016/j.freeradbiomed.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roberts A. C., Porter K. E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diabetes & Vascular Disease Research. 2013;10(6):472–482. doi: 10.1177/1479164113500680. [DOI] [PubMed] [Google Scholar]
- 112.Bastani A., Rajabi S., Daliran A., Saadat H., Karimi-Busheri F. Oxidant and antioxidant status in coronary artery disease. Biomedical Reports. 2018;9(4):327–332. doi: 10.3892/br.2018.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.El-Benna J., Dang P. M.-C., Gougerot-Pocidalo M.-A. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Seminars in Immunopathology. 2008;30(3):279–289. doi: 10.1007/s00281-008-0118-3. [DOI] [PubMed] [Google Scholar]
- 114.Zhang J., Wang M., Li Z., et al. NADPH oxidase activation played a critical role in the oxidative stress process in stable coronary artery disease. American Journal of Translational Research. 2016;8(12):5199–5210. [PMC free article] [PubMed] [Google Scholar]
- 115.Gramlich Y., Daiber A., Buschmann K., et al. Oxidative stress in cardiac tissue of patients undergoing coronary artery bypass graft surgery: the effects of overweight and obesity. Oxidative Medicine and Cellular Longevity. 2018;2018:13. doi: 10.1155/2018/6598326.6598326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Oakes S. A., Papa F. R. The role of endoplasmic reticulum stress in human pathology. Annual Review of Pathology. 2015;10(1):173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Seimon T. A., Nadolski M. J., Liao X., et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metabolism. 2010;12(5):467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang S., Binder P., Fang Q., et al. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. British Journal of Pharmacology. 2018;175(8):1293–1304. doi: 10.1111/bph.13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamaguchi O., Higuchi Y., Hirotani S., et al. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proceedings of the National Academy of Sciences of the United States of America. 2011;100(26):15883–15888. doi: 10.1073/pnas.2136717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qi Z., Chen L. Endoplasmic Reticulum Stress and Autophagy. Advances in Experimental Medicine and Biology. 2019;1206:167–177. doi: 10.1007/978-981-15-0602-4_8. [DOI] [PubMed] [Google Scholar]
- 121.Cominacini L., Mozzini C., Garbin U., et al. Endoplasmic reticulum stress and Nrf2 signaling in cardiovascular diseases. Free Radical Biology and Medicine. 2015;88(Part B):233–242. doi: 10.1016/j.freeradbiomed.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 122.Barbosa J. E., Stockler-Pinto M. B., da Cruz B. O., et al. Nrf2, NF-κB and PPARβ/δ mRNA expression profile in patients with coronary artery disease. Arquivos Brasileiros de Cardiologia. 2019;113(6):1121–1127. doi: 10.5935/abc.20190125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mozzini C., Pasini A. F., Garbin U., et al. Increased endoplasmic reticulum stress and Nrf2 repression in peripheral blood mononuclear cells of patients with stable coronary artery disease. Free Radical Biology and Medicine. 2014;68:178–185. doi: 10.1016/j.freeradbiomed.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 124.Zhu H., Jia Z., Zhang L., et al. Antioxidants and phase 2 enzymes in macrophages: regulation by Nrf2 signaling and protection against oxidative and electrophilic stress. Experimental Biology and Medicine. 2008;233(4):463–474. doi: 10.3181/0711-rm-304. [DOI] [PubMed] [Google Scholar]
- 125.Collins A. J., Foley R. N., Chavers B., et al. United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. American Journal of Kidney Diseases. 2012;59(1) Supplement 1 doi: 10.1053/j.ajkd.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 126.Visvikis-Siest S., Marteau J.-B., Samara A., Berrahmoune H., Marie B., Pfister M. Peripheral blood mononuclear cells (PBMCs): a possible model for studying cardiovascular biology systems. Clinical Chemistry and Laboratory Medicine. 2007;45(9):1154–1168. doi: 10.1515/cclm.2007.255. [DOI] [PubMed] [Google Scholar]
- 127.Shantsila E., Watson T., Lip G. Y. Endothelial progenitor cells in cardiovascular disorders. Journal of the American College of Cardiology. 2007;49:741–752. doi: 10.1016/j.jacc.2006.09.050. [DOI] [PubMed] [Google Scholar]
- 128.Watson T., Goon P. K. Y., Lip G. Y. H. Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension. Antioxidants & Redox Signaling. 2008;10(6):1079–1088. doi: 10.1089/ars.2007.1998. [DOI] [PubMed] [Google Scholar]
- 129.Ndrepepa G. Myeloperoxidase – a bridge linking inflammation and oxidative stress with cardiovascular disease. Clinica Chimica Acta. 2019;493:36–51. doi: 10.1016/j.cca.2019.02.022. [DOI] [PubMed] [Google Scholar]
- 130.Nielsen F., Mikkelsen B. B., Nielsen J. B., Andersen H. R., Grandjean P. Plasma malondialdehyde as biomarker for oxidative stress: reference interval and effects of life-style factors. Clinical Chemistry. 1997;43(7):1209–1214. doi: 10.1093/clinchem/43.7.1209. [DOI] [PubMed] [Google Scholar]
- 131.Mutlu-Türkoğlu U., Akalin Z., Ilhan E., et al. Increased plasma malondialdehyde and protein carbonyl levels and lymphocyte DNA damage in patients with angiographically defined coronary artery disease. Clinical Biochemistry. 2005;38(12):1059–1065. doi: 10.1016/j.clinbiochem.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 132.Abolhasani S., Shahbazloo S. V., Saadati H. M., Mahmoodi N., Khanbabaei N. Evaluation of serum levels of inflammation, fibrinolysis and oxidative stress markers in coronary artery disease prediction: a cross-sectional study. Arquivos Brasileiros de Cardiologia. 2019;113 doi: 10.5935/abc.20190214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yılmaz M., Altın C., Özyıldız A., Müderrisoğlu H. Are oxidative stress markers helpful for diagnosing the disease and determining its complexity or extent in patients with stable coronary artery disease? Türk Kardiyoloji Derneği Arşivi. 2017;45(7):599–605. doi: 10.5543/tkda.2017.80733. [DOI] [PubMed] [Google Scholar]
- 134.Ghaffarzadeh M., Ghaedi H., Alipoor B., et al. Association of MiR-149 (RS2292832) Variant with the Risk of Coronary Artery Disease. Journal of Medical Biochemistry. 2017;36(3):251–258. doi: 10.1515/jomb-2017-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Manuneedhi Cholan P., Cartland S. P., Dang L., et al. TRAIL protects against endothelial dysfunction in vivo and inhibits angiotensin-II-induced oxidative stress in vascular endothelial cells in vitro. Free Radical Biology and Medicine. 2018;126:341–349. doi: 10.1016/j.freeradbiomed.2018.08.031. [DOI] [PubMed] [Google Scholar]
- 136.Abaspour A. R., Taghikhani M., Parizadeh S. M. R., et al. HSP27 expression in the human peripheral blood mononuclear cells as an early prognostic biomarker in coronary artery disease patients. Diabetes and Metabolic Syndrome: Clinical Research and Reviews. 2019;13(3):1791–1795. doi: 10.1016/j.dsx.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 137.Khaper N., Kaur K., Li T., Farahmand F., Singal P. Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Molecular and Cellular Biochemistry. 2003;251:9–15. doi: 10.1007/978-1-4419-9238-3_2. [DOI] [PubMed] [Google Scholar]
- 138.Li X., Hou L., Cheng Z., Zhou S., Qi J., Cheng J. Overexpression of GAS5 inhibits abnormal activation of Wnt/β‐catenin signaling pathway in myocardial tissues of rats with coronary artery disease. Journal of Cellular Physiology. 2019;234(7):11348–11359. doi: 10.1002/jcp.27792. [DOI] [PubMed] [Google Scholar]
- 139.Wang X. B., Cui N. H., Zhang S., Liu Z. J., Ma J. F. Leukocyte telomere length, mitochondrial DNA copy number, and coronary artery disease risk and severity: a two-stage case-control study of 3064 Chinese subjects. Atherosclerosis. 2019;284:165–172. doi: 10.1016/j.atherosclerosis.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 140.Sarutipaiboon I., Settasatian N., Komanasin N., Kukongwiriyapan U., Sawanyawisuth K., Intraraphet P. Association of genetic variations in NRF2, NQO1, HMOX1, and MT with severity of coronary artery disease and related risk factors. Cardiovascular Toxicology. 2020;20(2):176–189. doi: 10.1007/s12012-019-09544-7. [DOI] [PubMed] [Google Scholar]
- 141.Xiao Y., Xia J., Cheng J., Huang H., Zhou Y. Inhibition of S-adenosylhomocysteine hydrolase induces endothelial dysfunction via epigenetic regulation of p66shc-mediated oxidative stress pathway. Circulation. 2019;139(19):2260–2277. doi: 10.1161/circulationaha.118.036336. [DOI] [PubMed] [Google Scholar]
- 142.Jorat M. V., Tabrizi R., Kolahdooz F., Akbari M., Salami M. The effects of coenzyme Q10 supplementation on biomarkers of inflammation and oxidative stress in among coronary artery disease: a systematic review and meta-analysis of randomized controlled trials. Inflammopharmacology. 2019;27(2):233–248. doi: 10.1007/s10787-019-00572-x. [DOI] [PubMed] [Google Scholar]
- 143.Lee B. J., Lin J. S., Lin Y. C., Lin P. T. Effects of L-carnitine supplementation on oxidative stress and antioxidant enzymes activities in patients with coronary artery disease: a randomized, placebo-controlled trial. Nutrition Journal. 2014;13(1) doi: 10.1186/1475-2891-13-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guby A. R. Nutrition treatments for acute myocardial infarction. Alternative Medicine Review. 2010;15:113–123. [PubMed] [Google Scholar]
- 145.Zhang Z., Jiang F., Zeng L., Wang X., Tu S. PHACTR1 regulates oxidative stress and inflammation to coronary artery endothelial cells via interaction with NF-κB/p65. Atherosclerosis. 2018;278:180–189. doi: 10.1016/j.atherosclerosis.2018.08.041. [DOI] [PubMed] [Google Scholar]
- 146.Zarzuelo M. J., Lopez-Sepulveda R., Sanchez M., et al. SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: implications for vascular aging. Biochemical Pharmacology. 2013;85(9):1288–1296. doi: 10.1016/j.bcp.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 147.Hung C. H., Chan S. H., Chu P. M., Tsai K. L. Homocysteine facilitates LOX-1 activation and endothelial death through the PKCβ and SIRT1/HSF1 mechanism: relevance to human hyperhomocysteinaemia. Clinical Science. 2015;129(6):477–487. doi: 10.1042/cs20150127. [DOI] [PubMed] [Google Scholar]
- 148.Chan S. H., Hung C. H., Shih J. Y., et al. SIRT1 inhibition causes oxidative stress and inflammation in patients with coronary artery disease. Redox Biology. 2017;13:301–309. doi: 10.1016/j.redox.2017.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Taqueti V. R., Di Carli M. F. Coronary microvascular disease pathogenic mechanisms and therapeutic options: JACC state-of-the-art review. Journal of the American College of Cardiology. 2018;72(21):2625–2641. doi: 10.1016/j.jacc.2018.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Padro T., Manfrini O., Bugiardini R., et al. ESC working group on coronary pathophysiology and microcirculation position paper on ‘coronary microvascular dysfunction in cardiovascular disease’. Cardiovascular Research. 2020;116(4):741–755. doi: 10.1093/cvr/cvaa003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Niccoli G., Scalone G., Crea F. Acute myocardial infarction with no obstructive coronary atherosclerosis: mechanisms and management. European Heart Journal. 2015;36(8):475–481. doi: 10.1093/cvr/cvaa003. [DOI] [PubMed] [Google Scholar]
- 152.Crea F., Camici P. G., Bairey Merz C. N. Coronary microvascular dysfunction: an update. European Heart Journal. 2014;35:1101–1111. doi: 10.1093/eurheartj/eht513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Smilowitz N. R., Mahajan A. M., Roe M. T., et al. Mortality of myocardial infarction by sex, age, and obstructive coronary artery disease status in the ACTION Registry–GWTG (Acute Coronary Treatment and Intervention Outcomes Network Registry–Get With the Guidelines) Circulation Cardiovascular Quality and Outcomes. 2017;10, article e003443 doi: 10.1161/circoutcomes.116.003443. [DOI] [PubMed] [Google Scholar]
- 154.Bradley S. M., Maddox T. M., Stanislawski M. A., et al. Normal coronary rates for elective angiography in the Veterans Affairs Healthcare System: insights from the VA CART program (veterans affairs clinical assessment reporting and tracking) Journal of the American College of Cardiology. 2014;63:417–426.10. doi: 10.1016/j.jacc.2013.09.055. [DOI] [PubMed] [Google Scholar]
- 155.Jespersen L., Hvelplund A., Abildstrom S. Z., et al. Stable angina pectoris with no obstructive coronary artery disease is associated with increased risks of major adverse cardiovascular events. European Heart Journal. 2012;33:734–744. doi: 10.1093/eurheartj/ehr331. [DOI] [PubMed] [Google Scholar]
- 156.Sharaf B., Wood T., Shaw L., et al. Adverse outcomes among women presenting with signs and symptoms of ischemia and no obstructive coronary artery disease: findings from the National Heart, Lung, and Blood Institute–sponsored Women's Ischemia Syndrome Evaluation (WISE) angiographic core laboratory. American Heart Journal. 2013;166:134–141. doi: 10.1016/j.ahj.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Meier P., Gloekler S., Zbinden R., et al. Beneficial effect of recruitable collaterals. A 10- year follow-up study in patients with stable coronary artery disease undergoing quantitative collateral measurements. Circulation. 2007;116:975–983. doi: 10.1161/circulationaha.107.703959. [DOI] [PubMed] [Google Scholar]
- 158.Elsman P., van’t Hof A. W., de Boer M. J., et al. Role of collateral circulation in the acute phase of ST-segment-elevation myocardial infarction treated with primary coronary intervention. European Heart Journal. 2004;25:854–858. doi: 10.1016/j.ehj.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 159.Loscalzo J. What we know and don’t know about Larginine and NO. Circulation. 2000;101:2126–2129. doi: 10.1161/01.cir.101.18.2126. [DOI] [PubMed] [Google Scholar]
- 160.Tritto I., Ambrosio G. The multi-faceted behavior of nitric oxide in vascular “inflammation”: catchy terminology or true phenomenon? Cardiovascular Research. 2004;63(1):1–4. doi: 10.1016/j.cardiores.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 161.Small H. Y., Migliarino S., Czesnikiewicz-Guzik M., Guzik T. J. Hypertension: focus on autoimmunity and oxidative stress. Free Radical Biology and Medicine. 2018;125:104–115. doi: 10.1016/j.freeradbiomed.2018.05.085. [DOI] [PubMed] [Google Scholar]
- 162.Corban M. T., Lerman L. O., Lerman A. Endothelin-1 in coronary microvascular dysfunction: a potential new therapeutic target once again. European Heart Journal. 2020;41(34):3252–3254. doi: 10.1093/eurheartj/ehz954. [DOI] [PubMed] [Google Scholar]
- 163.Bonderman D., Teml A., Jakowitsch J., et al. Coronary no-reflow is caused by shedding of active tissue factor from dissected atherosclerotic plaque. Blood. 2002;99:2794–2800. doi: 10.1182/blood.v99.8.2794. [DOI] [PubMed] [Google Scholar]
- 164.Lim N., Dubois M. J., De Backer D., Vincent J.-L. Do all nonsurvivors of cardiogenic shock die with a low cardiac index? Chest. 2003;124(5):1885–1891. doi: 10.1378/chest.124.5.1885. [DOI] [PubMed] [Google Scholar]
- 165.Joshi M. S., Mineo C., Shaul P. W., Bauer J. A. Biochemical consequences of the NOS3 Glu298Asp variation in human endothelium: altered caveolar localization and impaired response to shear. The FASEB Journal. 2007;21:2655–2663. doi: 10.1096/fj.06-7088com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shimasaki Y., Yasue H., Yoshimura M., et al. Association of the missense Glu298Asp variant of the endothelial nitric oxide synthase gene with myocardial infarction. Journal of the American College of Cardiology. 1998;31(7):1506–1510. doi: 10.1016/s0735-1097(98)00167-3. [DOI] [PubMed] [Google Scholar]
- 167.Nakayama M., Yasue H., Yoshimura M., et al. T −786→C mutation in the 5′-flanking region of the endothelial nitric oxide synthase gene is associated with coronary spasm. Circulation. 1999;99:2864–2870. doi: 10.1161/01.cir.99.22.2864. [DOI] [PubMed] [Google Scholar]
- 168.Ford T. J., Corcoran D., Padmanabhan S., et al. Genetic dysregulation of endothelin-1 is implicated in coronary microvascular dysfunction. European Heart Journal. 2020;41(34):3239–3252. doi: 10.1093/eurheartj/ehz915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dorobantu M., Badimon L. Microcirculation: From Bench to Bedside. Springer Nature; 2020. [Google Scholar]
- 170.Sorop O., van de Wouw J., Chandler S., et al. Experimental animal models of coronary microvascular dysfunction. Cardiovascular Research. 2020;116(4):756–770. doi: 10.1093/cvr/cvaa002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Badimon L., Bugiardini R., Cenko E., et al. Position paper of the European Society of Cardiology–working group of coronary pathophysiology and microcirculation: obesity and heart disease. European Heart Journal. 2017;38:1951–1958. doi: 10.1093/eurheartj/ehx181. [DOI] [PubMed] [Google Scholar]
- 172.Bagi Z., Feher A., Cassuto J. Microvascular responsiveness in obesity: implications for therapeutic intervention. British Journal of Pharmacology. 2012;165:544–560. doi: 10.1111/j.1476-5381.2011.01606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Grassi G., Seravalle G., Quarti-Trevano F., et al. Excessive sympathetic activation in heart failure with obesity and metabolic syndrome: characteristics and mechanisms. Hypertension. 2007;49:535–541. doi: 10.1161/01.hyp.0000255983.32896.b9. [DOI] [PubMed] [Google Scholar]
- 174.Kachur S., Morera R., Schutter A. D., Lavie C. J. Cardiovascular risk in patients with prehypertension and the metabolic syndrome. Current Hypertension Reports. 2018;20:p. 15. doi: 10.1007/s11906-018-0801-2. [DOI] [PubMed] [Google Scholar]
- 175.Kalinowski L., Dobrucki L. W., Szczepanska-Konkel M., et al. Third-Generation β-blockers stimulate nitric oxide release from endothelial cells through ATP Efflux. Circulation. 2003;107:2747–2752. doi: 10.1161/01.cir.0000066912.58385.de. [DOI] [PubMed] [Google Scholar]
- 176.Ford E. S., Ajani U. A., Croft J. B., et al. Explaining the decrease in U.S. deaths from coronary disease, 1980-2000. The New England Journal of Medicine. 2007;356:2388–2398. doi: 10.1056/NEJMsa053935. [DOI] [PubMed] [Google Scholar]
- 177.Stampfer M. J., Hu F. B., Manson J. E., Rimm E. B., Willett W. C. Primary Prevention of Coronary Heart Disease in Women through Diet and Lifestyle. The New England Journal of Medicine. 2000;343:16–22. doi: 10.1056/nejm200012143432415. [DOI] [PubMed] [Google Scholar]
- 178.Hill J. S., Hayden M. R., Frohlich J., Pritchard P. H. Genetic and environmental factors affecting the incidence of coronary artery disease in heterozygous familial hypercholesterolemia. Arteriosclerosis and Thrombosis: A Journal of Vascular Biology. 1991;11(2):290–297. doi: 10.1161/01.atv.11.2.290. [DOI] [PubMed] [Google Scholar]
- 179.Martino T., Arab S., Straume M., et al. Day/night rhythms in gene expression of the normal murine heart. Journal of Molecular Medicine. 2004;82:256–264. doi: 10.1007/s00109-003-0520-1. [DOI] [PubMed] [Google Scholar]
- 180.McNamara P., Seo S. B., Rudic R. D., Sehgal A., Chakravarti D., Fitz Gerald G. A. Regulation of CLOCK and MOP4 by nuclear hormone receptors in the vasculature. Cell. 2001;105:877–889. doi: 10.1016/s0092-8674(01)00401-9. [DOI] [PubMed] [Google Scholar]
- 181.Martino T. A., Sole M. J. Molecular time: an often overlooked dimension to cardiovascular disease. Circulation Research. 2009;105:1047–1061. doi: 10.1161/circresaha.109.206201. [DOI] [PubMed] [Google Scholar]
- 182.Muller J. E., Stone P. H., Turi Z. G., et al. Circadian variation in the frequency of onset of acute myocardial infarction. The New England Journal of Medicine. 1985;313:1315–1322. doi: 10.1056/NEJM198511213132103. [DOI] [PubMed] [Google Scholar]
- 183.Cohen M. C., Rohtla K. M., Lavery C. E., Muller J. E., Mittleman M. A. Meta-analysis of the morning excess of acute myocardial infarction and sudden cardiac death. The American Journal of Cardiology. 1997;79:1512–1516. doi: 10.1016/s0002-9149(97)00181-1. [DOI] [PubMed] [Google Scholar]
- 184.Knutson K. L., Ryden A. M., Mander B. A., Van Cauter E. Role of sleep duration and quality in the risk and severity of type 2 diabetes mellitus. Archives of Internal Medicine. 2006;166:1768–1774. doi: 10.1001/archinte.166.16.1768. [DOI] [PubMed] [Google Scholar]
- 185.Kohatsu N. D., Tsai R., Young T., et al. Sleep duration and body mass index in a rural population. Archives of Internal Medicine. 2006;166:1701–1705. doi: 10.1001/archinte.166.16.1701. [DOI] [PubMed] [Google Scholar]
- 186.Gangwisch J. E., Heymsfield S. B., Boden-Albala B., et al. Short sleep duration as a risk factor for hypertension: analyses of the first National Health and Nutrition Examination Survey. Hypertension. 2006;47:833–839. doi: 10.1161/01.HYP.0000217362.34748.e0. [DOI] [PubMed] [Google Scholar]
- 187.Marti-Soler H., Gubelmann C., Aeschbacher S., et al. Seasonality of cardiovascular risk factors: an analysis including over 230000 participants in 15 countries. Heart. 2014;100(19):1517–1523. doi: 10.1136/heartjnl-2014-305623. [DOI] [PubMed] [Google Scholar]
- 188.Tung P., Wiviott S. D., Cannon C. P., Murphy S. A., McCabe C. H., Gibson C. M. Seasonal variation in lipids in patients following acute coronary syndrome on fixed doses of pravastatin (40 mg) or atorvastatin (80 mg) (from the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis In Myocardial Infarction 22 [PROVE IT-TIMI 22] Study) The American Journal of Cardiology. 2009;103:1056–1060. doi: 10.1016/j.amjcard.2008.12.034. [DOI] [PubMed] [Google Scholar]
- 189.Laplaud P. M., Beaubatie L., Maurel D. A spontaneously seasonal hyper-cholesterolemic animal: plasma lipids and lipoproteins in the European badger (Meles meles L.) Journal of Lipid Research. 1980;21:724–738. [PubMed] [Google Scholar]
- 190.Marchant B., Ranjadayalan K., Stevenson R., Wilkinson P., Timmis A. D. Circadian and seasonal factors in the pathogenesis of acute myocar-dial infarction: the influence of environmental temperature. British Heart Journal. 1993;69:385–387. doi: 10.1136/hrt.69.5.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gold D. R., Mittleman M. A. New insights into pollution and the cardiovascular system: 2010 to 2012. Circulation. 2013;127:1903–1913. doi: 10.1161/circulationaha.111.064337. [DOI] [PubMed] [Google Scholar]
- 192.Yang L., Lof M., Veierød M. B., Sandin S., Adami H. O., Weiderpass E. Ultraviolet exposure and mortality among women in Sweden. Cancer Epidemiology, Biomarkers & Prevention. 2011;20:683–690. doi: 10.1158/1055-9965.epi-10-0982. [DOI] [PubMed] [Google Scholar]
- 193.Lindqvist P. G., Epstein E., Nielsen K., Landin-Olsson M., Ingvar C., Olsson H. Avoidance of sun exposure as a risk factor for major causes of death: a competing risk analysis of the Melanoma in Southern Sweden cohort. Journal of Internal Medicine. 2016;280:375–387. doi: 10.1111/joim.12496. [DOI] [PubMed] [Google Scholar]
- 194.Wang T. J. Vitamin D and cardiovascular disease. Annual Review of Medicine. 2016;67:261–272. doi: 10.1146/annurev-med-051214-025146. [DOI] [PubMed] [Google Scholar]
- 195.Pilz S., Verheyen N., Grübler M. R., Tomaschitz A., März W. Vitamin D and cardiovascular disease prevention. Nature Reviews Cardiology. 2016;13:404–417. doi: 10.1038/nrcardio.2016.73. [DOI] [PubMed] [Google Scholar]
- 196.De Mendoza S., Nucete H., Ineichen E., Salazar E., Zerpa A., Glueck C. J. Lipids and Lipoproteins in Subjects at 1,000 and 3, 500 Meter Altitudes. Archives of Environmental Health. 1979;34:308–311. doi: 10.1080/00039896.1979.10667422. [DOI] [PubMed] [Google Scholar]
- 197.Sharma S. Clinical, biochemical, electrocardiographic and noninvasive hemodynamic assessment of cardiovascular status in natives at high to ex-treme altitudes (3000m-5500m) of the Himalayan region. Indian Heart Journal. 1990;42:375–379. [PubMed] [Google Scholar]
- 198.Mohanna S., Baracco R., Seclén S. Lipid profile, waist circumference, and body mass index in a high altitude population. High Altitude Medicine & Biology. 2006;7:245–255. doi: 10.1089/ham.2006.7.245. [DOI] [PubMed] [Google Scholar]
- 199.Domínguez Coello S., Cabrera De León A., Bosa Ojeda F., Pérez Méndez L. I., Díaz González L., Aguirre-Jaime A. J. High density lipoprotein cholesterol increases with living altitude. International Journal of Epidemiology. 2000;29:65–70. doi: 10.1093/ije/29.1.65. [DOI] [PubMed] [Google Scholar]
- 200.Cabrera de Leon A., Gonzalez D. A., Mendez L. I., et al. Leptin and altitude in the cardiovascular diseases. Obesity Research. 2004;12:1492–1498. doi: 10.1038/oby.2004.186. [DOI] [PubMed] [Google Scholar]
- 201.Holick M. F., Chen T. C., Lu Z., Sauter E. Vitamin D and skin physiology: a D-lightful story. Journal of Bone and Mineral Research. 2007;22(S2):V28–V33. doi: 10.1359/jbmr.07s211. [DOI] [PubMed] [Google Scholar]
- 202.Dadvand P., Bartoll X., Basagaña X., et al. Green spaces and General Health: Roles of mental health status, social support, and physical activity. International Journal of Environmental Research and Public Health. 2017;14(11):p. 1411. doi: 10.1016/j.envint.2016.02.029. [DOI] [PubMed] [Google Scholar]
- 203.James P., Banay R. F., Hart J. E., Laden F. A review of the health benefits of greenness. Current Epidemiology Reports. 2015;2:131–142. doi: 10.1007/s40471-015-0043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lovasi G. S., Quinn J. W., Neckerman K. M., Perzanowski M. S., Rundle A. Children living in areas with more street trees have lower prevalence of asthma. Journal of Epidemiology and Community Health. 2008;62:647–649. doi: 10.1136/jech.2007.071894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Markevych I., Thiering E., Fuertes E., et al. A cross-sectional analysis of the effects of residential greenness on blood pressure in 10-year old children: results from the GINIplus and LISAplus studies. BMC Public Health. 2014;14, article 477 doi: 10.1186/1471-2458-14-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chaix B. Geographic life environments and coronary heart disease: a literature review, theoretical contributions, methodological updates, and a research agenda. Annual Review of Public Health. 2009;30:81–105. doi: 10.1146/annurev.publhealth.031308.100158. [DOI] [PubMed] [Google Scholar]
- 207.Hartung T. Toxicology for the twenty-first century. Nature. 2009;460:208–212. doi: 10.1038/460208a. [DOI] [PubMed] [Google Scholar]
- 208.Cosselman K. E., Navas-Acien A., Kaufman J. D. Environmental factors in cardiovascular disease. Nature Reviews. Cardiology. 2015;12:627–642. doi: 10.1038/nrcardio.2015.152. [DOI] [PubMed] [Google Scholar]
- 209.Bhatnagar A. Environmental cardiology: studying mechanistic links between pollution and heart disease. Circulation Research. 2006;99:692–705. doi: 10.1161/01.res.0000243586.99701.cf. [DOI] [PubMed] [Google Scholar]
- 210.Brook R. D., Rajagopalan S., Pope C. A., et al. Particulate matter air pollution and cardiovas-cular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–2378. doi: 10.1161/cir.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 211.Münzel T., Gori T., Babisch W., Basner M. Cardiovascular effects of envi-ronmental noise exposure. European Heart Journal. 2014;35:829–836. doi: 10.1093/eurheartj/ehu030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Said M. A., El-Gohary O. A. Effect of noise stress on cardiovascular system in adult male albino rat: implication of stress hormones, endothelial dysfunction and oxidative stress. General Physiology and Biophysics. 2016;35:371–377. doi: 10.4149/gpb_2016003. [DOI] [PubMed] [Google Scholar]
- 213.Schane R. E., Ling P. M., Glantz S. A. Health effects of light and intermittent smoking: a review. Circulation. 2010;121:1518–1522. doi: 10.1161/CIRCULATIONAHA.109.904235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Grundy S. M., Balady G. J., Criqui M. H., et al. Primary prevention of coronary heart disease: guidance from Framingham: a statement for healthcare professionals from the AHA Task Force on Risk Reduction. American Heart Association. Circulation. 1998;97:1876–1887. doi: 10.1161/01.cir.97.18.1876. [DOI] [PubMed] [Google Scholar]
- 215.O’Toole T. E., Conklin D. J., Bhatnagar A. Environmental risk factors for heart disease. Reviews on Environmental Health. 2008;23:167–202. doi: 10.1515/reveh.2008.23.3.167. [DOI] [PubMed] [Google Scholar]
- 216.Malakar A. K., Choudhury D., Halder B., Paul P., Uddin A., Chakraborty S. A review on coronary artery disease, its risk factors, and therapeutics. Journal of Cellular Physiology. 2019;234(10):16812–16823. doi: 10.1002/jcp.28350. [DOI] [PubMed] [Google Scholar]
- 217.Mudd S. H., Skovby F., Levy H. L., et al. The natural history of homocystinuria due tocystathionine β-synthase deficiency. American Journal of Human Genetics. 1985;37(1):1–31. [PMC free article] [PubMed] [Google Scholar]
- 218.McCully K. S. Vascular pathology of homocysteinemia: Implica-tions for the pathogenesis of arteriosclerosis. The American Journal of Pathology. 1969;56(1):111–128. [PMC free article] [PubMed] [Google Scholar]
- 219.Malinow M. R. Homocyst(e)ine and arterial occlusive diseases. Journal of Internal Medicine. 1994;236(6):603–617. doi: 10.1111/j.1365-2796.1994.tb00854.x. [DOI] [PubMed] [Google Scholar]
- 220.Dawson J., Quinn T., Walters M. Uric acid reduction: a new paradigm in the management of cardiovascular risk? Current Medicinal Chemistry. 2007;14(17):1879–1886. doi: 10.2174/092986707781058797. [DOI] [PubMed] [Google Scholar]
- 221.Montalcini T., Gorgone G., Gazzaruso C., Sesti G., Perticone F., Pujia A. Relation between serum uric acid and carotid intima-media thickness in healthy postmenopausal women. Internal and Emergency Medicine. 2007;2(1):19–23. doi: 10.1007/s11739-007-0004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gheorghiade M., Greene S. J., Butler J., et al. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction. JAMA. 2015;314(21):2251–2262. doi: 10.1001/jama.2015.15734. [DOI] [PubMed] [Google Scholar]
- 223.Flather M. D., Shibata M. C., Coats A. J. S., et al. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (seniors) European Heart Journal. 2005;26(3):215–225. doi: 10.1093/eurheartj/ehi115. [DOI] [PubMed] [Google Scholar]
- 224.Ambrosio G., Flather M. D., Bohm M., et al. β-Blockade with nebivolol for prevention of acute ischaemic events in elderly patients with heart failure. Heart. 2011;97(3):209–214. doi: 10.1136/hrt.2010.207365. [DOI] [PubMed] [Google Scholar]
- 225.Nishikimi T., Maeda N., Matsuoka H. The role of natriuretic peptides in cardioprotection. Cardiovascular Research. 2006;69(2):318–328. doi: 10.1016/j.cardiores.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 226.JJ M. M., Packer M., Desai A. S., et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. The New England Journal of Medicine. 2014;371(11):993–1004. doi: 10.1056/NEJMoa1409077. [DOI] [PubMed] [Google Scholar]
- 227.Solomon S. D., Zile M., Pieske B., et al. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. The Lancet. 2012;380(9851):1387–1395. doi: 10.1016/s0140-6736(12)61227-6. [DOI] [PubMed] [Google Scholar]
- 228.Pizzi C., Manfrini O., Fontana F., Bugiardini R. Angiotensin-converting enzyme inhibitors and 3-hydroxy-3-methylglutaryl coenzyme A reductase in cardiac Syndrome X: role of superoxide dismutase activity. Circulation. 2004;109(1):53–58. doi: 10.1161/01.CIR.0000100722.34034.E4. [DOI] [PubMed] [Google Scholar]
- 229.Ohyama K., Matsumoto Y., Takanami K., et al. Coronary adventitial and perivascular adipose tissue inflammation in patients with vasospastic angina. Journal of the American College of Cardiology. 2018;71(4):414–425. doi: 10.1016/j.jacc.2017.11.046. [DOI] [PubMed] [Google Scholar]
- 230.Keli S. O., Hertog M. G., Feskens E. J., Kromhout D. Dietary flavonoids, antioxidant vitamins, and incidence of stroke. Archives of Internal Medicine. 1996;156(6):637–642. doi: 10.1001/archinte.1996.00440060059007. [DOI] [PubMed] [Google Scholar]
- 231.Ness A. R., Powles J. W., Khaw K. T. J. Vitamin C and cardiovascular disease: a systematic review. Cardiovascular Risk. 1996;3:p. 513. [PubMed] [Google Scholar]
- 232.Wang H., Cao G., Prior R. L. Total Antioxidant Capacity of Fruits. Journal of Agricultural and Food Chemistry. 1996;44(3):701–705. doi: 10.1021/jf950579y. [DOI] [Google Scholar]
- 233.Kalt W., Forney C. F., Martin A., Prior R. L. Antioxidant capacity, vitamin C, phenolics, and anthocyanins after fresh storage of small fruits. Journal of Agricultural and Food Chemistry. 1999;47(11):4638–4644. doi: 10.1021/jf990266t. [DOI] [PubMed] [Google Scholar]
- 234.Sellappan S., Akoh C. C. Flavonoids and antioxidant capacity of Georgia-grown Vidalia onions. Journal of Agricultural and Food Chemistry. 2002;50(19):5338–5342. doi: 10.1021/jf020333a. [DOI] [PubMed] [Google Scholar]
- 235.Doré S. Unique properties of polyphenol stilbenes in the brain: more than direct antioxidant actions; gene/protein regulatory activity. Neurosignals. 2005;14(1-2):61–70. doi: 10.1159/000085386. [DOI] [PubMed] [Google Scholar]
- 236.Soleas G. J., Diamandis E. P., Goldberg D. M. Wine as a biological fluid: history, production, and role in disease prevention. Journal of Clinical Laboratory Analysis. 1997;11(5):287–313. doi: 10.1002/(SICI)1098-2825(1997)11:5<287::AID-JCLA6>3.0.CO;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kuroda S., Tsuchidate R., Smith M.-L., Maples K. R., Siesjö B. K. Neuroprotective effects of a novel nitrone, NXY-059, after transient focal cerebral ischemia in the rat. Journal of Cerebral Blood Flow & Metabolism. 1999;19(7):778–787. doi: 10.1097/00004647-199907000-00008. [DOI] [PubMed] [Google Scholar]
- 238.Sydserff S. G., Borelli A. R., Green A. R., Cross A. J. Effect of NXY-059 on infarct volume after transient or permanent middle cerebral artery occlusion in the rat; studies on dose, plasma concentration and therapeutic time window. British Journal of Pharmacology. 2002;135(1):103–112. doi: 10.1038/sj.bjp.0704449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Antonopoulos A. S., Margaritis M., Verheule S., et al. Mutual regulation of epicardial adipose tissue and myocardial redox state by PPAR-γ/Adiponectin signalling. Circulation Research. 2016;118(5):842–855. doi: 10.1161/CIRCRESAHA.115.307856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wójcicka G., Jamroz-Wiśniewska A., Atanasova P., Chaldakov G. N., Chylińska-Kula B., Bełtowski J. Differential effects of statins on endogenous H2S formation in perivascular adipose tissue. Pharmacological Research. 2011;63(1):68–76. doi: 10.1016/j.phrs.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 241.Samim A., Nugent L., Mehta P. K., Shufelt C., Bairey Merz C. N. Treatment of angina and microvascular coronary dysfunction. Current Treatment Options in Cardiovascular Medicine. 2010;12(4):355–364. doi: 10.1007/s11936-010-0083-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zhang X., Chen B., Wu J., et al. Aspirin enhances the protection of Hsp 90 from heat-stressed injury in cardiac microvascular endothelial cells Through PI3K-Akt and PKM2 pathways. Cells. 2020;9(1):p. 243. doi: 10.3390/cells9010243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Vilahur G., Gutiérrez M., Casani L., et al. Protective effects of ticagrelor on myocardial injury after infarction. Circulation. 2016;134(22):1708–1719. doi: 10.1161/circulationaha.116.024014. [DOI] [PubMed] [Google Scholar]
- 244.Bugiardini R., Borghi A., Pozzati A., Ottani F., Morgagni G. L., Puddu P. The paradox of nitrates in patients with angina pectoris and angiographically normal coronary arteries. The American Journal of Cardiology. 1993;72(3):343–347. doi: 10.1016/0002-9149(93)90683-4. [DOI] [PubMed] [Google Scholar]
- 245.Lerman A., Burnett J. C., Higano S. T., McKinley L. J., Holmes D. R. Long-term l-arginine supplementation improves small-vessel coronary endothelial function in humans. Circulation. 1998;97(21):2123–2128. doi: 10.1161/01.CIR.97.21.2123. [DOI] [PubMed] [Google Scholar]
- 246.Herrmann J., Kaski J. C., Lerman A. Coronary microvascular dysfunction in the clinical setting: from mystery to reality. European Heart Journal. 2012;33(22):2771–2783. doi: 10.1093/eurheartj/ehs246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Reriani M., Raichlin E., Prasad A., et al. Long-term administration of endothelin receptor antagonist improves coronary endothelial function in patients with early atherosclerosis. Circulation. 2010;122(10):958–966. doi: 10.1161/CIRCULATIONAHA.110.967406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Nasser S. A., El-Mas M. M. Endothelin ETA receptor antagonism in cardiovascular disease. European Journal of Pharmacology. 2014;737:210–213. doi: 10.1016/j.ejphar.2014.05.046. [DOI] [PubMed] [Google Scholar]
- 249.Fang D., Yang S., Quan W., Jia H., Quan Z., Qu Z. Atorvastatin suppresses Toll-like receptor 4 expression and NF-κB activation in rabbit atherosclerotic plaques. European Review for Medical and Pharmacological Sciences. 2014;18(2):242–246. [PubMed] [Google Scholar]
- 250.Yang S., Li R., Tang L., et al. TLR4-mediated anti-atherosclerosis mechanisms of angiotensin-converting enzyme inhibitor - Fosinopril. Cellular Immunology. 2013;285(1-2):38–41. doi: 10.1016/j.cellimm.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 251.Takahashi Y., Satoh M., Minami Y., Tabuchi T., Itoh T., Nakamura M. Expression of miR-146a/b is associated with the Toll-like receptor 4 signal in coronary artery disease: effect of renin-angiotensin system blockade and statins on miRNA-146a/b and Toll-like receptor 4 levels. Clinical Science (London, England) 2010;119(9):395–405. doi: 10.1042/CS20100003. [DOI] [PubMed] [Google Scholar]
- 252.Ji Y., Liu J., Wang Z., Liu N., Gou W. PPAR γ agonist, rosiglitazone, regulates angiotensin II-induced vascular inflammation through the TLR4-dependent signaling pathway. Laboratory Investigation. 2009;89(8):887–902. doi: 10.1038/labinvest.2009.45. [DOI] [PubMed] [Google Scholar]
- 253.Wang C.-Z., Zhang Y., Li X.-D., et al. PPARγ agonist suppresses TLR4 expression and TNF-α production in LPS stimulated monocyte leukemia cells. Cell Biochemistry and Biophysics. 2011;60(3):167–172. doi: 10.1007/s12013-010-9136-6. [DOI] [PubMed] [Google Scholar]
- 254.Ferreira A. E., Sisti F., Sônego F., et al. PPAR-γ/IL-10 axis inhibits MyD88 Expression and ameliorates murine polymicrobial sepsis. The Journal of Immunology. 2014;192(5):2357–2365. doi: 10.4049/jimmunol.1302375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Xu Y., Huang Y., Wu C., Zhang J., Liu Q. Effect of carvedilol on cardiomyocyte apoptosis in a rat model of myocardial infarction: a role for toll-like receptor 4. Indian Journal of Pharmacology. 2013;45(5):458–463. doi: 10.4103/0253-7613.117729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zhang D., Li Y., Liu Y., Xiang X., Dong Z. Paclitaxel ameliorates Lipopolysaccharide-Induced kidney injury by binding myeloid differentiation protein-2 to block Toll-Like receptor 4-mediated nuclear Factor-κB activation and cytokine production. Journal of Pharmacology and Experimental Therapeutics. 2013;345(1):69–75. doi: 10.1124/jpet.112.202481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Lucas K., Maes M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Molecular Neurobiology. 2013;48(1):190–204. doi: 10.1007/s12035-013-8425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Jia S. J., Niu P. P., Cong J. Z., Zhang B. K., Zhao M. TLR4 signaling: a potential therapeutic target in ischemic coronary artery disease. International Immunopharmacology. 2014;23(1):54–59. doi: 10.1016/j.intimp.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 259.Tsai K.-L., Hsieh P.-L., Chou W.-C., et al. IL-20 promotes hypoxia/reoxygenation-induced mitochondrial dysfunction and apoptosis in cardiomyocytes by upregulating oxidative stress by activating the PKC/NADPH oxidase pathway. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2020;1866(5):p. 165684. doi: 10.1016/j.bbadis.2020.165684. [DOI] [PubMed] [Google Scholar]
- 260.Samakova A., Gazova A., Sabova N., Valaskova S., Jurikova M., Kyselovic J. The pi 3k/Akt pathway is associated with angiogenesis, oxidative stress and survival of mesenchymal stem cells in pathophysiologic condition in ischemia. Physiological Research. 2019;68(2):S131–S138. doi: 10.33549/physiolres.934345. [DOI] [PubMed] [Google Scholar]
- 261.Friedenstein A., Chailakhjan R., Lalykina K. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell and Tissue Kinetics. 1970;3(4):393–403. doi: 10.1111/j.1365-2184.1970.tb00347.x. [DOI] [PubMed] [Google Scholar]
- 262.Friedenstein A. J., Chailakhyan R. K., Latsinik N. V., Panasyuk A. F., Keiliss-Borok I. V. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation. 1974;17(4):331–340. doi: 10.1097/00007890-197404000-00001. [DOI] [PubMed] [Google Scholar]
- 263.Rodius S., de Klein N., Jeanty C., et al. Fisetin protects against cardiac cell death through reduction of ROS production and caspases activity. Scientific Reports. 2020;10(1) doi: 10.1038/s41598-020-59894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ahmed S., Ahmed N., Rungatscher A., et al. Cocoa flavonoids reduce inflammation and oxidative stress in a myocardial schemia-reperfusion experimental model. Antioxidants. 2020;9(2):p. 167. doi: 10.3390/antiox9020167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Verma V. K., Malik S., Mutneja E., Sahu A. K., Bhatia J., Arya D. S. Attenuation of ROS-mediated myocardial ischemia–reperfusion injury by morin via regulation of RISK/SAPK pathways. Pharmacological Reports. 2020;72(4):877–889. doi: 10.1007/s43440-019-00011-2. [DOI] [PubMed] [Google Scholar]
- 266.Syeda M. Z., Fasae M. B., Yue E., et al. Anthocyanidin attenuates myocardial ischemia induced injury via inhibition of ROS-JNK-Bcl-2 pathway: New mechanism of anthocyanidin action. Phytotherapy Research. 2019;33(12):3129–3139. doi: 10.1002/ptr.6485. [DOI] [PubMed] [Google Scholar]
- 267.Cristina V. Oxidative stress and cardiovascular risk prediction: The long way towards a "radical" perspective. International Journal of Cardiology. 2018;273:252–253. doi: 10.1016/j.ijcard.2018.09.033. [DOI] [PubMed] [Google Scholar]
- 268.Vassalle C. An easy and reliable automated method to estimate oxidative stress in the clinical setting. Methods in Molecular Biology. 2008;477:31–39. doi: 10.1007/978-1-60327-517-0_3. [DOI] [PubMed] [Google Scholar]
- 269.Vassalle C., Pratali L., Boni C., Mercuri A., Ndreu R. An oxidative stress score as a combined measure of the pro-oxidant and anti- oxidant counterparts in patients with coronary artery disease. Clinical Biochemistry. 2008;41(14-15):1162–1167. doi: 10.1016/j.clinbiochem.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 270.Lubrano V., Pingitore A., Traghella I., et al. Emerging biomarkers of oxidative stress in acute and stable coronary artery disease: levels and determinants. Antioxidants. 2019;8(5):p. 115. doi: 10.3390/antiox8050115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Iamele L., Fiocchi R., Vernocchi A. Evaluation of an automated spectrophotometric assay for reactive oxygen metabolites in serum. Clinical Chemistry and Laboratory Medicine. 2002;40(7):673–676. doi: 10.1515/cclm.2002.115. [DOI] [PubMed] [Google Scholar]
- 272.Vassalle C., Maffei S., Boni C., Zucchelli G. C. Gender-related differences in oxidative stress levels among elderly patients with coronary artery disease. Fertility and Sterility. 2008;89:608–613. doi: 10.1016/j.fertnstert.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 273.Hirata Y., Yamamoto E., Tokitsu T., et al. Reactive oxygen metabolites are closely associated with the diagnosis and prognosis of coronary artery disease. Journal of the American Heart Association. 2015;4(2) doi: 10.1161/JAHA.114.001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Rodrigo R., Libuy M., Feliú F., Hasson D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Disease Markers. 2013;35(6):p. 790. doi: 10.1155/2013/974358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Chirinos J. A., Akers S. R., Trieu L., et al. Heart failure, left ventricular remodeling, and circulating nitric oxide metabolites. Journal of the American Heart Association. 2016;5(10) doi: 10.1161/JAHA.116.004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Akiyama K., Kimura A., Suzuki H., et al. Production of oxidative products of nitric oxide in infarcted human heart. Journal of the American College of Cardiology. 1998;32(2):373–379. doi: 10.1016/S0735-1097(98)00270-8. [DOI] [PubMed] [Google Scholar]
- 277.Higashino H., Tabuchi M., Yamagata S., et al. Serum nitric oxide metabolite levels in groups of patients with various diseases in comparison of healthy control subjects. Journal of Medical Sciences. 2009;10:1–11. doi: 10.3923/jms.2010.1.11. [DOI] [Google Scholar]
- 278.Cominacini L., Rigoni A., Pasini A. F., et al. The binding of oxidized low density lipoprotein (ox-LDL) to ox-LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. The Journal of Biological Chemistry. 2001;276(17):13750–13755. doi: 10.1074/jbc.M010612200. [DOI] [PubMed] [Google Scholar]
- 279.Mehta J. L., Chen J., Hermonat P. L., Romeo F., Novelli G. Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders. Cardiovascular Research. 2006;69(1):36–45. doi: 10.1016/j.cardiores.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 280.Kataoka H., Kume N., Miyamoto S., et al. Expression of Lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation. 1999;99(24):3110–3117. doi: 10.1161/01.CIR.99.24.3110. [DOI] [PubMed] [Google Scholar]
- 281.Hayashida K., Kume N., Murase T., et al. Serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are elevated in acute coronary syndrome: a novel marker for early diagnosis. Circulation. 2005;112(6):812–818. doi: 10.1161/CIRCULATIONAHA.104.468397. [DOI] [PubMed] [Google Scholar]
- 282.Ueda A., Kume N., Hayashida K., et al. ELISA for soluble form of lectin-like oxidized LDL receptor-1, a novel marker of acute coronary syndrome. Clinical Chemistry. 2006;52(6):1210–1211. doi: 10.1373/clinchem.2005.064808. [DOI] [PubMed] [Google Scholar]
- 283.Xuan Y., Gào X., Holleczek B., Brenner H., Schöttker B. Prediction of myocardial infarction, stroke and cardiovascular mortality with urinary biomarkers of oxidative stress: Results from a large cohort study. International Journal of Cardiology. 2018;273:223–229. doi: 10.1016/j.ijcard.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 284.Elesber A. A., Best P. J., Lennon R. J., et al. Plasma 8-iso-prostaglandin F2alpha, a marker of oxidative stress, is increased in patients with acute myocardial infarction. Free Radical Research. 2009;40(4):385–391. doi: 10.1080/10715760500539154. [DOI] [PubMed] [Google Scholar]
- 285.Vassalle C., Botto N., Andreassi M. G., Berti S., Biagini A. Evidence for enhanced 8-isoprostane plasma levels, as index of oxidative stress in vivo, in patients with coronary artery disease. Coronary Artery Disease. 2003;14(3):213–218. doi: 10.1097/01.mca.0000063504.13456.c3. [DOI] [PubMed] [Google Scholar]
- 286.Fierro-Fernández M., Miguel V., Lamas S. Role of redoximi Rs in fibrogenesis. Redox Biology. 2016;7:58–67. doi: 10.1016/j.redox.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Economou E. K., Oikonomou E., Siasos G., et al. The role of microRNAs in coronary artery disease: from pathophysiology to diagnosis and treatment. Atherosclerosis. 2015;241(2):624–633. doi: 10.1016/j.atherosclerosis.2015.06.037. [DOI] [PubMed] [Google Scholar]
- 288.Lee Y., Kim M., Han J., et al. MicroRNA genes are transcribed by RNA polymerase II. The EMBO Journal. 2004;23(20):4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Borchert G. M., Lanier W., Davidson B. L. RNA polymerase III transcribes human microRNAs. Nature Structural & Molecular Biology. 2006;13(12):1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 290.Okamura K., Ishizuka A., Siomi H., Siomi M. C. Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes & Development. 2004;18(14):1655–1666. doi: 10.1101/gad.1210204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Melak T., Baynes H. W. Circulating microRNAs as possible biomarkers for coronary artery disease: a narrative review. EJIFCC. 2019;30(2):179–194. [PMC free article] [PubMed] [Google Scholar]
- 292.Cheng X., Ku C.-H., Siow R. C. M. Regulation of the Nrf2 antioxidant pathway by microRNAs: New players in micromanaging redox homeostasis. Free Radical Biology & Medicine. 2013;64:4–11. doi: 10.1016/j.freeradbiomed.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 293.Mann G. E., Niehueser-Saran J., Watson A., et al. Nrf2/ARE regulated antioxidant gene expression in endothelial and smooth muscle cells in oxidative stress: implications for atherosclerosis and preeclampsia. Sheng Li Xue Bao. 2007;59(2):117–127. [PubMed] [Google Scholar]
- 294.Papageorgiou N., Tousoulis D., Charakida M., et al. Prognostic role of miRNAs in coronary artery disease. Current Topics in Medicinal Chemistry. 2013;13(13):1540–1547. doi: 10.2174/15680266113139990103. [DOI] [PubMed] [Google Scholar]
- 295.Zhang Y. H., Xia L. H., Jin J. M., Zong M., Chen M., Zhang B. Expression level of miR-155 in peripheral blood. Asian Pacific Journal of Tropical Medicine. 2015;8(3):214–219. doi: 10.1016/S1995-7645(14)60318-7. [DOI] [PubMed] [Google Scholar]
- 296.Huang R. S., Hu G. Q., Lin B., Lin Z. Y., Sun C. C. MicroRNA-155 silencing enhances inflammatory response and lipid uptake in oxidized low-density lipoprotein-stimulated human THP-1 macrophages. Journal of Investigative Medicine. 2015;58(8):961–967. doi: 10.231/JIM.0b013e3181ff46d7. [DOI] [PubMed] [Google Scholar]
- 297.Zhu G. F., Yang L. X., Guo R. W., et al. microRNA-155 is inversely associated with severity of coronary stenotic lesions calculated by the Gensini score. Coronary Artery Disease. 2014;25(4):304–310. doi: 10.1097/MCA.0000000000000088. [DOI] [PubMed] [Google Scholar]
- 298.Zhang M., Pan H., Xu Y., Wang X., Qiu Z., Jiang L. Allicin decreases lipopolysaccharide-induced oxidative stress and inflammation in human umbilical vein endothelial cells through suppression of mitochondrial dysfunction and activation of Nrf2. Cellular Physiology and Biochemistry. 2017;41(6):2255–2267. doi: 10.1159/000475640. [DOI] [PubMed] [Google Scholar]
- 299.Pulkkinen K. H., Yla-Herttuala S., Levonen A. L. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radical Biology & Medicine. 2011;51(11):2124–2131. doi: 10.1016/j.freeradbiomed.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 300.Reichard J. F., Motz G. T., Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Research. 2007;35:7074–7086. doi: 10.1093/nar/gkm638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Alam J., Cook J. L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? American Journal of Respiratory Cell and Molecular Biology. 2007;36:166–174. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 302.Igarashi K., Sun J. The heme-Bach 1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxidants & Redox Signaling. 2006;8:107–118. doi: 10.1089/ars.2006.8.107. [DOI] [PubMed] [Google Scholar]
- 303.Xiao W. Advances in NF-kappa B signaling transduction and transcription. Cellular & Molecular Immunology. 2004;1:425–435. [PubMed] [Google Scholar]
- 304.Zhang X., Yu Y., Lei H., et al. The Nrf-2/HO-1 signaling axis: a ray of hope in cardiovascular diseases. Cardiology Research and Practice. 2020;2020 doi: 10.1155/2020/5695723.5695723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Siow R. C., Sato H., Mann G. E. Heme oxygenase-carbon monoxide signalling pathway in atherosclerosis: anti-atherogenic actions of bilirubin and carbon monoxide? Cardiovascular Research. 1999;41:385–394. doi: 10.1016/s0008-6363(98)00278-8. [DOI] [PubMed] [Google Scholar]
- 306.Jin X., Xu Z., Cao J., et al. HO-1/EBP interaction alleviates cholesterol-induced hypoxia through the activation of the AKT and Nrf2/mTOR pathways and inhibition of carbohydrate metabolism in cardiomyocytes. International Journal of Molecular Medicine. 2017;39(6):1409–1420. doi: 10.3892/ijmm.2017.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Chen D., Jin Z., Zhang J., et al. HO-1 protects against hypoxia/reoxygenation-induced mitochondrial dysfunction in H9c2 cardiomyocytes. PLoS One. 2016;11(5) doi: 10.1371/journal.pone.0153587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Jin S. W., Hwang Y. P., Choi C. Y., et al. Protective effect of rutaecarpine against _t_ -BHP-induced hepatotoxicity by upregulating antioxidant enzymes via the CaMKII-Akt and Nrf2/ARE pathways. Food and Chemical Toxicology. 2017;100:138–148. doi: 10.1016/j.fct.2016.12.031. [DOI] [PubMed] [Google Scholar]
- 309.Bryan H. K., Olayanju A., Goldring C. E., Park B. K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochemical Pharmacology. 2013;85(6):705–717. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 310.Baker J. R., Vuppusetty C., Colley T., et al. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Scientific Reports. 2016;6(1) doi: 10.1038/srep35871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Tabuchi T., Satoh M., Itoh T., Nakamura M. MicroRNA-34a regulates the longevity-associated protein SIRT1 in coronary artery disease: effect of statins on SIRT1 and microRNA-34a expression. Clinical Science (London, England) 2012;123(3):161–171. doi: 10.1042/CS20110563. [DOI] [PubMed] [Google Scholar]
- 312.Bernardo B. C., Gao X.-M., Winbanks C. E., et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(43):17615–17620. doi: 10.1073/pnas.1206432109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Iekushi K., Seeger F., Assmus B., Zeiher A. M., Dimmeler S. Regulation of cardiac microRNAs by bone marrow mononuclear cell therapy in myocardial infarction. Circulation. 2012;125(14):1765–1773. doi: 10.1161/CIRCULATIONAHA.111.079699. [DOI] [PubMed] [Google Scholar]
- 314.Lin Y., Dan H., Lu J. Overexpression of microRNA-136-3p alleviates myocardial injury in coronary artery disease via the Rho A/ROCK signaling pathway. Kidney & Blood Pressure Research. 2020;45(3):477–496. doi: 10.1159/000505849. [DOI] [PubMed] [Google Scholar]
- 315.Wang H.-W., Huang T.-S., Lo H.-H., et al. Deficiency of the microRNA-31-microRNA-720 pathway in the plasma and endothelial progenitor cells from patients with coronary artery disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2014;34(4):857–869. doi: 10.1161/ATVBAHA.113.303001. [DOI] [PubMed] [Google Scholar]
- 316.Hulsmans M., Sinnaeve P., Van der Schueren B., Mathieu C., Janssens S., Holvoet P. Decreased miR-181a expression in monocytes of obese patients is associated with the occurrence of metabolic syndrome and coronary artery disease. The Journal of Clinical Endocrinology and Metabolism. 2012;97(7):E1213–E1218. doi: 10.1210/jc.2012-1008. [DOI] [PubMed] [Google Scholar]
- 317.Białek S., Górko D., Zajkowska A., et al. Release kinetics of circulating miRNA-208a in the early phase of myocardial infarction. Kardiologia polska. 2015;73(8):613–619. [PubMed] [Google Scholar]
- 318.Wang G.-K., Zhu J.-Q., Zhang J.-T., et al. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. European Heart Journal. 2010;31(6):659–666. doi: 10.1093/eurheartj/ehq013. [DOI] [PubMed] [Google Scholar]
- 319.Devaux Y., Vausort M., Goretti E., et al. Use of circulating microRNAs to diagnose acute myocardial infarction. Clinical Chemistry. 2012;58(3):559–567. doi: 10.1373/clinchem.2011.173823. [DOI] [PubMed] [Google Scholar]
- 320.Weber M., Baker M. B., Patel R. S., Quyyumi A. A., Bao G., Searles C. D. MicroRNA expression profile in CAD patients and the impact of ACEI/ARB. Cardiology Research and Practice. 2011;2011:5. doi: 10.4061/2011/532915.532915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Faccini J., Ruidavets J.-B., Cordelier P., et al. Circulating miR-155, miR-145 and let-7c as diagnostic biomarkers of the coronary artery disease. Scientific Reports. 2017;7(1) doi: 10.1038/srep42916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.D'Alessandra Y., Carena M. C., Spazzafumo L., et al. Diagnostic potential of plasmatic microRNA signatures in stable and unstable angina. PLoS ONE. 2013;8(11) doi: 10.1371/journal.pone.0080345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.D’Alessandra Y., Pompilio G., Capogrossi M. C. MicroRNAs and myocardial infarction. Current Opinion in Cardiology. 2012;27(3):228–235. doi: 10.1097/HCO.0b013e3283522052. [DOI] [PubMed] [Google Scholar]
- 324.Ren J., Zhang J., Xu N., et al. Signature of circulating microRNAs as potential biomarkers in vulnerable coronary artery disease. PLoS One. 2013;8(12, article e80738) doi: 10.1371/journal.pone.0080738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Li C., Fang Z., Jiang T., et al. Serum microRNAs profile from genome-wide serves as a fingerprint for diagnosis of acute myocardial infarction and angina pectoris. BMC Medical Genomics. 2013;6(1) doi: 10.1186/1755-8794-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Ward Nada Esa J. A. Circulating Cell and Plasma microRNA Profiles Differ between Non-STSegment and ST-Segment-Elevation Myocardial Infarction. Family medicine & medical science research. 2013;2(2) doi: 10.4172/2327-4972.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wang F., Long G., Zhao C., et al. Plasma microRNA-133a is a new marker for both acute myocardial infarction and underlying coronary artery stenosis. Journal of Translational Medicine. 2013;11(1):p. 222. doi: 10.1186/1479-5876-11-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.SN C. F. Piazza impact of bifurcation lesions on angiographic characteristics and procedural success in primary percutaneous coronary intervention for ST-segment elevation myocardial infarction. Archives of Cardiovascular Diseases. 2011;104(4):234–241. doi: 10.1016/j.acvd.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 329.Chen Z., Wen L., Martin M., et al. Oxidative stress activates endothelial innate immunity via sterol regulatory element binding protein 2 (SREBP2) transactivation of microRNA-92a. Circulation. 2015;131(9):805–814. doi: 10.1161/CIRCULATIONAHA.114.013675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Fan X., Wang E., Wang X., Cong X., Chen X. MicroRNA-21 is a unique signature associated with coronary plaque instability in humans by regulating matrix metalloproteinase-9 via reversion-inducing cysteine-rich protein with Kazal motifs. Experimental and Molecular Pathology. 2014;96(2):242–249. doi: 10.1016/j.yexmp.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 331.Soeki T., Yamaguchi K., Niki T., et al. Plasma microRNA-100 is associated with coronary plaque vulnerability. Circulation Journal. 2015;79(2):413–418. doi: 10.1253/circj.cj-14-0958. [DOI] [PubMed] [Google Scholar]
- 332.Jansen F., Yang X., Proebsting S., et al. MicroRNA expression in circulating microvesicles predicts cardiovascular events in patients with coronary artery disease. Journal of the American Heart Association. 2014;3(6):p. e001249. doi: 10.1161/JAHA.114.001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Schulte C., Molz S., Appelbaum S., et al. miRNA-197 and miRNA-223 predict cardiovascular death in a cohort of patients with symptomatic coronary artery disease. PLoS ONE. 2015;10(12, article e0145930) doi: 10.1371/journal.pone.0145930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Widera C., Gupta S. K., Lorenzen J. M., et al. Diagnostic and prognostic impact of six circulating microRNAs in acute coronary syndrome. Journal of Molecular and Cellular Cardiology. 2011;51(5):872–875. doi: 10.1016/j.yjmcc.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 335.He M., Gong Y., Shi J., et al. Plasma microRNAs as potential noninvasive biomarkers for instent restenosis. PLoS One. 2014;9(11, article e112043) doi: 10.1371/journal.pone.0112043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Fejes Z., Czimmerer Z., Szük T., et al. Endothelial cell activation is attenuated by everolimus via transcriptional and posttranscriptional regulatory mechanisms after drug-eluting coronary stenting. PLoS One. 2018;13(6, article e0197890) doi: 10.1371/journal.pone.0197890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Fichtlscherer S., De Rosa S., Fox H., et al. Circulating microRNAs in patients with coronary artery disease. Circulation Research. 2010;107(5):677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]