

Abstract
The development of next generation sequencing technology has dramatically improved our understanding of the genetic landscape of multiple myeloma. Several new drivers and recurrent events have been reported and linked to a potential driver role. This complex landscape is enhanced by intra-clonal mutational heterogeneity and variability introduced through the dimensions of time and space. The evolutionary history of multiple myeloma is driven by both the accumulation of different genomic drivers and by the activity of different mutational processes active overtime. In this review, we describe how these new findings and sequencing technologies have been progressively allowed to understand and re-shape our knowledge of the complexity of multiple myeloma at each of its developmental stages: premalignant, at diagnosis and in relapsed/refractory states. We discuss how these evolutionary concepts can be utilized in the clinic to alter evolutionary trajectories providing a framework for therapeutic intervention at early disease stages.
Keywords: Multiple Myeloma, Driver events, Mutational signatures, Cancer Prevention, Interception strategies
Introduction
Many blood cancers are associated with premalignant clonal expansions, the prevalence of which increases with age (1,2). These entities include monoclonal B-lymphocytosis (MBL) a precursor of chronic lymphocytic leukemia, clonal hematopoiesis (CH) a precursor of acute leukemia and importantly monoclonal gammopathy of uncertain significance (MGUS) and smoldering multiple myeloma (SMM) precursors of multiple myeloma (MM) (Fig. 1) (2–6). Clinical management of pre-cancer is becoming increasingly important and should be based on sound biological understandings. A unifying feature of all of these states is their heterogeneity at a mutational level comprising a variable set of gene mutations acquired by apparently stochastic processes that drive changes in biologic behavior. In this model multiple clonal propagating units compete for survival and clonal dominance resulting in either their suppression or selection and expansion over time leading to clonal diversification and metastatic seeding.(7,8) The model is credible because it can explain mutational differences at different sites (spatial variation), the acquisition or selection of mutations at relapse (temporal variation) and the development of resistance following treatment. Thus, before invasive cancer develops, many pre-malignant clones have been present for years within apparently normal cell populations. Importantly clinical intervention in this pre-expansion phase before the full repertoire of drivers are acquired is likely to offer the greatest potential to eradicate the clonal cells and result in long-term disease-free survival and potentially cure.
Fig. 1.
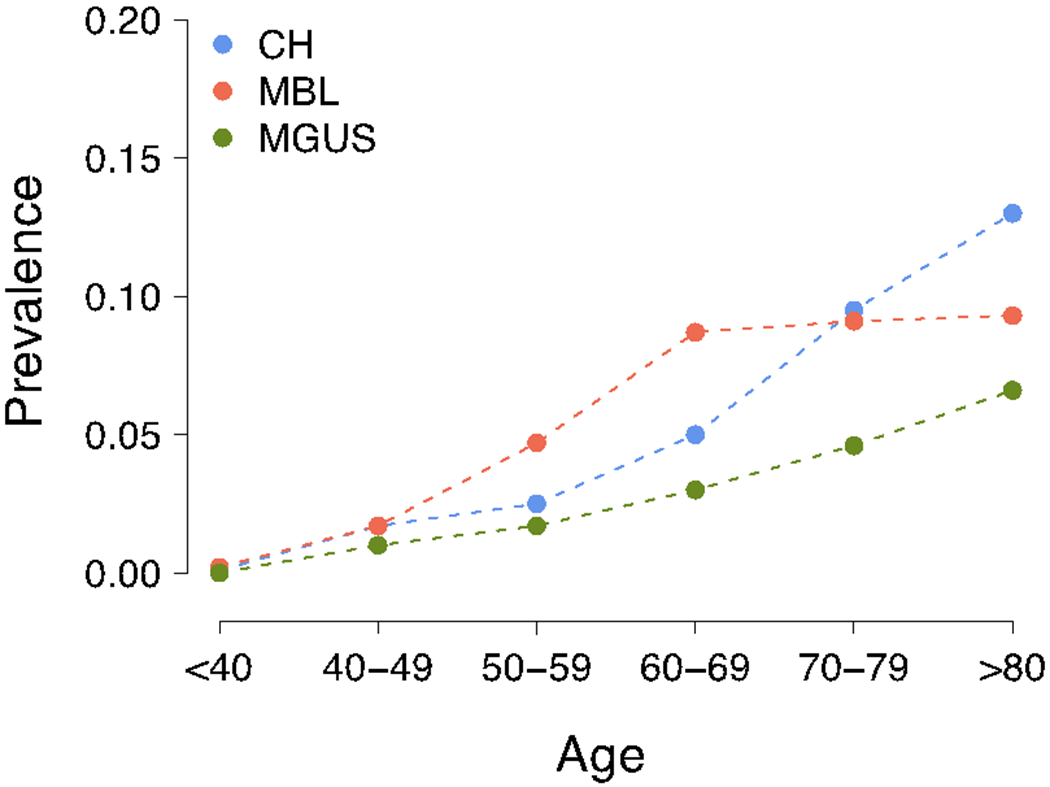
Increasing prevalence of MGUS, monoclonal B-cell lymphoproliferation (MBL) and clonal hemopoiesis (CH) with ageing.
The life history of MM is characterized by pre-malignant clinical phases (i.e. MGUS and SMM), which are recognized by the presence of a monoclonal immunoglobulin produced by the premalignant clone (6,7,9,10). According to historical models, the pre-malignant clone is immortalized by a limited number of key initiating genetic events in the germinal center (GC) (7,9) and then homes to the bone marrow, where it evolves, predominantly below the level of clinical detection until identified as either MGUS or SMM (11,12). Thus, before MM presents many pre-malignant clones have been present for decades and, within this ecosystem, multiple genetically diverse MM propagating units compete for survival. The clinical features of MM reflect the end result of evolutionary processes occurring as a result of the genetic diversification of the clone over time (13–16). While originally it was proposed that evolution proceeded steadily overtime, more recently it has been appreciated that it may also be driven by catastrophic events were multiple genes are deregulated simultaneously (i.e. “punctuated evolution”)(17,18), which can also result in rapid changes in behavior(5,19,20). Intercepting these processes to prevent transition to cancer has clinical relevance in MM because 5% of the over 60 year old population have a detectable monoclonal immunoglobulin that transforms to MM at 1–10% per annum a malignancy (5,19).
Multiple myeloma as a model to understand cancer initiation and progression
I. Insights from cytogenetic analyses
The use of metaphase cytogenetic analysis, Southern blotting, iFISH and gene expression analysis (7,9) identified two main initiating events: multiple trisomy’s of the odd numbered chromosomes (i.e. hyperdiploid) and recurrent chromosomal translocations involving the IGH locus that overexpressed key oncogenes including CCND1, CCND2, CCND3, MMSET, MAF and MAFB (Supplementary Fig. 1) (21–23). These events were identified as being etiologic because they are present in all tumor cells in all disease phases (24,25). The formation of these translocations is a consequence of AID activity in the GC, introducing DNA double strand breaks (DBS) in the IGH locus, which are then joined to another DBS occurring elsewhere in the genome. Distinct IGH-translocations and recurrent aneuploidies have been linked to poor outcome [e.g. t(4;14)(MMSET;IGH), amp 1q21 and del17p13 (TP53)] and have been integrated in clinical/biological prognostic scoring systems (26). The events on 1q21 and 17p were well know before SNP arrays, but clearly arrays and next generation sequencing have improved our understanding of their prevalence, evolution and the underlying mechanisms involved in their acquisition (Supplementary Fig. 1) (26–28). While the first 1q21 gain is an early event often occurring together with other large trisomies, it is clear that jumping translocations and additional amplifications of 1q21 and del17p13 are often late events.(17,28)
Ii. Insights from next generation sequencing
The initial genetic analyses of MM have allowed us to make a number of important conclusions about the contribution of genetic changes to disease progression. While superficially the acquired genetic features of MM appear complex, it is only a limited number of these events that are involved in “driving” the malignant process. Exome sequencing focused attention on approximately 80 recurrent “driver genes” mainly impacting RAS/MAPK and NFkB signaling, with some being prognostically important (13,14,16,17,29–34). Despite a significant fraction of these mutations in driver genes being selected, they are often subclonal suggesting that, they are late events occurring after other earlier clonal events.(32,35)
To fully understand the development of MM, it is critical to realize that most sequencing data has been generated from cases of newly diagnosed MM and that the genetic make-up of precursor phases is largely unknown. A number of different data sets including SNP array, exome and targeted sequencing data revealed that the frequency of recurrent driver mutations, copy number changes and structural events typical of MM occur at much lower frequencies in myeloma precursor disease (6,14,36–38). Further, the prognostic and clinical impact of any given lesion may be different i.e. RAS mutations lack prognostic importance in newly diagnosed MM but in SMM are emerging as a strong prognostic factor of transition to MM (39,40).
While exome sequencing has provided most data, it does not fully capture structural variants (SVs) and copy number abnormalities (CNAs) that occur in the non-coding sequences, which since the advent of whole genome sequencing have been shown to be pathologically crucial (6,17,18,41). Using CoMMpass, more than a 100 chromosomal regions of recurrent gain or loss involving amplified or deleted genes have been identified further expanding the compendium of known disease drivers (Supplementary Fig. 1) (18).
The SVs burden in MM is greater than CLL and AML, and is in the same order as B-NHL but is considerably less than in most solid cancers (17,42,43). Cataloguing SVs in MM, WGS identified three main complex SV events: chromothripsis, chromoplexy, and templated insertions (Fig. 2) (17,18,44). Chromothripsis occurs in a single catastrophic event where one or more chromosomes are shattered and randomly rejoined, leading to a pattern of oscillating copy number changes and localized clustering of breakpoints. This event was initially thought to be rare, but using WGS it can now be detected in 20–30% of MM and is associated with adverse survival. (18,45). Chromoplexy is a complex chained event detectable in 11% of patients based on concatenated rearrangements between different chromosomes causing copy number losses (17,18,44). A type of complex rearrangement termed “templated insertions” is associated with multiple focal gains and is seen in 20% of MM, a prevalence that is higher than other cancers (17,18,44). Templated insertions involve the immunoglobulin loci in 34% of cases and leads to the focal amplification of key drivers and immune-therapy targets such as MYC, CCND1, CCND2, TNFRSF17 (BCMA), SLAMF7 and KLF2 (Fig. 2) (18). Overall, from a structural perspective the genomic profile of MM is more stable than some other cancers, lacking evidence of the recently proposed complex events: rigma, thyphonas or double minute events.(18,46). These complex SVs are particularly important because they potentially involve multiple drivers simultaneously, suggesting that the full MM driver repertoire can be acquired in only few events. This concept is particularly important in the precursor settings and in its monitoring, where it could explain rapid changes in clinical behavior of these entities.
Fig. 2.
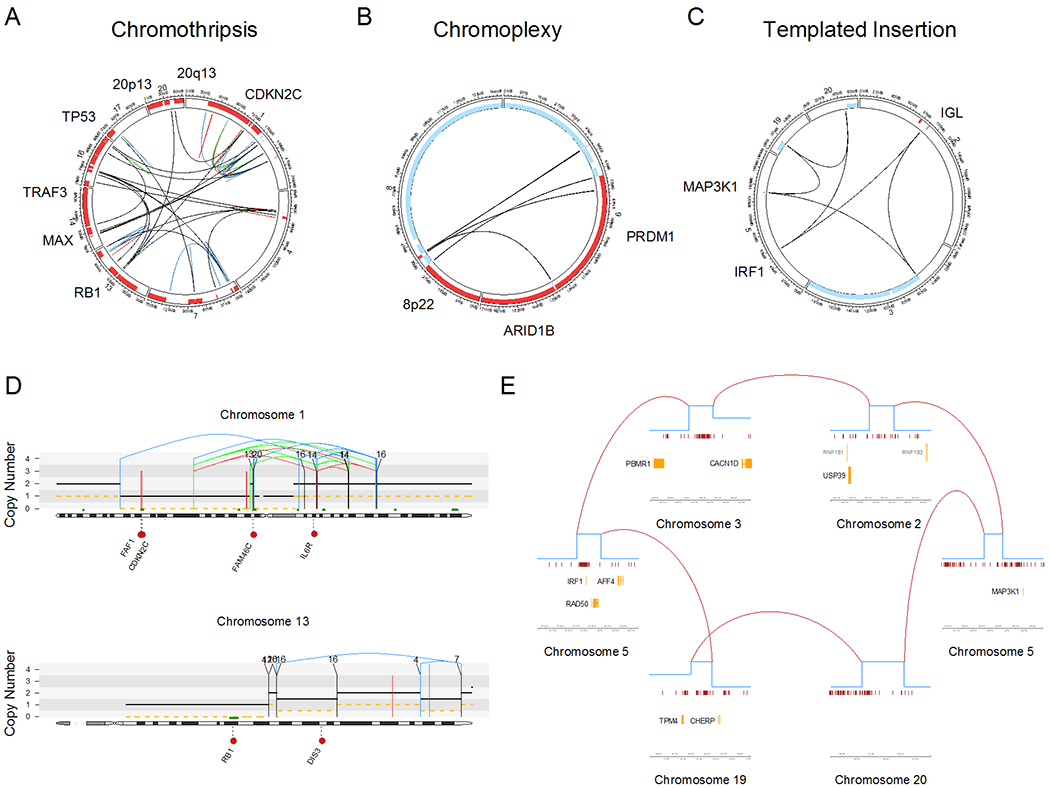
Types of complex structural variants identified in multiple myeloma patients enrolled in CoMMpass trial. A-C) Example of patients with chromothripsis (MMRF_2671_1_BM; A), chromoplexy (MMRF_2516_1_BM; b) and templated insertions (MMRF_1928_1_BM; C). All these complex events simultaneously involved multiple known driver events. D) Examples of chromothripsis (top) and chromoplexy (bottom) responsible for major copy number aberrations. E) Zoom in on each focal copy number gain and structural variants involved by a templated insertion (CoMMpass patient: MMRF_2330_1_BM).
Mutational signatures driving progression of early premalignant phases to multiple myeloma
The median number of mutations per MM exome and genome is 149 (range 3-6186) and 5885 (range 1511-15884), respectively and lies at the median of the genetically simpler acute leukemias and more complex tumors such as melanoma (Fig. 3A) (13,14,16,17,25,32,33,35,41,47). The mutational catalogue of each cancer represents the sum of the activity of multiple different mutational processes over time (25,48). Understanding the processes that drive these mutations active in early disease stages points the way towards active therapeutic intervention able to selectively influence clonal competition to suppress aggressive sub-clones. Signatures or “mutographs” of mutational prosses are based 96-mutational classes defined by the base on 5’ and 3’ side of each mutated nucleotide (i.e. 6 possible single nucleotide variants * 16 possible trinucleotide context) being readily generated for each tumor. Mutations in newly diagnosed MM are the result of 7 main mutational processes (Fig. 3B) (41). APOBEC deaminase signatures (single base substitution (SBS2 and SBS13) are seen in a significant fraction of MM (14,25,29,33,35,49). A high mutational burden with the features of APOBEC is associated with t(14;16), chromothripsis, bi-allelic TP53 and poor outcome (17,18,33). Two “clock-like” signatures have been identified: SBS1 and SBS5 that are characterized by constant rate over time in both normal and tumor cells (50). Three additional mutational processes; SBS8, SBS9 and SBS18, have recently been described in NDMM but their true relevance is unclear (14,25,41,49). SBS8 has been seen in breast and ovarian where it has been associated with BRCA1 and BRCA2 deficiency (51). However, in MM there has been no suggestion of a BRCA-null phenotype suggesting a different etiology (25). SBS18 is seen a small fraction of MM is linked to free radical oxygen stress (48). Of key importance to MM progression is SBS9, termed non-canonical activation-induced cytidine deaminase (AID) to differentiate it from the canonical AID signature (SBS84) and is directly related to the GC, somatic hyper mutation and AID exposure (25,48,52–54) The SBS9 mutational process doesn’t have the typical feature of AID-mediated somatic hyper mutation (C to T/G mutation at WRCY motifs, W=A or T, R=purine, Y=pyrimidine), but is enriched in T>G and T>C consistent with mismatch repair pathway repair of AID-related mutations. In contrast canonical AID (SBS84) is mostly involved in somatic hyper mutation or kataegis on distinct targets (e.g. IGH/IGK/IGL loci). While some of these canonical AID mutations are likely passenger events (e.g. intronic BCL6 mutations), others show some evidence of positive selection (25). Both AID mutational activities occurred are early, clonal and usually precede the gains of odd chromosomes leading to hyperdiploidy.
Fig. 3.
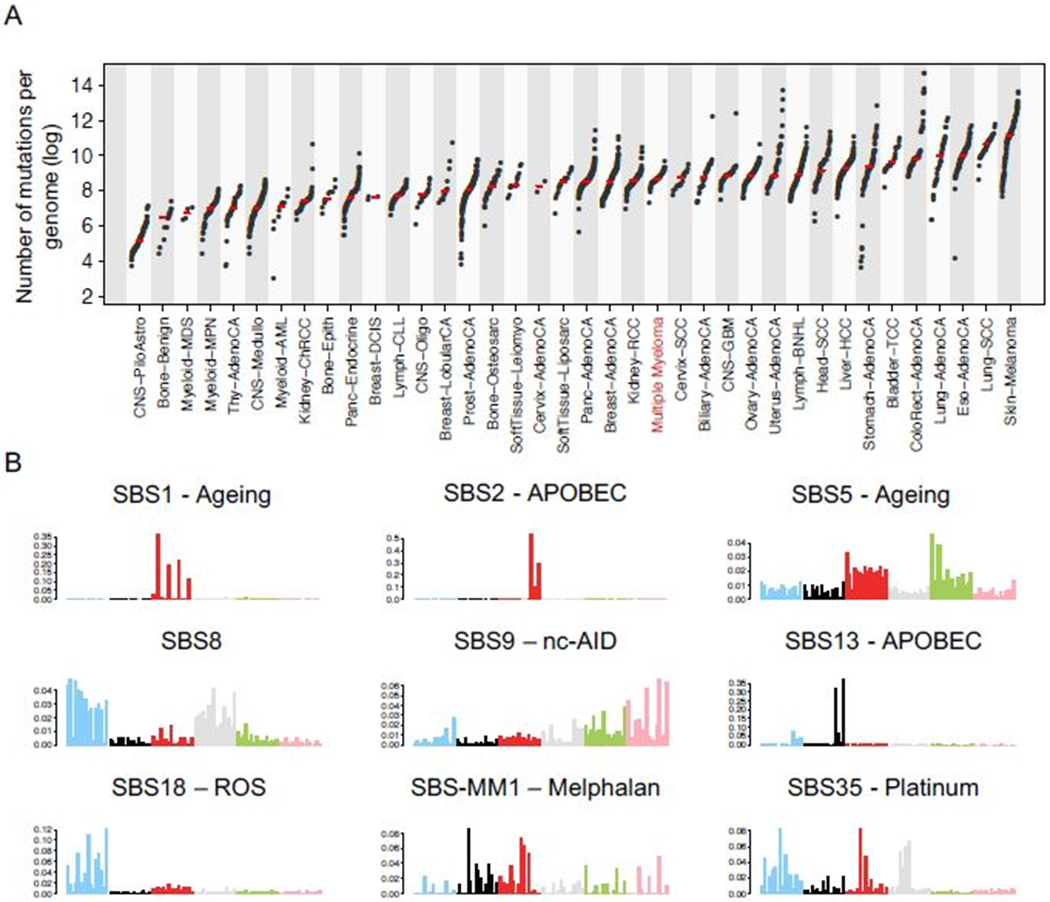
The multiple myeloma mutational signature landscape. a) The prevalence and median of somatic mutations across human cancer types evaluated by WGS. b) The nine mutational signatures extracted from WGS data in multiple myeloma.
At relapse two mutational signatures have been recently reported and involved in increasing the number of non-synonymous mutations: i) SBS35 and a new mutational signature, named SBS-MM1, as a consequence of exposure to the mutagenic activity of platinum and melphalan, respectively (Fig. 3B) (41,55).
The temporal relationship of mutational signatures and driver events
i. A timeline for the acquisition of driver events in multiple myeloma
Computational tools can de-convolute these temporally related events allowing the reconstruction of the life history of MM and an ability to build an encompassing disease model. Different approaches have been used to estimate the time point at which driver events are acquired during cancer life-history. The most common uses the cancer cell fraction (CCF; i.e. number of cells carrying a distinct alteration to define early and late time points based on their sub-clonal percentage) (17,32). Applying this to MM shows that the HRD trisomies and 1q21 gain have high CCF and are usually clonal, consistent with them being early events. In contrast, the recurrent deletions (e.g. del1p, del14q and del16q) and non-synonymous mutations have a heterogeneous distribution, consistent with them occurring later in the disease process.
Greater insights into the chronological order of driver acquisition can be gained by reconstructing the phylogenetic tree (17). The trunk of the tree being composed of clonal events acquired before the events identified in the branches (i.e. sub-clonal) (Supplementary Fig. 2). Phylogenetic tree reconstruction is based on few simple rules. If in one samples the fraction of events A and B within the cancer that are present at 75% and 40%, respectively, then according to the “pigeonhole principle” (56), there must be at least one tumor cell that contains both mutations, because the existence of two independent sub-clones with a cumulative cancer cell fraction > 100% is not possible (Supplementary Fig. 2A–B). Conversely, if B were present at a cell fraction of 20% this would be compatible with either the presence of independent cluster or with a clone carrying both mutations (Supplementary Fig. 2C). The resolution provided by the phylogenetic tree can be improved by the inclusion of multiple samples from the same individual collected at different time points or at different disease sites (17,55,57). Using this information, it is possible to reconstruct the chronological order of driver events and define patient specific “evolutionary trajectories” based on pairwise precedence. By taking the relative order of drivers in all patients, it is possible to define the order in which genes are mutated relative to one another.(17,41) This approach has shown that more than 50% of all mutations in MM occur as early events.
To refine temporal estimations further large chromosomal duplications can be used based on the fact that when an allele is duplicated all the genomic events acquired until that point are also duplicated (17,58). Thus, while their CCF will be always 100% the variant allele frequency (VAF) of the duplicated mutations will change from 50% to 66%, being present on two out of the three alleles. Conversely, clonal mutations acquired either before or after the gain on the minor allele will have a VAF of 33%. This technique has shown that the final karyotype, in a significant fraction of hyper diploid patients, reflects the sum of multiple independent events separated by time (17) By combining molecular time and CCF assessment for the timing of molecular events it is possible to define an evolutionary trajectory based on driver events that occur in the early (pre-gain), intermediate (post-gain and clonal) and late (sub-clonal) evolutionary phases of disease.
Despite their critical importance SVs have not been routinely used to reconstruct cancer evolutionary trees. To test the impact of SV on sub-clonal evolution, two main approaches can be used. The first links each SV to its corresponding CNA and uses the clonality of the latter. This approach cannot be used all the time because not all SV have copy change for accurate CCF estimation. The second approach requires multiple samples collected at different time points. In this analysis, conserved clonal events are acquired before unique selected events and a chronological hierarchy recreated. This analysis showed as expected that etiological IGH translocations are clonal. Chromothripsis was demonstrated to be an early event, was detected in the majority of clonal cells as were templated insertions. A fraction of complex events involves MYC as a late progression event. Chromoplexy is usually a late event and at relapse.
Applying this classification, IGH-translocations, copy number gains, AID-mediated mutations on driver genes, chromothripsis emerged as early events and detectable at the early precursor stage of disease. Templated insertions are also usually clonal and detectable at the early disease stage. However, a fraction of these complex events involves MYC a known driver of progression of SMM. In contrast the distribution of chromoplexy, focal and large chromosomal deletions and non-AID mutations on driver genes suggests they are usually acquired during late phases of cancer development and at relapse.
ii. A timeline for the acquisition of mutational signatures.
The same workflow has been used to define the timeline of each mutational process in MM and two main time windows were observed (Fig. 4) (14,41,49). The first is characterized by high AID and low/absent APOBEC, SBS18 and SBS8 mutational activity. During this phase the pre-malignant clone is chronically exposed to AID and the GC activity leading to the acquisition of key drivers. Different from what has historically been believed, these mutations are not acquired in one single passage through the GC, but are more consistent with evidence suggesting a prolonged exposure to AID and the GC of the pre-malignant clone. This chronic and prolonged AID activity is consistent with the recently proposed model wherein antigen stimulation results in clonal plasma cell expansion and M spike increase in mice (59,60).
Fig. 4.
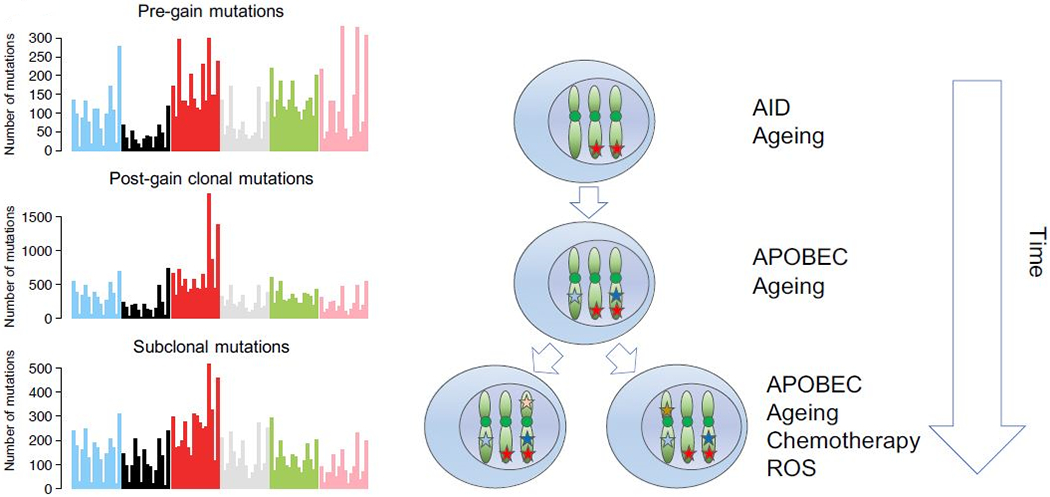
Illustration of how to reconstruct the mutational signature activity over time in multiple myeloma. When an allele is duplicated, all the mutations acquired since the fertilized egg will be duplicated and present on the two duplicated alleles. This will change their VAF from 50% to 66%. In contrast, all the mutations acquired after the duplication will be present only on one allele and have a 33% VAF. Differentiating pre and post gain mutations allows to explore the chronological order of clonal events. Combining this with data from phylogenic tree reconstruction (i.e. clonal vs subclonal) we can divide a faction of genomic events in early (pre gain), intermediate (post gain) and late (subclonal). Doing so on 52 WGS, we observed different patterns of mutations and signatures. Specifically, AID tends to dramatically decrease from pre to post gain (e.g. T>G peaks); APOBEC increase after gains (e.g. peaks in A[C>T]T and T[C>T]T); chemotherapy signatures are acquired later G[C>G]X, in line with their post-diagnosis acquisition.
The second time window is at a phase when the pre-malignant cell is independent of the GC-mediated AID mutational activity and at this stage; SBS8, SBS18 and APOBEC play major roles in ongoing mutation acquisition. In particular APOBEC has been shown to play a critical role in the acquisition of non-synonymous mutations and in the sub-clonal diversification after the emergence of the most common recent ancestor (35,41).This model does not apply only in the MAF/MAFB group that has a high APOBEC mutational burden.(29,33) Preliminary investigations suggest that in these patients the APOBEC signature has been active from the earliest disease phases, while AID and the GC contribution is barely detectable. In these patients the APOBEC mutational activity shows the same profile of APOBEC hyper-mutated solid cancers, with a major APOBEC3A/3B ratio compared to all other samples (41Chan, 2015 #6820,48,61).
These findings are important for intervention strategies with APOBEC emerging as a strong marker for risk of SMM progression (40), in re-defining high-risk(29,33) MM and in understanding clonal evolution (41). Importantly APOBEC is known to increase non-synonymous mutational burden, potentially introducing new drivers and enhancing clonal progression(41); APOBEC, in particular APOBEC3B, has emerged as the most important and dominant mutational process in a subset of MM characterized by MAF and MAFB translocations (41).
Insights from functional genomics.
Functional genomics can provide additional key insights into molecular targets that if manipulated could exert a potential therapeutic impact. If because of sub-clonal heterogeneity mutational targeted therapy is flawed, then biological based targeting aimed at essential cell functions offers a way forward. Furthermore, functional genomics can identify critical biological nodes that can be targeted. A number of functional screens have been performed in MM(62), but the approach has potential limitations because of introducing the guide sequences into a plasma cell background and because it is based on myeloma cell lines whose genomic and transcriptomic background doesn’t properly reflect the one observed in primary samples from patients diagnosed with MM. Despite this potential drawback this approach has successfully identified targets that can overcome therapeutic resistance for IMID drugs and proteasome inhibitors.(63,64) In the future a closer integration of genomic and function study will be critical to identify key drivers and therapeutic targets.
Building models to understand the bionetworks active in precursor disease phases
The pathway to MM is not direct and involves a long precursor phase where off target effects of the GC and AID are emerging as crucial for the first phase of cancer development. Dissecting the molecular processes active in the early natural history of MM can allow us to build credible models of the disease that could be build alternate therapeutic strategies appropriate for premalignant disease states.
During normal plasma cell ontology, B-cells with high-affinity receptors exit the GC as either memory B-cells or as plasma cells, which home to the bone marrow where they either produce antibody or become long-lived with appropriate survival signals from the bone marrow niches (65). Thus, B-cell maturation to a plasma cell is characterized by the potential for the acquisition of hundreds of mutations and structural variants as a consequence of AID-mediated DNA double breaks and by chance alone cells carrying these may be immortalized and have the potential to develop into malignancy. The observation of early and prolonged AID activity provides further evidence for the importance of the GC and implicates a cell of origin with the ability to re-enter a GC; potentially one with the features of a memory B-cell. The presence of this signature also implies a pre-MGUS phase before the full features of a BM plasma cell are developed and during which time sub-clonal diversity and selections are enhanced by exposure to the GC mutator-mechanism, (Fig. 5).
Fig. 5.
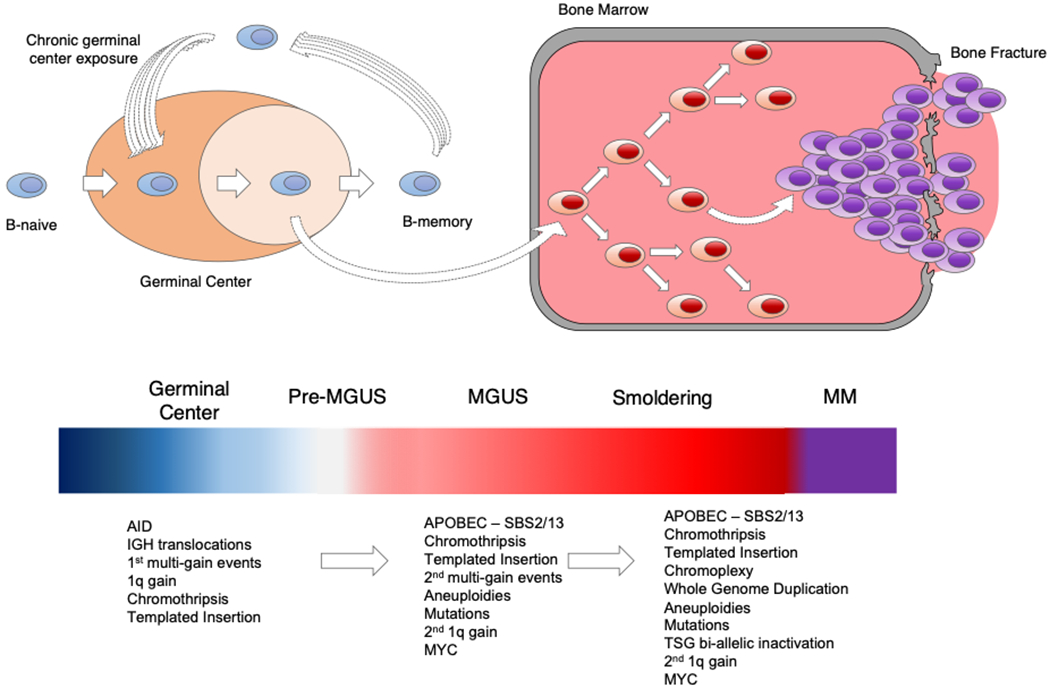
Model for the development of multiple myeloma based on the latest whole genome sequencing data. Compared to previous model we proposed here the existence of a pre-MGUS GC-phase. Recent data suggest that the pre-MGUS cell experienced a prolonged exposure to the GC. During these exposures several key drivers and AID mediated mutations are acquired. At certain point, this clone becomes GC-independent and move to the bone marrow, where it starts the MGUS>SMM>MM evolution.
After initiation and homing to the marrow, the plasma cell grows in its bone marrow niche where both intrinsic (i.e. cancer genomics) and extrinsic (i.e. microenvironment) factors influence the of rate progression. These factors may vary over time dependent upon the nature of acquired mutations but at this point there is little evidence of acquired genetic instability in MM. Despite MM is characterized by a higher cytogenetic complexity compared to other hematological malignancies, recent data showed that many of these aneuploidies are acquired in single catastrophic events (i.e. complex SVs), suggesting that that the full driver landscape of MM can be acquired in few events (17,18). The impact of different spontaneous mutational processes can be seen at the SMM-MM transition, which is comprised of at least 3 distinct patterns of transition to MM; a stable MGUS type disease, MM in the process of transition or an indolent subtype of MM (5,6). The speed of this transition is also variable some occurring rapidly while others occur slowly. Rapid changes may reflect punctuated evolution where a catastrophic event such as chromothripsis may deregulate more than one driver simultaneously, (Fig. 2).
Extrinsic factors take account of the natural selection acting on the clonal cells within their microenvironment (66–69). Such factors include the availability of resources, access to the MM niche and the impact of the immune microenvironment.
Manipulating multiple myeloma ecology to intercept disease transformation to multiple myeloma
Based on the evolutionary consideration we have outlined, the use of the same treatment strategies for premalignant disease states as for presenting MM may be inappropriate.(70) While using combinations of drugs with multiple modes of action has been successful at partially overcoming the diversity of newly diagnosed MM the potential for generating resistance, side effects and off target effects on normal tissue makes it difficult to justify the exposure of these patient to such risks in premalignant states. Such considerations are clearly relevant with the known mutagenic activity of melphalan and platinum, which has been demonstrated to increase the mutational burden after treatment.(41,55,71) Further, the long-term impact of cytotoxic chemotherapy on non-tumor cells can result in prolonged immunosuppression, which, in turn can facilitate MM genomic accelerated evolution and progression. These emerging data suggest that chemotherapy should be actively discouraged in the precursor settings (41,55,71). The evolutionary models of MM progression provide an alternate framework for considering the treatment of early cancers by modifying the evolutionary trajectory of the disease. For a successful interception approach, the assessment of disease will need to change to take account of both intrinsic (i.e. tumor cell) and extrinsic (i.e. microenvironment). This will require the development of evolutionary classifications that capture both of the genetics of the cancer as well as an assessment of the immune response to it. Using the combination of both these sets of data it will be possible to define the likelihood of progression and to personalize therapeutic decision-making.
The role of genetic assessment is supported by recent work suggesting that mutations in early disease stages (i.e. precursor disease) may have different clinical meaning to those seen in patients who fulfill the diagnostic criteria for MM. A key example of this is RAS mutations, which are predictive of a higher risk of SMM transformation in MM but have no clear prognostic value in newly diagnosed MM (40). Other emerging genetic features, such as APOBEC, have a clear prognostic role both in the precursors settings and in the newly diagnosed MM (29,33,40). The development of new single cell technologies able to more effectively interrogate the immune microenvironment will enhance the potential to fully determine the contribution of this to disease progression and to the likelihood of responding to immune therapies (69).
We believe that interception strategies will become critical features of cancer management in the future as the number potential interventions increases. Importantly one of the major goals of interception at a time before cancer symptomatic progression is the potential for cure. Targeted therapies aimed at key biological have good characteristics for clinical application in early disease stages but mutationally targeted therapy unless directed at initiating mutations such as t(11;14)(CCDN1;IGH) MM, which are present in all clonal cells will require novel application approaches to overcome the sub-clonal nature of mutation.(72)
For many reasons, immune interventions are preferable for early precursor stages of disease and the use of vaccine-based strategies offers a non-toxic and potentially ideal intervention especially if monitored by the stability of sub-clonal structure and immune response. However, clinical proof for the efficacy of such an approach is currently lacking. In contrast, there is evidence that other immune based approaches could prove useful in this setting. IMiD drugs either alone or in combination with anti-CD38 therapeutic are potentially relevant and a number of clinical trials shown to be relatively safe and potentially beneficial but their potential to cause resistant disease at future time points is currently unknown (6). More active interventions such as the bi-specific anti-BCMA antibodies used to direct T and NK cells or CAR T-cells offer a clear way forward. These agents are associated with dramatically increased response rates over prior agents and may deliver significant advances for interception if acute adverse reactions such as cytokine release syndrome can be overcome. What is clear for all such interventions is that effective clinical trial evaluation will be required. For this to be practical, new endpoints will have to be developed to give readouts of efficacy in a reasonable time period. Such endpoints are likely to be based on achieving deep responses where disease cannot be detected but in this setting, there will be a strong need to take account of the impact of the intervention on the immune microenvironment.
Supplementary Material
Acknowledgments
This study is supported by the MMRF and Perelman Foundation
Dr. Francesco Maura and Dr. Ola Landgren are supported by the Memorial Sloan Kettering Cancer Center NCI Core Grant (P30 CA 008748).
Dr. Francesco Maura is supported by the American Society of Hematology, the International Myeloma Foundation and The Society of Memorial Sloan Kettering Cancer Center.
Dr GJ Morgan is supported by Leukemia and Lymphoma Society
Footnotes
Disclosure of conflicts of interest: No conflict of interests to declare.
References
- 1.Ghobrial IM, Detappe A, Anderson KC, Steensma DP. The bone-marrow niche in MDS and MGUS: implications for AML and MM. Nat Rev Clin Oncol 2018;15(4):219–33 doi 10.1038/nrclinonc.2017.197 [DOI] [PubMed] [Google Scholar]
- 2.Landgren O, Albitar M, Ma W, Abbasi F, Hayes RB, Ghia P, et al. B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med 2009;360(7):659–67 doi 10.1056/NEJMoa0806122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371(26):2477–87 doi 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371(26):2488–98 doi 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med 2007;356(25):2582–90 doi 10.1056/NEJMoa070389. [DOI] [PubMed] [Google Scholar]
- 6.Maura F, Bolli N, Rustad EH, Hultcrantz M, Munshi N, Landgren O. Moving From Cancer Burden to Cancer Genomics for Smoldering Myeloma: A Review. JAMA Oncol 2019. doi 10.1001/jamaoncol.2019.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer 2012;12(5):335–48 doi 10.1038/nrc3257. [DOI] [PubMed] [Google Scholar]
- 8.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet 2012;13(11):795–806 doi 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol 2017;14(2):100–13 doi 10.1038/nrclinonc.2016.122. [DOI] [PubMed] [Google Scholar]
- 10.Rajkumar SV, Landgren O, Mateos MV. Smoldering multiple myeloma. Blood 2015;125(20):3069–75 doi 10.1182/blood-2014-09-568899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. Hematol Oncol Clin North Am 2007;21(6):1093–113, ix doi 10.1016/j.hoc.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood 2009;113(22):5412–7 doi 10.1182/blood-2008-12-194241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 2014;5:2997 doi 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun 2018;9(1):3363 doi 10.1038/s41467-018-05058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012;120(5):1067–76 doi 10.1182/blood-2012-01-405985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014;25(1):91–101 doi 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun 2019;10(1):3835 doi 10.1038/s41467-019-11680-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rustad E, Yellapantula V, Glodzik D, Maclachlan K, Diamond B, Boyle E, et al. Revealing the impact of recurrent and rare structural variants in multiple myeloma. Biorxiv 2019. doi 10.1101/2019.12.18.881086. [DOI] [Google Scholar]
- 19.Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, et al. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N Engl J Med 2018;378(3):241–9 doi 10.1056/NEJMoa1709974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landgren O, Hofmann JN, McShane CM, Santo L, Hultcrantz M, Korde N, et al. Association of Immune Marker Changes With Progression of Monoclonal Gammopathy of Undetermined Significance to Multiple Myeloma. JAMA Oncol 2019. doi 10.1001/jamaoncol.2019.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J Jr., Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood 2005;106(1):296–303 doi 10.1182/blood-2005-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chesi M, Bergsagel PL, Brents LA, Smith CM, Gerhard DS, Kuehl WM. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood 1996;88(2):674–81. [PubMed] [Google Scholar]
- 23.Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet 1997;16(3):260–4 doi 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barwick BG, Gupta VA, Vertino PM, Boise LH. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front Immunol 2019;10:1121 doi 10.3389/fimmu.2019.01121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maura F, Degasperi A, Nadeu F, Leongamornlert D, Davies H, Moore L, et al. A practical guide for mutational signature analysis in hematological malignancies. Nat Commun 2019;10(1):2969 doi 10.1038/s41467-019-11037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015;33(26):2863–9 doi 10.1200/JCO.2015.61.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorsteinsdottir S, Dickman PW, Landgren O, Blimark C, Hultcrantz M, Turesson I, et al. Dramatically improved survival in multiple myeloma patients in the recent decade: results from a Swedish population-based study. Haematologica 2018;103(9):e412–e5 doi 10.3324/haematol.2017.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aktas Samur A, Minvielle S, Shammas M, Fulciniti M, Magrangeas F, Richardson PG, et al. Deciphering the chronology of copy number alterations in Multiple Myeloma. Blood Cancer J 2019;9(4):39 doi 10.1038/s41408-019-0199-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maura F, Petljak M, Lionetti M, Cifola I, Liang W, Pinatel E, et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia 2017. doi 10.1038/leu.2017.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol 2015;33(33):3911–20 doi 10.1200/JCO.2014.59.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2018. doi 10.1038/s41375-018-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018;132(6):587–97 doi 10.1182/blood-2018-03-840132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun 2015;6:6997 doi 10.1038/ncomms7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolli N, Biancon G, Moarii M, Gimondi S, Li Y, de Philippis C, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia 2018;32(12):2604–16 doi 10.1038/s41375-018-0037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maura F, Rustad EH, Yellapantula V, Luksza M, Hoyos D, Maclachlan KH, et al. Role of AID in the temporal pattern of acquisition of driver mutations in multiple myeloma. Leukemia 2019. doi 10.1038/s41375-019-0689-0. [DOI] [PubMed] [Google Scholar]
- 36.Mailankody S, Kazandjian D, Korde N, Roschewski M, Manasanch E, Bhutani M, et al. Baseline mutational patterns and sustained MRD negativity in patients with high-risk smoldering myeloma. Blood Adv 2017;1(22):1911–8 doi 10.1182/bloodadvances.2017005934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mikulasova A, Smetana J, Wayhelova M, Janyskova H, Sandecka V, Kufova Z, et al. Genomewide profiling of copy-number alteration in monoclonal gammopathy of undetermined significance. Eur J Haematol 2016;97(6):568–75 doi 10.1111/ejh.12774. [DOI] [PubMed] [Google Scholar]
- 38.Mikulasova A, Wardell CP, Murison A, Boyle EM, Jackson GH, Smetana J, et al. The spectrum of somatic mutations in monoclonal gammopathy of undetermined significance indicates a less complex genomic landscape than that in multiple myeloma. Haematologica 2017;102(9):1617–25 doi 10.3324/haematol.2017.163766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Misund K, Keane N, Stein CK, Asmann YW, Day G, Welsh S, et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020;34(1):322–6 doi 10.1038/s41375-019-0543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bustoros M, Sklavenitis-Pistofidis R, Park J, Redd R, Zhitomirsky B, Dunford AJ, et al. Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. J Clin Oncol 2020:JCO2000437 doi 10.1200/JCO.20.00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rustad EH, Yellapantula V, Leongamornlert D, Bolli N, Ledergor G, Nadeu F, et al. Timing the initiation of multiple myeloma. Nat Commun 2020;11(1):1917 doi 10.1038/s41467-020-15740-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barwick BG, Neri P, Bahlis NJ, Nooka AK, Dhodapkar MV, Jaye DL, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat Commun 2019;10(1):1911 doi 10.1038/s41467-019-09555-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mikulasova A, Ashby C, Tytarenko RG, Qu P, Rosenthal A, Dent JA, et al. Microhomology-mediated end joining drives complex rearrangements and over expression of MYC and PVT1 in multiple myeloma. Haematologica 2019. doi 10.3324/haematol.2019.217927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020;578(7793):112–21 doi 10.1038/s41586-019-1913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Magrangeas F, Avet-Loiseau H, Munshi NC, Minvielle S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 2011;118(3):675–8 doi 10.1182/blood-2011-03-344069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hadi K, Yao X, Behr J, Deshpande A, Xanthopoulakis C, Rosiene J, et al. Novel patterns of complex structural variation revealed across thousands of cancer genome graphs. bioarxiv 2019. doi 10.1101/836296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dutta AK, Fink JL, Grady JP, Morgan GJ, Mullighan CG, To LB, et al. Subclonal evolution in disease progression from MGUS/SMM to multiple myeloma is characterised by clonal stability. Leukemia 2019;33(2):457–68 doi 10.1038/s41375-018-0206-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578(7793):94–101 doi 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoang PH, Cornish AJ, Dobbins SE, Kaiser M, Houlston RS. Mutational processes contributing to the development of multiple myeloma. Blood Cancer J 2019;9(8):60 doi 10.1038/s41408-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet 2015;47(12):1402–7 doi 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 2017;23(4):517–25 doi 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kasar S, Kim J, Improgo R, Tiao G, Polak P, Haradhvala N, et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat Commun 2015;6:8866 doi 10.1038/ncomms9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015;526(7574):519–24 doi 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 54.Hoang PH, Dobbins SE, Cornish AJ, Chubb D, Law PJ, Kaiser M, et al. Whole-genome sequencing of multiple myeloma reveals oncogenic pathways are targeted somatically through multiple mechanisms. Leukemia 2018. doi 10.1038/s41375-018-0103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Landau H, Yellapantula V, Diamond B, Rustad E, Maclachlan K, Gundem G, et al. Accelerated single cell seeding in relapsed multiple myeloma. Biorxiv 2020. doi 10.1101/2020.02.25.963272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell 2012;149(5):994–1007 doi 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun 2017;8(1):268 doi 10.1038/s41467-017-00296-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature 2020;578(7793):122–8 doi 10.1038/s41586-019-1907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nair S, Branagan AR, Liu J, Boddupalli CS, Mistry PK, Dhodapkar MV. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N Engl J Med 2016;374(6):555–61 doi 10.1056/NEJMoa1508808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nair S, Sng J, Boddupalli CS, Seckinger A, Chesi M, Fulciniti M, et al. Antigen-mediated regulation in monoclonal gammopathies and myeloma. JCI Insight 2018;3(8) doi 10.1172/jci.insight.98259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 2013;45(9):970–6 doi 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Matos Simoes R, Shirasaki R, Tang H, Sheffer M, Dashevsky O, Gandolfi S, et al. Functional Genomic Landscape of Genes with Recurrent Mutations in Multiple Myeloma. Blood 2018;132 (Supplement 1): 189. doi 10.1182/blood-2018-189. [DOI] [Google Scholar]
- 63.Liu J, Song T, Zhou W, Xing L, Wang S, Ho M, et al. A genome-scale CRISPR-Cas9 screening in myeloma cells identifies regulators of immunomodulatory drug sensitivity. Leukemia 2019;33(1):171–80 doi 10.1038/s41375-018-0205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi CX, Kortum KM, Zhu YX, Bruins LA, Jedlowski P, Votruba PG, et al. CRISPR Genome-Wide Screening Identifies Dependence on the Proteasome Subunit PSMC6 for Bortezomib Sensitivity in Multiple Myeloma. Mol Cancer Ther 2017;16(12):2862–70 doi 10.1158/1535-7163.MCT-17-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127(20):2375–90 doi 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Das R, Strowig T, Verma R, Koduru S, Hafemann A, Hopf S, et al. Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nat Med 2016;22(11):1351–7 doi 10.1038/nm.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manier S, Sacco A, Leleu X, Ghobrial IM, Roccaro AM. Bone marrow microenvironment in multiple myeloma progression. J Biomed Biotechnol 2012;2012:157496 doi 10.1155/2012/157496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bailur JK, McCachren SS, Doxie DB, Shrestha M, Pendleton K, Nooka AK, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight 2019;5 doi 10.1172/jci.insight.127807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oksana Z, Haradhvala NJ, Mouhieddine TH, Sklavenitis-Pistofidis R, Cai S, Reidy M, et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nature Cancer 2020(1):493–506 doi 10.1038/s43018-020-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghobrial IM, Landgren O. How I treat smoldering multiple myeloma. Blood 2014;124(23):3380–8 doi 10.1182/blood-2014-08-551549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pich O, Muinos F, Lolkema MP, Steeghs N, Gonzalez-Perez A, Lopez-Bigas N. The mutational footprints of cancer therapies. Nat Genet 2019;51(12):1732–40 doi 10.1038/s41588-019-0525-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bertamini L, Bonello F, Boccadoro M, Bringhen S. New drugs in early development for treating multiple myeloma: all that glitters is not gold. Expert Opin Investig Drugs 2020. doi 10.1080/13543784.2020.1772753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.