

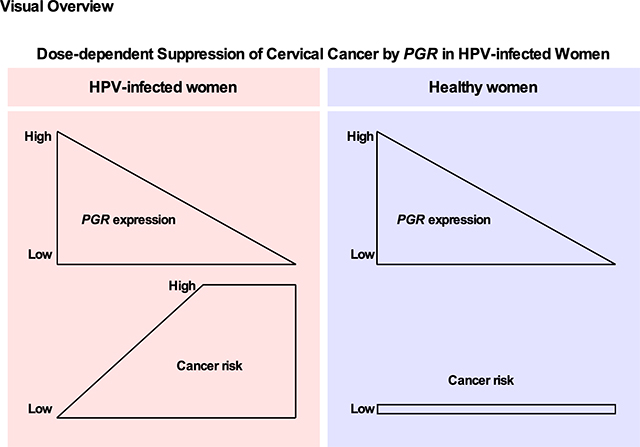
Abstract
Tumor suppressor genes (TSGs) are often deleted or transcriptionally suppressed in cancer. PGR codes for progesterone receptor (PR), a transcription factor whose function depends on its ligand. While PR expression is often undetectable in cervical cancer, its relevance to the endocrine-related etiology of this prevalent gynecological disease remains unclear. In this study, we show that the deletion of one Pgr allele in cervical epithelium promoted spontaneous cervical cancer in human papilloma viral oncogene-expressing transgenic mice as efficiently as the ablation of both Pgr alleles. We also show that tumors arising in the transgenic mice with one or both Pgr alleles did not express PR or expressed at the reduced levels compared to the normal epithelium. PR status correlated with estrogen receptor α (ERα) status in the mouse model and the TCGA dataset. TCGA data analyses revealed that PGR expression significantly decreased in cervical cancer and that the biallelic deletion of PGR was rare. Furthermore, low PGR expression was associated with poor prognosis in young cervical cancer patients. These discoveries point to PGR as a haploinsufficient TSG in the uterine cervix. They also raise the possibility that the restoration of PGR expression may improve the survival rate.
Keywords: Spontaneous cervical cancer, human papillomavirus, progesterone receptor, haploinsufficient tumor suppressor gene
Graphical Abstract
Introduction
Genetically engineered mouse models have been a powerful tool to discover novel tumor suppressor genes (TSGs) and define their molecular mechanisms. Functions of TSGs are frequently impaired in cancers by mutations or transcriptional repression. The removal of one allele of many haploinsufficient TSGs, such as Dicer1, Fbw7, and Pten, is sufficient to promote cancers in mice, and they have similar mono-allelic mutations in human cancers (1). Unlike TSGs with biallelic deletions, re-expression or reactivation of haploinsufficient TSGs may be exploited as therapeutic targets.
Cervical cancer remains the third most frequent cancer and the third leading cause of cancer deaths among women worldwide (2). While human papillomavirus (HPV) vaccines and the Pap test are effective in preventing cervical cancer, they are not readily available to women in low-income countries and those of low socioeconomic status in developed countries. Multiple full-term pregnancies and the use of oral contraceptives for longer than five years increases the risk of cervical cancer in HPV-infected women (3,4). While these results implicate female sex hormones in cervical cancer, retrospective studies attempting to determine individual roles of estrogen (E2) and progesterone (P4) have been inconclusive (5). E2 and P4 activate estrogen receptor α (ERα) and progesterone receptor (PR), respectively. Both receptors are ligand-dependent transcription factors that belong to the nuclear receptor superfamily.
HPV16 E6 and E7 oncoproteins are responsible for cervical cancer. HPV transgenic mice expressing these oncoproteins develop cervical cancer at high penetrance when treated with E2 for six months (6). We have previously shown that the cancer-promoting action of E2 depends on stromal ERα rather than epithelial ERα (7,8). In contrast, we discovered that P4 inhibits proliferation and induces apoptosis in the cervical epithelium in an epithelial PR-dependent manner (9). Activation of PR by synthetic progesterone medroxyprogesterone acetate (MPA) was effective in treating and preventing cervical cancer in the HPV transgenic mouse model (10). In the present study, we show that the deletion of one PR-coding Pgr allele sensitizes HPV transgenic mice to cervical cancer without E2 treatment. Our analyses of cervical cancer tissues from the mouse model and the Cancer Genome Atlas (TCGA) database reveal that PR is expressed at the lower level in cancer than normal epithelium. Furthermore, PGR heterozygosity is common in human cervical cancer, and low PGR expression is associated with poor overall survival in young cervical cancer patients. Our results support that PGR is a haploinsufficient TSG in the uterine cervix and a promising cervical cancer therapeutic target.
Materials and Methods
Animals and reagents
Experimental mice were generated by mating K14E7/Pgrf/f males with K14E6/Wnt7aCre/Pgrf/+ females. All mouse strains were previously described and summarized in Supplemental Table 1. Cervical tissues were harvested at 8–9 months of age without any treatment. The University of Houston Institutional Animal Care and Use Committee approved all procedures performed on mice. Antibodies and chemicals are described in Supplemental Table 2.
Tissue processing, histological staining, and analyses
Mouse cervical tissues were fixed in 4% paraformaldehyde, paraffin-embedded, and serially sectioned throughout the cervix at a 5 μm-thickness. Every tenth slide was stained with hematoxylin and eosin (H&E). Blinded histopathological analyses with the help of a pathologist and cancer measurement were carried out as described in Supplemental Materials and Methods.
Immunohistochemistry and apoptosis assay
Antigens were retrieved by incubating slides in pepsin solution for 5 min for p16Ink4a or microwaving sections for 20 min in 10 mM sodium citrate buffer (pH 6.0) for the others. Slides were incubated with primary antibodies diluted in the blocking buffer, as described in Supplemental Table 2. After extensive washes in phosphate-buffered saline, slides were subsequently incubated with a secondary antibody. Nuclei were stained with Hoechst 33258 solution (10 μg/mL) for 30 sec. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was carried out with ApopTag Fluorescein in situ apoptosis detection kit according to the manufacturer’s instruction. Slides were mounted with a gelvatol mounting medium.
Clinical data analysis
TCGA-Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma (CESC) data were used for survival analysis and the expression of PGR and ESR1 mRNA as described in Supplemental Materials and Methods. The Genotype-Tissue Expression (GTEx) Cervix cohort was also used for the latter.
Results
The deletion of one or both Pgr alleles sensitizes HPV transgenic mice to spontaneous cervical carcinogenesis.
While PR activation by MPA suppresses cervical cancer, E2 treatment results in similar cancer incidences in K14E7/Pgr+/+ and K14E7/Pgr−/− (10). We reasoned that it was due to the low level of P4 (i.e., minimal PR activation) because the treatment with exogenous E2 keeps mice at a continuous estrus-like stage (i.e., no P4 surges) (11). We sought to determine whether cervical cancer would arise at a high frequency without E2 treatment if the PR signaling pathway were inactive. We used the Wnt7aCre transgene (referred to as Cre hereafter), which deletes Pgr specifically in the cervical epithelium (9). We generated K14E6/K14E7 (E6/E7) double transgenic or K14E7 (E7) single transgenic mice on the Pgrf/+, Cre/Pgrf/+, and Cre/Pgrf/f background and aged them without any treatments. As expected, cervical cancer did not occur in non-transgenic (NTG)/Pgrf/+ and NTG/Cre/Pgrf/f control mice that did not express any HPV oncogenes (Table 1). Cervical cancer incidence was significantly higher in E7/Pgrf/+ (33.3%) and E6/E7/Pgrf/+ (28.6%) than NTG/Pgrf/+ control (P < 0.002). Notably, the incidence of cervical cancer significantly increased in E7/Cre/Pgrf/f (61.7%) and E6/E7/Cre/Pgrf/f (78.6%) compared to E7/Pgrf/+ and E6/E7/Pgrf/+, respectively (P < 0.009) (Table 1). Unexpectedly, the incidence of cervical cancer significantly increased in E7/Cre/Pgrf/+ (62.0%) and E6/E7/Cre/Pgrf/+ (64.3%) compared to E7/Pgrf/+ and E6/E7/Pgrf/+, respectively (P < 0.05) (Table 1). Also, cancer incidences in E7/Cre/Pgrf/+ and E6/E7/Cre/Pgrf/+ were not significantly different from those of E7/Cre/Pgrf/f and E6/E7/Cre/Pgrf/f (Table 1). These results indicated that the deletion of only one Pgr allele was sufficient to inhibit the tumor-suppressive activity of PR in the cervix.
Table 1.
Summary of the worst cervical neoplastic diseases
| Genotypes | HPV oncogenes | Epithelial Pgr status | Group size, n. | No Disease | CIN | Cancer | Cancer incidence (%) |
|---|---|---|---|---|---|---|---|
| E6/E7/Cre/Pgrf/f | E6 & E7 | null | 14 | 0 | 3 | 11 | 78.6* |
| E6/E7/Cre/Pgrf/+ | het | 14 | 0 | 5 | 9 | 64.3* | |
| E6/E7/Pgrf/+ | wt | 28 | 4 | 16 | 8 | 28.6** | |
| E7/Cre/Pgrf/f | E7 | null | 60 | 0 | 23 | 37 | 61.7* |
| E7/Cre/Pgrf/+ | het | 50 | 0 | 19 | 31 | 62.0* | |
| E7/Pgrf/+ | wt | 42 | 6 | 22 | 14 | 33.3** | |
| NTG/Cre/Pgrf/f | None | null | 22 | 22 | 0 | 0 | 0 |
| NTG/Cre/Pgrf/+ | het | 20 | 17 | 2 | 1 | 5 | |
| NTG/Pgrf/+ | wt | 29 | 28 | 1 | 0 | 0 | |
NTG/Pgrf/+ and NTG/Pgrf/f were pooled and shown as NTG/Pgrf/+.
E7/Cre/Pgrf/− and E7/Cre/Pgrf/f showed the similar disease burden. They were pooled and shown as E7/Cre/Pgrf/f.
E7/Pgr f/− and E7/Cre/Pgrf/+ showed the similar disease burden. They were pooled and shown as E7/Cre/Pgrf/+.
P < 0.05 compared to cancer incidence in E6/E7/Pgrf/+ or E7/Pgrf/+.
P < 0.002 compared to cancer incidence in NTG/Pgrf/+.
Het, heterozygote; wt, wild-type
PR is not expressed in the majority of cervical cancers arising in Pgr-sufficient mice.
Next, we asked whether the level of PR expression in cancers correlated with the Pgr genotype. We analyzed all cancers for PR expression by immunohistochemistry. As expected, PR was not expressed in all cancers in E7/Cre/Pgrf/f and E6/E7/Cre/Pgrf/f mice (Figure 1A). To our surprise, PR was undetectable in the majority of cancers arising in E7/Pgrf/+ (58.8%, n=17), E7/Cre/Pgrf/+ (56.9%, n=51), E6/E7/Pgrf/+ (61.5%, n=13), and E6/E7/Cre/Pgrf/+ (83.3%, n=18) (Figure 1A; see PR− cancer). The percentage of PR-negative cancer was highest in E6/E7/Cre/Pgrf/+ mice, but the difference did not reach statistical significance. The remaining cancers expressed PR (Figure 1A; see PR+ cancer). Cancer-associated stroma was positive for PR in all mice. Like cancers arising in E2-treated mice (6), all cervical cancers were microscopic and well-differentiated regardless of genotypes and PR status (Supplemental Figure 1A). Dysplastic epithelia adjacent to PR-negative cancers did not express PR in both E7/Pgrf/+ and E7/Cre/Pgrf/+ (Figure 1B). PR expression was similar in PR-positive cancer and nearby dysplastic epithelium, but it was reduced compared to the normal epithelium distant from cancer (Figure 1B). H&E staining of corresponding epithelia is shown in Supplementary Figure 1B. These results supported that PR expression was lost or downregulated in a precancer stage. ERα is necessary for the expression PR in the cervix (9). ERα expression was undetectable in 29 out of 33 (87.9%) PR-negative cancers (Figure 1C). The remaining PR-negative cancers and all PR-positive cancers (n=20) expressed ERα (Figure 1C). There was a strong positive correlation between the PR and ERα status (rφ = 0.86, P = 5.25 × 10−11). These results suggested that ERα−dependent and −independent mechanisms were involved in PR downregulation.
Figure 1. PR is not expressed in most cervical cancers.

(A) The majority of cancers developed on the Pgrf/+ and Cre/Pgrf/+ backgrounds are negative for PR. Cervical cancer sections were stained for PR (green). Nuclei are pseudo-colored red. Dotted lines separate cervical cancer (cc) and epithelium (ep) from stroma (st). Numbers are percentages of PR− and PR+ cancer in each genotype. A Pgr−/− cervical section was used as a negative control. Scale bar, 50 μm. (B) Cancer-associated epithelium has lower PR expression than normal epithelium. Cervical cancer-containing sections were stained for PR (green) and nuclei (red). H&E staining of corresponding regions are shown in Supplementary Figure 1B. Scale bar, 50 μm. (C) ERα is undetectable in most PR− cancers. Randomly selected PR− (33 out of 62) and PR+ cancers (20 out of 37) from Pgrf/+ and Cre/Pgrf/+ genotypes were stained for ERα (green) and nuclei (red). Dotted lines separate cervical cancer (cc) from stroma (st). An Esr1−/− cervical section was used as a negative control. Scale bar, 50 μm.
PR-negative cancers are larger than PR-positive cancers.
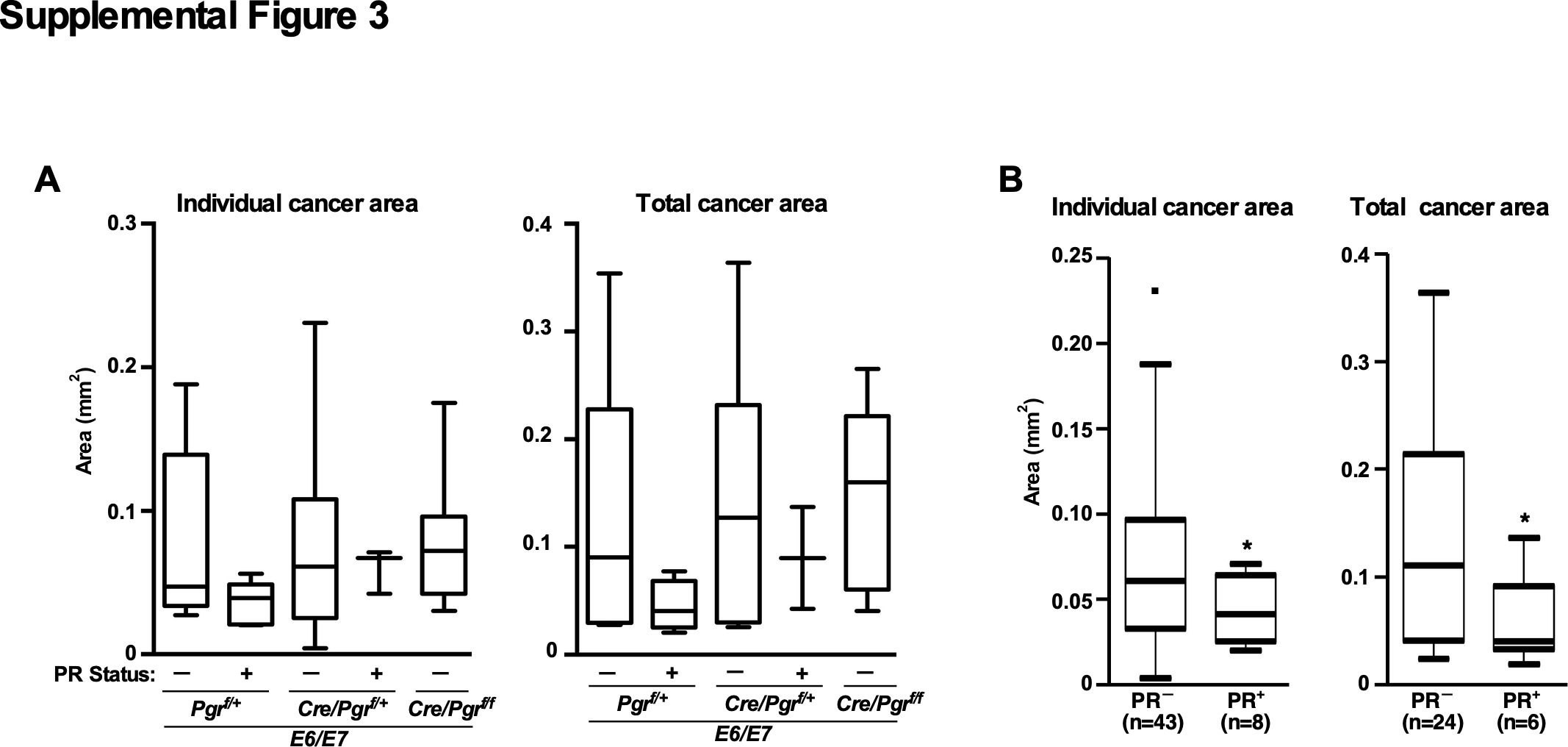
The expression of p16Ink4a and Mcm7, biomarkers for HPV+ cervical cancer, was increased in both PR− and PR+ cervical cancer in E7 and E6/E7 mice compared to a cancer in NTG/Cre/Pgrf/+ (Supplemental Figure 2A–B). To assess an effect of PR on cancer growth, we divided cancers in each genotype according to the PR status. In both E7/Pgrf/+ and E7/Cre/Pgrf/+, the largest and total cancer areas were significantly larger for PR− than PR+ cervical cancer (Figure 2A). The size of PR− cancer in these mice was not different from that in E7/Cre/Pgrf/f, indicating that the PR status rather than the Pgr genotype was predictive of cancer size. We observed a similar trend in E6/E7/Pgrf/+ and E6/E7/Cre/Pgrf/+ mice, but the difference did not reach statistical significance (Supplemental Figure 3A). When pooled from all genotypes, however, PR− cancers had significantly greater individual and total cancer areas than PR+ cancers (Supplemental Figure 3B). Larger cancers could be due to either earlier development (i.e., growth for a longer period) and/or faster growth. We could not evaluate the former possibility because diseases were diagnosed at end points. To test the latter, we determined proliferation and apoptosis rate in PR− and PR+ cancer. To control for hormonal conditions, we compared a PR− carcinoma with a PR+ cancer in the same mouse. A fraction of Ki67+ cells was always higher in PR− than PR+ cancer (Figure 2B). In all five paired comparisons, the rate of TUNEL+ cells was lower in PR− than PR+ cervical cancer (Figure 2C). These findings suggested that, albeit the decreased expression in PR+ cancers and failure to inhibit cancer development, PR was still active in suppressing tumor growth.
Figure 2. The low level of PR expression inhibits the growth of cervical cancer.

(A) PR− cancers are larger than PR+ cancers. The largest cancer area and total cancer area in each mouse are shown in box–and–whisker plots. The group sizes were as the following: PR− (n=8) and PR+ cancer (n=7) in Pgrf/+, PR− (n=23) and PR+ cancer (n=20) in Cre/Pgrf/+, and PR− cancer (n=37) in Cre/Pgrf/f. *P < 0.05. **P < 0.01 (one-sided Wilcoxon rank-sum test). (B) PR− cancer is more proliferative than PR+ cancer. Five cervical tissues bearing both PR− and PR+ cancer were selected from K14E7/Cre/Pgrf/+ mice. Upper panel: Cancer sections were stained for Ki67 (green) and nuclei (red). Dotted lines separate cancers (cc) from stroma (st). Scale bar, 50 μm. Lower panel: Results shown in the upper panel were quantified. Approximately 400–600 cells per cancer were counted. A pair of cancers from the same mouse are shown in the same color. *P = 0.006 (paired one–sided t–test). (C) PR− cancer is less apoptotic than PR+ cancer. Cancers shown in (B) were subjected to TUNEL assay. TUNEL+ cells are green, and nuclei are pseudocolored red. White dotted lines separate cancers (cc) from stroma (st). Scale bar, 50 μm. Results shown in the images were quantified and analyzed as described in (B). *P = 0.03 (paired one–sided t–test).
The level of PGR expression decreases in cervical cancer.
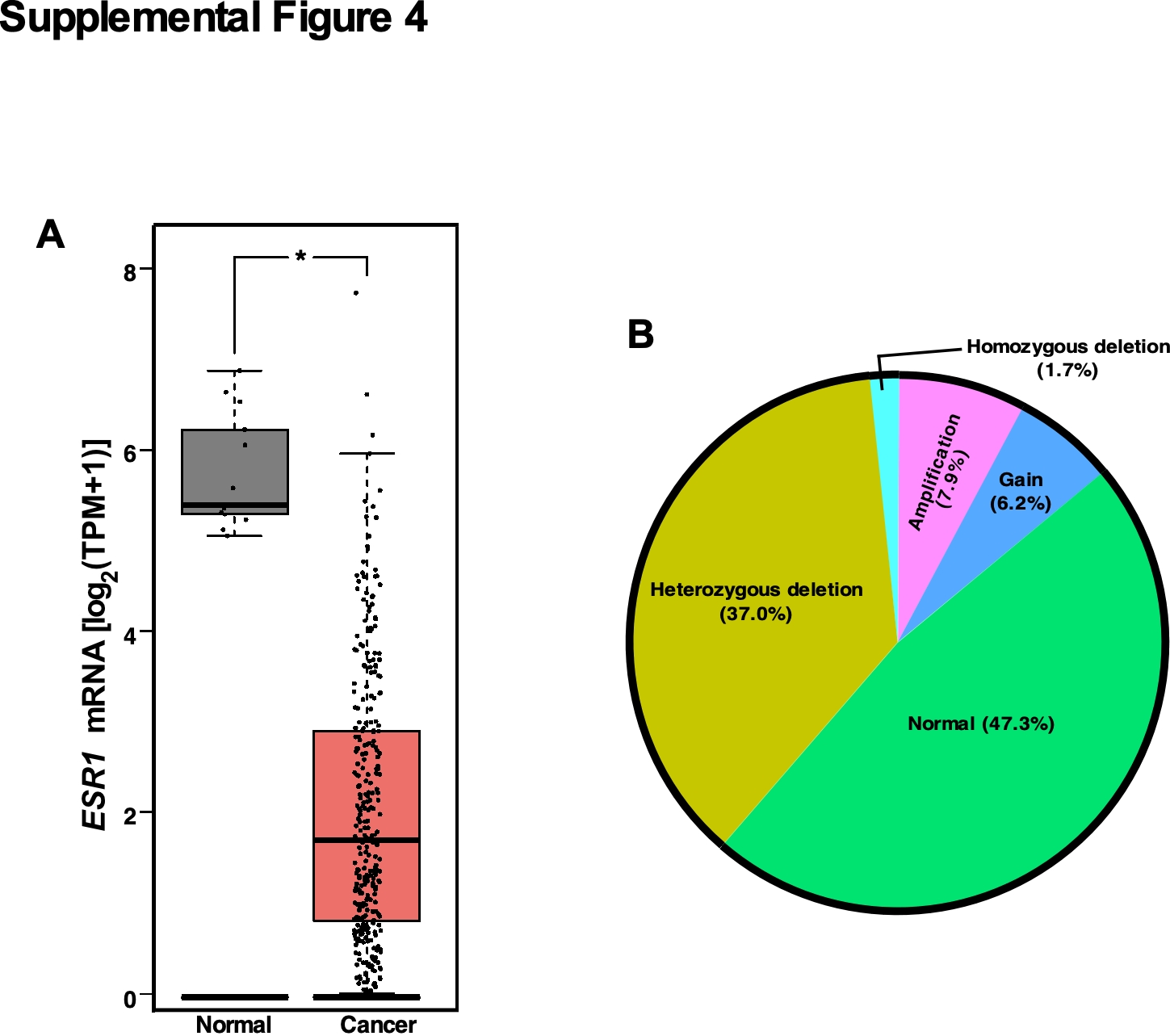
We sought to determine whether our findings in the mouse model were relevant to patients. Using publicly available GTEx and TCGA datasets, we first analyzed PGR expression in cancerous and normal cervical tissues. The mean log-normalized transcripts per million (TPM) value of PGR indicated an approximately 55-fold reduction in the cancer tissue compared to the normal tissue (Figure 3A). A similar analysis showed an 11-fold decrease in ESR1 mRNA levels in cervical cancer compared to the normal cervix (Supplemental Figure 4A). Decreased expression of ESR1 and PGR in cervical cancer has been observed in an independent cohort (12). There was a modest, significant positive correlation between transcript levels of ESR1 and PGR in the TCGA dataset (Figure 3B). To determine whether PGR copy number correlated with the expression level, we analyzed GISTIC-thresholded copy number variation (CNV) data from TCGA. Notably, PGR expression levels were not significantly different among all CNV groups (Figure 3C). While homozygous deletion was infrequent (5 out of 292 cancers), 47.3% and 37.0% had no copy number change and heterozygous PGR deletion, respectively, and 41 cancers (14.1%) had an increased copy number (Supplemental Figure 4B).
Figure 3. PGR downregulation is associated with poor prognosis in cervical cancer patients.

(A) PGR is downregulated in cervical cancer. PGR transcript levels were compared between cervical cancer (n = 306) and normal cervical tissues (n = 13). *P = 1.5 × 10−6 (Welch’s t-test). TPM, transcripts per million. (B) Expression of ESR1 and PGR are positively correlated in cervical cancer patients. Pearson’s correlation coefficient was 0.65. P = 2.2 × 10−16. (C) Copy number variation (CNV) does not correlate with PGR expression levels. A box-and-whisker plot was used to show PGR mRNA levels in each CNV group. (D) Low PGR expression is associated with poor prognosis in young patients. Patients in TCGA dataset were divided into young (31–50 years old) and old (50+ years old) age groups. They are further grouped based on the median expression levels of PGR and analyzed for overall survival using the Kaplan–Meier method. P = 0.024 (log-rank test), high vs. low PGR in the young age group; P = 0.38, high vs. low PGR in the old age group.
Low PGR expression is associated with poor prognosis in cervical cancer.
Menopause is diagnosed after women have gone one year without a menstrual period. The average age of menopause is 51 in the United States. Because progesterone levels are low without a menstrual cycle, we divided TCGA patients into two age groups, 31 to 50 years of age (young) and 50+ years of age (old) for survival analyses. In the young cohort, patients with high PGR expression had a better 18-month overall survival than those with low PGR expression (hazard ratio = 0.203; Figure 3D). On the contrary, the survival benefit of high PGR status was absent in the old patient group (hazard ratio = 0.639). These results suggest that a progesterone surge during the menstrual cycle protects cervical cancer patients with high PGR expression.
Discussion
We showed that the deletion of one Pgr allele promoted cervical cancer (Table 1) and that more than one-third of TCGA patient samples had heterozygous deletion of PGR (Figure 3C). Regardless of the PR-coding gene copy number, levels of PR decreased in cervical cancer compared to the normal epithelium in both mouse models and clinical samples (Figure 1A–B and Figure 3C). These results strongly support that PGR is a dose-dependent, haploinsufficient TSG in cervical cancer. PR similarly suppresses endometrial cancer (13). It would be interesting to see whether PGR is also haploinsufficient in this malignancy.
Dose-dependent TSGs fail to suppress tumorigenesis when expression drops below a threshold level, and nullizygotes for most of them are more susceptible to cancer than heterozygotes (1). However, Pgr null and heterozygotes had similar cancer incidence (Table 1). In this regard, Pgr was similar to a handful of haploinsufficient TSGs. Dmp1+/− and Dmp1−/− mice display similarly accelerated Kras-driven lung tumorigenesis (14). The deletion of either one or both Trp53bp1 alleles augments tumorigenesis to a similar degree in a glioma mouse model (15). In E7/Cre/Pgrf/+ (i.e., epithelial Pgr heterozygote) mice, PR expression was lower in cancer than the normal epithelium (Figure 1A–B). We postulate that a threshold level for Pgr is just below 50% of the normal level.
The expression of PR is undetectable in 60–80% of cervical cancer (10,16,17). In our spontaneous cervical cancer models, 57–83% of cancers arising in Pgr-sufficient mice did not express PR (Figure 1A). TCGA data analyses showed that PGR expression decreased in cervical cancer (Figure 3A). Multiple mechanisms could be responsible for the reduced expression of PR. The promoter of PGR is hypermethylated in cervical cancer tissues compared to normal tissues (18). PGR is a direct transcriptional target of ERα (19), and the expression of ESR1 and PGR are correlated (Figure 1C and Figure 3B). Overexpression of ERα restores the expression of PGR mRNA in HeLa cervical cancer cells (20). These observations support that transcriptional repression contributes, at least in part, to the reduced PGR expression in cervical cancer. Although we cannot rule out post-transcriptional and post-translational mechanisms, we do not favor the mechanism of loss of heterozygosity because percentages of PR-negative cancers were similar between Pgrf/+ and Cre/Pgrf/+ mice (Figure 1A) and because one or more PGR copies were retained in 98.4% of human cancers (Figure 3C). High stromal PR expression is associated with better survival of cervical cancer patients (21), suggesting that stromal PR also has the anti-cervical cancer activity and that PR signaling in cervical cancer is more complex than currently appreciated.
In summary, our results demonstrate that PGR is downregulated in cervical cancer and suggest that reactivation of PGR expression may improve the survival rate of cervical cancer patients. Our spontaneous cervical cancer model demonstrated the development of PR-negative cervical cancer for the first time. Further studies are warranted to better understand the mechanism of PGR downregulation during cervical carcinogenesis in vivo, and our mouse model provides a valuable tool for such studies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Implications:
The decreased expression of PR may increase the risk of cervical cancer in HPV-infected women.
Acknowledgments
We thank Dr. Roger E. Price for consultation on histopathology. We also thank Drs. Richard Behringer and John Lydon for providing us with Wnt7aCre and Pgrf/f mice, respectively. This work was supported in part by National Institutes of Health grant R01 CA188646 (S. Chung), Cancer Prevention and Research Institute of Texas grant RP180275 (S. Chung), and University of Houston Large Core Equipment grants (S. Chung).
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature 2011;476:163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019;144:1941–53. [DOI] [PubMed] [Google Scholar]
- 3.Moreno V, Bosch FX, Munoz N, Meijer CJ, Shah KV, Walboomers JM, et al. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: the IARC multicentric case-control study. Lancet 2002;359:1085–92. [DOI] [PubMed] [Google Scholar]
- 4.Munoz N, Franceschi S, Bosetti C, Moreno V, Herrero R, Smith JS, et al. Role of parity and human papillomavirus in cervical cancer: the IARC multicentric case-control study. Lancet 2002;359:1093–101. [DOI] [PubMed] [Google Scholar]
- 5.Chung SH. Targeting female hormone receptors as cervical cancer therapy. Trends Endocrinol Metab 2015;26:399–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res 2003;63:4862–71. [PubMed] [Google Scholar]
- 7.Chung SH, Shin MK, Korach KS, Lambert PF. Requirement for stromal estrogen receptor alpha in cervical neoplasia. Horm Cancer 2013;4:50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Son J, Park Y, Chung SH. Epithelial oestrogen receptor alpha is dispensable for the development of oestrogen-induced cervical neoplastic diseases. J Pathol 2018;245:147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta FF, Son J, Hewitt SC, Jang E, Lydon JP, Korach KS, et al. Distinct functions and regulation of epithelial progesterone receptor in the mouse cervix, vagina, and uterus. Oncotarget 2016;7:17455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoo YA, Son J, Mehta FF, DeMayo FJ, Lydon JP, Chung SH. Progesterone signaling inhibits cervical carcinogenesis in mice. Am J Pathol 2013;183:1679–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elson DA, Riley RR, Lacey A, Thordarson G, Talamantes FJ, Arbeit JM. Sensitivity of the cervical transformation zone to estrogen-induced squamous carcinogenesis. Cancer Res 2000;60:1267–75. [PubMed] [Google Scholar]
- 12.den Boon JA, Pyeon D, Wang SS, Horswill M, Schiffman M, Sherman M, et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc Natl Acad Sci U S A 2015;112:E3255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang S, Thiel KW, Leslie KK. Progesterone: the ultimate endometrial tumor suppressor. Trends Endocrinol Metab 2011;22:145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mallakin A, Sugiyama T, Taneja P, Matise LA, Frazier DP, Choudhary M, et al. Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell 2007;12:381–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Squatrito M, Vanoli F, Schultz N, Jasin M, Holland EC. 53BP1 is a haploinsufficient tumor suppressor and protects cells from radiation response in glioma. Cancer Res 2012;72:5250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fonseca-Moutinho JA, Cruz E, Carvalho L, Prazeres HJ, de Lacerda MM, da Silva DP, et al. Estrogen receptor, progesterone receptor, and bcl-2 are markers with prognostic significance in CIN III. Int J Gynecol Cancer 2004;14:911–20. [DOI] [PubMed] [Google Scholar]
- 17.Kwasniewska A, Postawski K, Gozdzicka-Jozefiak A, Kwasniewski W, Grywalska E, Zdunek M, et al. Estrogen and progesterone receptor expression in HPV-positive and HPV-negative cervical carcinomas. Oncol Rep 2011;26:153–60. [DOI] [PubMed] [Google Scholar]
- 18.Widschwendter A, Muller HM, Fiegl H, Ivarsson L, Wiedemair A, Muller-Holzner E, et al. DNA methylation in serum and tumors of cervical cancer patients. Clin Cancer Res 2004;10:565–71. [DOI] [PubMed] [Google Scholar]
- 19.Lim E, Palmieri C, Tilley WD. Renewed interest in the progesterone receptor in breast cancer. Br J Cancer 2016;115:909–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang ER, Lim SJ, Lee ES, Jeong G, Kim TY, Bang YJ, et al. The histone deacetylase inhibitor trichostatin A sensitizes estrogen receptor alpha-negative breast cancer cells to tamoxifen. Oncogene 2004;23:1724–36. [DOI] [PubMed] [Google Scholar]
- 21.Hong MK, Wang JH, Su CC, Li MH, Hsu YH, Chu TY. Expression of estrogen and progesterone receptor in tumor stroma predicts favorable prognosis of cervical squamous cell carcinoma. Int J Gynecol Cancer 2017;27(6):1247–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.