

Abstract
The definition of epigenetics refers that molecular modifications on DNA that can regulate gene activity are independent of DNA sequence and mitotically stable. Notably, epigenetics studies have grown exponentially in the past few years. Recent progresses that lead to exciting discoveries and groundbreaking nature of this area demand thorough methodologies and advanced technologies to move epigenetics to the forefront of molecular biology. The most recognized epigenetic regulations are DNA methylation, histone modifications, and non-coding RNAs (ncRNAs). This review will discuss the modern techniques that are available to detect locus-specific and genome-wide changes for all epigenetic codes. Furthermore, updated analysis of technologies, newly developed methods, recent breakthroughs and bioinformatics pipelines in epigenetic analysis will be presented. These methods, as well as many others presented in this specific issue, provide comprehensive guidelines and advanced technologies in the area of epigenetics that facilitate further developments in this promising and rapidly developing field.
Keywords: Epigenetics, methods, advanced technologies, DNA methylation, histone modifications, non-coding RNAs
1. Introduction
Epigenetics encompasses heritable structural and biochemical alterations of the chromatin without changing DNA sequence [1]. Epigenetic mechanisms manipulate various physiological and pathological processes through regulations of relevant gene expressions via changing the accessibility of epigenetic codes to the chromatin locally and globally [2–4]. There are three primary epigenetic codes that have been well studied including DNA methylation, histone modifications and non-coding RNAs (ncRNAs).
As the field of epigenetics is rapidly expanding, interest in exploration of novel technologies to decode the epigenetic landmarks that are related to health and disease status has dramatically increased. Notably, a number of novel technologies has been developed as the epigenetics field moves forward. These epigenetic techniques are capable of detecting chromatin states at multiple dimensions from locus-specific analysis to genome-wide sequencing. The advancement of epigenetics methodologies utilizes multiple strategies through, for example, high-quality antibodies, chromatin functional assays, imaging tools, high-throughput sequencing technologies and integrated bioinformatics pipelines [5].
DNA methylation is the most important epigenetic mechanism that has been intensively investigated. There are different DNA methylation modifications such as 5-methylcytosine (5mC), N6-methyladenine (6mA) and 4-methylcytosine (4mC) [6,7]. While 6mA and 4mC are commonly found in prokaryotic genome, 5mC is the most widely distributed methylation type in eukaryotes, and the most studied and understood DNA modification pattern overall [8]. There are many conventional techniques to analyze the approximate or exact methylation contents of DNA. Bisulfite modification is the foundation for the majority of DNA methylation assays, which converts cytosine to uracil in single-stranded DNA but does not affect 5mC [9]. Other techniques are based on digestion of genomic DNA by specific endonucleases with different methylation sensitivities for obtaining a rough estimate of the totality of DNA methylation [10]. Through these modifications, DNA methylation status in specific loci or global contents can be evaluated through multiple techniques. The methods for DNA methylation analysis have been greatly improved over the past decades. For example, recently emerged third-generation sequencing-based technologies allow long sequence reading, which offer exciting opportunities to study a wide range of base modifications including 5mC, 6mA and 4mC without bisulfite treatment [11]. Furthermore, the techniques that can identify the oxidized forms of 5mC such as 5-hydroxymethylcytosine (5hmC) have been developed [12].
As another important epigenetic code, a number of technologies have been developed to study functions and dynamics of histone modifications. Most of these technologies are developed based on the platform of chromatin immunoprecipitation (ChIP) assay [13,14]. The specific antibody-directed ChIP assay is a particularly useful technique in studying DNA-protein interactions that allows the chromatin structure surrounding specific DNA sequence to be analyzed. ChIP assay can also be integrated with other techniques in elucidating the interaction of histone modifications with other chromatin regulators at the specific loci [15]. The three-dimensional (3D) spatial structure of chromatin that links chromatin organization to biological functions can be determined using chromosome conformation capture (3C) technology [16]. Combined with ChIP-sequencing and 3C-based methodologies such as Hi-C, epigenetic landscapes in 3D chromatin architecture can be predicted that underline the relationships between histone modifications patterns and the 3D spatial arrangement of chromosomes in regulation of biological functions in eukaryotic cells [17].
ncRNAs represent a group of RNA transcripts that do not encode proteins like mRNAs [18]. ncRNAs have been considered as by-products of protein transcription with less biological functions. As a matter of fact, ncRNAs have been implicated as important epigenetic regulators that actively participate in multiple physiological and pathological processes [19]. The most important and well-studied regulatory ncRNAs that have been identified in regulation of gene expression in transcriptional and post-transcriptional levels include microRNA (miRNA), small interfering RNA (siRNA), piwi-interacting RNA (piRNA) and long ncRNA (lncRNA) etc. The advanced development of high-throughput technologies and bioinformatics promotes identification of novel transcripts and better understanding the diverse roles of ncRNAs in gene regulation. As ncRNAs regulate gene expression network through interplay between the other biomolecules such as other coding and non-coding RNAs, DNAs and proteins [20], novel methodologies have been developed to identify chromatin occupancy sites with a specific ncRNA or protein binding affinity using immunoprecipitation-based approaches followed by Next-Generation Sequencing (NGS) [21,22].
The expanding epigenetics area will probably lead to a surge in high-throughput sequencing technologies, which will provide a promising opportunity for decoding the nature of the epigenome at the systematic level. Thus, bioinformatic and biostatistic tools/pipelines are critical for processing of large volumes of datasets and identifying useful information in this “omic” era [23]. Several commonly used epigenomic pipelines and bioinformatic tools are introduced in this review.
The important breakthrough in the field of epigenetics promotes advanced technology development in support of finely designed tools and strategies to detect, quantify, and visualize the dynamics of chromatin state. This review summarizes the most contemporary methods as well as novel technologies in the field of epigenetics.
2. Epigenetic regulations and functions
DNA methylation is the most thoroughly studied epigenetic mechanism. As the most common DNA methylation pattern, 5mC involves the transfer of a methyl group to the C5 position of cytosine within CpG dinucleotides through an enzymatic process carried out by a group of DNA methyltransferases (DNMTs) [6]. Many CpG dinucleotides are often clustered together in a certain regulatory region of a gene such as the promoter region to form CpG islands, which frequently participates in gene transcriptional regulation [24]. DNA methylation plays a pivotal role in regulating various physiological and pathological processes and aberrant DNA methylation is often associated with multiple disease development [25]. The most common way in influencing biological procedures through DNA methylation is gene expression control via dynamic regulation of methylation status of CpG islands in the regulatory region of a specific gene [24,25]. Hypermethylation of CpG islands in the gene regulatory region such as the promoter is normally associated with a compacted or closed chromatin structure resulting in transcriptional silencing of the affiliated gene. By contrast, hypomethylation of CpG islands leads to an open chromatin structure, which is generally associated with gene transcriptional activation. Moreover, DNA methylation is also important in modulating various biological processes such as embryonic development, genomic imprinting, X inactivation, cellular differentiation and proliferation [1,26]. Thus, aberrations of DNA methylation occurred within the promoter regions of critical tumor-related genes could result in dysregulation of gene expression such as tumor suppressor gene silencing and/or oncogenic activation, ultimately leading to tumorigenesis [2,4,25]. Therefore, a precise and efficient method for detection of the exact DNA methylation contents is critical to elucidate the essential roles of DNA methylation in biological procedures and to enhance development of novel diagnostic and therapeutic targets.
In addition to DNA methylation, chromatin modification is considered another important epigenetic mechanism that plays a key role in controlling gene transcription [27]. Chromatin remodeling regulates gene expression through posttranslational modifications of histone proteins by a number of chemical processes including, but not limited to, acetylation and methylation normally on the specific lysine residues of core histone tails [3]. Different from DNA methylation, histone modification conveys a unique identity to the nucleosome that regulates transcriptional activity in a more robust and dynamic pathway during different biological stages. For example, increased histone acetylation is correlated with transcriptional activation, whereas histone deacetylation normally leads to gene transcription silencing [3,27]. Histone methylation-induced gene transcriptional regulation is more complicated showing both site-specific and methylation pattern-dependent index. For instance, methylation of lysine residue K4 of histone H3 results in transcriptional activation of chromatin regions, whereas methylation of lysine residue K9 of histone H3 usually leads to transcriptional repression [3,27]. The mechanisms by which histone modifications regulate gene transcription have been well documented. The most acceptable concept is that chromatin structure changes caused by histone modifications can directly change the spatial conformation of the DNA polymer leading to accessibility changes of key transcriptional factors and/or epigenetic modulators to the core gene regulatory region of DNA, and subsequently alter transcriptional activity [13]. Multiple enzymes have been reported to actively catalyze the processes of histone modifications such as the histone acetyltransferases (HATs), histone deacetylases (HDACs) and histone methyltransferases (HMTs), etc [28]. These enzymes play major roles not only in regulating histone modification patterns through reversible enzymatic activities but also in maintaining chromatin configurations that allow faithful epigenetic inheritance.
Recent studies indicate that different classes of ncRNAs can modify chromatin structure leading to either active or silenced gene transcription in eukaryotic cells [19]. These important ncRNAs have shown critical roles in control of both normal cellular functions and disease progresses. In particular, many small regulatory ncRNAs can assemble with the Argonaute proteins and form RNA-induced silencing complexes (RISCs) leading to transcriptional silencing processes [18,19]. Importantly, small ncRNAs have been shown to induce RNA-mediated transcriptional gene silencing through RNA-directed DNA methylation (RdDM) mechanism, indicating that ncRNAs can participate in regulation of other epigenetic modification patterns and influence mapping of epigenetic landscapes [29]. lncRNAs have been shown to play vital roles in regulating gene expression during developmental and differentiation processes [30]. Thus, understanding of the roles of ncRNA-mediated epigenetic regulation provides a different layer of epigenetic events in regulation of gene transcription.
With rapidly expanding in the epigenetics field, exploration of novel and advanced technologies for better illumination of biological consequences of individual epigenetic traits will be revolutionary in advancing scientific area.
3. Modern epigenetics technologies
The rapid progress in the field of epigenetics requires advanced technologies. Except for the conventional epigenetic technologies such as bisulfite sequencing for analysis of DNA methylation status and ChIP assay for detection of chromatin modifications, a number of novel tools and techniques have been developed on the basis of traditional platforms that allow scientists to make significant discoveries [5,31]. These epigenetic technologies include new approaches that can access to single-cell level, high-throughput genomic and epigenomic profiles with increasingly high resolution, which greatly benefit advancement in epigenetics area. In this review, we will discuss the conventional methodologies as well as the advanced technologies in analyzing epigenetic codes, which provide a comprehensive overview in the field of epigenetics methodology (Figure 1 and Table 1).
Figure 1.
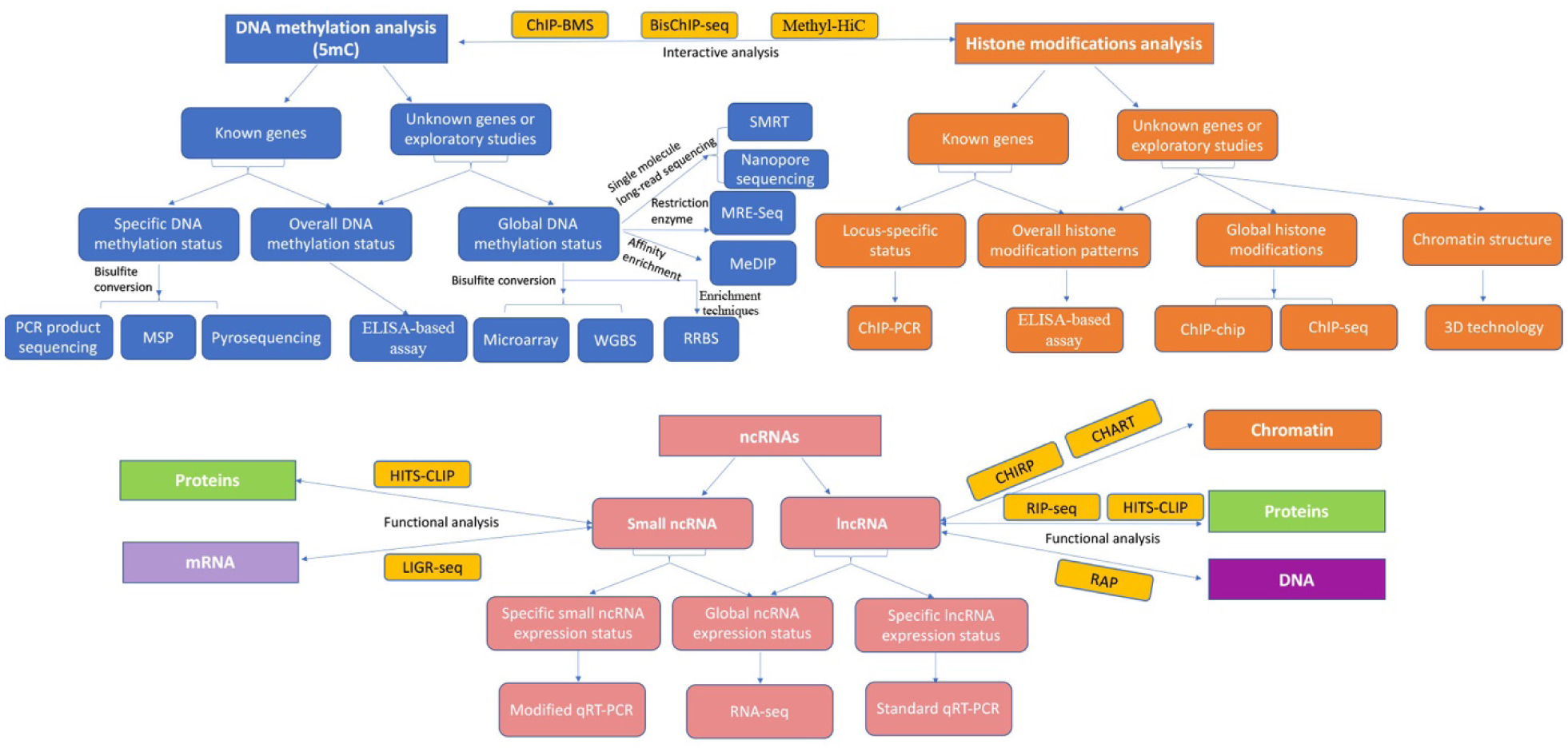
Algorithmic representation for appropriate method selections of different epigenetic codes. Different methods are chosen for DNA methylation analysis on 5mC (blue), histone modification (orange) and ncRNAs (red) depended on the purpose of the study design.
Table 1.
Comparison of epigenetics methods
| Method | Suitable project | Strength | Weakness | References |
|---|---|---|---|---|
| DNA methylation | ||||
| PCR-based bisulfite sequencing | The candidate gene and/or the targeted region are known. |
|
|
34 |
| MSP | The candidate gene and/or the targeted CpGs are known. |
|
|
35, 37 |
| Pyrosequencing | The candidate gene and/or the targeted region are known. |
|
|
36 |
| WGBS | De novo DNA methylation exploration |
|
|
38, 42 |
| HumanMet hylation450 | De novo DNA methylation (exploration |
|
|
38, 40, 41 |
| RRBS | De novo DNA methylation exploration |
|
|
43, 44 |
| MRE-Seq | De novo DNA methylation exploration |
|
|
46 |
| MeDIP | De novo DNA methylation exploration |
|
|
47 |
| ELISA-based assay | Broad prediction of global DNA methylation changes |
|
|
32, 48 |
| Single-cell bisulfite sequencing | De novo DNA methylation exploration in single-cell level |
|
Same weaknesses in WGBS or RRBS assays | 39, 49, 50 |
| SMRT sequencing | De novo DNA methylation exploration |
|
|
38, 39, 54 |
| Nanopore sequencing | De novo DNA methylation exploration |
|
|
38, 39, 55 |
| OxBS-seq | De novo DNA methylation exploration for 5hmC |
|
|
65 |
| Histone modifications | ||||
| ChIP-PCR | Confirmatory studies |
|
|
14 |
| ChIP-chip | De novo exploratory studies |
|
|
68 |
| ChIP-seq | De novo exploratory studies |
|
|
69 |
| ELISA-based assay | Broad prediction of histone modification changes |
|
|
48 |
| ncRNAs | ||||
| qRT-PCR | Confirmatory studies or small-scale experiments |
|
|
18, 19, 84 |
| RNA-Seq | De novo exploratory studies |
|
|
84, 88 |
| HITS-CLIP | Genome-wide functional analysis |
|
|
22 |
| Integrative analysis | ||||
| ChIP-BMS | Confirmatory studies |
|
|
71 |
| BisChIP-seq | Confirmatory studies and de novo studies |
|
|
72 |
| Methyl-HiC | Confirmatory studies and de novo studies | Combining in situ Hi-C and WGBS to simultaneously capture chromosome conformation and DNA methylome in a single assay |
|
79 |
3.1. Epigenetics technologies in DNA methylation
Conventional techniques in detecting DNA methylation status at specific loci
As the field of DNA methylation has extraordinarily progressed during the past few decades, many established techniques have been well adopted to determine the DNA methylation status in various levels and different dimensions. Thus, choosing an appropriate method that is best suitable to answering a particular scientific question will be critically important [32].
Invented in early 90’s, DNA bisulfite treatment is considered the fundamental technology for detection of DNA methylation status [9]. As 5mC is the most frequent type of DNA methylation [6,24], this method is based on the treatment of genomic DNA with sodium bisulfite resulting in deamination of unmethylated cytosines to uracil, leaving methylated cytosines intact that allows 5mCs to be distinguished from unmethylated cytosines throughout the genomic DNA [33]. Bisulfite genomic sequencing is regarded as a gold standard for DNA methylation detection and many methodologies that are based on analysis of bisulfite-treated DNA are therefore developed. Thus, bisulfite treatment-based DNA methylation analysis provides a highly quantitative and efficient approach to identify 5mC at single base-pair resolution.
For locus-specific analysis, several methods are available such as direct bisulfite PCR product sequencing or sub-cloning sequencing [34], methylation-specific PCR (MSP) [35] and pyrosequencing analyses [36]. In the MSP assay, bisulfite-converted DNA is used as the template for subsequent PCR processes to determine the methylation status in the specific loci of interest by using specific primers for recognizing methylated or unmethylated DNA template [37]. Pyrosequencing is a sequencing-based method that can quantitatively monitor the real-time incorporation of nucleotides through bioluminometric detection of pyrophosphate [36]. Pyrosequencing-based DNA methylation analysis that combines bisulfite conversion with pyrosequencing protocol can provide reproducible and accurate quantification for methylation status of the specific gene of interest with high quantitative resolution after PCR amplification.
Moving forward to genome-wide DNA methylation profiling
DNA methylation regulates gene expression in a macroscopic-controlled manner by coordinating with other message changes of DNA methylation along with the whole genome. Understanding the global profiling of DNA methylation distribution highlights the significance of these complex patterns and their roles in biological functions.
Followed by bisulfite conversion, global DNA methylation status can be readily determined by hybridization-based microarray such as Illumina HumanMethylation450 (HM450K) or high-throughput NGS analysis such as whole genome bisulfite sequencing (WGBS) [38,39]. Using predesigned probes for methylated and unmethylated CpGs, HM450K can interrogate more than 450,000 methylation sites that cover most CpG islands, which has been widely used for human methylomic studies [40,41]. To investigate global DNA methylation status that theoretically covers all the CpG information, WGBS is the standard profiling method to assess comprehensive methylation state, including low CpG-density regions such as intergenic regions, partially methylated domains and distal regulatory elements [42]. Although WGBS provides the most comprehensive and unbiased survey of DNA methylation status, the large amount of dataset obtained from WGBS requires sophisticated bioinformatics analysis. Moreover, WGBS faces high budgetary issues when multiple replicative samples are tested [38]. Since only a small fraction of the genome has the potential to be differentially methylated such as heavily CpG-rich regulatory regions, methods have migrated to low cost-associated enrichment techniques followed by bisulfite sequencing such as reduced representation bisulfite sequencing (RRBS) [43,44]. Integrated with restriction enzyme digestion, bisulfite conversion and NGS, RRBS can cover 85% of CpG islands, mostly located in the CpG-enriched regulatory regions such as the promoters, which compose only 1–3% of the mammalian genome [44]. RRBS has been widely applied in analysis of the DNA methylome in various species including non-human mammals, fishes, insects and plants [38]. Thus, RRBS is a more cost-effective and recommended method for large-scale or exploratory profiling studies than WGBS analysis.
Although sodium bisulfite conversion-based DNA methylation detections prevail, these methods have some drawbacks that need to be considered. For example, the bisulfite treated-DNA template is not stable and tends to degradation resulting in difficulties and mistakes occurring during PCR amplification [45]. Other alternative methylation sequencing analyses based on restriction enzyme digestion or affinity enrichment have been developed such as methylation-sensitive restriction enzymes (MRE) digestion [46] and methylated DNA immunoprecipitation (MeDIP) [47] followed by high-throughput sequencing, respectively. These methods typically yield a low resolution and low coverage of the genome, and in most cases, individual methylation context could not be discriminated. However, these alternative methods can be cost-effective and complement drawbacks of bisulfite treatment as a harsh chemical treatment on DNA when single-base resolution is not desired.
If broad global DNA methylation status is of interest, enzyme-linked immunosorbent assay (ELISA)-based assays, primarily for detecting global 5mC, are recommended [32,48]. Many commercially-available biological companies provide such ready-to-use kits that enable quick assessment of DNA methylation status. These assays are only suitable for the rough estimation of global DNA methylation status due to their high variability and low quantitative properties.
Novel technologies in DNA methylation
Novel technology advancement in the biological and engineering areas provides great opportunities for the field of epigenetics. Single-cell bisulfite sequencing utilizes the platform of single-cell genomics that offer unprecedented insight into individual cells [49,50]. In addition to the persisting matter of DNA degradation due to bisulfite treatment, single-cell bisulfite sequencing suffers from low input and a restricted ability to assess methylation heterogeneity between individual cells [50,51]. These disadvantages have been corrected by coupling post-bisulfite adaptor tagging (PBAT) through performing bisulfite treatment ahead of adapter tagging that enables the use of a low input of DNA to proceed to PCR amplification followed by deep sequencing at the single-cell level [52]. Both conventional methylomic profiling techniques such as single-cell WGBS (scWGBS) and single-cell RRBS (scRRBS) [50,51] are well adopted for single-cell methylation analysis. Single-cell related techniques have been widely applied to investigate cellular functions during development and tumorigenesis. Importantly, advanced technologies that integrate single-cell DNA methylation analysis with other single-cell omics approaches such as single-cell transcriptome and metabolome provide an insight of the functional link and dynamic network between multi-omics contexts at single-cell level, which eventually facilitate elucidation of the biological functions of epigenetics in complex biological systems [51,53].
Benefitted from third-generation long-reading sequencing technologies [11], single-molecule real-time sequencing (SMRT) from Pacific Biosciences (PacBio) [54] and nanopore sequencing from Oxford Nanopore Technologies (ONT) [55] have been recently adopted in epigenetics research. Both strategies can sequence native DNA and DNA degradation due to bisulfite conversion is therefore avoided. Because PCR amplification is not available, these approaches are best suited for bulk samples rather than small amounts of DNA.
PacBio SMRT sequencing can directly detect base modifications by monitoring the kinetics of the polymerase during synthesis of different fluorescently labeled nucleotides into DNA double-strands [56]. As compared to WGBS or HM450K, the biggest advantage of SMRT sequencing is to detect both nucleotide sequence and major types of DNA methylation patterns such as 5mC, 5hmC, 6mA and 4mC simultaneously [57]. However, because SMRT identifies base modifications via kinetic perturbations, the different sensitivities due to the signal-to-noise ratios specific to each modification type may lead to inaccurate reading. For example, 6mA and 4mC produce high confident kinetics signals, whereas 5mC signal is relatively low [58]. Thus, SMRT sequencing is particularly recommended to detect bacterial genomes, where 6mA and 4mC are frequent and often concentrated on specific motifs [59]. In a recent study, SMRT has been applied to identify a novel DNA methylation motif that may play a role in the regulation of gene expression in Bacillus pumilus BA06 [60].
Nanopore sequencing can identify DNA methylation patterns when a single-stranded DNA is ratcheted through a biological nanopore, and the ion current deviations due to specific base modifications through the pore are recorded [55]. To distinguish the current for methylated and unmethylated bases, the hidden Markov model (HMM) is used for basecalling the observed signal measurements. In 2017, Rand et al. developed a variable-order HMM with hierarchical Dirichlet process (HMM-HDP) to learn the ionic distributions, which can distinguish three cytosine variants (C, 5mC and 5hmC) and two adenine variants (A and 6mA) in Escherichia coli (E. coli) on single molecules of synthetic oligonucleotides in multiple motifs [61]. Because the performances of both SMAT and nanopore sequencing are largely dependent on established algorithms and training data, applying machine learning in long-read sequencing approaches will likely improve the throughput and reading accuracy. For example, Ni et al. have developed DeepSignal, a deep-learning method, to detect DNA methylation states of 6mA and 5mC at a higher performance level from Nanopore sequencing reads in human, bacteria (E. coli) and plasmid (pUC19) [62].
Evaluation of oxidized forms of DNA methylation
Likewise, DNA demethylation is mediated by a group of enzymes called the Ten-Eleven Translocation enzymes (TET1, 2, and 3), which remove the methyl group from 5mC and form several oxidized forms of 5mC including 5hmC, 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) [12,63]. Among all these oxidized forms of 5mC, 5hmC is the most abundant and now is widely accepted as the sixth base in mammalian genome, while its precursor, 5mC, is considered as the fifth base [64]. In addition to its role as the most prevalent demethylation intermediate of 5mC, 5hmC has been found to play key roles in regulation of multiple physiological and pathological processes such as embryogenesis and carcinogenesis. A modified bisulfite sequencing technique, oxidative bisulfite sequencing (OxBS-seq) can identify 5hmC via specific chemical oxidation of 5hmC to 5fC, whereas 5mC remains intact from bisulfite-induced deamination [65]. However, noticeable degradation of DNA due to multiple bisulfite treatments requires high amount of input DNA and high sequencing depths for confident determination of scarcely abundant modifications. In 2020, Gibas et al. have developed a high-resolution bisulfite-free method for analysis of 5hmC, named hmTOP-seq (5hmC-specific tethered oligonucleotide-primed sequencing) [66]. This novel method is designed based on direct sequence readout primed at covalently labeled 5hmC sites from an in situ tethered DNA oligonucleotide. The hmTOP-seq offers a cost-efficiency, high robustness and single-base resolution approach in mapping whole-genome 5hmC content that is superior to routine WGBS.
Taken together, the advanced technologies in epigenetics provide scientists with more accurate and affordable options to meet their particular research purposes. Better understanding of the distinct strengths and limitations of each technique that influence their respective cases will assist the scientists to choose the best suitable methods in their epigenetics studies (Figure 1 and Table 1).
3.2. Epigenetics technologies in histone modifications
Histone modification is one of the most important epigenetic regulations that has been found to frequently affect chromatin-based reactions and gene expression through influencing transcriptional programs [3,13]. Determining the function of histone modifications replies on two important factors, the modification abundance and interacting factors in a locus-specific manner such as transcriptional factors binding to the regulatory regions. Figure 1 provides a graphical guide to choose suitable methods to determining histone modification patterns according to different study designs.
Conventional techniques in evaluating histone modifications
The most commonly used method to determine and quantify chromatin modifications and interaction patterns is chromatin immunoprecipitation (ChIP) technique [67]. ChIP assay relies on specific antibodies that can recognize particular histone modification markers or epigenetic modulators in conjunction with specific DNA fragments, which allows for assigning locus-specific functions of histone modifications or transcriptional factor complexes that may directly or indirectly influence chromatin structure and subsequent transcriptional machinery efficiency [14]. Like bisulfite treatment, ChIP assay is considered as a fundamental procedure for studying histone modifications and chromatin structures. If the target histone modification and DNA regulatory region are specific, ChIP followed by conventional PCR or quantitative real-time PCR (qRT-PCR) will reveal the enrichments of specific histone modifications or binding ability of a remodeling complex to the specific DNA region [14]. However, if the specific modification patterns are undefined, sequencing-based ChIP methods such as ChIP-chip or ChIP-seq enable the analysis of protein-DNA binding events and histone modification enrichment at large numbers of loci simultaneously [68, 69]. ChIP-seq has been widely employed in several studies such as studies in the association of transcription factors and the architecture of nucleosomes that may have a wide effect on chromatin dynamics.
Integrative DNA methylation and histone modification analysis
Epigenetic events such as DNA methylation and histone modification working in conjunction on gene transcription primarily rely on a dynamic equilibrium between these two mechanisms on conformation change of DNA or chromatin cooperatively [70]. Analysis of the dynamic interactions between methylation and chromatin is a challenging task. To determine the complex interactions between different epigenomic states, several methods have been developed to interrogate allele-specific DNA methylation at base-resolution marked with a specific histone modification. We have developed a ChIP-bisulfite methylation sequencing (ChIP-BMS) approach which has been used to determine the methylation status of ChIP DNA pulled-down by a specific antibody (histone markers or transcription factors) [71]. Statham et al. have developed a technique based on high-throughput sequencing of bisulfite-treated chromatin immunoprecipitated DNA (BisChIP-seq), which can directly interpret the relationship between DNA methylation and important epigenetic regulators at the genome-wide scale [72]. Although the theoretic principle of ChIP-BMS and BisChIP-seq are similar, ChIP-BMS has been used for locus-specific methylation detection and BisChIP-seq focuses on global profiling. Moreover, the combination of epigenetic modification-specific antibodies and single-molecule imaging techniques, SCAN (Single Chromatin molecule Analysis in Nanochannels), enables co-localization high-throughput fluorescent assessment of combinatorial epigenomic states of histone modifications and DNA methylation on single nucleosomes [73]. These methods could be useful in determination of the cross-talk between DNA methylation and histone modifications or methylation-sensitive transcription factors in regulation of gene transcription.
Advanced methods for characterizing three-dimensional chromatin organization and the epigenome
Mammalian chromosomes are generally organized together into distinct spatial conformations that are of the essential for maintaining chromatin functions [3,13]. Structural biology plays a critical role in elucidating the epigenetic regulation of three-dimensional (3D) organization of chromatin structure [74]. As technologies advance that are capable of providing atomic images of molecular recognition, powerful imaging systems such as macromolecular X-ray crystallography [75], PET and MRI [76] provide insights to the mechanistic understanding of 3D chromosomal organization of epigenetics phenomena in atomic details [77]. The 3D chromatin structure can be captured by a novel technology, Hi-C, that is able to generate high-resolution contact maps of the genome at a large scale [78]. Although histone modifications and chromatin architecture are determined in different assays, one can interrogate the correlation of chromatin organization such as chromatin interaction compartments and topologically associated domain with specific histone modifications by integration of ChIP-seq and Hi-C data [16,17]. This predictive model system reveals important networks between chromatin structure and epigenetic mechanisms underlying dynamic gene expression regulation. Remarkably, Li and Ren et al. have recently developed a novel molecular assay, Methyl-Hi-C, that can simultaneously capture the chromosome conformation and DNA methylome at the single-cell level [79]. This novel technology enables simultaneous characterization of cell type-specific chromatin organization and epigenome in complex tissues demonstrating a big leap that advances studies in chromosome structural changes and their biomedical functions in the single-cell perspective. The epigenetics field with respect to histone modifications has significantly expanded in recent years and advanced technologies in this area will allow better understanding of their functional interactions with surrounding histone modifications and transcriptional machinery as well as the roles in biology (Table 1).
3.3. Epigenetics technologies in ncRNAs
ncRNAs have been implicated as important epigenetic regulators of gene expression through direct and indirect actions on chromatin [19]. ncRNAs can be divided into two categories based on their regulatory roles: the housekeeping ncRNAs such as ribosomal RNA (rRNA) and transfer RNA (tRNA) that primarily regulate generic cellular functions, and the regulatory ncRNAs that actively mediate gene expression during complex molecular and cellular processes. The most important regulatory ncRNAs include miRNA, siRNA, piRNA and lncRNA [18,19]. miRNA, siRNA and piRNA are considered as small ncRNAs that are normally less than 200 nucleotides, whereas lncRNA exceeds 200 nucleotides in lengths. Although the roles of the majority of ncRNAs are not well defined, research has made extraordinary advance in this exciting area demonstrating key regulatory functions of ncRNAs in shaping cellular activity and the potential as novel biomarkers or therapies, for example, in cancer research [80]. Here, we discuss some emerging methodologies in evaluating several important ncRNAs (Figure 1 and Table 1).
Methods for small ncRNAs
The role of ncRNAs is best understood for miRNAs with approximately 22 nucleotides long. As one of the most important ncRNAs, miRNAs are known to dynamically regulate post-transcriptional gene expression in a broad range of normal physiological processes such as organismal development and differentiation [19]. Notably, miRNAs have been found to play a key role in disease etiology such as cancer [80,81]. Dysregulation of miRNA expression profiles are highly correlated with human cancer initiation and development, where these aberrant transcripts are commonly accompanied with epigenetic disruption [81]. siRNAs are a class of double-stranded RNA molecules processed in a Dicer-dependent manner. Similar to miRNA, siRNAs mediate post-transcriptional silencing through RNA interference (RNAi) processes when forming RISC bound to the 3’UTR of mRNA [19]. piRNAs are a group of small ncRNAs with approximately 21–35 nucleotides in length. piRNA is known to associate with PIWI proteins and form piRNA-induced silencing complexes during germline development [82]. Studies have shown that these small regulatory ncRNAs can interact with mRNA leading to gene transcriptional silencing through RISCs-induced mRNA degradation [19,20,83].
The methods for small ncRNAs profiling are currently well established including quantitative reverse transcription PCR (qRT-PCR)-based analysis, hybridization-based microarrays and high-throughput RNA sequencing [84]. Until today, many commercially available kits offer convenient, sensitive, precise quantification and profiling of small ncRNAs using modified RT-PCR-based analysis such as the utilization of stem-loop reverse transcription primers for 3’ extended templates during RT-PCR. Small ncRNAs can interact with different levels of molecules such as RNA, DNA and proteins, and therefore, in turn, regulate gene expression through various molecular mechanisms [20]. Functional analysis of small ncRNAs and their targets can be greatly improved by the development of the tools using in silico predictors and network analysis. Many experimental approaches are employed for the investigation of ncRNA-based targets. For example, high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP) has been widely used for identification of functional protein-RNA interaction sites [22], and ligation of interacting RNA followed by high-throughput sequencing (LIGR-seq) enables the profiling of miRNA-mRNA interaction [85]. These advanced evaluation platforms and methodologies enable small ncRNAs as an attractive biomarker for pathological screening and therapeutic applications [86].
Methods for lncRNAs
lncRNAs are defined as non-coding transcripts greater than 200 base pairs. They are transcribed by RNA Pol II from independent promoters and usually have common characteristics of mature transcripts such as caps and poly(A) tails. In contrast to small ncRNAs that normally mediate RNA silencing processes, lncRNAs exhibit extensive mechanistic diversity through interaction with RNA binding proteins (RBPs) at specific DNA regions [87]. As lncRNAs have poly(A) tail, they can be detected by standard qRT-PCR through a poly-A tailing method. Moreover, to better understand the global profiling of lncRNA, many high-throughput technologies have been developed [88]. These techniques are designed to suit different study purposes. For example, ChIRP (Chromatin isolation by RNA purification) [89] and CHART (Capture Hybridization Analysis of RNA Targets) [90] are developed to investigate lncRNA binding sites on chromatin genome-wide; RAP (RNA antisense purification) can map the localization of a given lncRNA across the genome [91]; RIP (RNA Immunoprecipitation)-associated array or sequencing such as RIP-chip and RIP-seq have been widely used for protein-RNA interaction [92]; and RNA pull-down followed by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) techniques are used to identify the interaction proteome of a target lncRNA [93]. Combined with these methods-generated genome-wide datasets and bioinformatics applications, scientists are able to better characterize and predict the functions of lncRNAs. In summary, advances in ncRNAs methodologies enhance our understanding of the functional ncRNAs in regulation of a variety of biological processes and benefit further applications of these promising approaches in clinical practice.
4. Advances in epigenetic bioinformatics
Advances in high-throughput sequencing technologies provide great opportunities to access comprehensive epigenome-wide biological data at the systematic level. The development of accurate computational approaches is essential to interpreting large datasets, fishing out useful information and identifying candidate genes that may underlie the intricate interaction networks between DNA methylation, chromatin modifications and ncRNAs in control of gene expression [23,94].
The general pipeline for the bioinformatics analysis of genome-wide DNA methylation data includes data processing and quality control, alignment to the reference genome, quantification and visualization of DNA methylation, general profiling, validation and interpretation [95]. Array-based bisulfite sequencing data such as those from Illumina HM450K can be quantified by fluorescence intensities of the relative abundance of methylated and unmethylated loci. For restriction enzyme- and enrichment affinity-based data from MRE-seq and MeDIP-seq, data are usually analyzed by comparing the relative abundance of the fragments. For bisulfite sequencing-based data from WGBS and RRBS, methylation status is determined at single-nucleoside residues by comparing differential methylation loci (DML) and differential methylation regions (DMRs) among different samples [96]. Many useful tools have been developed to analyze various types of DNA methylation sequencing data during different processing steps. For example, Cutadapt can remove adapters for quality trimming; BS Seeker and Bismark are used for bisulfite sequencing read alignment [97,98]; BSmooth is a comprehensive tool that has been used for most of the abovementioned analyses as well as identifications of DMLs and DMRs for WGBS and RRBS data [99]; and web-based genome browser such as UCSC Genome Browser or Ensembl can be used for data visualization.
Computational pipeline for analysis of ChIP-seq data includes data processing, reading mapping, peak calling and visualization, quantitative assessment of binding or enrichment changes and functional analysis [100]. Many ChIP-seq tools are designed for calling peak reliably in the entire genome and MACS2 is the most commonly used for peak-calling tool [101]. Several web servers such as UCSC genome browser and WashU Epigenome Browser can integrate ChIP-seq data with other annotation data. An updated DROMPAplus (Draw and Observe Multiple enrichment Profiles and Annotation) is a universal ChIP-seq pipeline tool that can perform quality check, normalization, statistical analysis and visualization of multiple ChIP-seq samples in one package [102]. ncRNA-related sequencing analyses mostly rely on RNA-seq pipeline. However, bioinformatics tools designed for functional analysis and target prediction are important for identification of novel ncRNAs and their potential targets that may link to their biological roles. Notably, novel computational methods or technologies for analyses of the complex and integrative genome-wide or “multi-omics” studies including genome, epigenome, transcriptome, proteome, metabolome and microbiome have been developed. For example, pipelines Allelome.PRO [103] and WASP [104] can accommodate both RNA- and ChIP-seq datasets, and pipeline MEA (methylomic and epigenomic analysis) can integrate DNA methylation sequencing data with ChIP- and RNA-seq data at allelic resolution [105]. These novel bioinformatics pipelines facilitate identification of biomarkers of disease and physiology and finding novel and coherently matching geno-phenotype relationship between biological entities at the single cell level in multicellular organisms [106,107].
5. Conclusion
With rapid development in the epigenetics field, many powerful techniques have been discovered to access epigenetic functions and mechanisms of the epigenome and its associated proteins that have driven the remarkable advances in epigenetics. Until today, numerous methods have been developed that help to provide better, precise, accurate and systematic understanding of biological functions of epigenetic regulations. It is speculated that the advances in the epigenetics field will continually illuminate unresolved problems. The quick advances in a broad range of overall biological technologies will likely improve interpretation of a massive amount of information in epigenomic mapping. Thus, further developments in bioinformatics and computational sciences are critical in handling sophisticated datasets and interpreting epigenomics functions as well as multi-omics data derived from combinatorial integration analyses. This special issue will be useful to provide the reader with a set of contemporary and advanced technologies in the rapidly developing field of epigenetics.
Highlights.
Review both conventional and advanced technologies in profiling DNA methylation, histone modifications and ncRNAs.
Provide practical guide to assist the scientist in choosing the suitable methods in the epigenetics studies.
Review integrative genome-wide methodologies in multiple dimensions.
Provide useful bioinformatics pipelines in analyzing epigenomic data.
Acknowledgments
This work was supported by the grant from the National Institutes of Health/National Center for Complementary and Integrative Health (K01 AT009373).
Abbreviations
- ChIP
Chromatin immunoprecipitation
- DNMTs
DNA methyltransferases
- HATs
Histone acetyltransferases
- HDACs
Histone deacetylases
- HMTs
Histone methyltransferases
- 5mC
5-methylcytosine
- 5hmC
5-hydroxymethylcytosine
- 6mA
N6-methyladenine
- 4mC
4-methylcytosine
- MSP
methylation-specific PCR
- NSG
next-generation sequencing
- WGBS
Whole genome bisulfite sequencing
- RRBS
Reduced representation bisulfite sequencing
- MRE-Seq
Methylation-sensitive restriction enzymes-sequencing
- MeDIP
Methylated DNA immunoprecipitation
- ELISA
Enzyme-linked immunosorbent assay
- PBAT
post-bisulfite adaptor tagging
- SMRT
Single-molecule real-time sequencing
- 5fC
5-formylcytosine
- 5caC
5-carboxylcytosine
- hmTOP-seq
5hmC-specific tethered oligonucleotide-primed sequencing
- ChIP-Seq
ChIP-sequencing
- ChIP-BMS
ChIP-bisulfite methylation sequencing
- BisChIP-seq
bisulfite-treated ChIP DNA-sequencing
- ncRNAs
non-coding RNAs
- miRNA
microRNA
- siRNA
short interfering RNA
- piRNA
piwi-interacting RNA
- lncRNA
long non-coding RNA
- RISCs
RNA-induced silencing complexes
- qRT-PCR
quantitative reverse transcription PCR
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Holliday R, Epigenetics: a historical overview, Epigenetics 1(2) (2006) 76–80. [DOI] [PubMed] [Google Scholar]
- [2].Baylin SB, Ohm JE, Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction?, Nat Rev Cancer 6(2) (2006) 107–16. [DOI] [PubMed] [Google Scholar]
- [3].Kouzarides T, Chromatin modifications and their function, Cell 128(4) (2007) 693–705. [DOI] [PubMed] [Google Scholar]
- [4].Egger G, Liang G, Aparicio A, Jones PA, Epigenetics in human disease and prospects for epigenetic therapy, Nature 429(6990) (2004) 457–63. [DOI] [PubMed] [Google Scholar]
- [5].Zheng YG, Bernstein AI, Epigenetic technological applications, Elsevier Science & Technology, (2015). [Google Scholar]
- [6].Chen K, Zhao BS, He C, Nucleic Acid Modifications in Regulation of Gene Expression, Cell Chem Biol 23(1) (2016) 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, Li C, Liu B, Luo Y, Zhu Y, Zhang N, He S, He C, Wang H, Chen D, N6-methyladenine DNA modification in Drosophila, Cell 161(4) (2015) 893–906. [DOI] [PubMed] [Google Scholar]
- [8].Jones PA, Takai D, The role of DNA methylation in mammalian epigenetics, Science 293(5532) (2001) 1068–70. [DOI] [PubMed] [Google Scholar]
- [9].Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL, A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands, Proc Natl Acad Sci U S A 89(5) (1992) 1827–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Melnikov AA, Gartenhaus RB, Levenson AS, Motchoulskaia NA, Levenson Chernokhvostov VV, MSRE-PCR for analysis of gene-specific DNA methylation, Nucleic Acids Res 33(10) (2005) e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schadt EE, Turner S, Kasarskis A, A window into third-generation sequencing, Hum Mol Genet 19(R2) (2010) R227–40. [DOI] [PubMed] [Google Scholar]
- [12].Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, Min JH, Jin P, Ren B, He C, Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome, Cell 149(6) (2012) 1368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bannister AJ, Kouzarides T, Regulation of chromatin by histone modifications, Cell Res 21(3) (2011) 381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gade P, Kalvakolanu DV, Chromatin immunoprecipitation assay as a tool for analyzing transcription factor activity, Methods Mol Biol 809 (2012) 85–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Davies JO, Oudelaar AM, Higgs DR, Hughes JR, How best to identify chromosomal interactions: a comparison of approaches, Nat Methods 14(2) (2017) 125–134. [DOI] [PubMed] [Google Scholar]
- [16].Qi Y, Zhang B, Predicting three-dimensional genome organization with chromatin states, PLoS Comput Biol 15(6) (2019) e1007024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Di Pierro M, Cheng RR, Lieberman Aiden E, Wolynes PG, Onuchic JN, De novo prediction of human chromosome structures: Epigenetic marking patterns encode genome architecture, Proc Natl Acad Sci U S A 114(46) (2017) 12126–12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li J, Liu C, Coding or Noncoding, the Converging Concepts of RNAs, Front Genet 10 (2019) 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Holoch D, Moazed D, RNA-mediated epigenetic regulation of gene expression, Nat Rev Genet 16(2) (2015) 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang P, Wu W, Chen Q, Chen M, Non-Coding RNAs and their Integrated Networks, J Integr Bioinform 16(3) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T, Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP, Cell 141(1) (2010) 129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chi SW, Zang JB, Mele A, Darnell RB, Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps, Nature 460(7254) (2009) 479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Angarica VE, Del Sol A, Bioinformatics Tools for Genome-Wide Epigenetic Research, Adv Exp Med Biol 978 (2017) 489–512. [DOI] [PubMed] [Google Scholar]
- [24].Bird AP, CpG-rich islands and the function of DNA methylation, Nature 321(6067) (1986) 209–13. [DOI] [PubMed] [Google Scholar]
- [25].Robertson KD, Wolffe AP, DNA methylation in health and disease, Nat Rev Genet 1(1) (2000) 11–9. [DOI] [PubMed] [Google Scholar]
- [26].Bird A, Perceptions of epigenetics, Nature 447(7143) (2007) 396–8. [DOI] [PubMed] [Google Scholar]
- [27].Berger SL, The complex language of chromatin regulation during transcription, Nature 447(7143) (2007) 407–12. [DOI] [PubMed] [Google Scholar]
- [28].Marmorstein R, Trievel RC, Histone modifying enzymes: structures, mechanisms, and specificities, Biochim Biophys Acta 1789(1) (2009) 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Matzke MA, Mosher RA, RNA-directed DNA methylation: an epigenetic pathway of increasing complexity, Nat Rev Genet 15(6) (2014) 394–408. [DOI] [PubMed] [Google Scholar]
- [30].Fatica A, Bozzoni I, Long non-coding RNAs: new players in cell differentiation and development, Nat Rev Genet 15(1) (2014) 7–21. [DOI] [PubMed] [Google Scholar]
- [31].Tollefsbol TO, Advances in epigenetic technology, Methods Mol Biol 791 (2011) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kurdyukov S, Bullock M, DNA Methylation Analysis: Choosing the Right Method, Biology (Basel) 5(1) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jones PA, Functions of DNA methylation: islands, start sites, gene bodies and beyond, Nat Rev Genet 13(7) (2012) 484–92. [DOI] [PubMed] [Google Scholar]
- [34].Li Y, Tollefsbol TO, DNA methylation detection: bisulfite genomic sequencing analysis, Methods Mol Biol 791 (2011) 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rand K, Qu W, Ho T, Clark SJ, Molloy P, Conversion-specific detection of DNA methylation using real-time polymerase chain reaction (ConLight-MSP) to avoid false positives, Methods 27(2) (2002) 114–20. [DOI] [PubMed] [Google Scholar]
- [36].Delaney C, Garg SK, Yung R, Analysis of DNA Methylation by Pyrosequencing, Methods Mol Biol 1343 (2015) 249–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Derks S, Lentjes MH, Hellebrekers DM, de BruÔne AP, Herman JG, van Engeland M, Methylation-specific PCR unraveled, Cell Oncol 26(5–6) (2004) 291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yong WS, Hsu FM, Chen PY, Profiling genome-wide DNA methylation, Epigenetics & Chromatin 9(26) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gouil Q, Keniry A, Latest techniques to study DNA methylation, Essays Biochem 63(6) (2019) 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, Fan JB, Shen R, High density DNA methylation array with single CpG site resolution, Genomics 98(4) (2011) 288–95. [DOI] [PubMed] [Google Scholar]
- [41].Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F, Evaluation of the Infinium Methylation 450K technology, Epigenomics 3(6) (2011) 771–84. [DOI] [PubMed] [Google Scholar]
- [42].Kernaleguen M, Daviaud C, Shen Y, Bonnet E, Renault V, Deleuze JF, Mauger F, Tost J, Whole-Genome Bisulfite Sequencing for the Analysis of Genome-Wide DNA Methylation and Hydroxymethylation Patterns at Single-Nucleotide Resolution, Methods Mol Biol 1767 (2018) 311–349. [DOI] [PubMed] [Google Scholar]
- [43].Masser DR, Berg AS, Freeman WM, Focused, high accuracy 5-methylcytosine quantitation with base resolution by benchtop next-generation sequencing, Epigenetics Chromatin 6(1) (2013) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R, Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis, Nucleic Acids Res 33(18) (2005) 5868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Genereux DP, Johnson WC, Burden AF, Stger R, Laird CD, Errors in the bisulfite conversion of DNA: modulating inappropriate- and failed-conversion frequencies, Nucleic Acids Res 36(22) (2008) e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bonora G, Rubbi L, Morselli M, Ma F, Chronis C, Plath K, Pellegrini M, DNA methylation estimation using methylation-sensitive restriction enzyme bisulfite sequencing (MREBS), PLoS One 14(4) (2019) e0214368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jacinto FV, Ballestar E, Esteller M, Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylome, Biotechniques 44(1) (2008) 35, 37, 39 passim. [DOI] [PubMed] [Google Scholar]
- [48].The enzyme-linked immunosorbent assay (ELISA), Bull World Health Organ 54(2) (1976) 129–39. [PMC free article] [PubMed] [Google Scholar]
- [49].Schwartzman O, Tanay A, Single-cell epigenomics: techniques and emerging applications, Nat Rev Genet 16(12) (2015) 716–26. [DOI] [PubMed] [Google Scholar]
- [50].Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, Kelsey G, Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity, Nat Methods 11(8) (2014) 817–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Karemaker ID, Vermeulen M, Single-Cell DNA Methylation Profiling: Technologies and Biological Applications, Trends Biotechnol 36(9) (2018) 952–965. [DOI] [PubMed] [Google Scholar]
- [52].Miura F, Enomoto Y, Dairiki R, Ito T, Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging, Nucleic Acids Res 40(17) (2012) e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu X, Wen L, Tang F, Huang Y, Peng J, Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas, Cell Res 26(3) (2016) 304–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kingan SB, Heaton H, Cudini J, Lambert CC, Baybayan P, Galvin BD, Durbin R, Korlach J, Lawniczak MKN, A high-quality de novo genome assembly from a single mosquito using PacBio sequencing, Genes (Basel) 10(1) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Laszlo AH, Derrington IM, Brinkerhoff H, Langford KW, Nova IC, Samson JM, Bartlett JJ, Pavlenok M, Gundlach JH, Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA, Proc Natl Acad Sci U S A 110(47) (2013) 18904–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S, Real-time DNA sequencing from single polymerase molecules, Science 323(5910) (2009) 133–8. [DOI] [PubMed] [Google Scholar]
- [57].Roberts RJ, Carneiro MO, Schatz MC, The advantages of SMRT sequencing, Genome Biol 14(7) (2013) 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schadt EE, Banerjee O, Fang G, Feng Z, Wong WH, Zhang X, Kislyuk A, Clark TA, Luong K, Keren-Paz A, Chess A, Kumar V, Chen-Plotkin A, Sondheimer N, Korlach J, Kasarskis A, Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases, Genome Res 23(1) (2013) 129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Beaulaurier J, Zhang XS, Zhu S, Sebra R, Rosenbluh C, Deikus G, Shen N, Munera D, Waldor MK, Chess A, Blaser MJ, Schadt EE, Fang G, Single molecule-level detection and long read-based phasing of epigenetic variations in bacterial methylomes, Nat Commun 6 (2015) 7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liu G, Jiang YM, Liu YC, Han LL, Feng H, A novel DNA methylation motif identified in Bacillus pumilus BA06 and possible roles in the regulation of gene expression, Appl Microbiol Biotechnol 104(8) (2020) 3445–3457. [DOI] [PubMed] [Google Scholar]
- [61].Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, Paten B, Mapping DNA methylation with high-throughput nanopore sequencing, Nat Methods 14(4) (2017) 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ni P, Huang N, Zhang Z, Wang DP, Liang F, Miao Y, Xiao CL, Luo F, Wang J, DeepSignal: detecting DNA methylation state from Nanopore sequencing reads using deep-learning, Bioinformatics 35(22) (2019) 4586–4595. [DOI] [PubMed] [Google Scholar]
- [63].Wu X, Zhang Y, TET-mediated active DNA demethylation: mechanism, function and beyond, Nat Rev Genet 18(9) (2017) 517–534. [DOI] [PubMed] [Google Scholar]
- [64].Song CX, He C, The hunt for 5-hydroxymethylcytosine: the sixth base, Epigenomics 3(5) (2011) 521–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Booth MJ, Ost TW, Beraldi D, Bell NM, Branco MR, Reik W, Balasubramanian S, Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine, Nat Protoc 8(10) (2013) 1841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gibas P, Narmontė M, Staševskij Z, Gordevičius J, Klimašauskas S, Kriukienė E, Precise genomic mapping of 5-hydroxymethylcytosine via covalent tether-directed sequencing, PLoS Biol 18(4) (2020) e3000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Collas P, The current state of chromatin immunoprecipitation, Mol Biotechnol 45(1) (2010) 87–100. [DOI] [PubMed] [Google Scholar]
- [68].Pillai S, Chellappan SP, ChIP on chip assays: genome-wide analysis of transcription factor binding and histone modifications, Methods Mol Biol 523 (2009) 341–66. [DOI] [PubMed] [Google Scholar]
- [69].Furey TS, ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions, Nat Rev Genet 13(12) (2012) 840–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kondo Y, Epigenetic cross-talk between DNA methylation and histone modifications in human cancers, Yonsei Med J 50(4) (2009) 455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Li Y, Tollefsbol TO, Combined chromatin immunoprecipitation and bisulfite methylation sequencing analysis, Methods Mol Biol 791 (2011) 239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Statham AL, Robinson MD, Song JZ, Coolen MW, Stirzaker C, Clark SJ, Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) directly informs methylation status of histone-modified DNA, Genome Res 22(6) (2012) 1120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Murphy PJ, Cipriany BR, Wallin CB, Ju CY, Szeto K, Hagarman JA, Benitez JJ, Craighead HG, Soloway PD, Single-molecule analysis of combinatorial epigenomic states in normal and tumor cells, Proc Natl Acad Sci U S A 110(19) (2013) 7772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zhou K, Gaullier G, Luger K, Nucleosome structure and dynamics are coming of age, Nat Struct Mol Biol 26(1) (2019) 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Cramer P, A tale of chromatin and transcription in 100 structures, Cell 159(5) (2014) 985–994. [DOI] [PubMed] [Google Scholar]
- [76].Mishra A, Hawkins RD, Three-dimensional genome architecture and emerging technologies: looping in disease, Genome Med 9(1) (2017) 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zentner GE, Henikoff S, High-resolution digital profiling of the epigenome, Nat Rev Genet 15(12) (2014) 814–27. [DOI] [PubMed] [Google Scholar]
- [78].Mota-Gómez I, Lupiáñez DG, A (3D-Nuclear) Space Odyssey: Making Sense of Hi-C Maps, Genes (Basel) 10(6) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Li G, Liu Y, Zhang Y, Kubo N, Yu M, Fang R, Kellis M, Ren B, Joint profiling of DNA methylation and chromatin architecture in single cells, Nat Methods 16(10) (2019) 991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Slack FJ, Chinnaiyan AM, The Role of Non-coding RNAs in Oncology, Cell 179(5) (2019) 1033–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bracken CP, Scott HS, Goodall GJ, A network-biology perspective of microRNA function and dysfunction in cancer, Nat Rev Genet 17(12) (2016) 719–732. [DOI] [PubMed] [Google Scholar]
- [82].Siomi MC, Sato K, Pezic D, Aravin AA, PIWI-interacting small RNAs: the vanguard of genome defence, Nat Rev Mol Cell Biol 12(4) (2011) 246–58. [DOI] [PubMed] [Google Scholar]
- [83].Pasquinelli AE, MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship, Nat Rev Genet 13(4) (2012) 271–82. [DOI] [PubMed] [Google Scholar]
- [84].Pritchard CC, Cheng HH, Tewari M, MicroRNA profiling: approaches and considerations, Nat Rev Genet 13(5) (2012) 358–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sharma E, Sterne-Weiler T, O’Hanlon D, Blencowe BJ, Global Mapping of Human RNA-RNA Interactions, Mol Cell 62(4) (2016) 618–26. [DOI] [PubMed] [Google Scholar]
- [86].Brown BD, Naldini L, Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications, Nat Rev Genet 10(8) (2009) 578–85. [DOI] [PubMed] [Google Scholar]
- [87].Long Y, Wang X, Youmans DT, Cech TR, How do lncRNAs regulate transcription?, Sci Adv 3(9) (2017) eaao2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Cao M, Zhao J, Hu G, Genome-wide methods for investigating long noncoding RNAs, Biomed Pharmacother 111 (2019) 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Chu C, Qu K, Zhong FL, Artandi SE, Chang HY, Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions, Mol Cell 44(4) (2011) 667–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, Borowsky ML, Kuroda MI, Kingston RE, The genomic binding sites of a noncoding RNA, Proc Natl Acad Sci U S A 108(51) (2011) 20497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Engreitz J, Lander ES, Guttman M, RNA antisense purification (RAP) for mapping RNA interactions with chromatin, Methods Mol Biol 1262 (2015) 183–97. [DOI] [PubMed] [Google Scholar]
- [92].Zhang Y, Feng Y, Hu Z, Hu X, Yuan CX, Fan Y, Zhang L, Characterization of Long Noncoding RNA-Associated Proteins by RNA-Immunoprecipitation, Methods Mol Biol 1402 (2016) 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Giambruno R, Mihailovich M, Bonaldi T, Mass Spectrometry-Based Proteomics to Unveil the Non-coding RNA World, Front Mol Biosci 5 (2018) 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lim SJ, Tan TW, Tong JC, Computational Epigenetics: the new scientific paradigm, Bioinformation 4(7) (2010) 331–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bock C, Analysing and interpreting DNA methylation data, Nat Rev Genet 13(10) (2012) 705–19. [DOI] [PubMed] [Google Scholar]
- [96].Rauluseviciute I, Drabløs F, Rye MB, DNA methylation data by sequencing: experimental approaches and recommendations for tools and pipelines for data analysis, Clin Epigenetics 11(1) (2019) 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Krueger F, Andrews SR, Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications, Bioinformatics 27(11) (2011) 1571–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Huang KYY, Huang YJ, Chen PY, BS-Seeker3: ultrafast pipeline for bisulfite sequencing, BMC Bioinformatics 19(1) (2018) 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hansen KD, Langmead B, Irizarry RA, BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions, Genome Biol 13(10) (2012) R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Nakato R, Sakata T, Methods for ChIP-seq analysis: A practical workflow and advanced applications, Methods (2020). [DOI] [PubMed] [Google Scholar]
- [101].Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MACS), Genome Biol 9(9) (2008) R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Nakato R, Itoh T, Shirahige K, DROMPA: easy-to-handle peak calling and visualization software for the computational analysis and validation of ChIP-seq data, Genes Cells 18(7) (2013) 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Andergassen D, Dotter CP, Kulinski TM, Guenzl PM, Bammer PC, Barlow DP, Pauler FM, Hudson QJ, Allelome.PRO, a pipeline to define allele-specific genomic features from high-throughput sequencing data, Nucleic Acids Res 43(21) (2015) e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].van de Geijn B, McVicker G, Gilad Y, Pritchard JK, WASP: allele-specific software for robust molecular quantitative trait locus discovery, Nat Methods 12(11) (2015) 1061–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Richard Albert J, Koike T, Younesy H, Thompson R, Bogutz AB, Karimi MM, Lorincz MC, Development and application of an integrated allele-specific pipeline for methylomic and epigenomic analysis (MEA), BMC Genomics 19(1) (2018) 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Hu Y, An Q, Sheu K, Trejo B, Fan S, Guo Y, Single Cell Multi-Omics Technology: Methodology and Application, Front Cell Dev Biol 6 (2018) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Subramanian I, Verma S, Kumar S, Jere A, Anamika K, Multi-omics Data Integration, Interpretation, and Its Application, Bioinform Biol Insights 14 (2020) 1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]