

Abstract
Background:
Irinotecan/5-FU (FOLFIRI) or oxaliplatin/5-FU (FOLFOX), combined with bevacizumab or cetuximab, are approved, first-line treatments for metastatic colorectal cancer (mCRC). We aimed at identifying germline variants associated with survival in mCRC patients treated with these regimens in CALGB/SWOG 80405.
Methods:
Patients with mCRC receiving either FOLFOX or FOLFIRI were randomized to either cetuximab or bevacizumab. DNA from peripheral blood was genotyped for ~700,000 single-nucleotide polymorphisms (SNPs). The association between SNPs and overall survival (OS) was tested in 613 patients of genetically-estimated European ancestry using Cox proportional hazards models.
Results:
The four most significant SNPs associated with OS were three haplotypic SNPs between MGST1 and LMO3 (representative hazard ratios (HR) 1.56, p-value 1.30x10−6), and rs11644916 in AXIN1 (HR 1.39, p-value 4.26x10−6). AXIN1 is a well-established tumor suppressor gene in CRC, and rs11644916 (G>A) conferred shorter OS. Median OS for patients with the AA, AG or GG genotypes was 18.4, 25.6, or 36.4 months, respectively. In 90 stage IV CRC patients from TCGA, rs11649255 in AXIN1 (in almost complete linkage disequilibrium (LD) with rs11644916), was associated with shorter OS (HR 2.24, p-value 0.0096). Using rs11648673 in AXIN1 (in very high LD with rs11644916 and with functional evidence), luciferase activity in three CRC cell lines was reduced.
Conclusions:
This is the first large GWAS ever conducted in mCRC patients treated with first-line standard treatment in a randomized phase III trial. A common SNP in AXIN1 confers worse OS and the effect is replicated in TCGA. Further studies in CRC experimental models are required.
Keywords: CALGB/SWOG 80405, GWAS, overall survival, AXIN1
INTRODUCTION
Cancer and Leukemia Group B (CALGB)/SWOG 80405 was designed to test whether cetuximab or bevacizumab or both added to 5-fluorouracil/leucovorin with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) lead to superior overall survival (OS) as first-line therapy in advanced or metastatic colorectal cancer (mCRC). CALGB is now part of the Alliance for Clinical Trials in Oncology (Alliance). In the primary cohort patients, such as patients with KRAS wild-type for codon 12/13 and treated with either cetuximab or bevacizumab, there was no statistically significant difference in OS between the bevacizumab arm and the cetuximab arm (1). Over the years from the start of the trial, the combinations of those biologics with chemotherapy became the current standard of care treatment in the first-line setting of mCRC.
As part of the protocol, prospective collection of germline DNA from peripheral blood has been obtained from consented patients. The analysis of the common germline variation of cancer patients can allow discoveries of new biological determinants of outcome. Although most of the attention of biomarker discovery efforts in oncology has been focused on the somatic genome, many hallmarks of cancer lie outside of the cancer genome. They include angiogenesis, inflammation, adaptive immunity, and others (2,3).
We hypothesized that novel germline variants that drive tumor progression and/or resistance to therapy might affect patient outcome. Outcome data and DNA from peripheral blood collected from this relatively large study might provide an unprecedented opportunity for identifying germline DNA markers that affect OS. In this study, the germline DNA of patients has been scanned using high-density single-nucleotide polymorphism (SNP) chips that detect hundreds of thousands of SNPs. We used this approach to identify novel genes associated with OS in mCRC patients. In addition, we have used TCGA data and in vitro experiments to corroborate the clinical findings from CALGB/SWOG 80405.
METHODS
CALGB/SWOG 80405
The trial was designed to compare either cetuximab, bevacizumab, or cetuximab/bevacizumab combined with FOLFOX or FOLFIRI as first-line treatment of advanced and mCRC. Within 3 years, interim analysis of outcome data led to a restriction of eligibility to patients with KRAS WT (codon 12 and 13) and to closure of the dual antibody arm. A revised 2-arm trial (chemotherapy plus either cetuximab or bevacizumab) became the “primary cohort”. Randomization was stratified by chemotherapy, prior adjuvant chemotherapy, and prior pelvic radiation. Eligibility criteria can be found in the original publication (1).
In this genomic study, the primary endpoint of OS was defined as time of randomization until death due to any cause. Patients without reported deaths were censored at their last known follow-up. The median follow-up is 60.4 months (95% CI 56.3-64.3).
Table 1 reports demographics and clinical characteristics of patients in this study, who belong to a subset of the primary cohort. Institutional review board approval was obtained for this study and all participating patients provided written informed consent to this analysis. Patient registration, data collection, and data analysis were performed by the Alliance Statistics and Data Center. Data quality was ensured by careful review of data by Alliance Statistics and Data Center staff and the study chair of the clinical trial. This study was conducted in accordance with the ethical guidelines from the Declaration of Helsinki.
Table 1.
Clinical characteristics and demographics of patients genotyped from the primary cohort of CALGB/SWOG 80405.
| GWAS (N=613) | Primary Cohort (N=1137) | |
|---|---|---|
| Age (years) | ||
| Median | 59.2 | 59.1 |
| Range | (21.8-84.3) | (20.8-89.5) |
| Arm | ||
| Chemotherapy/Bevacizumab | 312 (50.9%) | 559 (49.2%) |
| Chemotherapy/Cetuximab | 301 (49.1%) | 578 (50.8%) |
| Chemotherapy | ||
| FOLFOX | 459 (74.9%) | 835 (73.4%) |
| FOLFIRI | 154 (25.1%) | 302 (26.6%) |
| Prior Adjuvant Chemotherapy | ||
| No | 534 (87.1%) | 977 (85.9%) |
| Yes | 79 (12.9%) | 160 (14.1%) |
| Prior Pelvic Radiation | ||
| No | 554 (90.4%) | 1035 (91.0%) |
| Yes | 59 (9.6%) | 102 (9.0%) |
| Gender | ||
| Male | 380 (62.0%) | 697 (61.3%) |
| Female | 233 (38.0%) | 440 (38.7%) |
| Race | ||
| More than one race | 0 | 5 |
| Unknown | 2 (0.3%) | 24 (2.1%) |
| White | 611 (99.8%) | 934 (82.1%) |
| African American | 0 (0.0%) | 129 (11.3%) |
| Asian | 0 (0.0%) | 35 (3.1%) |
| Native Hawaiian or Pacific Islander | 0 (0.0%) | 3 (0.3%) |
| American Indian or Alaska Native | 0 (0.0%) | 6 (0.5%) |
| Not Reported | 0 (0.0%) | 1 (0.1%) |
| ECOG PS | ||
| 0 | 363 (59.2%) | 657 (57.8%) |
| 1 | 250 (40.8%) | 478 (42.0%) |
| 2 | 0 (0.0%) | 2 (0.2%) |
| Number of Metastatic Sites | ||
| Missing | 18 | 42 |
| 1 | 344 (57.8%) | 635 (55.8%) |
| 2 | 190 (31.9%) | 355 (31.2%) |
| 3+ | 61 (10.3%) | 105 (9.2%) |
| Tumor Location | ||
| Left | 375 (61.2%) | 689 (60.6%) |
| Right | 154 (25.1%) | 280 (24.6%) |
| Transverse | 36 (5.9%) | 62 (5.5%) |
| Multiple | 0 (0.0%) | 1 (0.1%) |
| Unknown | 48 (7.8%) | 105 (9.2%) |
| Liver Metastases Only | ||
| Missing | 4 | 9 |
| No | 367 (60.3%) | 683 (60.1%) |
| Yes | 242 (39.7%) | 445 (39.1%) |
| Overall Survival (months) | ||
| Median (95% CI) | 29.5 (26.7-32.6) | 29.4 (27.6-31.4) |
| Progression-Free Survival (months) | ||
| Median (95% CI) | 10.6 (9.7-11.3) | 10.6 (9.8-11.1) |
Genotyping data
DNA was extracted from peripheral blood. The first batch of genotyping was performed on the Illumina HumanOmniExpress-12v1 platform at the Riken Institute, Japan, and included 731,412 SNPs. The second batch of genotyping was performed on the Illumina HumanOmniExpress-8v1 and included 964,193 SNPs. 719,461 SNPs from HapMap from batch 1 were also on the chip from batch 2. 1,178 of these were excluded for having mismatched annotation between the two platforms. Among the 718,283 SNPs in common between the two platforms, 70,108 SNPs were excluded due to call rates less than 99%, 107,761 SNPs with minor allele frequencies (MAF) less than 0.05 were removed, and 77 SNPs with strong evidence for departure from Hardy-Weinberg equilibrium (HWE, p-value<10−8) were removed. Among the 540,337 SNPs passing the filter, 540,021 were autosomal, and were used in the association analyses. Patients whose genotypic data were identified as unintentional duplicates, or whose genotypically estimated sex did not match their self-reported gender were also excluded. Genetic ancestral origin of patients was estimated using principal components analysis prior to filtering the variants (4). This resulted in 613 patients of European ancestry in the primary cohort (Supplementary Figure 1).
Bioinformatic analysis of SNP function
Putative functional effects of the four most significant SNPs associated with OS and variants in high LD (r2 > 0.8 in Europeans) were investigated using SNPnexus (5) which integrates resources from ENCODE and the Roadmap Epigenomics Consortium to map and annotate non-coding variants onto different classes of regulatory regions and predict their functional impact using 8 non-coding variant scoring algorithms and computational methods. Variants were also examined for annotation of putative function using RegulomeDB (www.regulomeDB) and HaploReg v4.1 (www.broadinstitute.org/mammals/haploreg/haploreg.php).
Statistical considerations
Our primary analysis determined the association between SNPs and OS in both arms combined, in patients of genetically-estimated European ancestry in the primary cohort (N=613). Patients with genetically-estimated ancestries other than European were not included in this study. The Cox score (log-rank) test was used, and the analysis was conducted under an additive genetic model. To assess potential confounding of baseline risk factors, we conducted an adjusted analysis within the framework of a multivariable additive log-linear Cox proportional-hazards model (6). These covariates were age (log base 10 transformed), sex, and tumor location (left vs. right/transverse). In an alternative adjusted analysis, tumor location was replaced by BRAF V600E mutation status, microsatellite instability status (MSI), and consensus molecular subtype (CMS). For the unadjusted analyses, the hazard ratio (HR) of the genetic effect and a 95% confidence interval (CI) were reported, along with the p-value for the score test. For the adjusted analyses, the Wald p-value of the SNP effect was reported instead. As an exploratory analysis, an interaction model was used to test for differences in SNP effect between the two treatment arms (cetuximab and bevacizumab). The interaction model was adjusted for age (log base 10 transformed), sex, and tumor location (left vs. right/transverse) and stratified by chemotherapy. The Wald p-value of the interaction term was reported, along with the HR and 95% CI of the genetic effect in each treatment arm (estimated separately). The interaction model was repeated using progression-free survival (PFS, measured as previously reported [7]) as the outcome. The interaction between SNP and chemotherapy was also tested, stratified by treatment arm.
As secondary analyses, selected SNPs from the genome-wide analysis were tested for independence from tumor location (left vs. right/transverse), BRAF V600E, and MSI using generalized linear models, and independence from CMS using the chi-squared test. An analysis of deviance was also conducted to determine if tumor location contributed additional information to the survival model beyond that of each SNP alone. Analyses involving covariates were limited to the subset of patients with data available. The p-values and confidence intervals were not adjusted for multiple comparisons, as they were used as a feature selection for additional testing in TCGA and in vitro experiments. Linkage disequilibrium (LD) r2 values were reported based on the European population from the 1,000 Genomes Project, unless otherwise noted.
A quantile-quantile (QQ) plot was generated to illustrate the distribution of p-values from the primary analysis (Supplementary Figure 2). Kaplan-Meier and LocusZoom (8) plots were generated to illustrate the association of selected SNPs with survival. The R software environment for statistical computing and graphics (9) and the survival (10) extension package was used to conduct the statistical analyses and generate the figures. The analyses were carried out with adherence to the principles of reproducible analysis using the knitr package (11) for generation of dynamic reports and Duke’s gitlab (https://gitlab.oit.duke.edu) for source code management. The code for replicating the statistical analysis was made accessible through a public source code repository (https://gitlab.oit.duke.edu/dcibioinformatics/pubs/calgb-80405-gwas).
TCGA CRC replication analysis
We used OS and germline genetic data from stage IV TCGA colon adenocarcinoma (COAD) and rectum adenocarcinoma (READ) patients (n=90) (12). rs11644916 in AXIN1 was not genotyped in TCGA. Hence, we used a proxy, such as rs11649255 (A>G) in AXIN1, which is in almost complete LD with rs11644916 (r2=0.97). The association between rs11649255 and OS was assessed using a Cox’s proportional hazards model. Since there were only three patients that were homozygous for the rs11649255 variant allele, a resampling was performed using 100,000 permutations without replacement. rs10772941 was also tested for association with OS, as a proxy for rs10846370 (r2=0.87) (intergenic between MGST1 and LMO3).
Cell culture, plasmids, and luciferase assay
HCT116, SW480, and RKO CRC cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). HCT116 and SW480 cells were grown in McCoy’s 5A (Mediatech) supplemented with 10% fetal bovine serum (Omega Scientific, Inc.), and 1% penicillin/ streptomycin, and incubated at 37°C and 5% CO2. RKO cells were grown in DMEM (Mediatech) supplemented with 10% fetal bovine serum (Omega Scientific, Inc.), and 1% penicillin/streptomycin, and incubated at 37°C and in 5% CO2.
To test the effects of the AXIN1 variant on transcriptional efficiency, DNA fragments corresponding to the candidate enhancer region in AXIN1 (chr16:327363-328800, hg38) were amplified by PCR from normal human genomic DNA using the following forward and reverse primers, respectively: 5’-GGGCAAGAGAATGAGTCACC-3’ and 5’-CATGAGTGTTTGGGTTCCTG-3’. PCR fragments were subcloned into the Sac II restriction enzyme site in both directions, upstream of a thymidine kinase (TK) minimal promoter-firefly-luciferase vector obtained courtesy of Dr. G. A. Coetzee (Van Andel Research Institute). Fragments were cloned using the In-Fusion HD cloning kit (Takara). rs11648673 (in LD with rs11644916, r2=0.87) was used instead of rs11644916 in the assays because of stronger functional prediction based upon the bioinformatic analysis (see in the Results section). The AXIN1 functional variant was created using site-directed mutagenesis. Plasmid clones were sequenced by Sanger sequencing to confirm the presence of the candidate variants and the absence of any PCR amplification-induced mutations. A region of Chr8q24 previously shown to have no activity in any of the cell lines served as the negative control, and a region of Chr8q24 that was previously shown to have enhancer activity in all of the cell lines served as the positive control. For enhancer assays, SW480 (10x104 cells/well), HCT116 and RKO cells (6x104 cells/well) were seeded into 96-well plates. Cells were co-transfected with reporter plasmids and constitutively active pRL-TK Renilla luciferase plasmid (Promega) using Lipofectamine 2000 Reagent (Life Technologies) according to the manufacturer’s instructions. After 24 hours, cells were harvested, and extracts were assayed for luciferase activity using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions and measured using a Tecan Infinite F200Pro Microplate Reader. The ratio of normalized luminescence from the experimental sample to the negative control reporter was calculated for each sample and defined as the relative luciferase activity. Luciferase activity was tested in a minimum of three independent clones for each allele. The data are presented as mean +/- SD of four independent transfection experiments. The differences in activity between the two alleles was calculated using a two-sided student’s paired t-test.
Gene editing by CRISPR
To delete an intronic fragment surrounding rs11648673 in AXIN1, RNA sequences were designed using Benchling (oligonucleotide sequences in Supplementary Table 1). Complementary oligonucleotides for each target sequence were incubated at 37°C for 30 minutes, followed by 95°C for 5 minutes, and annealed by decreasing 5°C/minute to 25°C using a PCR thermocycler (Eppendorf). The annealed fragments were then ligated into the Cas9-T2A-GFP-Puro plasmid linearized by the BsaI restriction enzyme site. The plasmids were transformed into DH5α cells and were grown on ampicillin plates at 37°C overnight. Bacterial colonies were selected and expanded. Plasmid DNA was extracted using the HiSpeed Plasmid Maxi kit (Qiagen). The gRNA inserts were verified through Sanger sequencing. HCT-116 cells were transfected with Cas-T2A-GFP-Puro plasmid containing gRNA insert of interest using Lipofectamine 2000 to generate the AXIN1 knockout cell lines. HCT-116 cells were electroporated with Cas-T2A-GFP-Puro plasmid containing gRNA insert of interest and donor oligo using the Nucleofector (Lonza) to generate a deletion spanning the AXIN1 intronic SNP, rs11648673. Cells were grown in T25 flask for 48 hours prior to single cell sorting by dilution in 96-well plates. Cells were expanded and deletion spanning rs11648673 was verified through PCR and Sanger sequencing. The AXIN1 knockouts (Supplementary Figure 3) were verified by complete abrogation of protein through western blot, while AXIN1 with 32-bp intronic deletion (Supplementary Figure 3) preserved protein expression (results not shown).
TCF/LEF reporter assay
To test the effect of the AXIN1 mutant cell lines on WNT signaling activity, HCT-116 cells were plated at the density of 10,000 cells/well in a 96-well plate 5 hours prior to infection of 10 μl of Cignal Lenti TCF/LEF Reporter (Qiagen). After 24 hours at 37°C, the media was replaced and a final concentration of 10 μM CT99021 was added to the cells. A Dual-Luciferase Reporter assay (Promega) was performed after 24 hours and luminescence was read on a GloMax (Promega). Firefly luciferase was normalized to Renilla luminescence. The data are presented as mean +/− SD of three independent experiments. The differences between constructs was calculated using a two-sided student’s paired t-test.
RESULTS
Genomic analysis of OS
The top three ranked SNPs according to their corresponding unadjusted p-values for association with OS are rs7300446, rs12426370, and rs10846370. They are intergenic between MGST1 (microsomal glutathione S-transferase 1) and LMO3 (LIM domain only 3) and belong to a haplotype block (r2>0.85 among them). The next highly ranked SNP is rs11644916, located in intron 4 of AXIN1 (Table 2) (for a LocusZoom plot of these variants, see Supplementary Figure 4). Among these three genes, i.e. MGST1, LMO3, and AXIN1, we selected AXIN1 for further evaluations and mechanistic studies, as the biology of AXIN1 is directly involved in the pathogenesis of CRC and progression of disease (13–15). AXIN1 is a cytoplasmic protein that negatively regulates the WNT signaling pathway by forming a β-catenin destruction complex with APC and GSK-3β. The WNT signaling pathway is highly dysregulated in CRC, making the SNP in AXIN1 (rs11644916, G>A) a relevant candidate to investigate further.
Table 2. Top ten SNPs ranked by p-value for association with OS in CALGB/SWOG 80405 of the primary cohort.
Chr, chromosome; LCL and UCL, lower and upper confidence limit, respectively.
| RSID | Chr | Gene | Alleles | Feature | MAF | HR | 95% LCL | 95% UCL | P-value |
|---|---|---|---|---|---|---|---|---|---|
| rs7300446 | 12 | MGST1/LMO3 | A>G | Intergenic | 0.15 | 1.56 | 1.30 | 1.87 | 1.30x10−6 |
| rs12426370 | 12 | MGST1/LMO3 | A>G | Intergenic | 0.15 | 1.55 | 1.29 | 1.86 | 1.76x10−6 |
| rs10846370 | 12 | MGST1/LMO3 | A>G | Intergenic | 0.15 | 1.50 | 1.27 | 1.78 | 2.29x10−6 |
| rs11644916 | 16 | AXIN1 | G>A | Intronic | 0.30 | 1.40 | 1.20 | 1.59 | 4.26x10−6 |
| rs2461035 | 8 | STMN2/HEY1 | A>G | Intergenic | 0.33 | 0.72 | 0.62 | 0.83 | 4.65x10−6 |
| rs16985284 | 2 | KCNS3/RDH14 | A>C | Intergenic | 0.12 | 1.56 | 1.29 | 1.89 | 5.12x10−6 |
| rs11246159 | 11 | ANO9 | A>G | Intronic | 0.17 | 1.47 | 1.24 | 1.75 | 6.38x10−6 |
| rs11649255 | 16 | AXIN1 | A>G | Intronic | 0.29 | 1.38 | 1.20 | 1.58 | 6.83x10−6 |
| rs898838 | 2 | MSGN1/KCNS3 | A>G | Intergenic | 0.45 | 0.74 | 0.65 | 0.85 | 7.51x10−6 |
| rs2467754 | 8 | STMN2/HEY1 | A>G | Intergenic | 0.33 | 0.72 | 0.63 | 0.84 | 8.42x10−6 |
Association between AXIN1 rs11644916 and OS in CALGB/SWOG 80405
The A allele of rs11644916 (G>A) was associated with shorter OS with treatment arms combined (p-value=4.26x10−6, HR=1.39, 95% CI 1.20-1.59) (Figure 1A). The median OS for patients with the AA, AG or GG genotypes was 18.4 (95% CI 14.2-27.6), 25.6 (95% CI 23.6-30.4), or 36.4 (95% CI 32.9-40.6) months, respectively.
Figure 1.
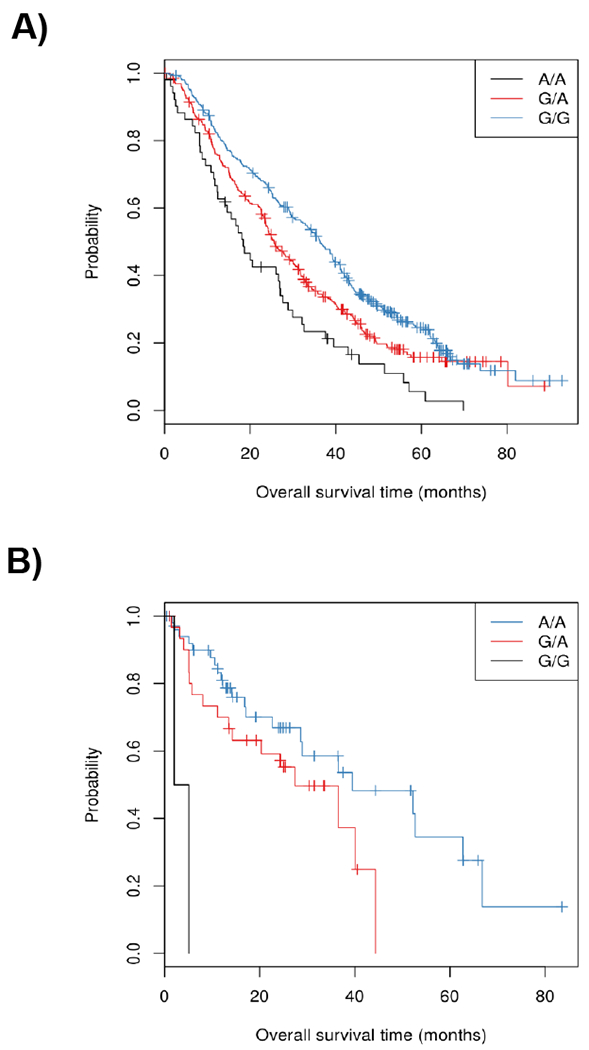
Kaplan-Meier plots of OS for rs11644916 (G>A) in AXIN1 in CALGB/SWOG 80405 (A) and for rs11644916 (A>G) in AXIN1 in TCGA (B).
In a model adjusted for covariates and stratified by chemotherapy, there was no evidence of an interaction between rs11644916 and treatment (bevacizumab versus cetuximab) in the association with OS (p-value=0.23). HRs estimated separately in the two arms were 1.52 (95% CI 1.24-1.85) and 1.16 (95% CI 0.94-1.44) in the bevacizumab and cetuximab arms, respectively (Figure 2). For the PFS outcome, there was no evidence of interaction between the SNP and treatment arm (bevacizumab versus cetuximab) (p-value=0.87). Similarly, no evidence of interaction between the SNP and chemotherapy (FOLFOX versus FOLFIRI) was observed (p-value=0.68).
Figure 2.
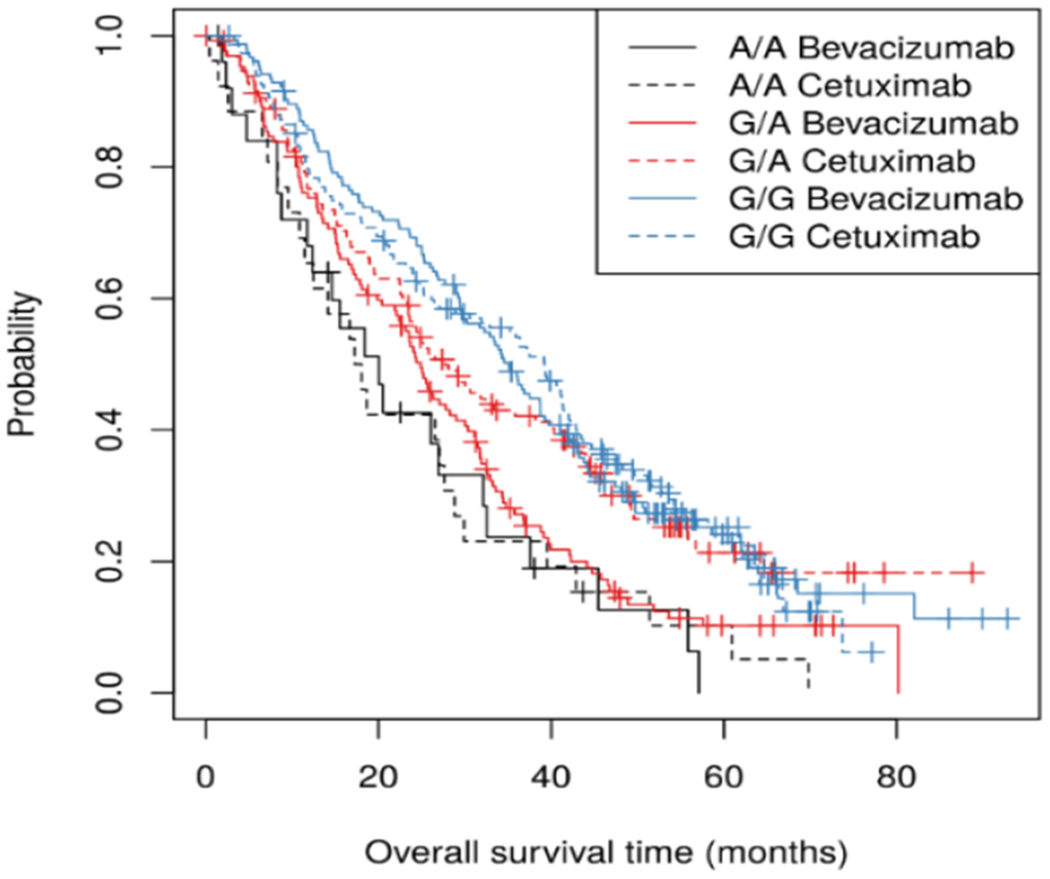
Kaplan-Meier plots of OS for rs11644916 by arm (bevacizumab versus cetuximab) in CALGB/SWOG 80405.
Association between AXIN1 rs11644916 and OS in TCGA
In TCGA, because of the difference in the genotyping platform used, AXIN1 rs11649255 (A>G) was used for analysis with OS instead of rs11644916 (G>A). The two variants are almost in complete LD (r2=0.99). In 90 stage IV CRC patients from TCGA, the AXIN1 rs11649255 (A>G) was associated with shorter survival (HR 2.24, 95% CI 1.20-4.19, p-value 0.0096, one-degree of freedom, Figure 1B). The direction of the observed effect is concordant with that of rs11644916 (G>A) in CALGB/SWOG 80405 (p-value=4.26x10−6, HR=1.39, 95% CI 1.20-1.59).
The median OS for patients with the AA, AG, and GG genotypes was 44.4 (95% CI, 36.5 to not yet reached), 22.7 (95% CI, 13.4 to 44.4), and 9.7 (95% CI, 5.1 to not yet reached) months. Given that only three patients were GG, a resampling permutation test was used as a sanity check for the asymptotic p-value (p-value=0.011 of the permutation test). For a direct comparison, the association of AXIN1 rs11649255 with OS was tested in CALGB/SWOG 80405, with similar results (p-value=6.83x10−6, HR=1.38, 95% CI 1.20-1.58) (Table 2).
Although we selected the AXIN1 variant for replication, we also evaluated the association of rs10772941 with OS as a proxy for rs10846370 (intergenic between MGST1 and LMO3, r2=0.87, Table 2). In the same 90 stage IV CRC patients (which included only two variant homozygotes, and 23 heterozygotes), there was no evidence that rs10772941 was associated with OS (p-value=0.9802, HR=1.01, 95% CI 0.54-1.87).
Tumor location analysis
As expected, patients with left-sided tumors were associated with longer OS than right/transverse-sided tumors (p-value=1.37x10−7, HR=0.59, 95% CI 0.49-0.72). Both AXIN1 rs11644916 (associated with OS) and rs11648673 (functional variant selected by bioinformatics) were associated with an increased odds of having the tumor on the right/transverse side of the colon (p-values=1.85x10−3 and 2.21x10−3, respectively) (Figures 3A,B). Both SNPs were associated with worse OS in a model adjusted for age, sex, and tumor location (p-value=1.80x10−4, HR=1.32, 95% CI 1.14-1.53, and p-value=3.28x10−3, HR=1.25, 95% CI 1.08-1.46, respectively, Figure 3C,D).
Figure 3.
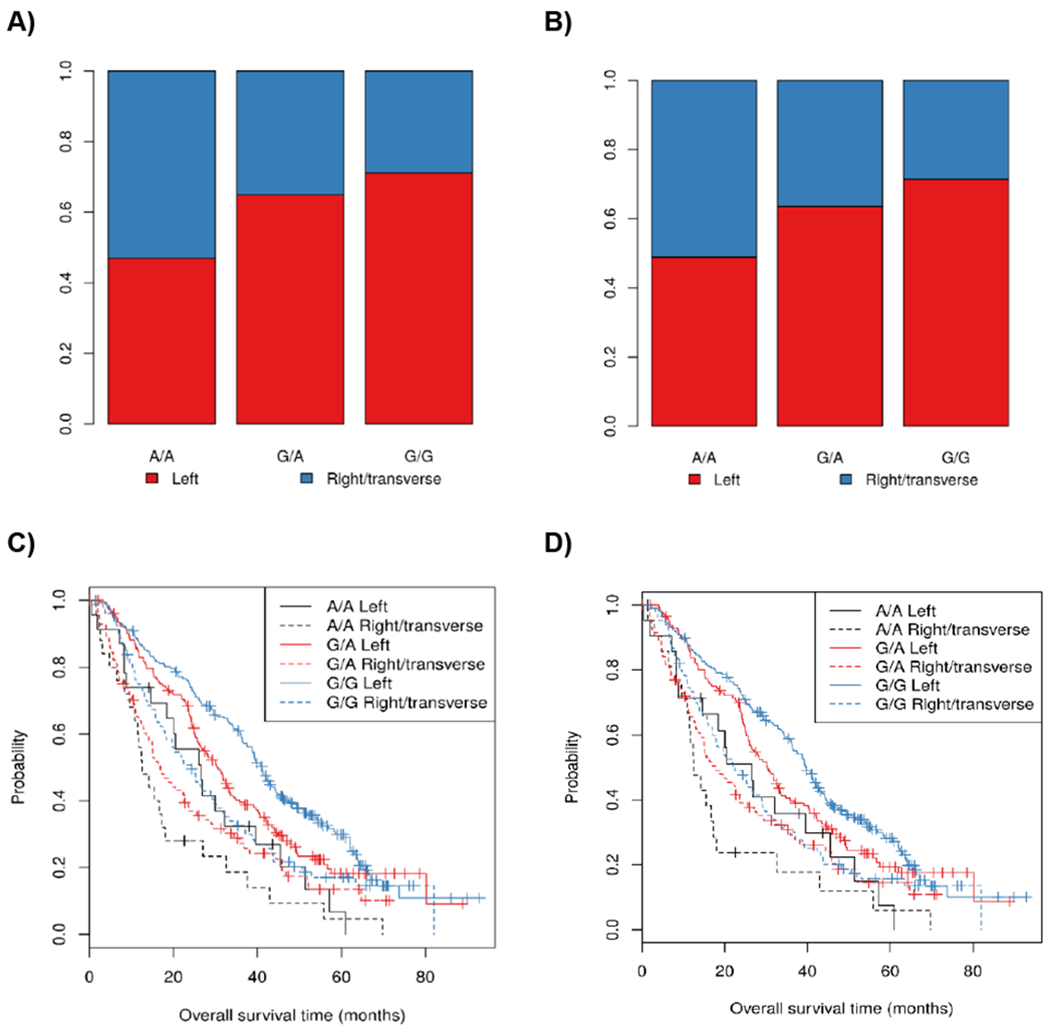
Tumor location by genotype for rs11644916 (A) and rs11648673 (B) in AXIN1. Kaplan-Meier plots of OS overall survival by tumor location and rs11644916 (C) and rs11648673 (D).
Supplementary Table 2 shows the results of the top ten SNPs, ranked by p-value from the unadjusted analysis of OS in CALGB/SWOG 80405, adjusted for age (log10 transformed), sex, and tumor location (left vs. right/transverse). Supplementary Table 3 shows the results for the same SNPs as Supplementary Table 2, when tumor location was replaced in the model by BRAF V600E, MSI, and CMS, for the 289 patients for which those data were available.
Bioinformatics assessment of SNPs associated with OS
For the three intergenic SNPs between MGST1 and LMO3, none of them appear to be located in regulatory regions or have enhancer functions (Supplementary Table 4). Similarly, AXIN1 rs11644916, located in intron 4, does not appear to be located in a regulatory region or affect transcription factor binding. The analysis of HaploReg for other SNPs in high LD with AXIN1 rs11644916 selected rs11648673 (in intron 2 of AXIN1) as the functional SNP (LD r2=0.81). This SNP is located in a region containing several regulatory elements, and RegulomeDB indicates that it is more likely to affect transcription factor binding (score=1f) (Supplemental Table 4). The A allele of rs11648673 was associated with worse OS (p-value=1.86x10−4, HR=1.31, 95% CI 1.14-1.52, Supplementary Figure 5). Due to the lack of bioinformatics evidence for the three SNPs located between MGST1 and LMO3 (Supplemental Table 4), we performed functional assays on the AXIN1 SNP.
Luciferase assay of AXIN1 rs11648673
Due to the lack of functionality of rs11644916 through bioinformatics, further functional validation was performed using the SNP in high LD with it, rs11648673 (Supplementary Table 4). A luciferase reporter assay was used to determine differences in enhancer activity between the reference and variant allele of rs11648673 in AXIN1. The forward constructs did not show any enhancer activity. The reverse constructs showed enhancer activity in both alleles, with the G allele showing higher activity than the A allele in all the three CRC cell lines tested (Figure 4A).
Figure 4.
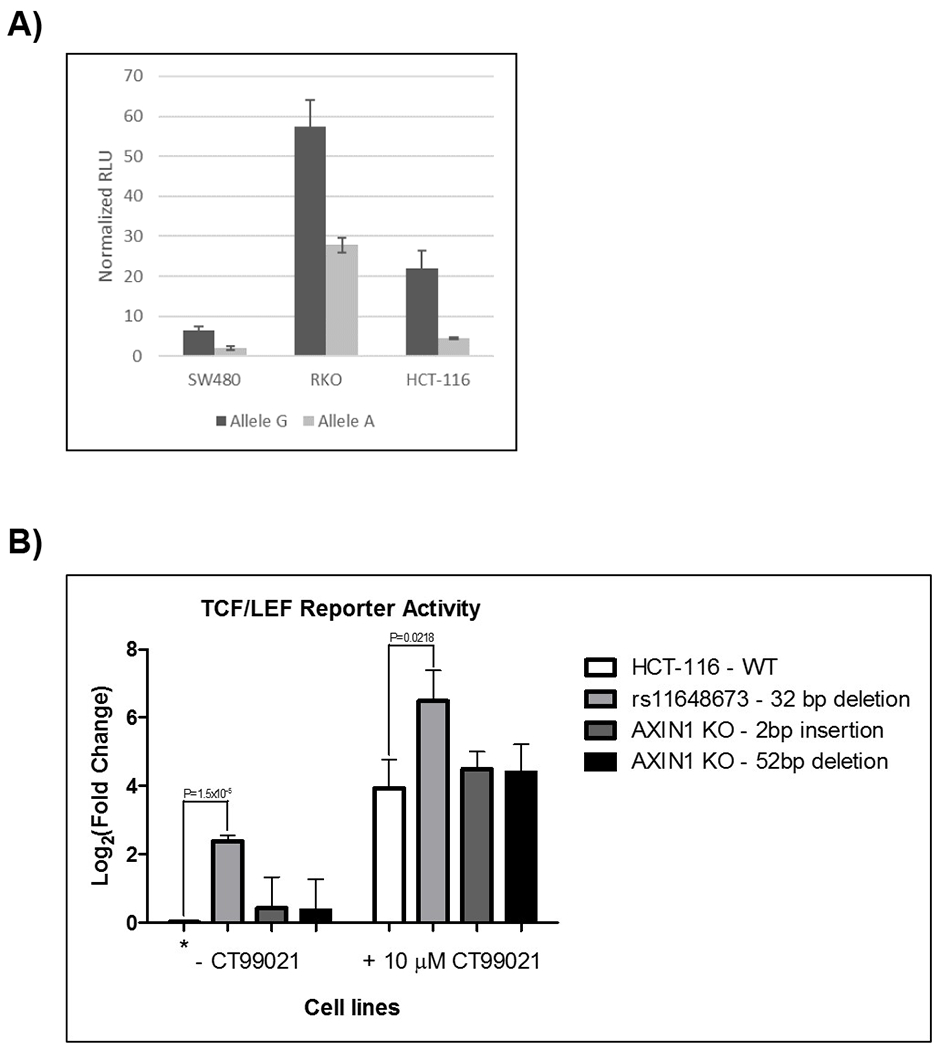
A) Luciferase assay of AXIN1 rs11648673. The G allele had statistically greater enhancer activity than the A allele of rs11648673 in SW480 (P-value=0.0008), HCT-116 (P-value=0.0042), and RKO (P-value=0.0016) human CRC cell lines. RLU, relative luminometer units. B) TCF/LEF reporter assay. Cells were treated with 10 μM CT99021 for WNT activation. Luciferase activity for each mutated HCT-116 cell line +/− CT99021 was normalized to WT HCT-116 (control) cell line at baseline (shown with an *). The log2(fold change) is reported. N=3 independent replicates.
TCF/LEF reporter assay
We generated two AXIN1 knockouts HCT-116 cell lines through the creation of frameshift mutations in exon 2, as well as a mutant HCT-116 cell line with a 32-bp deletion surrounding rs11648673 in intron 2. A TCF/LEF reporter assay measures β-catenin binding to its transcriptional domain, allowing for visualization of the effect of this deletion on WNT/β-catenin signaling. There was no change in TCF/LEF reporter activity in the AXIN1 knockout cell lines compared to wild type (WT). In the mutant cell line containing the 32-bp deletion spanning rs11648673, there was a significant increase in luciferase activity from the WNT reporter compared to the WT cell line, with and without treatment of a WNT activator (CT99021) (Figure 4B).
DISCUSSION
A genome-wide association study (GWAS) of OS in mCRC patients treated with standard of care therapies has selected a common SNP in AXIN1 as a novel prognostic factor in mCRC. This result has been supported by a concordant effect on OS in mCRC patients from TCGA, as well as bioinformatics and experimental evidence of functionality of the SNP in CRC cell lines. AXIN1 is a known tumor suppressor gene in the WNT pathway but this is the first germline variation in this gene to be reported as associated with reduced survival in mCRC.
AXIN1 is negative regulator of the WNT/β-catenin pathway. The WNT/β-catenin pathway is central to CRC tumorigenesis and progression (13–15) and is highly dysregulated in more than 90% of CRCs (16). In the WNT pathway, AXIN1 acts with APC, GSK3β, and CK1 to form a β-catenin destruction complex, which prevents transcriptional activation of targets of β-catenin, the ultimate effector of the pathway (13). In cell lines and transgenic mice, overexpression of AXIN1 leads to increased β-catenin degradation and attenuation of WNT signaling, supporting its tumor suppressor activity (17, 18).
The luciferase assays in three human CRC cell lines clearly indicate a reduction in the enhancer activity in the variant construct versus WT (Figure 4A). This result is consistent with the biological function of AXIN1 because a potential reduction in the ability of AXIN1 to efficiently activate a destruction complex would result in a more aggressive tumor phenotype, and eventually, worse survival. Moreover, despite the fact that WNT is constitutively activated in the HCT-116 CRC cell line due to a deletion in the β-catenin gene (19), the deletion of a 32-bp region surrounding the AXIN1 SNP in HCT-116 CRC cells increases WNT activation (measured by the TCF/LER reporter activity assay), both at baseline and after stimulation with a WNT activator (Figure 4B). These results indicate that the AXIN1 variant might interact with a functional element in intron 2 of AXIN1. The bioinformatic analysis (Supplementary Table 4) already indicates the presence of a few regulatory transcription factors, and we hypothesize that their activity is modulated by the presence of the AXIN1 variant.
While the experimental evidence provided above is in line with the clinical effect on OS, it does not exactly recapitulate the biological mechanism by which a SNP in AXIN1 alters OS. Our study provides the rationale for developing animal models to test the underlying mechanism. At the same time, it should be considered that alterations of the WNT pathway in cancer cells might alter the function of other cell systems. In addition to its central role in CRC oncogenesis and progression, WNT/β-catenin was the first pathway causally linked to T-cell inflammation (20). WNT/β-catenin signaling promotes cancer progression by regulating the tumor-immune cycle in most of its nodes, including dendritic, T, and tumor cells (21). Recent publications in CRC have shown that the activation of tumor-intrinsic WNT/β-catenin signaling was correlated with the absence of T-cell infiltration in CRC (22). This mechanism is a pan-tumor effect observed in 90% of tumor types from TCGA, including CRC (23). Tumor, immunocompetent xenografts should be used for testing the function of genetic manipulations of the AXIN1 gene.
The potential tumor suppressor function of AXIN1 has long been hypothesized based largely on their roles in the β-catenin destruction complex, but the in vivo importance of AXIN1 proteins for the inhibition of tumor development or progression is not well established, and the functional consequences of AXIN1 mutations in cancer remain largely undefined (24).
The present study provides further evidence of the tumor suppressor role of AXIN1, indicating that germline alterations in the AXIN1 gene affect the survival of patients with mCRC. With all the caveats of using survival data from TCGA, the effect of the AXIN1 SNP is concordant between TCGA and CALGB/SWOG 80405 (Figure 1A,B), even after a resampling, permutation test in TCGA (due its smaller sample size). The results of this study suggest that this could be a prognostic effect, as it seems independent from the biologic used (bevacizumab or cetuximab). In this regard, an uncertainty remains in relation to establishing differences between FOLFIRI and FOLFOX, as only one third of patients were treated with FOLFIRI. Cytotoxicity experiments with 5-FU, irinotecan, SN-38 or oxaliplatin in a CRC cell line rule out that this AXIN1 variant can change the efficacy of these agents in vitro (results not shown).
None of the SNP-OS associations had a p-value less than 10−8, the conventional genome-wide statistical significance. Although the lack of adjustment for genome-wide testing can increase the likelihood of false positivity, as stated in the methods of this article, the p-values were used for feature selection to direct further validation and experimental studies. When a GWAS was not been pre-planned and was not the primary endpoint of the clinical study, as in this case, losses in power and/or statistical significance should be compensated by triangulation of evidence from other results. This limitation is therefore in part mitigated by the well-established role of AXIN1 in CRC, the replication of this signal in TCGA, and the functional assessment of the effect of the AXIN1 variants in CRC cell lines.
Interestingly, the AXIN1 variant seems to co-localize in part with the right/transverse primary tumors, which are characterized by worse OS as compared to left tumors (7). However, both the AXIN1 variant and right/transverse primary tumors contribute, independently, to worse OS (Figure 3C, D). If further confirmed in additional studies, AXIN1 germline variants could represent a modifier predisposing CRC patients to have right/transverse-sided and more aggressive disease at presentation.
Supplementary Material
Statement of Translational Relevance.
This paper demonstrates, for the first time, that common germline variation in AXIN1 in patients with metastatic colorectal cancer may affect patient survival. This paper also provides new results about the clinical role of the entire genomic variation in patients. If confirmed in additional studies, these results can be used for reaching decisions to obtain optimized treatment in patients with metastatic colorectal cancer.
Acknowledgements:
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers U10CA180821, U10CA180882, and U24CA196171 (to the Alliance for Clinical Trials in Oncology), UG1CA233253, U10CA180820 (ECOG-ACRIN) and U10CA180888 (SWOG) and NIH R01CA143237. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. https://acknowledgments.alliancefound.org. Preliminary results were presented at the ASCO and ESMO 2015 Annual Meetings.
Conflict of Interest: Howard McLeod is on the Board of Directors of Cancer Genetics Inc and is a Co-Founder of Interpares Biomedicine and ClariFI. He is on a non-branded speakers bureau for Genentech. He is also a scientific advisor to eviCORE and Pharmazam. Heinz-Josef Lenz is on the advisory board and/or consultant for Bayer, Merck KG and Roche. Mark Ratain has received research funding (clinical trial) from Genentech/Roche. Alan Venook has received a grant from Genentech and Roche.
Footnotes
All other authors declare no potential conflicts of interest.
References
- 1).Venook AP, Niedzwiecki D, Lenz HJ, Innocenti F, Fruth B, Meyerhardt JA, et al. Effect of First-Line Chemotherapy Combined With Cetuximab or Bevacizumab on Overall Survival in Patients With KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA 2017;317(23):2392–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Innocenti F, Owzar K, Cox NL, Evans P, Kubo M, Zembutsu H, et al. A genome-wide association study of overall survival in pancreatic cancer patients treated with gemcitabine in CALGB 80303. Clin Cancer Res 2012;18(2):577–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 4).Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38(8):904–9. [DOI] [PubMed] [Google Scholar]
- 5).Ullah AZD, Oscanoa J, Wang J, Nagano A, Lemoine NR, Chelala C. SNPnexus: assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res 2018;46(Web Server issue):W109–W113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Therneau TM and Grambsch PM. Modeling Survival Data: Extending the Cox Model. New York: Springer; 2000. [Google Scholar]
- 7).Innocenti F, Ou FS, Qu X, Zemla TJ, Niedzwiecki D, Tam R, et al. Mutational Analysis of Patients With Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J Clin Oncol 2019;37(14):1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 2010; 26(18): 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).R Core Team (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/ [Google Scholar]
- 10).Therneau T (2015). A Package for Survival Analysis in S. version 2.38, https://CRAN.R-project.org/package=survival
- 11).Xie Y Dynamic Documents with R and knitr. 2nd edition Chapman and Hall/CRC Boca Raton, Florida: Taylor and Francis Group; 2015. [Google Scholar]
- 12).The Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012;487(7407):330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T, Clevers H. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell 2012;149(6):1245–1256. [DOI] [PubMed] [Google Scholar]
- 14).Flanagan DJ, Vincan E, Phesse TJ. Wnt Signaling in Cancer: Not a Binary ON:OFF Switch. Cancer Res 2019;79(23):5901–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene 2017;36(11):1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Silva AL, Dawson SN, Arends MJ, Guttula K, Hall N, Cameron EA, et al. Boosting Wnt activity during colorectal cancer progression through selective hypermethylation of Wnt signaling antagonists. BMC Cancer 2014;14:891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Picco G, Petti C, Centonze A, Torchiaro E, Crisafulli G, Novara L, et al. Loss of AXIN1 drives acquired resistance to WNT pathway blockade in colorectal cancer cells carrying RSPO3 fusions. EMBO Mol Med 2017;9(3): 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Hsu W, Shakya R, Costantini F. Impaired mammary gland and lymphoid development caused by inducible expression of Axin in transgenic mice. J Cell Biol 2001;155(6):1055–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Miyamoto M, Hayashi T, Kawasaki Y, Akiyama T. Sp5 negatively regulates the proliferation of HCT116 cells by upregulating the transcription of p27. Oncol Lett 2018:15(3):4005–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015;523(7559):231–235. [DOI] [PubMed] [Google Scholar]
- 21).Wang B, Tian T, Kalland KH, Ke X, Qu Y. Targeting Wnt/beta-Catenin Signaling for Cancer Immunotherapy. Trends Pharmacol Sci 2018;39(7):648–658. [DOI] [PubMed] [Google Scholar]
- 22).Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov 2018;8(6):730–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/beta-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin Cancer Res 2019;25(10):3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Mazzoni SM, Fearon ER. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett 2014;355(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.