

Abstract
A human microfluidic four-cell liver acinus microphysiology system (LAMPS), was evaluated for reproducibility and robustness as a model for drug pharmacokinetics and toxicology. The model was constructed using primary human hepatocytes or human induced pluripotent stem cell (iPSC)-derived hepatocytes and 3 human cell lines for the endothelial, Kupffer and stellate cells. The model was tested in two laboratories and demonstrated to be reproducible in terms of basal function of hepatocytes, Terfenadine metabolism, and effects of Tolcapone (88 μM), Troglitazone (150 μM), or caffeine (600 μM) over 9 days in culture. Additional experiments compared basal outputs of albumin, urea, lactate dehydrogenase (LDH) and tumor necrosis factor (TNF)α, as well as drug metabolism and toxicity in the LAMPS model, or 2D cultures seeded with either primary hepatocytes or iPSC-hepatocytes. Further experiments to study the effects of Terfenadine (10 μM), Tolcapone (88 μ), Trovafloxacin (150 μ with or without 1 μg/mL lipopolysaccharide), Troglitazone (28 μM), Rosiglitazone (0.8 μM), Pioglitazone (3 μM), and caffeine (600 μM) were carried out over 10 days. We found that both primary human hepatocytes and iPSC-derived hepatocytes in 3D culture maintained excellent basal liver function and Terfenadine metabolism over 10 days compared the same cells in 2D cultures. In 2D, non-overlay monolayer cultures, both cell types lost hepatocyte phenotypes after 48 hours. With respect to drug effects, both cell types demonstrated comparable and more human-relevant effects in LAMPS, as compared to 2D cultures. Overall, these studies show that LAMPS is a robust and reproducible in vitro liver model, comparable in performance when seeded with either primary human hepatocytes or iPSC-derived hepatocytes, and more physiologically and clinically relevant than 2D monolayer cultures.
Keywords: hepatotoxicity, in vitro, drug toxicity
1. Introduction
Development of experimental models that can enable studies of the liver in cell-based systems is an active area of scientific inquiry (Soldatow et al. 2013; Taylor et al. 2019; Weaver et al. 2017). On the one hand, pre-clinical animal models are widely regarded as limited predictors of the human liver effects of drugs (Monticello et al. 2017) even though liver toxicity is consistently among the top concerns in drug development (Weaver et al. 2020). On the other hand, liver toxicity is a common “critical effect” that is used to determine human cancer hazard of, and derive safe exposure limits for, environmental chemicals (Krewski et al. 2019). Most existing cell-based models for studies of acute and sub-chronic effects of xenobiotics on the liver have important limitations in terms of their ability to sustain metabolic capacity and replicate the microphysiological environment of the multi-cellular organ (Shah et al. 2015; Sistare et al. 2016). Among many general directions to improve cell-based models’ performance in studies of hepatotoxicity are important efforts to (i) create multi-cellular microphysiological models (Griffith et al. 2014; Khetani and Bhatia 2008; Vernetti et al. 2016), and (ii) replace cancer-derived cell lines or primary hepatocytes with a persistent supply of human hepatocytes by way of differentiating induced pluripotent stem cells (iPSC) (Zhao et al. 2003).
Traditional static (i.e., no directional media movement) in vitro experiments with hepatocytes are conducted in the form of cell suspensions, in monolayers or sandwiches in extracellular matrix, are mono- or co-cultures with other cell types, and also can include assembly of cells into spheroids (LeCluyse et al. 2012). The development of microphysiological test systems, or tissue chips, has accelerated in the past decade and resulted in a dramatic increase in the number of organ-specific and multi-organ models (Marx et al. 2020). Creation of liver-on-chip models has been among the most active areas of research in biomedical engineering (Ribeiro et al. 2019; Underhill and Khetani 2018). The “fit for purpose” perspective has been an important element of the developments with a balance between throughput/cost and optimal model functioning (Taylor et al. 2019). Research directions in this area are focused on creating improved models for drug safety evaluation (Cavero et al. 2019), pharmacokinetics (Oleaga et al. 2018), liver disease modeling (Hassan et al. 2020), and connecting the liver with other organs (Edington et al. 2018; Vernetti et al. 2017b). Even though these models have much potential and hold promise to recreate the complex cellular and molecular milieu of the liver in the dish, it is widely acknowledged, by both the researchers and prospective users of these models, that many challenges remain. Among them are the relatively low throughput, high complexity, and cost; compared to the higher throughput and much lower physiological complexity and cost of the simpler models. In addition, there is a lack of consistency in the optimization and characterization of the more complex models by the developers, all of which makes it a challenge to transition these models from research laboratories to application in the practice of drug and chemical safety testing, or disease modeling (Baudy et al. 2020).
Equally large is the challenge of creating a sufficient and reproducible supply of human cells for in vitro studies of the liver. For hepatocytes alone, dozens of options exist for human-based models – cancer cell lines, primary cells from individual donors or donor pools, and iPSC-derived cells (Godoy et al. 2013; Lauschke et al. 2016; Soldatow et al. 2013). While primary human hepatocytes are a model of choice for most investigators, their cost, availability, and donor-to-donor variability are well recognized, prompting studies of more reproducible cell types, such as human iPSC-derived hepatocytes (Grimm et al. 2015; Guguen-Guillouzo et al. 2010; Sirenko et al. 2014) and immortalized human hepatic cell lines (Ramboer et al. 2015). Despite the challenges of their largely fetal-like phenotype, they can be a reliable and reproducible model for drug metabolism upon in vitro maturation (Bulutoglu et al. 2020).
The current study aimed to determine if a recently developed human liver acinus microphysiological system (LAMPS) platform is a reproducible model that can be replicated in another laboratory. In addition, we compare performance of LAMPS seeded with primary human hepatocytes or human iPSC-derived hepatocytes and conclude that iPSC-derived hepatocyte-constructed LAMPS is a robust model for studies of basic liver functions, drug pharmacokinetics, drug metabolism, and chemical safety.
2. Materials and Methods
2.1. Cells:
Primary human hepatocytes were obtained from ThermoFisher (HMCPTS, lot#HU1838, Waltham, MA), and iPSC-derived hepatocytes were purchased from FujiFilm-Cellular Dynamics International (CDI, 01279, lot#103934, Santa Ana, CA). These hepatocytes were seeded directly into chips or plates from thaw. Supporting cells were cultured (37°C, 5% CO2) and expanded in flasks prior to seeding as follows. EA.hy926 (CRL-2922, lot 63396642, ATCC, Manassas, VA) cells were cultured in DMEM (11965–092, Invitrogen, Waltham, MA) supplemented with 10% FBS (35010CV, Corning, Corning, NY), and 1% pen-strep (SV30010, Hyclone, Logan, UT); LX-2 (SCC064, lot# 2924839, Millipore Sigma, Burlington, MA) cells in DMEM with 2% FBS, and 1% pen-strep; and THP-1 (TIB-202, lot# 70005912, ATCC) cells were grown in RPMI (SH3009601, Hyclone) with 10% FBS, 1% pen-strep, and 2 mM L-glutamine (SH3003401, Hyclone). THP-1 monocyte cells were differentiated to adherent macrophages via treatment with 200 ng/mL PMA (Phorbol-12-myristate-13-acetate; 524400, Millipore Sigma, St. Louis, MO) 48 hours prior to seeding into chips or plates (Lee-Montiel et al. 2017). Finally, the 20-donor pool mix of human hepatocytes qualified for cell suspension testing were obtained from Lonza (Basel, Switzerland).
2.2. Chemicals:
Terfenadine, Caffeine, LPS, Rosiglitazone, Pioglitazone, Troglitazone, Tolcapone, Trovafloxacin and Mifepristone were all purchased from Millipore Sigma.
2.3. Tissue Chips and Cell Culture Plates:
The microfluidic tissue chips used in this study were purchased from Nortis Bio (SCC-001, Seattle, WA). This PDMS, plastic and glass platform contains a central growth area where cells and ECM are sequentially layered to establish the LAMPS model (Vernetti et al. 2016). Black-walled, clear-bottom, tissue culture-treated 96 well plates (3603, Corning) were used for monolayer studies.
2.4. Cell Culture in Tissue Chips:
Device chambers were first coated with a mixed solution of 50 μg/mL fibronectin (F1141, Sigma-Aldrich, St. Louis, MO) and 200 μg/mL collagen I (CB354249, Corning Inc.) in physiologically-buffered saline (PBS), and incubated for 1 hr at room temperature. Once the devices were coated, hepatocytes were injected at a density of 2.75×106 cells/mL (150 μL/chip) in hepatocyte plating media [HPM-primary: 93% William’s E medium (A12176–01, Invitrogen), 5% fetal bovine serum (FBS), 1% pen-strep, 2 mM L-glutamine; or HPM-iPSC: RPMI, B27 (17504044, Invitrogen), 20 ng/mL Oncostatin M (295-OM-010, R&D Systems), 0.1 μM Dexamethasone (ICN19456125, MP Biomedicals, Santa Ana, CA), 25 μg/mL Gentamicin (15750060, Invitrogen), and iCell Hepatocytes 2.0 Medium Supplement (M1024, FujiFilm Cellular Dynamics)] and incubated overnight (37°C, 5% CO2). The following morning, THP-1 (0.8×106 cells/mL ending) and EaHy926 (1.5×106 cells/mL ending) were pooled in 10% DMEM, and injected into devices (150 μL/chip), then incubated for 2 hrs at 37°C. After this incubation period to allow for cell attachment, LX-2 cells were re-suspended in a 2.5 mg/mL solution of pH 7.2 rat tail collagen/10 mM HEPES/HBSS at 0.2×106 cells/mL, and injected into devices (150 μL/chip). At this time, chips were inverted and incubated for 1 hr at 37°C so that LX-2 cells would settle and adhere to the top layer of the chip. After this short incubation, chips were re-inverted and incubated overnight at 37°C.
The next morning, device tubing was prepared as follows: inlet tubing was prepared by cutting 10 inch sections of PEEK tubing (1569XL, IDEX, Lake Forest, IL), and fitting 1 inch of c-flex tubing (06422–00, Cole-Parmer, Vernon Hills, IL) to each end with a catheter blunt (SC 20/15, Instech, Plymouth Meeting, PA) on one end to interface with the inlet of the chip (the other c-flex section would fit over a needle blunt on the media syringe). Outlet tubing was prepared by cutting 2-inch sections of PEEK tubing, and fitting a 1-inch section of c-flex tubing to one end with a catheter blunt to interface with the outlet of the chip. The other end of the PEEK tubing was placed into a glass vial with a pre-slit cap. All tubing was autoclaved and primed with cell culture media prior to chip connection. Hepatocyte maintenance media [HMM-primary: 95% William’s E medium, 4% cocktail B (CM4000, Invitrogen), 100 nM dexamethasone, and 1% FBS; or HMM-iPSC: RPMI, B27, 0.1 μM dexamethasone, 25 μg/mL gentamicin, and iCell Hepatocytes 2.0 medium supplement] was perfused through the use of syringe pumps (Fusion 200, Chemyx, Stafford, TX) at a rate of 15 μL/hr for up to 10 days. Each day, 360 μL of effluent was collected for downstream analyses.
2.5. Cell Culture in Monolayer (96-well Plates):
The 96-well plates were first coated with a mixed solution of 50 μg/mL fibronectin and 200 μg/mL collagen I in PBS, and incubated for 1 hr at room temperature. After this incubation, hepatocytes were plated at a density of 2.75×106 cells/mL (30 μL/well) and incubated overnight (37°C, 5% CO2). The following morning, differentiated THP-1 (0.8×106 cells/mL ending) and EaHy926 (1.5×106 cells/mL ending) were pooled, and plated (30 μL/well) on top of the hepatocyte layer, then incubated for 2 hrs at 37°C. After this incubation period to allow for cell attachment, LX-2 cells were re-suspended in HMM at 0.2×106 cells/mL, and plated over top of the other cell types in the plate (30 μL/well). Plates were then incubated overnight at 37°C. The next morning, media was removed, and 175 μL of fresh HMM was added to the wells. Media was collected and exchanged daily.
2.6. Drug Treatments in Tissue Chips and in Monolayer Cultures:
Chemical treatments were performed on cells that were grown in plates or devices (15 μL/hour flow rate) for up to 10 days. Treatments were added to syringes (3D cultures) or cell culture media (2D cultures) on the first day of perfusion (d0). Culture media containing vehicle (0.1% DMSO), 10 μM Terfenadine, 600 μM Caffeine, 150 μM Trovafloxacin, 28 μM Troglitazone, 88 μM Tolcapone, 0.8 μM Rosiglitazone, 3 μM Pioglitazone, 1 μg/mL LPS, or a combination of LPS + Trovafloxacin was perfused (3D), or exchanged daily (2D) and effluent samples were collected throughout the exposure period for analytical chemistry and biomarker testing.
2.7. Imaging Assays:
Phase contrast images were collected on the first (d0) and last (d10) day of chip perfusion for both 3D experiments, as well as the parallel 2D 96-well plate experiments. These images were collected using an inverted VWR microscope, fitted with a Leica DFC3000G Monochrome camera at 10× magnification.
2.8. Biochemical Assays:
Urea nitrogen (SB-0580–250, Stanbio, Boerne, TX) and lactate dehydrogenase (LDH; G1780, Promega, Madison, WI) levels were measured from 2D and 3D effluent/media samples following manufacture’s protocols. Cell viability (A13261, PrestoBlue Cell Viability Reagent, Invitrogen) was measured on day 10 of culture. Briefly, a 1x solution of PrestoBlue reagent in HMM was added to syringes and perfused through chips at a rate of 50 μL/hr for 2 hours at 37°C. The perfusion rate in the experiments with PrestoBlue was temporarily increased to enable collection of the sufficient volume of the effluent as we found that the reagents are unstable in the incubator over periods of longer perfusion (data not shown). In 96-well plates, 100 μL of 1X solution of PrestoBlue reagent in HMM was added to each well for 2 hours at 37°C. After incubation, media was collected from chips and wells, and fluorescence was measured at 560/590 nm on a plate reader (SpectraMax iD3, Molecular Devices). Values for all assays were normalized to vehicle (0.1% DMSO).
2.9. Protein Assays:
Daily albumin levels were measured in chip effluents and 2D supernatants using a Human Albumin ELISA Quantitation Set (E80–129, Bethyl Labs, Montgomery, TX) following manufacturer’s protocols. Samples were diluted 1:100 in HMM prior to testing. TNF-α was measured in chip effluents and 2D supernatants on days 1, 2, 4, 6, and 10 using a TNF alpha Human ELISA Kit (KHC3011, Invitrogen) following manufacturer’s protocols.
2.10. Determination of free fraction of drugs in cell medium:
Binding of drugs to proteins in cell culture media was evaluated using a rapid equilibrium dialysis (RED) assay (89809 and 89811, Thermo Scientific, Rockford, IL) according to the manufacturer’s protocol. Nonspecific binding of each drug in the RED experiment was further assessed by incorporating protein-free controls (i.e., PBS buffer) in sample chambers. Twenty microliters of working stock solution of each drug (100 μM) was spiked in 180 μL of cell medium or PBS buffer to reach a final concentration of 10 μMin sample chambers. After 4-hour incubation at 37°C, 50 μL of cell medium or PBS buffer sample was mixed with an equal volume of PBS buffer or cell medium, respectively, to reduce the matrix effect. All samples were spiked with 10 μL of Mifepristone (1 μM), and then protein precipitation was performed with 200 μL of chilled acetonitrile followed by centrifugation at 10,000×g for 10 mins. Two hundred microliters of the supernatant was transferred to a 2 mL tube, dried under vacuum, and reconstituted with 100 μL of distilled water for subsequent LC-MS/MS analyses. Free fraction of a drug was calculated by comparing the response ratios of analytes and Mifepristone (internal standard, IS) between PBS buffer and medium buffer chamber, based on the following formula:
2.11. Determination of drug concentrations using liquid chromatography (LC) - tandem mass spectrometry (MS/MS) analyses:
Samples (10 μL) were chromatographed on a ZORBAX SSHD Eclipse Plus C18 column (3×50 mm, 1.8 μm, 959757–302; Agilent, Santa Clara, CA) with a guard column (2.1×5 mm, 1.8 μm, 821725–901; Agilent) using 1290 Infinity II LC (Agilent). Column temperature and the LC flow rate were set at 40°C and 0.4 ml/min. Initial chromatographic condition was maintained at 90% mobile phase A (water with 0.1% formic acid, v/v) and 10% mobile phase B (acetonitrile with 0.1% formic acid, v/v) for one min, then increased to 80% B by 3 min, then to 95% B by 4 min, and then returned to initial condition at 5 min until 8 min for sufficient equilibrium. Unmodified mobile phases A (water) and B (acetonitrile) were used for the analysis of Troglitazone.
MS/MS analyses were performed in positive ion mode (all test drugs except Troglitazone) or negative ion mode (Troglitazone) with an electrospray ionization (ESI) source using 6470 triple-quadruple MS (Agilent). The capillary voltage was set at 3500 V. The nebulizer gas pressure and gas temperature were set at 35 psi and 350°C, respectively. The MS/MS parameters for each test compound are summarized in Supplemental Table 1.
2.12. Terfenadine Metabolism Studies:
Terfenadine was administered to LAMPS models constructed in the Nortis microfluidic device, to 2D monolayer cultures, to the static LAMPS model constructed in a 96-well microtiter plate, and to hepatocyte suspensions as a 10 μM (1% DMSO) solution in HMM media for primary hepatocytes or in proprietary CDI maintenance media for the iPSC-derived hepatocytes. Media efflux was collected daily for 10 days in the constant infusion models for testing. Media was collected and replenished with fresh 10 μM Terfenadine media daily for 9 or 10 days in metabolism experiments conducted in the 2D monolayer and 3D LAMPS models in the 96 well microtiter plate. All media samples were prepared for mass spectroscopy analysis by adding a 2× volume of acetonitrile (depending on the sample volume) followed by vigorous vortexing for 30 seconds. The media/acetonitrile solutions were centrifuged in an Eppendorf centrifuge for 2.5 minutes at 15,000 rpm to pellet the insoluble material. A 20 μl aliquot of the supernatant was added to 180 μL of acetonitrile/H2O (20/80 v/v) solution and submitted to a mass spectroscopy facility. For experiments using hepatocyte suspensions, 35,000 primary hepatocytes or iPSC hepatocytes were suspended in 50 μL of HMM or CDI proprietary media supplemented with 10 μM Terfenadine. Metabolic activity for the T0 minute time point was immediately stopped by the addition of 2× acetonitrile followed by vortexing. Additional tubes were capped and placed on a rocking platform at 37°C. Metabolic activity was stopped in tubes at 10, 30, 60 or 120 minutes by adding a 2× volume of acetonitrile followed by vortexing. The final sample preparations for mass spectroscopy measurements were as described above. Terfenadine was measured using a Waters Acquity UPLC (Milford, MA) equipped with a C18, 1.7 μm, 2.1×100 mm reversed-phase column. Detection and quantification were performed in the positive ion mode with a TSQ Quantum Ultra Mass Spectrometer (ThermoFisher Scientific), interfaced via an electrospray ionization (ESI) probe.
2.13. Terfenadine Intrinsic Clearance Measurements:
The PBPK analytics module in the Microphysiology Systems database (MPS-Db, mps.csb.pitt.edu) (Schurdak et al. 2020) was used to calculate the intrinsic clearance (CLint) of Terfenadine when tested as a bolus dose in cell suspension, under continuous perfusion in the LAMPS device or constructed in a 96 well microtiter plate as a static suspension.
2.14. Bolus Calculation:
Terfenadine CLint was calculated from 0–2 hours when incubated in a suspension of CDI iPSC derived hepatocytes or a 20 pool mix primary hepatocytes. The clearance calculation in the bolus condition requires t 1/2 calculation:
where t1/2 is the half-life of Terfenadine, Conc. t0 is the measured concentration of Terfenadine at time = 0, and Conc. t is the measured concentration Terfenadine at two hours. CLint is then calculated as:
In these experiments, the volume of incubation (Vinc) = 50 μl, the number of cells exposed to the compound (NDcells) = 35,000, the estimated as number of hepatocytes in one gram of human liver (Ncells) = 130 × 106, and the typical mass of a human liver (g liver) = 1400 g.
2.15. Steady State Calculation:
Terfenadine clearance in microfluidic and 96-well microtiter plate LAMPS models constructed from CDI iHeps or single donor primary hepatocytes was calculated from 0–10 days under constant infusion when the compound achieved steady state, i.e., when Terfenadine concentration in the efflux did not vary with time. The clearance calculation at steady state requires extraction ratio (ER):
which is used to calculate the intrinsic clearance as follows:
Where the flow rate Q = 15 μl/hour, the number of cells exposed to the compound (NDcells) = 80,000, the estimated as number of hepatocytes in one gram of human liver (Ncells) = 130×106, and the typical mass of a human liver = 1,400 g. CLint is reported as mL/min, which is the volume of blood cleared of measurable Terfenadine in one minute.
2.16. Reproducibility Measurements:
Reproducibility measurements were calculated using the Reproducibility Analytics module found in the Microphysiology Systems database (MPS-Db, https://mps.csb.pitt.edu/). The method for determining inter- and intra-laboratory study reproducibility was previously published (Schurdak et al. 2020). Briefly, single time point experiment reproducibility is calculated from coefficient of variation (CV) and multiple time point experiments are calculated by Max CV or intra-class correlation coefficient (ICC). Max CV is the maximum of CVs for all time points. The intra-class correlation coefficient (ICC) is a measure of agreement in magnitude and trend among data replicates in sequential data (e.g. time or concentration sequences) (Shoukri et al. 2008). The criteria to classify reproducibility is as follows. Single time point: CV≤5% is “Excellent”; 5%<CV≤15% is “Acceptable”; CV >15% is “Poor”. Multiple time points: Max CV≤5% or ICC≥0.8 is ‘Excellent’; 5%<Max CV<15%, or 0.2≤ ICC<0.8 is “Acceptable”; and ICC<0.2 is “Poor”.
2.17. Statistical analyses:
Statistical significance (p<0.05 was selected as a threshold) was tested with either pairwise t-test or one-way ANOVA followed by Dunnett’s multiple comparisons test as indicated in Figure and Table legends.
2.18. Data availability:
The experimental protocols, raw data for each Figure and Table, and analysis tools including calculations used to determine inter- and intra-study reproducibility and clearance can be found in MPS-Db (https://mps.csb.pitt.edu/) using links included in Supplemental Table 2.
3. Results
The adoption of tissue chip technology into basic research and drug or chemical safety evaluation requires rigorous testing to build confidence in the performance of these models (Baudy et al. 2020; Sakolish et al. 2018). Therefore, the major goals of this study were to (i) determine the degree of intra- and inter-laboratory reproducibility of a complex microfluidic four-cell LAMPS, (ii) compare performance of LAMPS seeded with human primary hepatocytes to that seeded with iPSC-derived hepatocytes, and (iii) evaluate performance of LAMPS as compared to traditional monolayer cultures of human primary or iPSC-derived hepatocytes.
To accomplish these goals, we conducted a series of experiments (Figure 1A) in two laboratories that included a comparison of LAMPS and monolayer cultures, as well as different hepatocyte cell sources (primary vs iPSC-derived). The overall dataset from these studies allowed for quantitative assessment of the reproducibility, reliability, robustness, and relevance of the LAMPS tissue chips. First, we evaluated intra-laboratory reproducibility of the experiments (Figure1B). The pie charts provide a visual summary representation of intra-study reproducibility metrics between replicate chips or wells for each assay from 8 separate single and multiple time point studies in Laboratory 1 for 2D vs 3D models and primary hepatocytes vs iPSC hepatocytes; Laboratory 2 reproducibility is presented for 2 separate multiple time point studies in 3D LAMPS with primary hepatocytes. Supplemental Figure 1 shows the reproducibility for each assay type from each laboratory. For example, reproducibility of PrestoBlue assay in 13 – 60 MPS devices is shown for 2D and 3D, and for both primary or iPSC derived hepatocytes (Laboratory 1 only). Inter-study reproducibility is shown for multiple time points of 4 assays between Laboratory 1 and 2 in 3D primary hepatocytes. Intra-study reproducibility in most cases is dominated by acceptable (yellow) and excellent (green) correlation within studies and between cells, tissue organization (2D vs 3D) and testing laboratories. The Flowrate had poor (red) reproducibility in some studies (see Discussion).
Figure 1.
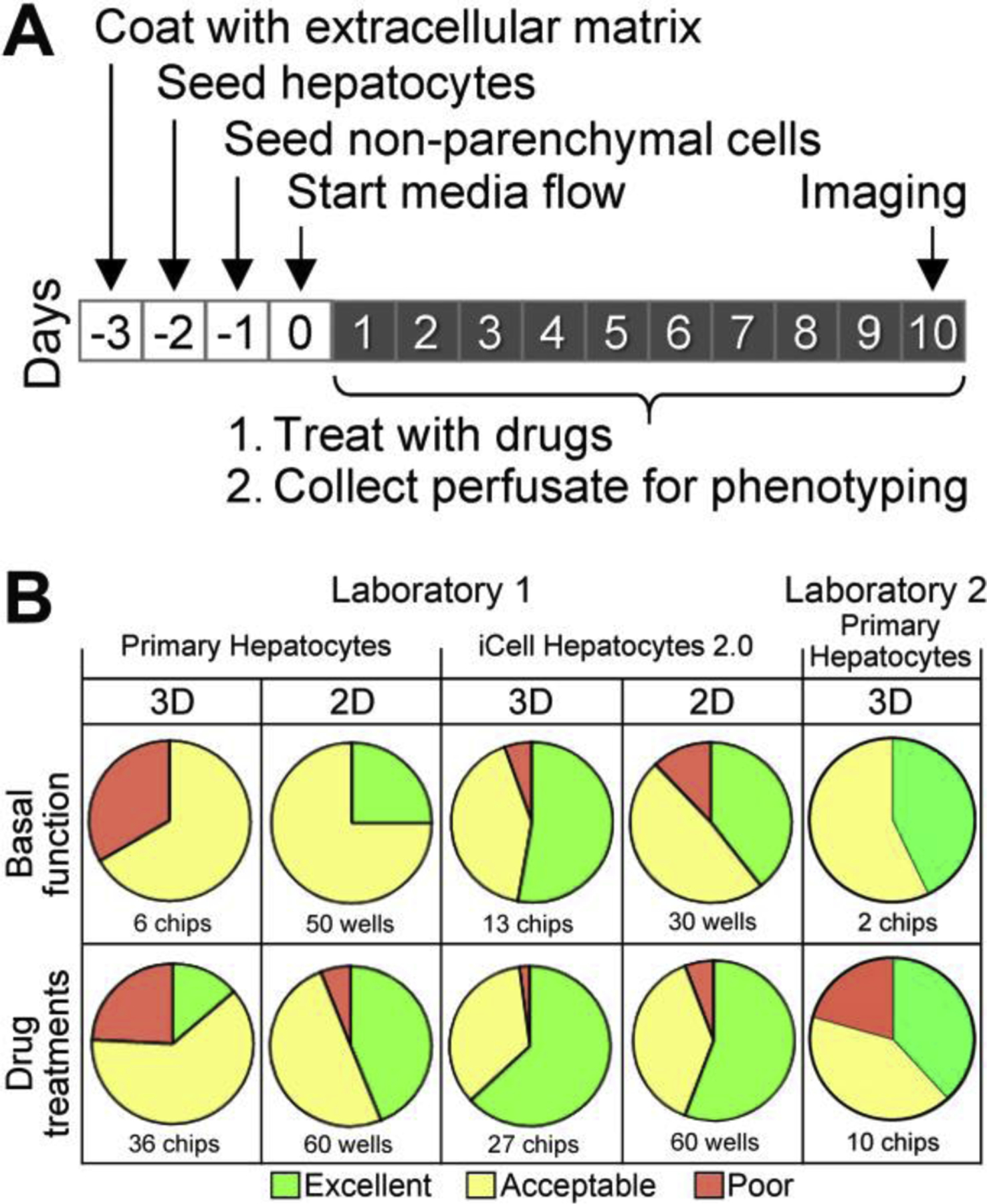
Experimental design and study reproducibility. (A) Schematic of the overall experimental workflow for LAMPS experiments. Tissue chips were prepared over 4 days by coating with extracellular matrix, sequential seeding with four cell types and testing of the media flow through the assembled device. (B) Pie charts showing study reproducibility for each type of the experiment (laboratory, cell type, or study type: 2D=hepatocyte monolayer cultures, 3D=LAMPS chips). Colors depict overall study reproducibility score (green=excellent, yellow=acceptable, red=poor). See details of each experiment and study reproducibility for each parameter in Supplemental Figure 1.
3.1. Intra- and Inter-Laboratory Testing of LAMPS
A single lot of single donor primary human hepatocytes was procured by both laboratories and the experimental protocols and overall study design were coordinated (Figure 1A). First, we conducted a series of experiments to test the basal function of, and drug effects on, the primary human hepatocytes in LAMPS in two separate laboratories. LAMPS tissue chips were seeded with 4 cell types as detailed in Methods and cultured without treatments for 10 days (Figure 2A). In both laboratories, over the course of the experiment the device perfusate contained stable levels of albumin (~15–20 μg/day/million hepatocytes) and urea (~60–70 μg/day/million hepatocytes). Because secretion of albumin and urea varies in different lots of primary hepatocytes, we used the same lot of hepatocytes in both laboratories for this comparison. The amount of albumin secreted was about 2-fold higher than in a previous LAMPS study using a different lot of primary human hepatocytes (Vernetti et al. 2016), about the same as in the human primary hepatocyte seeded Hepatopac® micro-patterned device (Kang et al. 2020), and about 2–5 fold lower than human liver in vivo estimates (Baudy et al. 2020). The amount of urea secreted was also about 2-fold higher than the previous LAMPS study (Vernetti et al. 2016), about 2.5-fold lower than in the primary human hepatocyte-seeded Hepatopac® micro-patterned device (Kang et al. 2020), and in the lower range of human liver in vivo estimates (Baudy et al. 2020). LDH, initially high from the thawing of primary cells, returned to basal levels over 24 hours. LDH release was generally low in both laboratories, and either stayed low (Laboratory 2) or steadily increased after day 5 (Laboratory 1) over the course of the study. While in this particular experiment basal LDH values in Laboratory 1 were above those in Laboratory 2, in subsequent experiments in laboratory 1 (see links to the raw data for all studies in Supplemental Table 2) LDH baseline levels were 0.3–0.5 μg/day/million, and those in drug-treated experiments were ~2.5–3 μg/day/million. Clear increase in LDH after drug treatments can be deduced. TNFα release from LAMPS decreased after day 1 (Laboratories 1&2) and remained low over the course of the study (Laboratory 1).
Figure 2.
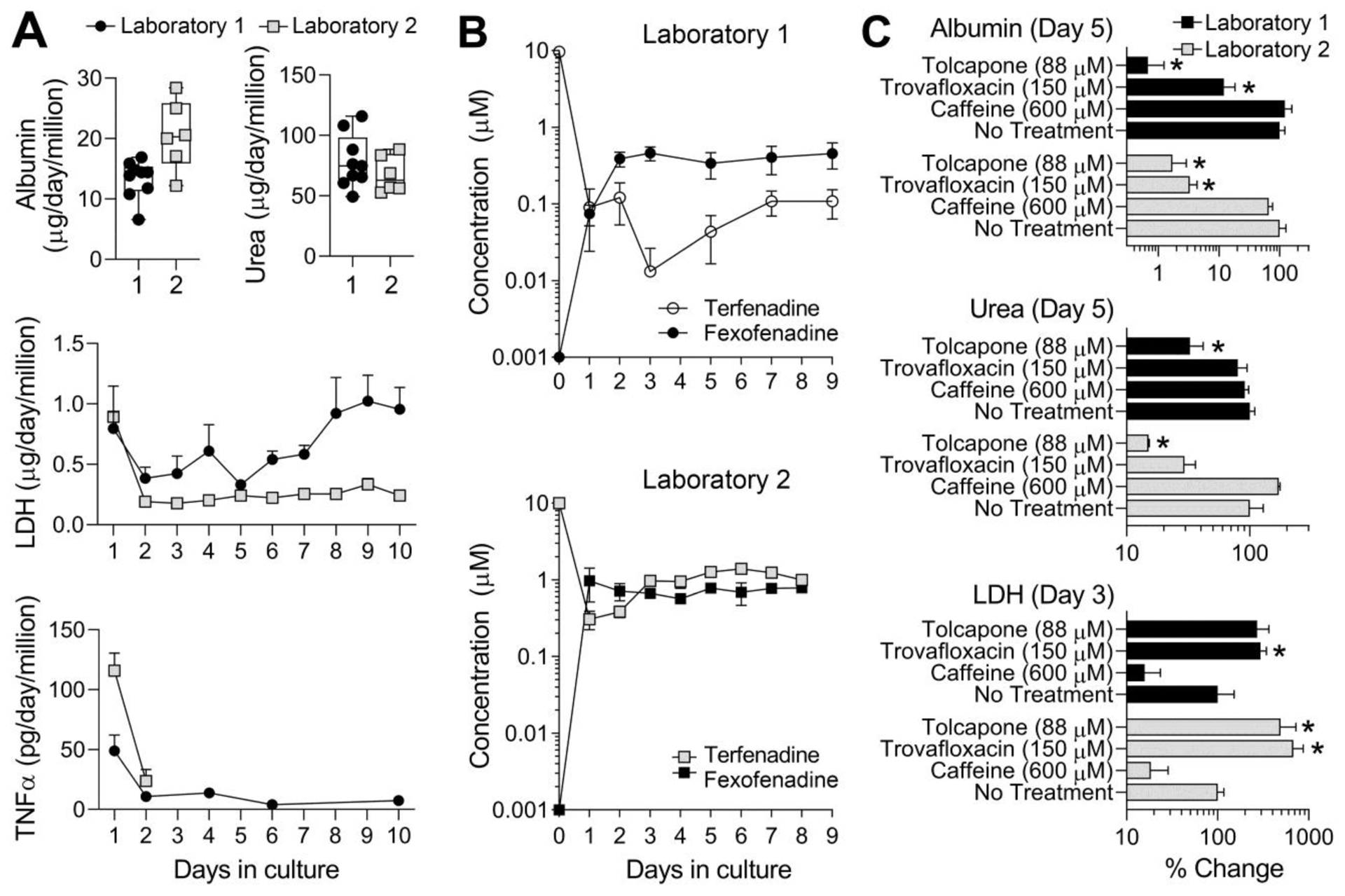
LAMPS reproducibility with respect to the (A) functional performance, (B) metabolic capacity, and (C) drug-induced effects across different laboratories. Experiments were performed in parallel in two laboratories (1=circles and 2=squares) independently, but using the same protocols and the same batch of primary human hepatocytes. For albumin in panel A, daily (days 3–9) average values (from 4–6 chips) are shown as individual symbols. Box and whiskers plots show the median (line), interquartile range (box), and minimal and maximal values (whiskers). All other data shown are mean±SEM (n=3–8 per condition). In panel B, “day 0” drug concentrations are either infused concentration (for Terfenadine) or the limit of quantitation (for Fexofenadine). Other day’s concentrations are based on the instrumental analysis of each drug’s concentration in the effluent. In panel C, laboratory 1 data are black bars and laboratory 2 data are gray bars. All data were normalized to controls (“No Treatment”) at the same day as the treatments (days indicated for each phenotype). Asterisks (*) indicate statistically significant (p<0.05) difference from “No treatment” group within each center using one-way ANOVA with Dunnett’s multiple comparisons test.
Next, we examined the metabolic capacity of the primary human hepatocytes in LAMPS over time (Figure 2B). For this, Terfenadine (10 μM) was infused into the chips and the concentrations of Terfenadine and its metabolite through CYP3A4 oxidation pathway, Fexofenadine (Ling et al. 1995), were measured in the effluent. In both laboratories, about 100-fold reduction in the concentration of Terfenadine and concurrent increase in the formation of Fexofenadine were observed over 1–2 days. Metabolism of Terfenadine to Fexofenadine was stable over the remaining course of the experiment (9 days for Laboratory 1 and 8 days for Laboratory 2).
In addition, we sought to test the effects of two drugs that were withdrawn from the market due to hepatotoxicity, and a substance that is a negative control. For the former, we used the idiosyncratic liver toxicants Trovafloxacin (Senior 2014; Waring et al. 2006) and Tolcapone (Watkins 2000); for the latter, we used caffeine (Alferink et al. 2018). Tolcapone was tested at 40, 88 and 220 μM in a 14-day LAMPS study at Laboratory 2. Figure 2C shows that in both laboratories, Tolcapone exposure for 5 days at 88 μM resulted in a statistically significant decrease in albumin and urea production. LDH elevation (2–4 fold) was observed on day 3 in both laboratories, but was only statistically significant in Laboratory 2 studies. Treatment with 150 μM Trovafloxacin (Schurdak et al. 2020) produced significant effects on albumin and LDH in both laboratories. In a previous study in Laboratory 2, Caffeine, tested at 600 μM and different donor hepatocytes (Vernetti et al. 2016), was without significant effect on albumin and urea and reduced LDH levels. These compound effects were similar between laboratories.
3.2. Comparison of basal liver function parameters in LAMPS seeded with human primary hepatocytes or human iPSC-derived hepatocytes
It has been suggested that reproducibility and scalability of in vitro organ models can be improved through the use of human iPSC-derived cells (D’Costa et al. 2020). To compare performance of LAMPS seeded with primary human hepatocytes or commercially-available and widely used iCell human hepatocytes (Sirenko et al. 2014), we conducted a series of experiments in either traditional 2D monolayer cultures, or in LAMPS chips. First, we conducted morphological and functional characterization of both cell types. Supplemental Figure 2 shows representative transmitted light images of primary human hepatocytes and iCell hepatocytes after 1 and 10 days of culture in LAMPS (the images of all 2D and 3D cultures can be accessed through the MPS-Db, see Supplemental Table 2 for access information). While the imaging of cells in LAMPS is more challenging because of the reduced optical access through the device and presence of other cell types, binucleation and bile canaliculi are still clearly evident in all images, with day 10 images showing an improved morphological organization of the cell layer.
Next, we evaluated basic function and metabolic capacity of LAMPS with human primary and iPSC-derived hepatocytes, and as compared to the 2D monolayer cultures (Figure 3). In the experiments with primary human hepatocytes (Figure 3A), albumin and urea production were 2–5-fold greater in LAMPS as compared to 2D monolayer cultures, while no differences were seen in LDH levels and TNFα release. In cells treated with Terfenadine at non-toxic dose (10 μM), these parameters were largely indistinguishable from untreated conditions. In the experiments with human iCell hepatocytes (Figure 3B), we observed a similar improvement in functionality (high albumin and urea, low LDH and TNFα) in LAMPS as compared to 2D monolayer cultures. Treatment with Terfenadine led to consistent reduction in production of albumin and urea, but was without effect on LDH and TNFα. The functional ability of iCell hepatocytes, as well as primary hepatocytes, appeared to have improved over time in LAMPS, as evidenced from a steady increase in production of albumin and urea, concordant with the observations that fluidic microenvironment improves functional maturity of iPSC-derived hepatocytes (Danoy et al. 2019).
Figure 3.

Basic function and drug metabolism in monolayer cultures (circles) or LAMPS chips (squares) that were seeded with (A) primary hepatocytes, or (B) iCell hepatocytes 2.0. Amounts of albumin, urea, LDH and TNFα were evaluated daily in the static cell culture media or perfusate collected from the devices. Control wells/chips (filled symbols) or wells/chips treated with daily additions with Terfenadine (10 μM, open symbols) were used in these experiments. (C) Drug metabolism capacity was evaluated by monitoring concentrations through addition of Terfenadine (10 μM, open symbols) to cell culture media or perfusate and formation of Fexofenadine (filled diamonds). Cell type and testing conditions are indicated in each chart. Data shown are mean±SEM (n=3–9 per condition).
Figure 3C shows that the metabolic function of human primary hepatocytes, as determined by the metabolism of Terfenadine to Fexofenadine, was sustained over time (up to 9 days) in LAMPS, as compared to 2D monolayer cultures where CYP3A4 function was completely lost by day 3 and Fexofenadine release into the media ceased by day 5. Figure 3D shows the same outcome of the experiments with iCell hepatocytes, albeit the amount of Fexofenadine formed in LAMPS was about 10-fold less than that in experiments with primary human hepatocytes. In agreement with the data on albumin, Fexofenadine formation also gradually increased over time in LAMPS seeded with iCell hepatocytes, further suggesting a gradual functional maturation under flow. We used the PBPK analytics module in the MPS-Db to compare the intrinsic clearance of Terfenadine in primary human hepatocytes and iPSC-derived human hepatocytes under 10-day continuous perfusion in the 3D LAMPS model, in 48 hr bolus treated 2D monolayer cultures, and in 4 hr bolus treated suspension cultures (Table 1). We found that short-term suspension cultures, a traditional choice for pre-clinical pharmacokinetic studies, yielded the highest values for intrinsic clearance of Terfenadine. Interestingly, both primary human hepatocytes (20 donor pool) and iPSC-derived iCell Hepatocytes 2.0 performed equally well under these conditions indicative of the preserved CYP 3A4 activity. Experiments in the 3D LAMPS and 2D plate models, yielded clearance values about 40% lower than those in suspension cultures. The data obtained with primary human hepatocytes (single donor) or iCell Hepatocytes 2.0 were largely indistinguishable. Finally, it is noted that metabolism of Terfenadine using the iCells cultured as a 2D monolayer decreased after 48 hrs, similar to primary hepatocytes (Figures 3C–D).
Table 1.
Comparison of predicted human Terfenadine clearance in primary human hepatocytes and iPSC derived iCell hepatocytes determined from continuous perfusion over 10 days in the microfluidic 3D LAMPS, 48 hr bolus treatment in static 2D plate culture and 4 hr bolus treatment in suspension culture. All values represent mean ± SD.
| Cell Type | 3D LAMPS (ml/min) | 2D Plate Culture (ml/min) | Suspension Culture (ml/min) |
|---|---|---|---|
| Primary human hepatocytes# | 482 ±7 (n=4) | 351 ± 6 (n=4) | 1,172 ± 175 (n=2) |
| Human iCell Hepatocytes 2.0 | 516 ± 1* (n=3) | 273 ± 10* (n=4) | 771 ± 90 (n=2) |
For 3D LAMPS and 2D plate studies, human hepatocytes from the same batch of a single donor were used. For the 4 hr suspension cultures, human hepatocytes from a pool of 20 donors were used. All models used the same lot of iCell iPSC derived hepatocytes.
Significantly different (p<0.05) from primary human hepatocytes in the same model by unpaired t-test (two-tailed).
3.3. Comparison of drug toxicity and pharmacokinetics in LAMPS seeded with human primary hepatocytes and human iPSC-derived hepatocytes
In these experiments, we aimed to study both short- (days 1 and 3) and long-term (day 10) effects of several test compounds. These included Tolcapone and Trovafloxacin that were tested in inter-laboratory comparison studies. Lipopolysaccharide (LPS) alone or in combination with Trovafloxacin, and the ‘glitazone’ series of compounds (Troglitazone, Rosiglitazone and Pioglitazone) were tested in Laboratory 1. Caffeine was used as a negative control. Figure 4 provides the data for cell viability, albumin and urea production on day 10, and LDH and TNFα release on days 3 and 1, respectively. In 2D cultures with primary hepatocytes, cell viability was the most informative endpoint showing pronounced effects of Tolcapone (88 μM), Trovafloxacin (150 μM) or Trovafloxacin and LPS (1 μg/mL) combination treatment. Data for albumin and urea were less informative and more variable because of very low levels of these functional markers in 2D cultures on day 10. Early effects on LDH and TNFα release were also largely concordant with known in vivo hepatotoxic effects, although there was low consistency among the markers and we did not observe potentiation of the effect of Trovafloxacin by the addition of LPS (Waring et al. 2006). In 2D cultures with iCell hepatocytes, similar effects were observed, with cell viability and TNFα release being the most concordant with expected changes; however, production of albumin, but not urea, also followed the expected trends.
Figure 4.
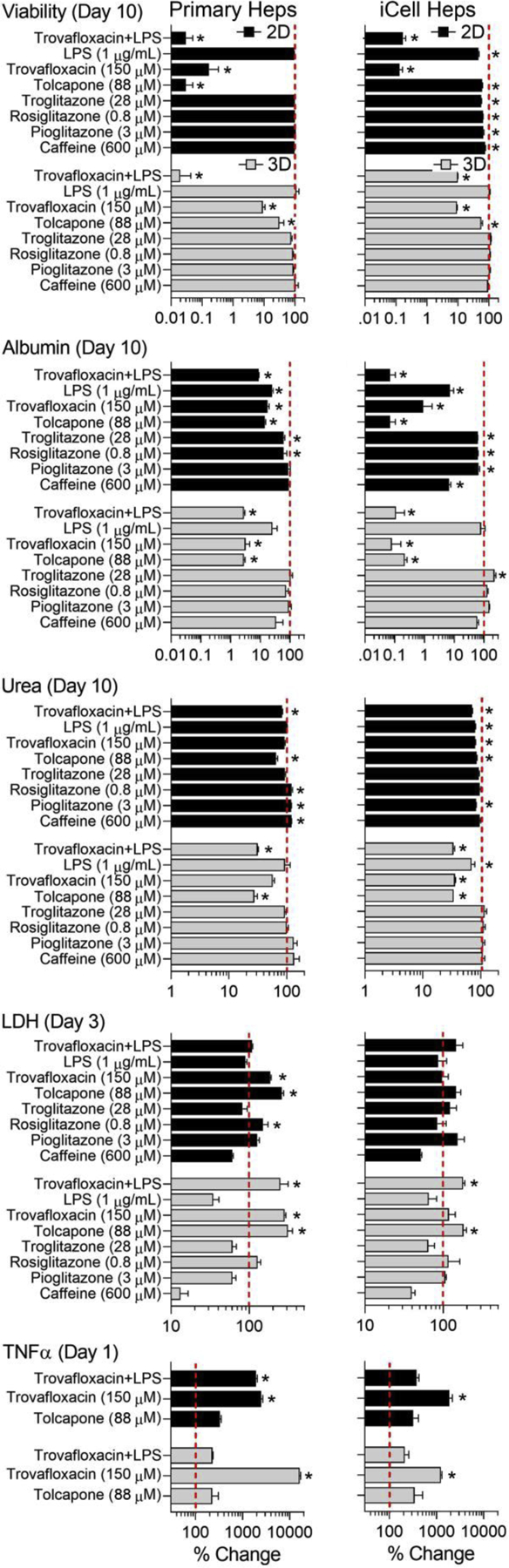
Effects of various drugs (see y-axis legends for concentrations) on viability and functional parameters of human primary hepatocytes (left panel) or iCell hepatocytes 2.0 (right panel) cultured in monolayer monolayers (black bars) or in LAMPS chips (gray bars). All data are normalized to controls (vertical dotted line=100%) at the same day as the treatments (days indicated for each phenotype). Data shown are mean±SEM (n=3–9 per condition). Asterisks (*) indicate statistically significant (p<0.05) difference from control groups (not shown) within each cell type and culture condition (2D and 3D) using one-way ANOVA with Dunnett’s multiple comparisons test.
In LAMPS chips, drug effects on all parameters were greatly concordant between primary human hepatocytes and iCell hepatocytes. Long-term effects on the viability, albumin and urea were consistent across all markers and between cell types. The short-term effects on LDH and TNFα release were also concordant between the two cell types. Overall, drug effects in primary human hepatocytes were more pronounced than in iCell hepatocytes, except for urea at day 10, where iCell hepatocytes responded in a similar manner. It should be noted, however, that this experiment did not reveal the expected difference among the ‘glitazone’ drugs at the tested concentrations, which were selected to be mostly representative of the human Cmax levels (Supplemental Table 3).
We also conducted a series of experiments to evaluate drug recovery from the chips and 2D cultures and drug binding to cell culture media proteins in both primary and iCell hepatocyte media preparations (Figure 5). Rapid equilibrium dialysis was used to characterize free fraction of each drug in the primary hepatocyte or iCell hepatocyte culture media formulations. The bioavailability of all compounds was comparable between media formulations (this experiment was necessary because iCell media formulation is proprietary) and was dependent on the LogP of the tested compounds, as expected. Low LogP substances (Trovafloxacin and Caffeine) were essentially 100% free, while the highest LogP compound in this test, Troglitazone, was largely bound with a free fraction of about 1%.
Figure 5.
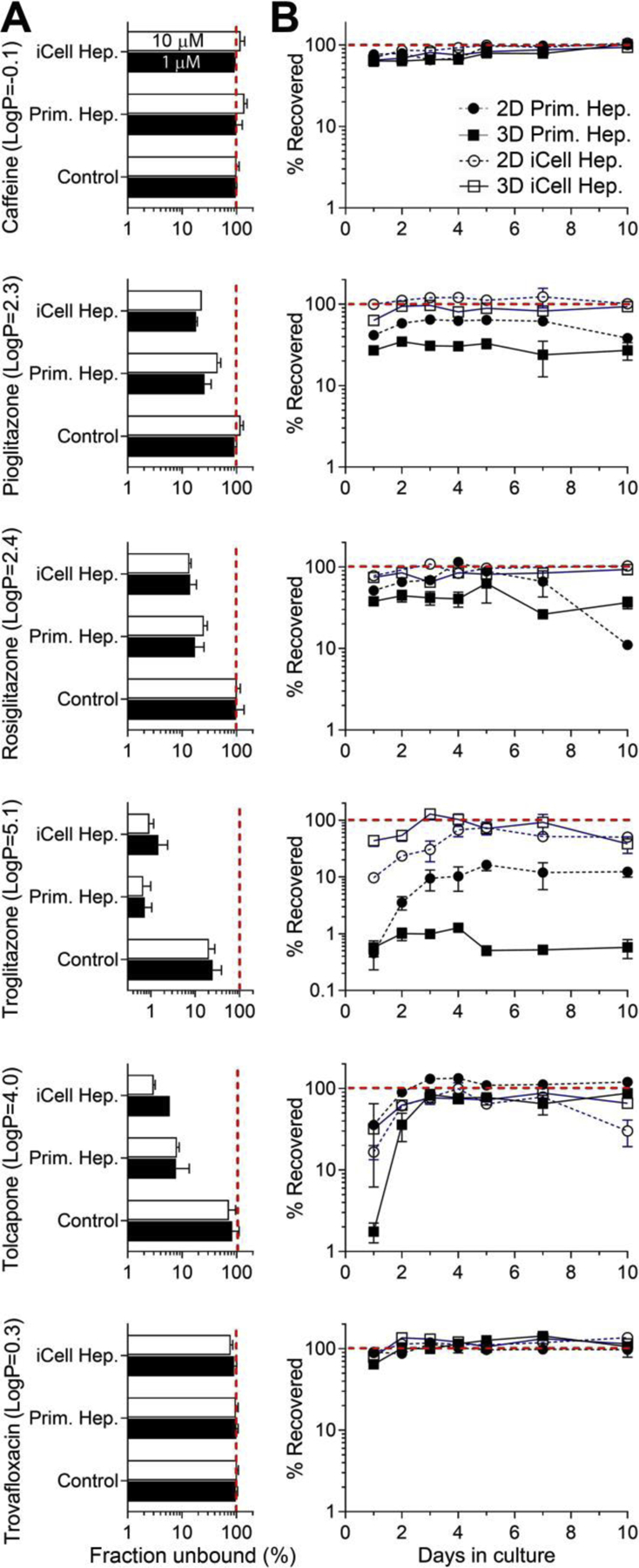
Pharmacokinetic parameters for tested drugs. (A) Fraction unbound for each drug was determined using rapid equilibrium dialysis method and is shown for the physiological saline (Control) of cell-specific culture media (for primary hepatocytes or iCell hepatocytes 2.0). Each drug was tested at 1 μM (black bars) and 10 μM (white bars). (B) The amount of drug recovered from either monolayer culture (circles) or perfusate through LAMPS chips (squares) seeded with primary hepatocytes (filled symbols) or iCell hepatocytes 2.0 (open symbols) and cultured for up to 10 days. Data shown are mean±SEM (n=3–9 per condition) and were normalized to starting concentration (red dotted line=100%).
Next, we tested nonspecific binding of drugs in 2D cultures, or in LAMPS chips, seeded with either primary or iCell hepatocytes. The concentration of each drug in the effluent (for LAMPS chips) or in cell culture media after a period of time is a function of both non-specific binding and metabolism. Therefore, the results of these experiments cannot be interpreted solely in the context of the potential non-specific binding to the polydimethylsiloxane. We found that caffeine and Trovafloxacin, both low LogP compounds, were recoverable at close to 100% of test concentration over this 10-day experiment, regardless of the hepatocyte type or culture conditions.
Other tested compounds (Tolcapone, Pioglitazone, Rosiglitazone and Troglitazone) showed nearly 100% recovery only in experiments with iCell hepatocytes; lower recoveries (10–40%) were observed only in the first 3–5 days and there were differences between LAMPS chips and in 2D monolayer cultures. Experiments with primary human hepatocytes demonstrated that LAMPS chips had lower recovery than 2D monolayer cultures. This effect was most pronounced for Troglitazone and Pioglitazone. For Troglitazone, 2D monolayer-cultured primary human hepatocytes showed similar low recovery (<1%) to the LAMPS chips only on day 1, recovery gradually increased (to over 10%) over time in culture.
4. Discussion
This study used a liver-on-chip model comprised of a co-culture of human hepatocytes and non-parenchymal cells that were exposed to regulated fluidic flow (Vernetti et al. 2016). This configuration of an in vitro liver model has been shown to enhance hepatocyte function because it includes microfluidic media transport and non-parenchymal cells (Vernetti et al. 2017b; Vernetti et al. 2016). LAMPS is one type of MPS for liver studies in vitro; strengths and weaknesses of various categories of liver MPS with respect to different “fit for purpose or “context of use”” scenarios for biomedical research or drug/chemical safety evaluation have been recently reviewed (Baudy et al. 2020; Taylor et al. 2019). Much attention in the field of biomedical engineering of liver models is devoted to demonstration of the physiological relevance of various MPS and establishing their performance with industry-relevant compounds (Sistare et al. 2016; Soldatow et al. 2013). As MPS are transferred from research laboratories to prospective end-users, additional important consideration is given to the models that are robust and reasonably easy to use, where both cells and devices are readily available, and for which the performance and reproducibility of the MPS have been established (Ewart et al. 2017; Marx et al. 2020; Sakolish et al. 2018). Accordingly, this study focused on demonstrating the application of LAMPS to drug safety assessment, and aimed to address a number of the following practical questions. How well does LAMPS perform in different laboratories when the same cells and similar experimental protocols are used? What is the performance of LAMPS with iPSC-derived human hepatocytes as compared to primary human hepatocytes? And finally, what are the advantages of LAMPS as compared to 2D monolayer cultures of hepatocytes?
The technology transfer has been a general critical bottleneck in the process of adoption of MPS by the end-users (Ewart et al. 2017; Low and Tagle 2017; Sakolish et al. 2018). The complexity of some of the MPS, the need for specialized equipment, and the unique technical skills needed to familiarize the end-users with the experimental procedures create challenges for adoption (Marx et al. 2020). At the same time, most prospective end-users posit that independent review and testing of the complex in vitro models is needed to establish confidence in their performance (Leite et al. 2018). Therefore, this study’s focus on providing quantitative and qualitative data on (i) intra-lab technical reproducibility for each model and phenotype (Figure 1B and Supplemental Figure 1), (ii) inter-lab concordance in both basal hepatocyte function and drug-induced effects (Figure 2), and (iii) intra-lab concordance in the outcomes obtained using primary of iPSC-induced human hepatocytes in 2D and 3D cultures (Figure 3), is very informative. Specifically, we found that function of human hepatocytes was maintained over a 10-day period in LAMPS, but not in monolayer cultures, and was comparable in the experiments conducted by two independent laboratories. Importantly, both albumin production and urea synthesis rates were comparable to those in human liver (Baudy et al. 2020), or to those in other liver MPS such as a micro-patterned co-culture of hepatocytes and fibroblasts (Kang et al. 2020; Khetani and Bhatia 2008). CYP3A4-mediated metabolism and toxic effects of Tolcapone and Trovafloxacin were also replicated in LAMPS that were seeded and maintained in two different laboratories that used the same lot of primary human hepatocytes. Standardization of the experimental protocols and the use of the same cells were necessary to demonstrate the repeatability of the outcomes and instill confidence in the future use of this model in other laboratories. Importantly, all of the raw and processed data and images from these studies in both laboratories are publicly available through a database and data analysis platform (Gough et al. 2016; Schurdak et al. 2020), a step that will further increase confidence in the reliability and replicability of LAMPS.
The use of iPSC-derived hepatocytes as an alternative to primary human hepatocytes or liver-derived cell lines is increasing in both research laboratories and by the industry (Sistare et al. 2016). The long-term consistent supply of liver parenchymal and non-parenchymal cells in general, as well as the ability to study a specific patient population or disease, the ability to induce gene mutations, and the ability to produce isogenic liver cell types are the most attractive features of iPSC-derived hepatocytes. Several studies have compared stem cell-derived hepatocytes to other cell types as models for hepatotoxicity, either as monolayer cultures or spheroids (Bell et al. 2017; Sirenko et al. 2014). These studies used primary human hepatocytes as a “bar” for the comparison with iPSC-derived hepatocytes. They concluded that iPSC-derived hepatocytes have important limitations, primarily in their immature xenobiotic metabolism phenotype. More recent studies showed that culture of iPSC-derived hepatocytes under fluidic conditions does improve their maturity (Danoy et al. 2019). Indeed, our study demonstrated that performance of iPSC-derived human hepatocytes improves under microfluidic conditions and in presence of non-parenchymal cells. This was evident in both their basal function and in drug-mediated toxicity, indicating great promise in the utility of these cells, because they are readily available from commercial suppliers, are tightly quality controlled during differentiation steps, and are consistent in their genetic makeup. Still, these cells are somewhat inferior in terms of their basal function and drug metabolism, and their routine use in combination with MPS may need additional consideration such as testing of higher concentrations of drugs and detailed analytical characterization of the major metabolites. The small perfusion volumes and enclosed configuration of the chip make the traditional endpoints of gene expression or fluorescence-based metabolism more challenging.
Another comparison that is often not included in the publications of new MPS data is a comparison with a conventional, non-overlay 2D cell culture models. While it may be difficult to recapitulate the complex cell-cell interactions of the MPS in multi-well plate-configured monolayer cultures, the latter are not only more familiar to the end-users, they are also far less technically challenging and higher in throughput. A direct comparison of the 2D and 3D models, including same cell source, drugs and preferably concurrent execution, is usually most convincing to the prospective end-users with respect to understanding the advantages and limitations of the more complex MPS. Several previous studies have shown that the advantages of the MPS over traditional 2D cultures are nuanced and that the “context of use” is of great importance. For example, our experiments found 2D and 3D models were approximately equivalent in discriminating hepatotoxicity of Trovafloxacin and Tolcapone from Caffeine and 3 glitazone-family drugs using 5 measured viability and functional phenotypes. However, the drug metabolism experiments with Terfenadine show a clear advantage in the 3D constant flow through LAMPS compared to 2D cultures with periodic media changes, consistent with improved mRNA levels of the major drug metabolizing genes and higher cytochrome P450 activity levels in hepatocytes maintained in 3D microfluidic cultures compared to static 2D cultures (Vivares et al. 2015). It is important to note, that the small number of compounds tested, limited to one or two concentrations is too limited of a dataset to make definitive conclusions on value of 3D MPS over traditional 2D models for toxicity and drug metabolism comparison. Such comparisons will require testing larger compound sets and a wider range of concentrations.
In our comparison of 2D and 3D models with primary and iPSC-derived hepatocytes, we found Trovafloxacin ± LPS and Tolcapone, used at about 30× and 9× Cmax blood levels, respectively, produced significant toxicity in 2D and 3D models for most of the viability and functional phenotypes. These findings are concordant with clinical reports of liver-related adverse events (Supplemental Figure 3). Of particular interest is our finding that Trovafloxacin did not induce higher levels of TNFα release on Day 1 than when combined with LPS (Waring et al. 2006). As it is standard practice for the in vitro immune-mediated liver toxicity studies to include LPS to release TNFα from the typical innate immune cells, including primary Kupffer cells or differentiated macrophages from THP-1 or U937 monoblast cell lines (Lee-Montiel et al. 2017; Sarkar et al. 2017; Vernetti et al. 2016), this finding warrants additional assessment.
Three antihyperglycemic drugs were chosen for testing included Troglitazone, Rosiglitazone and Pioglitazone screened at 10×, 1×, and 1× Cmax levels, respectively. Based on reported clinical liver associated adverse events (Supplemental Figure 3), we anticipated toxicity ranking to follow that in vivo: Troglitazone>>Rosiglitazone>>Pioglitazone. The Troglitazone tested at 10× Cmax level had previously been reported to induce toxicity in the LAMPS model using a different donor lot of primary human hepatocytes and U-937 differentiated Kupffer-like macrophages (Vernetti et al. 2016). However, none of the viability and function indicators discriminated hepatotoxicity of the glitazone-family drugs in these experiments. This result is not, however, surprising for opportunistic DILI compounds for which clinical hepatotoxicity occurs is highly patient-dependent. The results serve to underscore why preclinical in vitro toxicity studies typically expand the testing range up to 100× of expected Cmax (Godoy et al. 2013). Indeed, when tested at 30× Cmax in the LAMPS model with primary human hepatocytes, Troglitazone induced acute, significant hepatotoxicity in <5 days compared to significant toxicity at ≥10 days with Rosiglitazone (Schurdak et al. 2020; Vernetti et al. 2016).
PBPK analysis of compounds is one of the key optimization tools used by drug discovery programs to help select drug candidates for clinical evaluation. The use of suspension cultures of primary human hepatocytes is a widely-used in vitro approach to predicting in vivo human liver clearance (Lancett et al. 2018) even though clearance is only part of the data necessary for estimating human PK. However, the decline of phase 1 and phase 2 metabolic activity of hepatocytes in suspension limit the assay to 2–4 hours, which is insufficient to study PK of low clearance compounds. The low clearance compounds are typically favored because they improve in vivo exposure, half-life and limit the potential for reactive metabolites with possible off-target toxicity liabilities. Thus, alternative in vitro PBPK approaches to the cell suspension designed to prolong metabolic activity are actively pursued. These include a variety of static 2D and 3D models of primary or iPSC-derived hepatocytes, hepatocytes co-cultured with additional cell types and more recently, the inclusion of continuous flow through, multi-cellular microphysiological models of the liver.
Another important consideration is that the use of iPSC-derived hepatocytes and non-parenchymal liver cell types is widely anticipated to surpass the use of donor-isolated primary cells. The advantages of iPSC-derived cells are numerous: (i) renewable supply, (ii) the ability to create isogenic multiple cell types, (iii) application of gene editing tools to knock in/knock out polymorphisms to induce disease or DILI susceptibility, and (iv) 28 day culture longevity reported for iPSC derived hepatocytes compared to <15 days for primary hepatocytes (Godoy et al. 2013; Walker et al. 2020; Wills and Rajagopalan 2020). However, at the current state of iPSC technology, iPSC-derived hepatocytes do not yet possess all of the terminally differentiated functions of primary hepatocytes. The iCell Hepatocytes 2.0 were evaluated as an alternative for primary hepatocytes in the LAMPS model. We found the CYP 3A4 activity in iPSC-derived hepatocytes produced intrinsic clearance of Terfenadine equal to that of primary hepatocytes in both LAMPS model under constant infusion over 10 days, and in short-term (4 hrs) suspension culture model (Table 1). These data provide additional evidence for the utility of iPSC-derived hepatocytes as a reproducible and useful in vitro model for studies of liver toxicity.
In addition, a number of recent publications pointed out that the application of tissue chips to drug safety evaluation needs to take into consideration the potential non-specific binding to PDMS, of which many tissue chips are made (Auner et al. 2019; Vernetti et al. 2017a). Estimation of the free fraction in cell culture is an important part of the overall experimental design that enables more precise extrapolation from in vitro to in vivo (Wetmore 2015). In this regard, the use of iPSC-derived hepatocytes presents unique challenges as the media formulations are often proprietary. This challenge can be overcome by the experiments, as demonstrated in this study. We found, as expected, that higher LogP (>2) compounds were only partially (1–10%) free in cell culture media and that little difference in fraction unbound was found between two media formulations. An even more instructive observation was that recovery of the drugs from the perfused LAMPS cannot be explained by device binding. All of the drugs were recovered in the amounts close to those infused, after protein binding in the cell cultured media was accounted for. The only major differences in recovery were observed between 2D and 3D studies, where in 3D drug recovery was generally lower, indicative of either metabolism or binding. However, the prevalence of metabolism as the cause of the disappearance of the substrate is a more plausible explanation because for several drugs the recovery in the 3D experiments with primary hepatocytes was lower than that in the experiments with iPSC-derived hepatocytes. Therefore, we conclude that device binding to PDMS in LAMPS, or any MPS model containing PDMS, needs to be verified experimentally for each compound.
Supplementary Material
Acknowledgements
These experiments were funded, in part, by the grants from the National Center for Advancing Translational Sciences (University of Pittsburgh: R01 DK001881; U24 TR002632; U24 TR001935; UH2 TR000503; UH3 TR000503; Texas A&M University: U24 TR001950, U24 TR002633), the National Center for Advancing Translational Sciences/National Institute for Diabetes and Digestive and Kidney Disease (University of Pittsburgh: UG3 DK119973) the US Environmental Protection Agency (University of Pittsburgh: RD83573601; Texas A&M University: RD84003201), National Institute of Health (University of Pittsburgh: S10 OD012269) and the University of Pittsburgh Alternatives Research and Development Foundation (ARDF) grant 713390. Courtney Sakolish was supported, in part by a training grant from the National Institute of Environmental Health Sciences (T32 ES026568). The views expressed in this manuscript do not reflect those of the funding agencies. The use of specific commercial products in this work does not constitute endorsement by the funding agencies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors declare no competing interests.
References
- Alferink LJM, Kiefte-de Jong JC and Darwish Murad S (2018) Potential Mechanisms Underlying the Role of Coffee in Liver Health. Semin Liver Dis 38, 193–214. [DOI] [PubMed] [Google Scholar]
- Auner AW, Tasneem KM, Markov DA, McCawley LJ and Hutson MS (2019) Chemical-PDMS binding kinetics and implications for bioavailability in microfluidic devices. Lab Chip 19, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudy AR, Otieno MA, Hewitt P, Gan J, Roth A, Keller D, Sura R, Van Vleet TR and Proctor WR (2020) Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip 20, 215–225. [DOI] [PubMed] [Google Scholar]
- Bell CC, Lauschke VM, Vorrink SU, Palmgren H, Duffin R, Andersson TB and Ingelman-Sundberg M (2017) Transcriptional, Functional, and Mechanistic Comparisons of Stem Cell-Derived Hepatocytes, HepaRG Cells, and Three-Dimensional Human Hepatocyte Spheroids as Predictive In Vitro Systems for Drug-Induced Liver Injury. Drug Metab Dispos 45, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulutoglu B, Rey-Bedon C, Mert S, Tian L, Jang YY, Yarmush ML and Usta OB (2020) A comparison of hepato-cellular in vitro platforms to study CYP3A4 induction. PLoS One 15, e0229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavero I, Guillon JM and Holzgrefe HH (2019) Human organotypic bioconstructs from organ-on-chip devices for human-predictive biological insights on drug candidates. Expert Opin Drug Saf 18, 651–677. [DOI] [PubMed] [Google Scholar]
- D’Costa K, Kosic M, Lam A, Moradipour A, Zhao Y and Radisic M (2020) Biomaterials and Culture Systems for Development of Organoid and Organ-on-a-Chip Models. Ann Biomed Eng 48, 2002–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danoy M, Bernier ML, Kimura K, Poulain S, Kato S, Mori D, Kido T, Plessy C, Kusuhara H, Miyajima A, Sakai Y and Leclerc E (2019) Optimized protocol for the hepatic differentiation of induced pluripotent stem cells in a fluidic microenvironment. Biotechnol Bioeng 116, 1762–1776. [DOI] [PubMed] [Google Scholar]
- Edington CD, Chen WLK, Geishecker E, Kassis T, Soenksen LR, Bhushan BM, Freake D, Kirschner J, Maass C, Tsamandouras N, Valdez J, Cook CD, Parent T, Snyder S, Yu J, Suter E, Shockley M, Velazquez J, Velazquez JJ, Stockdale L, Papps JP, Lee I, Vann N, Gamboa M, LaBarge ME, Zhong Z, Wang X, Boyer LA, Lauffenburger DA, Carrier RL, Communal C, Tannenbaum SR, Stokes CL, Hughes DJ, Rohatgi G, Trumper DL, Cirit M and Griffith LG (2018) Interconnected Microphysiological Systems for Quantitative Biology and Pharmacology Studies. Sci Rep 8, 4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewart L, Fabre K, Chakilam A, Dragan Y, Duignan DB, Eswaraka J, Gan J, Guzzie-Peck P, Otieno M, Jeong CG, Keller DA, de Morais SM, Phillips JA, Proctor W, Sura R, Van Vleet T, Watson D, Will Y, Tagle D and Berridge B (2017) Navigating tissue chips from development to dissemination: A pharmaceutical industry perspective. Exp Biol Med (Maywood) 242, 1579–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Bottger J, Braeuning A, Budinsky RA, Burkhardt B, Cameron NR, Camussi G, Cho CS, Choi YJ, Craig Rowlands J, Dahmen U, Damm G, Dirsch O, Donato MT, Dong J, Dooley S, Drasdo D, Eakins R, Ferreira KS, Fonsato V, Fraczek J, Gebhardt R, Gibson A, Glanemann M, Goldring CE, Gomez-Lechon MJ, Groothuis GM, Gustavsson L, Guyot C, Hallifax D, Hammad S, Hayward A, Haussinger D, Hellerbrand C, Hewitt P, Hoehme S, Holzhutter HG, Houston JB, Hrach J, Ito K, Jaeschke H, Keitel V, Kelm JM, Kevin Park B, Kordes C, Kullak-Ublick GA, LeCluyse EL, Lu P, Luebke-Wheeler J, Lutz A, Maltman DJ, Matz-Soja M, McMullen P, Merfort I, Messner S, Meyer C, Mwinyi J, Naisbitt DJ, Nussler AK, Olinga P, Pampaloni F, Pi J, Pluta L, Przyborski SA, Ramachandran A, Rogiers V, Rowe C, Schelcher C, Schmich K, Schwarz M, Singh B, Stelzer EH, Stieger B, Stober R, Sugiyama Y, Tetta C, Thasler WE, Vanhaecke T, Vinken M, Weiss TS, Widera A, Woods CG, Xu JJ, Yarborough KM and Hengstler JG (2013) Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 87, 1315–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough A, Vernetti L, Bergenthal L, Shun TY and Taylor DL (2016) The Microphysiology Systems Database for Analyzing and Modeling Compound Interactions with Human and Animal Organ Models. Appl In Vitro Toxicol 2, 103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith LG, Wells A and Stolz DB (2014) Engineering liver. Hepatology 60, 1426–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm FA, Iwata Y, Sirenko O, Bittner M and Rusyn I (2015) High-Content Assay Multiplexing for Toxicity Screening in Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Hepatocytes. Assay Drug Dev Technol 13, 529–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guguen-Guillouzo C, Corlu A and Guillouzo A (2010) Stem cell-derived hepatocytes and their use in toxicology. Toxicology 270, 3–9. [DOI] [PubMed] [Google Scholar]
- Hassan S, Sebastian S, Maharjan S, Lesha A, Carpenter AM, Liu X, Xie X, Livermore C, Zhang YS and Zarrinpar A (2020) Liver-on-a-Chip Models of Fatty Liver Disease. Hepatology 71, 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Podtelezhnikov AA, Tanis KQ, Pacchione S, Su M, Bleicher KB, Wang Z, Laws GM, Griffiths TG, Kuhls MC, Chen Q, Knemeyer I, Marsh DJ, Mitra K, Lebron J and Sistare FD (2020) Development and Application of a Transcriptomic Signature of Bioactivation in an Advanced In Vitro Liver Model to Reduce Drug-induced Liver Injury Risk Early in the Pharmaceutical Pipeline. Toxicol Sci. [DOI] [PubMed] [Google Scholar]
- Khetani SR and Bhatia SN (2008) Microscale culture of human liver cells for drug development. Nat Biotechnol. 26, 120–126. [DOI] [PubMed] [Google Scholar]
- Krewski D, Rice JM, Bird M, Milton B, Collins B, Lajoie P, Billard M, Grosse Y, Cogliano VJ, Caldwell JC, Rusyn II, Portier CJ, Melnick RL, Baan RA, Little J and Zielinski JM (2019) Concordance between sites of tumor development in humans and in experimental animals for 111 agents that are carcinogenic to humans. J Toxicol Environ Health B Crit Rev 22, 203–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancett P, Williamson B, Barton P and Riley RJ (2018) Development and Characterization of a Human Hepatocyte Low Intrinsic Clearance Assay for Use in Drug Discovery. Drug Metab Dispos 46, 1169–1178. [DOI] [PubMed] [Google Scholar]
- Lauschke VM, Hendriks DF, Bell CC, Andersson TB and Ingelman-Sundberg M (2016) Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chem Res Toxicol 29, 1936–1955. [DOI] [PubMed] [Google Scholar]
- LeCluyse EL, Witek RP, Andersen ME and Powers MJ (2012) Organotypic liver culture models: meeting current challenges in toxicity testing. Crit Rev Toxicol 42, 501–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Montiel FT, George SM, Gough AH, Sharma AD, Wu J, DeBiasio R, Vernetti LA and Taylor DL (2017) Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp Biol Med (Maywood) 242, 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite SB, Cipriano M, Carpi D, Whelan M and Barroso J (2018) Establishing the Scientific Validity of Complex in vitro Models: A crowdsource of opinions to improve their acceptance. European Commision Joint Research Centre, Ispra, Italy. [Google Scholar]
- Ling KH, Leeson GA, Burmaster SD, Hook RH, Reith MK and Cheng LK (1995) Metabolism of terfenadine associated with CYP3A(4) activity in human hepatic microsomes. Drug Metab Dispos 23, 631–636. [PubMed] [Google Scholar]
- Low LA and Tagle DA (2017) Organs-on-chips: Progress, challenges, and future directions. Exp Biol Med (Maywood) 242, 1573–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx U, Akabane T, Andersson TB, Baker E, Beilmann M, Beken S, Brendler-Schwaab S, Cirit M, David R, Dehne EM, Durieux I, Ewart L, Fitzpatrick SC, Frey O, Fuchs F, Griffith LG, Hamilton GA, Hartung T, Hoeng J, Hogberg H, Hughes DJ, Ingber DE, Iskandar A, Kanamori T, Kojima H, Kuehnl J, Leist M, Li B, Loskill P, Mendrick DL, Neumann T, Pallocca G, Rusyn I, Smirnova L, Steger-Hartmann T, Tagle DA, Tonevitsky A, Tsyb S, Trapecar M, Van de Water B, Van den Eijnden-van Raaij J, Vulto P, Watanabe K, Wolf A, Zhou X and Roth A (2020) Biology-inspired microphysiological systems to advance patient benefit and animal welfare in drug development. ALTEX 37, 365–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticello TM, Jones TW, Dambach DM, Potter DM, Bolt MW, Liu M, Keller DA, Hart TK and Kadambi VJ (2017) Current nonclinical testing paradigm enables safe entry to First-In-Human clinical trials: The IQ consortium nonclinical to clinical translational database. Toxicol Appl Pharmacol 334, 100–109. [DOI] [PubMed] [Google Scholar]
- Oleaga C, Riu A, Rothemund S, Lavado A, McAleer CW, Long CJ, Persaud K, Narasimhan NS, Tran M, Roles J, Carmona-Moran CA, Sasserath T, Elbrecht DH, Kumanchik L, Bridges LR, Martin C, Schnepper MT, Ekman G, Jackson M, Wang YI, Note R, Langer J, Teissier S and Hickman JJ (2018) Investigation of the effect of hepatic metabolism on off-target cardiotoxicity in a multi-organ human-on-a-chip system. Biomaterials 182, 176–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramboer E, Vanhaecke T, Rogiers V and Vinken M (2015) Immortalized Human Hepatic Cell Lines for In Vitro Testing and Research Purposes. Methods Mol Biol 1250, 53–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro AJS, Yang X, Patel V, Madabushi R and Strauss DG (2019) Liver Microphysiological Systems for Predicting and Evaluating Drug Effects. Clin Pharmacol Ther 106, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakolish C, Weber EJ, Kelly EJ, Himmelfarb J, Mouneimne R, Grimm FA, House JS, Wade T, Han A, Chiu WA and Rusyn I (2018) Technology Transfer of the Microphysiological Systems: A Case Study of the Human Proximal Tubule Tissue Chip. Sci Rep 8, 14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar U, Ravindra KC, Large E, Young CL, Rivera-Burgos D, Yu J, Cirit M, Hughes DJ, Wishnok JS, Lauffenburger DA, Griffith LG and Tannenbaum SR (2017) Integrated Assessment of Diclofenac Biotransformation, Pharmacokinetics, and Omics-Based Toxicity in a Three-Dimensional Human Liver-Immunocompetent Coculture System. Drug Metab Dispos 45, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurdak M, Vernetti L, Bergenthal L, Wolter QK, Shun TY, Karcher S, Taylor DL and Gough A (2020) Applications of the microphysiology systems database for experimental ADME-Tox and disease models. Lab Chip 20, 1472–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior JR (2014) Evolution of the Food and Drug Administration approach to liver safety assessment for new drugs: current status and challenges. Drug Saf 37 Suppl 1, S9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah F, Leung L, Barton HA, Will Y, Rodrigues AD, Greene N and Aleo MD (2015) Setting Clinical Exposure Levels of Concern for Drug-Induced Liver Injury (DILI) Using Mechanistic in vitro Assays. Toxicol Sci 147, 500–514. [DOI] [PubMed] [Google Scholar]
- Shoukri MM, Colak D, Kaya N and Donner A (2008) Comparison of two dependent within subject coefficients of variation to evaluate the reproducibility of measurement devices. BMC Med Res Methodol 8, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirenko O, Hesley J, Rusyn I and Cromwell EF (2014) High-content assays for hepatotoxicity using induced pluripotent stem cell-derived cells. Assay Drug Dev Technol 12, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistare FD, Mattes WB and LeCluyse EL (2016) The Promise of New Technologies to Reduce, Refine, or Replace Animal Use while Reducing Risks of Drug Induced Liver Injury in Pharmaceutical Development. ILAR J 57, 186–211. [DOI] [PubMed] [Google Scholar]
- Soldatow VY, Lecluyse EL, Griffith LG and Rusyn I (2013) In vitro models for liver toxicity testing. Toxicol Res (Camb) 2, 23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DL, Gough A, Schurdak ME, Vernetti L, Chennubhotla CS, Lefever D, Pei F, Faeder JR, Lezon TR, Stern AM and Bahar I (2019) Harnessing Human Microphysiology Systems as Key Experimental Models for Quantitative Systems Pharmacology. Handb Exp Pharmacol 260, 327–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill GH and Khetani SR (2018) Bioengineered Liver Models for Drug Testing and Cell Differentiation Studies. Cell Mol Gastroenterol Hepatol 5, 426–439 e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernetti L, Gough A, Baetz N, Blutt S, Broughman JR, Brown JA, Foulke-Abel J, Hasan N, In J, Kelly E, Kovbasnjuk O, Repper J, Senutovitch N, Stabb J, Yeung C, Zachos NC, Donowitz M, Estes M, Himmelfarb J, Truskey G, Wikswo JP and Taylor DL (2017a) Corrigendum: Functional Coupling of Human Microphysiology Systems: Intestine, Liver, Kidney Proximal Tubule, Blood-Brain Barrier and Skeletal Muscle. Sci Rep 7, 44517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernetti L, Gough A, Baetz N, Blutt S, Broughman JR, Brown JA, Foulke-Abel J, Hasan N, In J, Kelly E, Kovbasnjuk O, Repper J, Senutovitch N, Stabb J, Yeung C, Zachos NC, Donowitz M, Estes M, Himmelfarb J, Truskey G, Wikswo JP and Taylor DL (2017b) Functional Coupling of Human Microphysiology Systems: Intestine, Liver, Kidney Proximal Tubule, Blood-Brain Barrier and Skeletal Muscle. Sci Rep 7, 42296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernetti LA, Senutovitch N, Boltz R, DeBiasio R, Shun TY, Gough A and Taylor DL (2016) A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp Biol Med (Maywood) 241, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivares A, Salle-Lefort S, Arabeyre-Fabre C, Ngo R, Penarier G, Bremond M, Moliner P, Gallas JF, Fabre G and Klieber S (2015) Morphological behaviour and metabolic capacity of cryopreserved human primary hepatocytes cultivated in a perfused multiwell device. Xenobiotica 45, 29–44. [DOI] [PubMed] [Google Scholar]
- Walker PA, Ryder S, Lavado A, Dilworth C and Riley RJ (2020) The evolution of strategies to minimise the risk of human drug-induced liver injury (DILI) in drug discovery and development. Arch Toxicol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring JF, Liguori MJ, Luyendyk JP, Maddox JF, Ganey PE, Stachlewitz RF, North C, Blomme EA and Roth RA (2006) Microarray analysis of lipopolysaccharide potentiation of trovafloxacin-induced liver injury in rats suggests a role for proinflammatory chemokines and neutrophils. J Pharmacol Exp Ther 316, 1080–1087. [DOI] [PubMed] [Google Scholar]
- Watkins P (2000) COMT inhibitors and liver toxicity. Neurology 55, S51–52; discussion S53–56. [PubMed] [Google Scholar]
- Weaver RJ, Betts C, Blomme EAG, Gerets HHJ, Gjervig Jensen K, Hewitt PG, Juhila S, Labbe G, Liguori MJ, Mesens N, Ogese MO, Persson M, Snoeys J, Stevens JL, Walker T and Park BK (2017) Test systems in drug discovery for hazard identification and risk assessment of human drug-induced liver injury. Expert Opin Drug Metab Toxicol 13, 767–782. [DOI] [PubMed] [Google Scholar]
- Weaver RJ, Blomme EA, Chadwick AE, Copple IM, Gerets HHJ, Goldring CE, Guillouzo A, Hewitt PG, Ingelman-Sundberg M, Jensen KG, Juhila S, Klingmuller U, Labbe G, Liguori MJ, Lovatt CA, Morgan P, Naisbitt DJ, Pieters RHH, Snoeys J, van de Water B, Williams DP and Park BK (2020) Managing the challenge of drug-induced liver injury: a roadmap for the development and deployment of preclinical predictive models. Nat Rev Drug Discov 19, 131–148. [DOI] [PubMed] [Google Scholar]
- Wetmore BA (2015) Quantitative in vitro-to-in vivo extrapolation in a high-throughput environment. Toxicology 332, 94–101. [DOI] [PubMed] [Google Scholar]
- Wills LR and Rajagopalan P (2020) Advances in Human Induced Pluripotent Stem Cell-Derived Hepatocytes for Use in Toxicity Testing. Ann Biomed Eng 48, 1045–1057. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Glesne D and Huberman E (2003) A human peripheral blood monocyte-derived subset acts as pluripotent stem cells. Proc Natl Acad Sci U S A 100, 2426–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The experimental protocols, raw data for each Figure and Table, and analysis tools including calculations used to determine inter- and intra-study reproducibility and clearance can be found in MPS-Db (https://mps.csb.pitt.edu/) using links included in Supplemental Table 2.