

Abstract
Mantle Cell Lymphoma (MCL) is a non-Hodgkin lymphoma with a median survival rate of five years. Standard treatment with high-dose chemotherapy plus rituximab (anti-CD20 antibody) has extended overall survival although, the disease remains incurable. Histone deacetylases (HDAC) are a family of enzymes that regulate multiple proteins and cellular pathways through post-translational modification. Broad spectrum HDAC inhibitors have shown some therapeutic promise, inducing cell cycle inhibition and apoptosis in leukemia and non-Hodgkin’s lymphoma. However, the therapeutic effects of these broad-spectrum HDAC inhibitors can detrimentally dampen Natural Killer (NK) cell cytotoxicity, reduce NK viability, and downregulate activation receptors important for NK mediated anti-tumor responses. Impairment of NK function in MCL patients during therapy potentially limits therapeutic activity of rituximab. Thus, there is an unmet need to decipher specific roles of individual HDACs in order to preserve and/or enhance NK function, while, directly impairing MCL viability. We investigated the impact of HDAC8 in MCL cell lines. Inhibition or genetic loss of HDAC8 caused MCL cells to undergo apoptosis. In contrast, exposure of primary human NK cells to an HDAC8 inhibitor does not alter viability, receptor expression, or antibody dependent cellular cytotoxicity (ADCC). However, an increase in effector cytokine interferon-gamma (IFNγ) producing NK cells was observed in response to HDAC8 inhibition. Taken together these data suggest that selective HDAC8 inhibitors may simultaneously preserve NK functional activity, while impairing MCL tumor growth, establishing a rationale for future clinical evaluation.
Keywords: Mantle cell lymphoma, Histone deacetylase 8, human Natural Killer cells, interferon-gamma, apoptosis, antibody dependent cellular cytotoxicity
Introduction
Mantle cell lymphoma (MCL) is a subtype of B cell lymphoma, representing about 5–10% of all Non-Hodgkin Lymphoma (NHL) diagnosis in the US, with an overall survival of 4–5 years. Despite progress with current treatments, the disease still remains incurable. The cytogenetic hallmark of MCL is the chromosomal translocation t(11;14)(q13;q32), causing constitutive cyclin D1 gene expression from the immunoglobulin heavy chain locus promoter [1]. Immunotherapeutic treatment using Rituximab (anti-CD20 monoclonal antibody) combined with chemotherapy has shown significant improvements in complete response with up to 87% for newly diagnosed aggressive MCL, while modest responses have been observed with single-agent Rituximab treatment [2–4]. Despite these advances, patients have a high occurrence of relapse with a more aggressive and resistant disease. Thus, there is an unmet need for MCL treatment options. Clinical trials have led to FDA approval of the broad-spectrum histone deacetylase (HDAC) inhibitors; belinostat, vorinostat (SAHA), and romidepsin (FK228) for the treatment of peripheral or cutaneous T cell lymphoma [5–7]. Trichostatin (TSA) and SAHA decrease cyclin D1 expression and upregulate p21 (CDKN1A) expression in lymphomas, an indicator of apoptosis [8, 9]. Valproic acid (VPA) and FK228 increase surface expression of CD20, thought to potentially improve response to Rituximab [10]. However, there are significant toxicity issues with broad-spectrum HDAC inhibitors including: cardiotoxicities, anemia, leucopenia, lymphopenia, neutropenia, and adverse neurological and gastrointestinal effects [11, 12]. Broad-spectrum inhibitors also have a negative effect on immune cell responses, critical for response to antibody therapy such as Rituximab. TSA, sodium butyrate, and VPA are detrimental to Natural Killer Cell (NK) viability and cytolytic responses as they cause downregulation of the activation receptors NKG2D, NKp44, NKp46, and CD25 [13, 14]. Furthermore, SAHA and TSA can stimulate production of immunosuppressive Foxp3+ T-regulatory cells [15]. There are eleven HDAC proteins, thus targeting select HDACs may provide an opportunity to maintain therapeutic benefit while avoiding confounding adverse effects. The biological impact of inhibiting selective HDACs in primary NK cells is not well defined. Therefore, we investigated both the effect of selectively inhibiting HDACs in MCL cell lines and their impact on primary human NK cells, as functional NK cells are critical for response to Rituximab treatment.
Materials and Methods
Cell culture and inhibitors
MCL cell lines were obtained from ATCC. The HDAC8 inhibitor, PCI-34051 (#S2012, Selleckchem) was solubilized in DMSO. Primary Natural Killer cells were isolated from human peripheral blood mononuclear cells using the EasySep Human NK Cell Enrichment kit (STEMCELL Technologies Inc.). NK purity was verified by flow cytometry, and routinely found to be >95% CD3−/CD56+. Isolated NK cells were grown in Stem Cell Growth media (CellGenix) supplemented with 10% FBS and 1% penicillin-streptomycin. PCI-34051 or vehicle treatment was for 1 hour followed by stimulation with interleukin 2 (IL2) (100U/ml; PeproTech), interleukin 12 (IL12) (20ng/ml; PeproTech), and interleukin 15 (IL15) (50ng/ml; PeproTech). Lymphokine activated killer cells were generated per manufactures protocol (#130-094-483, Miltenyl Biotec) and expanded for 9 days with 500U/mL IL2. After expansion, cells were either rested in low dose IL2 (100U/ml) or high dose IL2 (500U/ml) for 24 hours prior to treatment.
Real-time PCR, Immunoblotting, and Viability assays
Samples were prepared and analyzed as previously described [16]. Primers pairs were generously provided by Dr. Edward Seto (George Washington University) (Supplemental Table 1). Immunoblotting antibodies are detailed in (Supplemental Table 2) Viability was measured using the CellTiter-Glo assay (Promega) using a standard luminometer. Apoptosis was measured using the Celigo Cytometer (Nexcelom Biosciences) after labeling with 5μM Incucyte® Caspase-3/7 Reagent per 1×10^4 cells according to the manufacturer’s protocol (Essen Biosciences).
Lenti-viral shRNA constructs
shRNA knockdown was performed using MISSION® TRC pLKO.5-puro HDAC8shRNA (HDAC8sh-1, TRC0000350469; HDAC8sh-2, TRC0000314872) or non-targeting control (NTsh; SHC202; Sigma Aldrich). mCherry-tagged shRNA vectors were generated by replacing the puromycin resistance gene with the mCherry gene from the pLVmCherry vector (Addgene). Lenti-virus particles were produced and transduced as previously reported [17].
Flow cytometry
All antibodies used are provided in Supplemental Table 2. Cell death was assessed by staining with Annexin V and propidium iodide (#556463) (BD Biosciences). NK receptor staining was done per manufacturer protocol and Zombie NIR fixable dye was used to distinguish live cell populations, (#423106, BioLegend). For accurate gating, fluorescence minus one (FMO) controls were used. Experimental standardization was performed using, Rainbow Fluorescent Particles, 3.0–3.4μm (mid-range FL1 fluorescence) (Sphere, BD Biosciences). For compensation; cells were used for live/dead and BD compensation beads for all antibodies were used at the optimal titration (#552843, BD Biosciences). Intracellular IFNγ was assessed by permeabilizing the cell membrane using BD cytoperm/cytofix buffer, followed by staining with anti-IFNγ. Flow cytometric detection was performed using the LSRII (BD Biosciences, San Jose, CA) and analyzed using FlowJo V10 cytometry software.
ELISA
Primary human NK cells were treated with DMSO or PCI-34051 for 1 hour, followed by stimulation with IL2, IL12, and IL15, supernatant was collected at 24 hours post inhibitor treatment and secretion of IFNγ was measured using the human IFN gamma ELISA Ready-Set-Go!® kit per the manufacturer’s instructions (#88–7316, Affymetrix eBiosciences). ELISAs were performed in triplicate.
NK cytotoxicity assays
NK cells were treated with 20mM of PCI-34051 for 1 hour prior to cytokine stimulation. Cells were washed and resuspended in RPMI with 20% FBS, followed by co-culture with 5000 K562 cells or Jeko-1 cells at 1:10 effector to target ratios and cytolytic function assessed at 4 hour post co-culture. For antibody dependent cellular cytotoxicity (ADCC) experiments, Jeko-1 cells were pretreated with 10μg of Rituximab for 1 hour (#A1049, BioVision), washed twice to remove excess Rituximab, and co-cultured with stimulated NK cells for 4 hours. Cr-51 labeling and release was measured as previously described [16].
Statistical analysis
Two-tailed paired t tests were used for statistical analyses; p<0.05 were considered significant.
Results
HDAC8 inhibition induces cell death in B Cell Lymphoma cell lines
MCL cell line dependence on HDAC8 function was assessed in response to the selective HDAC8 inhibitor, PCI-34051. In six MCL lines the EC50 values were 3.4, 4.4, 7.2, 19.2, 20.1, and 26.9μM (Fig. 1A). These concentrations are well below the previously described concentration, 50μM-100μM, needed to increase histone acetylation in Jurkat cells [18]. HDAC8 is similarly expressed in all six MCL cell lines tested (Fig. 1B). Cell death in response to HDAC8 inhibition was measured by Annexin V and propidium iodide staining. Five of the six MCL cell lines showed significant levels of apoptosis at 5μM of PCI-34051 (Fig. 1C). The most resistant cell line, JVM-2, is notable for the absence of SOX11 expression. While approximately 95% of all MCL express SOX11 [19], in vitro genetic knockdown of SOX11 in MCL cell lines increases proliferation and was associated with an aggressive MCL phenotype in vivo [20, 21]. PCI-34051 treatment did not change HDAC8 expression levels (Fig. 1D). Structural maintenance of chromosome 3 (SMC3) is the best characterized HDAC8 substrate [22]. Deacetylation of SMC3 enables proper release from the cohesion complex on the sister chromatid, resulting in correct segregation during division [23]. Failure of this process can lead to DNA damage. The on-target effect of PCI-34051 was confirmed by the observed hyperacetylation of SMC3 and is also indicated by a significant increase in the DNA damage associated phosphorylated Histone-H2AX (γH2AX) occurring within 24–48 hours of PCI-34501 treatment (Fig. 1D). Thus, HDAC8 inhibition causes DNA damage and loss in viability of MCL cell lines.
Figure 1. HDAC8 selective inhibition induces DNA damage and cell death in MCL cell lines.
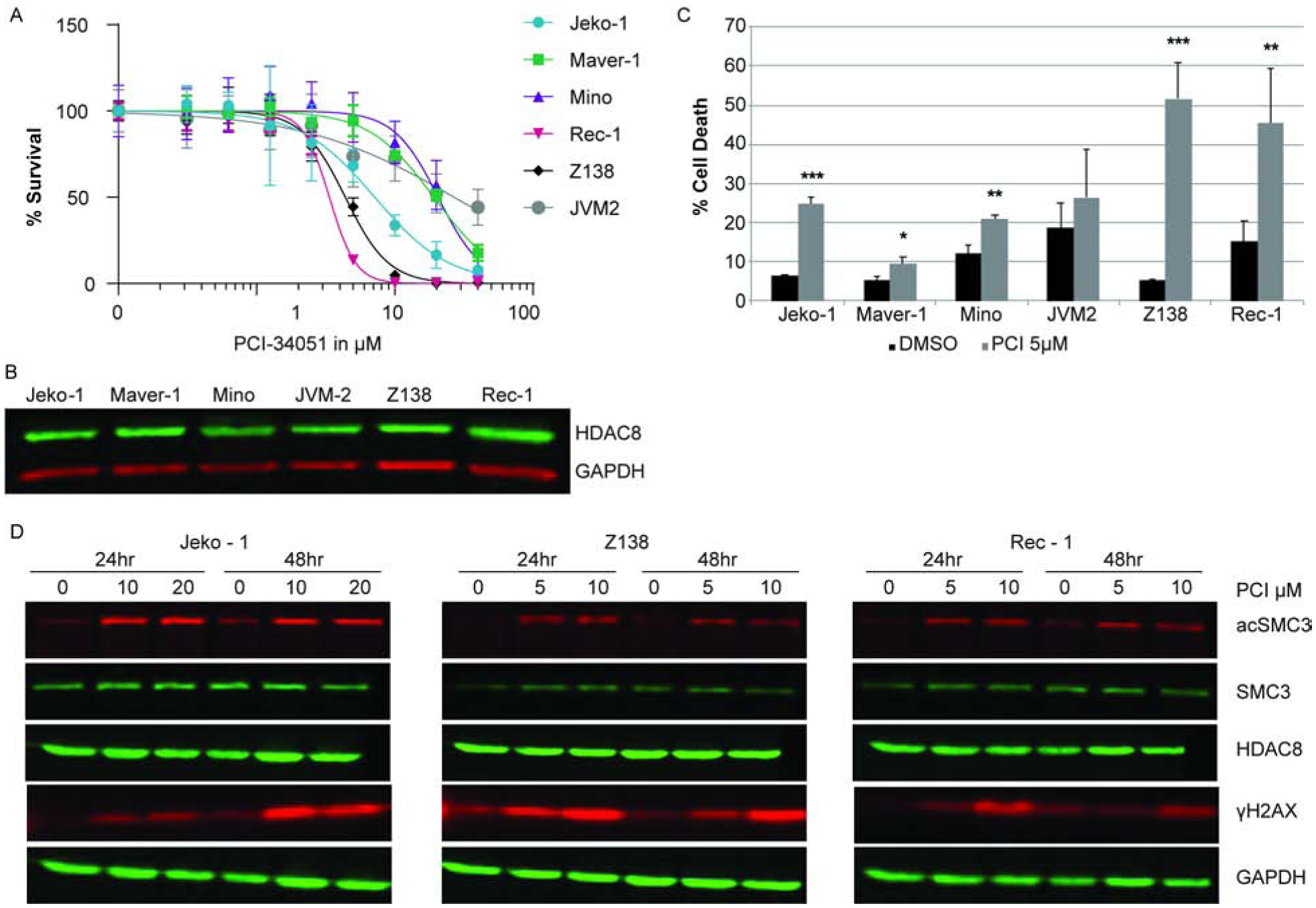
(A) Dose response to HDAC8 inhibition. MCL cell lines were treated with increasing concentrations of PCI-34051 as indicated. Viability was assessed at 72 hours. (B) HDAC8 expression. Immunoblot of HDAC8 expression in MCL cell lines. (C) Cell death during HDAC8 inhibition. Cell death was measured by Annexin V and PI staining. Cells were treated with vehicle (DMSO) or 5μM PCI-34051 at the time of seeding and 24 hours later. Cell death was assessed after 72 hours. (D) Specificity of PCI-34051 to HDAC8 inhibition. Immunoblot analysis of acetylated SMC3, total SMC3, phosphorylated H2AX, HDAC8 expression, and the loading control GAPDH. The MCL cell lines are indicated above each panel and were treated within the EC50 concentration as indicated. Each lane represents 0.5×106 cell equivalent of whole cell lysate. All data represent 3 independent experiments and error bars represent standard deviation. *p<0.05, **p<0.001, *** p<0.0001.
Inhibition or genetic loss of HDAC8 induces caspase-dependent apoptosis
Caspase 3/7 activation was monitored to evaluate the mechanism of cell death during HDAC8 inhibition. PCI-34051 significantly induced caspase-dependent apoptosis at 48 hours in Jeko-1, Z138, and Rec-1 MCL cell lines (Fig. 2A). This was validated by shRNA knockdown of HDAC8 expression which also revealed a significant induction of caspase activity in the absence of HDAC8 expression (Fig. 2B). In conclusion, blocking HDAC8 activity or knockdown of HDAC8 expression leads to caspase-dependent apoptosis in MCL cell lines.
Figure 2. HDAC8 inhibition or genetic loss induces caspase-dependent apoptosis of MCL cell lines.
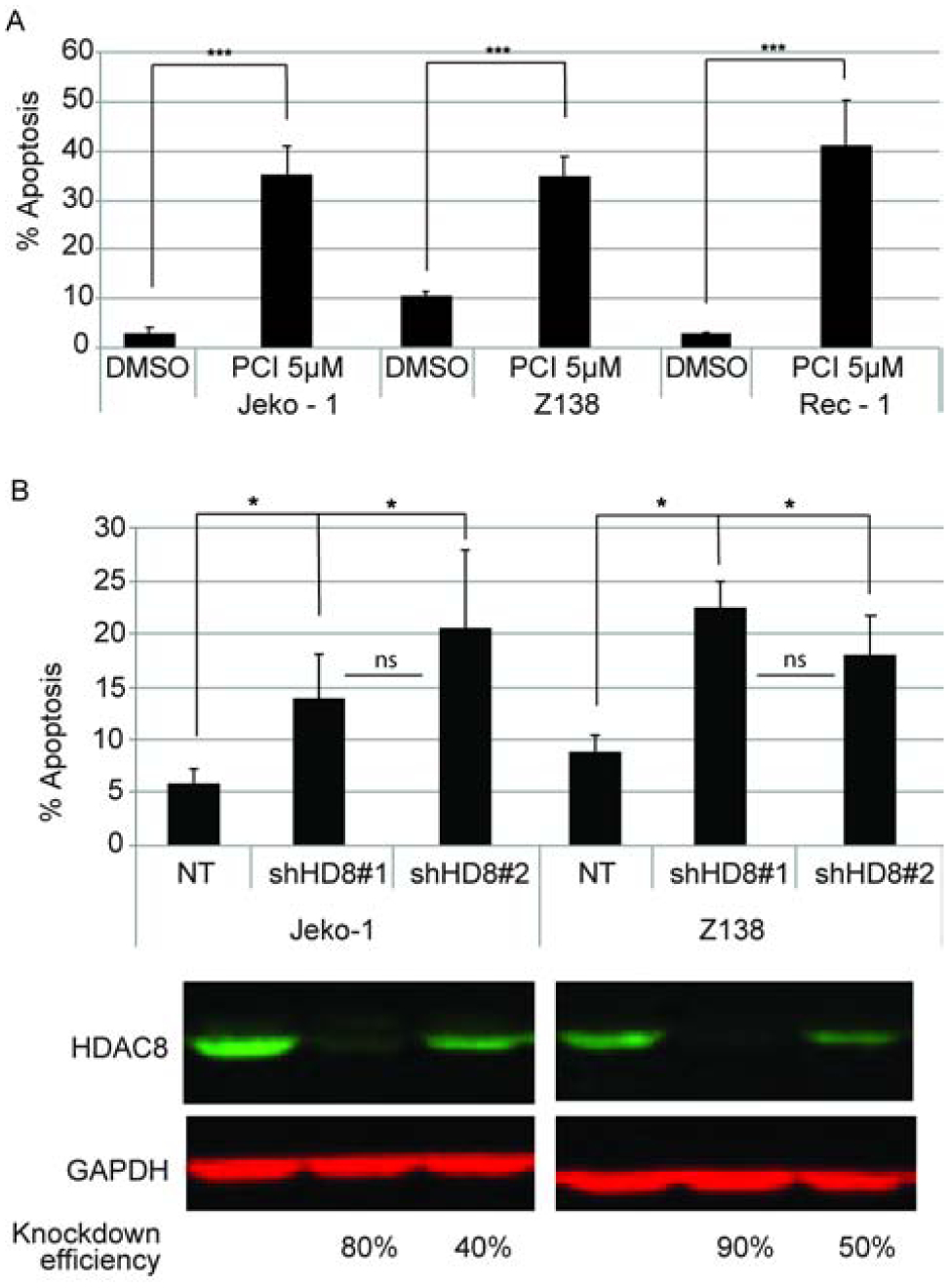
(A) HDAC8 inhibitor induced apoptosis. MCL cells were treated with 5μM PCI-34051 at seeding and 24 hours post seeding and Caspase3/7 activity was measured at 48 hours. (B) Genetic loss of HDAC8 induces apoptosis. Jeko-1 and Z138 cell lines were transduced with either non-targeting shRNA, or shRNAs specific for HDAC8 (#1 or #2). Apoptosis was measured 96 hours post transduction. The efficiency of HDAC8 knockdown is revealed by immunoblotting. Data is representative of 3 independent experiments and error bars represent the standard deviation. p-values (*p<0.05, *** p< 0.0001) represent significance compared to the NT control. The difference between shHD8#1 and shHD8#2 was not significant.
Primary human Natural Killer Cells upregulate HDAC8
Primary NK cells were isolated from peripheral blood of healthy donors by negative selection and cultured in the presence of cytokines [16]. Transcriptional expression of the eleven HDACs was assessed after 24 hours. HDAC1, HDAC2, and HDAC8 are upregulated during stimulation, whereas, HDAC4, HDAC9, and HDAC10 are downregulated (Fig. 3A). At the protein level, primary NK cells display an increase in HDAC8 expression (Fig. 3B). High expression of HDAC8 is also observed in NKL and NK92 cell lines, which represent an activated NK cell phenotype. Viability was determined by a dose response and assessed over 3 days. Human primary NK cell viability is not affected by HDAC8 inhibition, even at the highest concentration of 20μM over 3 days (Fig. 3C). However, the broad-spectrum inhibitors, Vorinostat and Panobinostat (LBH-589), severely impair NK viability at low concentrations, 1 μM and 0.05μM respectively. Lymphokine-activated killer cells (LAKs) were generated to evaluate if HDAC8 inhibition impacts primary NK cell viability during proliferative expansion. Abrogating HDAC8 activity does not impact viability of rested or activated LAKs (Fig. 3D).
Figure 3. HDAC8 is upregulated in stimulated NK cells but not required for viability.
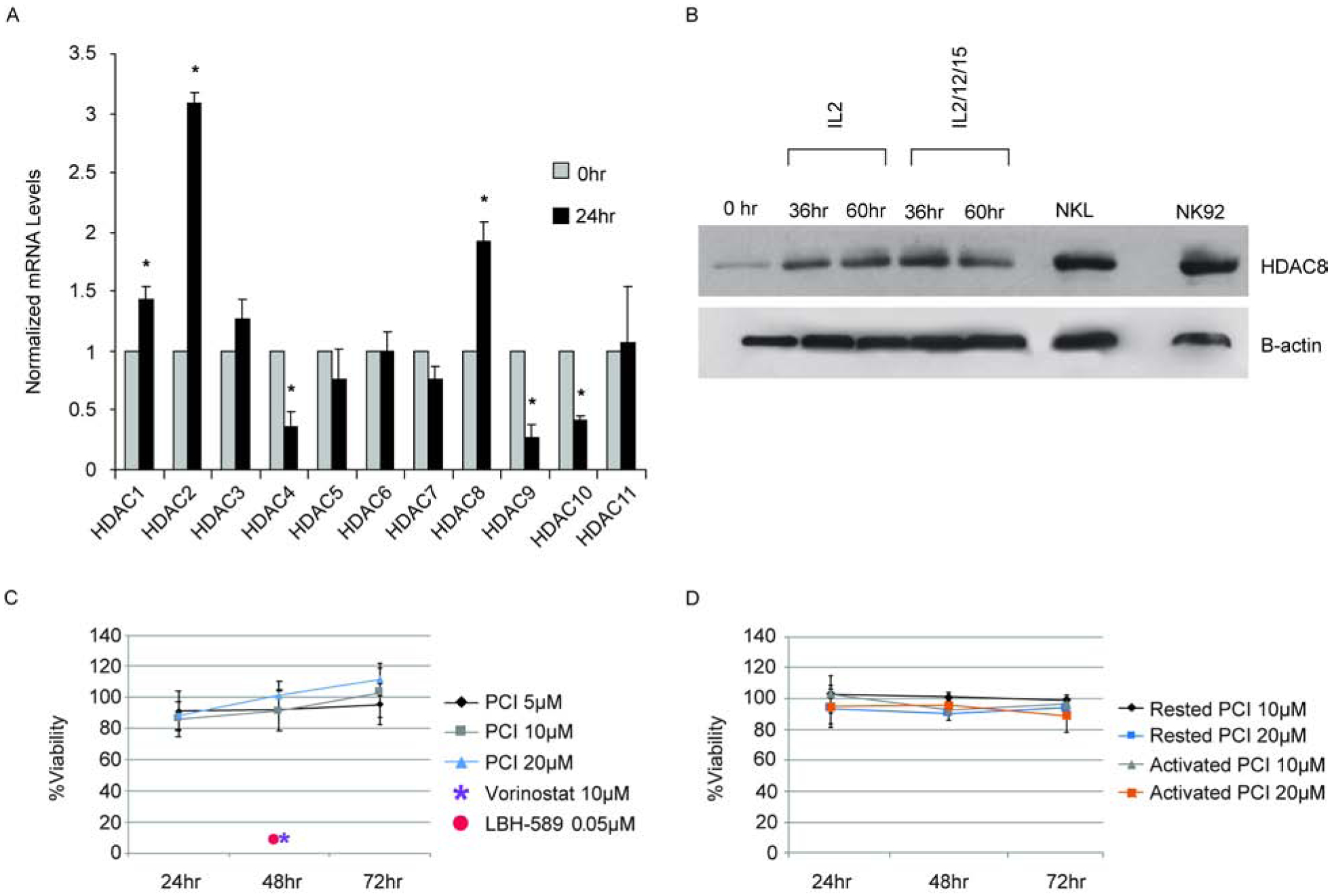
(A) Transcriptional expression of HDACs during cytokine stimulation of primary human NK cells. RNA from freshly isolated human NK cells or NK cells stimulated with IL2, IL12, and IL18 for 24 hours were assessed by RT-qPCR. The data represents the average of 3 independent experiments with the error bars representing the standard deviation. *p<0.05. (B) Immunoblot of HDAC8 protein expression in primary NK and NK cell lines as indicated. (C) Viability of primary NK cells during HDAC8 inhibition. NK cells were treated with PCI-34051, vorinostat, or LBH-589 at the indicated concentration, followed by stimulation with IL2, IL12, and IL15. Viability was assessed by CTG at the times indicated and normalized to the vehicle (DMSO) control. Data represents the average of 3 independent experiments and error bars represent the standard deviation. (D) Viability of ex vivo expanded NK cells during HDAC8 inhibition. Lymphokine activated killer cells were expanded for 9 days, rested (100U/ml IL2) or subsequently reactivated (500U/ml IL2) overnight, followed by exposure to PCI-34051 at the indicated concentration. Viability was assessed as in panel C. Data represents the average of 2 independent experiments with the error bars representing the standard deviation.
HDAC8 inhibition preserves NK functional responses
NK cell anti-tumor responses are governed by a balance of inhibitory and activation receptor signaling [24]. Activation receptors include NKG2C, NKG2D, NKp30, NKp44 and NKp46. NKG2A and KIR2DL3 are inhibitory receptors, while CD94 is required for both NKG2C and NKG2A signaling. The low affinity Fc binding receptor, CD16 (FcγRIIIa), is fundamental for NK antibody-dependent cellular cytotoxicity (ADCC). Tumor targets coated with antibodies are recognized by NK cells through the CD16 receptor. CD16 activation polarizes NK cells and initiates the release of perforin and granzymes, inducing death of the target cell. A multicolor flow cytometry panel was used to assess these key receptors in response to HDAC8 inhibition. The expression levels remained unchanged compared to the vehicle control (Fig. 4A). Furthermore, the percentage of cells expressing these receptors were not altered by HDAC8 inhibition (Fig. 4B)
Figure 4. Blocking HDAC8 activity preserves NK phenotype and cytolytic function.
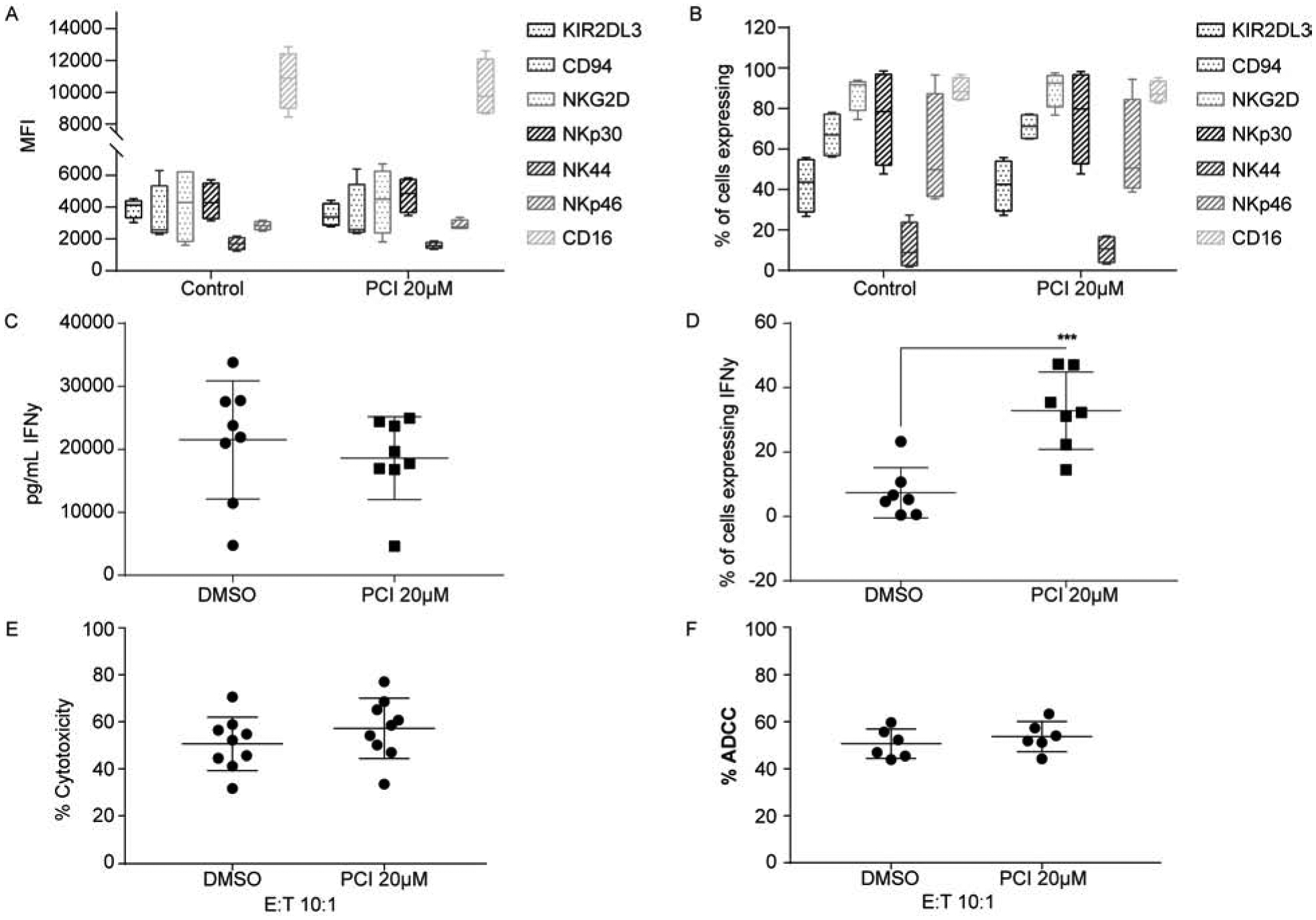
Analysis of NK receptor expression as median fluorescence intensity (MFI) (A) and the percentage of expressing cells (B) was assessed 24hours post PCI-34051 treatment. Human primary NK cells were isolated from PBMCs and treated with PCI-34051 for 1 hour prior to cytokine stimulation for 24 hours. Data represents four independent experiments with standard deviation indicated. (C) IFNγ secretion in response to HDAC8 inhibition. NK cells were treated as in panels A and B, and IFNγ was assessed by ELISA. Eight independent experiments are shown with the average and standard deviation indicated. (D) Percentage of IFNγ positive NK cells assessed by intracellular IFNγ flow cytometry. Cells were treated as indicated above. Each of the individual independent experiments are shown with the average and standard deviation indicated. *** p<0.0001 (E) NK cytolytic activity after HDAC8 inhibitor treatment. NK cells were co-cultured with K562 cells at an effector:target (E:T) ratio of 10:1. Each of the individual independent experiments are shown with the average and standard deviation indicated. (F) NK ADCC cytolytic activity. NK cells were co-cultured at an E:T ratio of 10:1 with Jeko-1 cells precoated with 10μg of Rituximab. Each of the individual independent experiments are shown with the average and standard deviation indicated.
IFNγ is a key effector molecule secreted by NK cells and is capable of increasing DC and T cell functional anti-tumor responses [25, 26]. To determine if HDAC8 inhibition impacts secretion of IFNγ in primary NK cells, cells were treated with PCI-34051 for 24 hours and secretion was assessed. No change in IFNγ secretion was observed in response to PCI-34051 (Fig. 4C). However, there was a significant increase in the percentage of IFNγ producing NK cells (Fig. 4D). We next assessed changes in NK cytolytic activity against MHC-I negative target cells, K562, and against an MCL cell line precoated with 10μg Rituximab antibody. Inhibiting HDAC8 in NK cells does not interfere with their ability to elicit either direct cytolytic activity or antibody-dependent cellular cytotoxicity (ADCC) responses (Fig. 4E and 4F). These results indicate that HDAC8 inhibition preserves NK viability, phenotype, and increases IFNγ producing cells.
Discussion
In this report, we establish that HDAC8 is functionally important for MCL survival. Knocking down or abrogating HDAC8 activity in MCL cells induces caspase-dependent apoptosis. This finding is consistent with the observations seen in the T cell leukemia cell line, Jurkat, in which PCI-34051 induces caspase dependent apoptosis [18]. HDAC8 is transcriptionally regulated by the transcription factor SOX4 in adult T-cell leukemia (ATL) and lymphoma [27]. Abolishing SOX4 expression in ATL reduces HDAC8 expression and suppresses growth. In our experiments, all MCL cell lines examined express HDAC8 and expression does not change during PCI-34051 treatment. The MCL cell lines sensitive to HDAC8 inhibition were SOX11 positive and approximately 95% of all MCL tumors express SOX11, a neuronal transcription factor [19]. There is a positive correlation associated with SOX11 expression, with respect to overall survival and event free survival [28, 29]. It is important to note, as a prognostic marker, SOX11 expression is still being investigated due to a lack of uniformity in correlative data. This may be due to genetic differences that were not accounted for in these studies, such as TP53 mutations [28]. However, the significance of SOX11 expression on patient overall survival remains uncertain [19].
Genetic knockout of HDAC8 in mice is embryonically lethal due to skull instability [30, 31]. In humans, mutations in HDAC8 are linked to Cornelia de Lange syndrome, and are classified as an X-linked dominant disorder that causes facial malformations and developmental issues [22, 30, 32]. HDAC8 expression is present in multiple cancers types, contributing to survival, growth, or invasiveness. In neuroblastoma, high expression of HDAC8 is associated with poor overall survival [33]. Knocking down or inhibiting HDAC8 in neuroblastoma cell lines significantly reduces growth, blocks proliferation, and enhances retinoic acid-mediated differentiation [33, 34]. As described previously, blocking HDAC8 activity in T cell lymphoma causes activation of Phospholipase C and Ca+2 induced caspase-dependent apoptosis [18]. In our studies, HDAC8 is expressed across all MCL cell lines tested and 5 out of 6 are sensitive to HDAC8 inhibition. Furthermore, genetic knockdown or inhibition of HDAC8 induces caspase-dependent apoptosis.
NK cell function is critical for MCL patient response to the immunotherapeutic drug Rituximab. Newer agents have shown exceptional clinical response, such as treatment with Ibrutinib, a Bruton’s tyrosine kinase (BTK) inhibitor. BTK is an integral kinase in B cell receptor (BCR) signaling. However, in some cases there is clinical evidence of initial and acquired resistance to treatment with Ibrutinib resulting in an accelerated proliferative state and poor clinical outcome [35, 36]. The factors associated with resistance to Ibrutinib are likely due to specific genetic abnormalities contributing to MCL heterogeneity, such as augmentation of the NFκB signaling pathway due to mutations in CARD11 and BIRC3; or loss of SMARCA4 and/or chromosome 9p21.1-p24.3 deletion; or activation of the PI3K/AKT/mTOR pathway [37, 38]. Thus, combinatorial approaches using Rituximab with a selective HDAC inhibitor may provide the needed benefit to improve OS and quality of life in MCL patients. Our data demonstrates that selective HDAC8 inhibition in NK cells preserves their ADCC function, secretion of IFNγ, survival, and increases the population of IFNγ producing NK cells, while inducing apoptosis in MCL cell lines.
Taken together these results indicate MCL cell lines are sensitive to HDAC8 inhibition, while preserving primary NK phenotype and ADCC response. Thus, targeting HDAC8 in MCL patients warrants further investigation as a potential therapeutic approach in combination with Rituximab. Interestingly, the percent of IFNγ producing NK cells significantly increases upon HDAC8 inhibition, nonetheless, this does not increase cytolytic responses in vitro. Although, an increase in IFNγ producing NK cells may have important biological significance in vivo, as increases in IFNγ producing NK cells have been shown to increase NK persistence [39].
Supplementary Material
Highlights:
HDAC8 inhibition or ablation impairs Mantle Cell Lymphoma (MCL) viability
Natural Killer cell function against MCL is preserved upon HDAC8 inhibition
HDAC8 inhibition increases interferon-gamma producing Natural Killer cells
Acknowledgements
This work has been supported in part by the Flow Cytometry Core Facility at the Moffitt Cancer, an NCI designated Comprehensive Cancer Center (P30-CA076292) and by a research grant from FORMA Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Swerdlow SH, Campo E, Pileri SA, et al. , The 2016 revision of the World Health Organization classification of lymphoid neoplasms, Blood, 127 (2016) 2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Forstpointner R, Unterhalt M, Dreyling M, et al. , Maintenance therapy with rituximab leads to a significant prolongation of response duration after salvage therapy with a combination of rituximab, fludarabine, cyclophosphamide, and mitoxantrone (R-FCM) in patients with recurring and refractory follicular and mantle cell lymphomas: Results of a prospective randomized study of the German Low Grade Lymphoma Study Group (GLSG), Blood, 108 (2006) 4003–4008. [DOI] [PubMed] [Google Scholar]
- [3].Ghielmini M, Schmitz SF, Cogliatti S, et al. , Effect of single-agent rituximab given at the standard schedule or as prolonged treatment in patients with mantle cell lymphoma: a study of the Swiss Group for Clinical Cancer Research (SAKK), J Clin Oncol, 23 (2005) 705–711. [DOI] [PubMed] [Google Scholar]
- [4].Romaguera JE, Fayad L, Rodriguez MA, et al. , High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine, J Clin Oncol, 23 (2005) 7013–7023. [DOI] [PubMed] [Google Scholar]
- [5].Lee HZ, Kwitkowski VE, Del Valle PL, et al. , FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma, Clin Cancer Res, 21 (2015) 2666–2670. [DOI] [PubMed] [Google Scholar]
- [6].Mann BS, Johnson JR, Cohen MH, et al. , FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma, Oncologist, 12 (2007) 1247–1252. [DOI] [PubMed] [Google Scholar]
- [7].Whittaker SJ, Demierre MF, Kim EJ, et al. , Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma, J Clin Oncol, 28 (2010) 4485–4491. [DOI] [PubMed] [Google Scholar]
- [8].Hrgovic I, Doll M, Kleemann J, et al. , The histone deacetylase inhibitor trichostatin a decreases lymphangiogenesis by inducing apoptosis and cell cycle arrest via p21-dependent pathways, BMC Cancer, 16 (2016) 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sakajiri S, Kumagai T, Kawamata N, et al. , Histone deacetylase inhibitors profoundly decrease proliferation of human lymphoid cancer cell lines, Exp Hematol, 33 (2005) 53–61. [DOI] [PubMed] [Google Scholar]
- [10].Shimizu R, Kikuchi J, Wada T, et al. , HDAC inhibitors augment cytotoxic activity of rituximab by upregulating CD20 expression on lymphoma cells, Leukemia, 24 (2010) 1760–1768. [DOI] [PubMed] [Google Scholar]
- [11].Chun P, Histone deacetylase inhibitors in hematological malignancies and solid tumors, Arch Pharm Res, 38 (2015) 933–949. [DOI] [PubMed] [Google Scholar]
- [12].Minucci S, Pelicci PG, Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer, Nat Rev Cancer, 6 (2006) 38–51. [DOI] [PubMed] [Google Scholar]
- [13].Ni L, Wang L, Yao C, et al. , The histone deacetylase inhibitor valproic acid inhibits NKG2D expression in natural killer cells through suppression of STAT3 and HDAC3, Sci Rep, 7 (2017) 45266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rossi LE, Avila DE, Spallanzani RG, et al. , Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression, J Leukoc Biol, 91 (2012) 321–331. [DOI] [PubMed] [Google Scholar]
- [15].Tao R, de Zoeten EF, Özkaynak E, et al. , Deacetylase inhibition promotes the generation and function of regulatory T cells, Nature Medicine, 13 (2007) 1299–1307. [DOI] [PubMed] [Google Scholar]
- [16].Smith MA, Maurin M, Cho HI, et al. , PRDM1/Blimp-1 controls effector cytokine production in human NK cells, J Immunol, 185 (2010) 6058–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Alexander LMM, Watters J, Reusch JA, et al. , Selective expression of the transcription elongation factor ELL3 in B cells prior to ELL2 drives proliferation and survival, Mol Immunol, 91 (2017) 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Balasubramanian S, Ramos J, Luo W, et al. , A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas, Leukemia, 22 (2008) 1026–1034. [DOI] [PubMed] [Google Scholar]
- [19].Mozos A, Royo C, Hartmann E, et al. , SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype, Haematologica, 94 (2009) 1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Conrotto P, Andreasson U, Kuci V, et al. , Knock-down of SOX11 induces autotaxin-dependent increase in proliferation in vitro and more aggressive tumors in vivo, Mol Oncol, 5 (2011) 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gustavsson E, Sernbo S, Andersson E, et al. , SOX11 expression correlates to promoter methylation and regulates tumor growth in hematopoietic malignancies, Mol Cancer, 9 (2010) 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Deardorff MA, Bando M, Nakato R, et al. , HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle, Nature, 489 (2012) 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li S, Yue Z, Tanaka TU, Smc3 Deacetylation by Hos1 Facilitates Efficient Dissolution of Sister Chromatid Cohesion during Early Anaphase, Mol Cell, 68 (2017) 605–614 e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Morvan MG, Lanier LL, NK cells and cancer: you can teach innate cells new tricks, Nat Rev Cancer, 16 (2016) 7–19. [DOI] [PubMed] [Google Scholar]
- [25].Adam C, King S, Allgeier T, et al. , DC-NK cell cross talk as a novel CD4+ T-cell-independent pathway for antitumor CTL induction, Blood, 106 (2005) 338–344. [DOI] [PubMed] [Google Scholar]
- [26].Morandi B, Bougras G, Muller WA, et al. , NK cells of human secondary lymphoid tissues enhance T cell polarization via IFN-gamma secretion, Eur J Immunol, 36 (2006) 2394–2400. [DOI] [PubMed] [Google Scholar]
- [27].Higuchi T, Nakayama T, Arao T, et al. , SOX4 is a direct target gene of FRA-2 and induces expression of HDAC8 in adult T-cell leukemia/lymphoma, Blood, 121 (2013) 3640–3649. [DOI] [PubMed] [Google Scholar]
- [28].Nordstrom L, Sernbo S, Eden P, et al. , SOX11 and TP53 add prognostic information to MIPI in a homogenously treated cohort of mantle cell lymphoma--a Nordic Lymphoma Group study, Br J Haematol, 166 (2014) 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nygren L, Baumgartner Wennerholm S, Klimkowska M, et al. , Prognostic role of SOX11 in a population-based cohort of mantle cell lymphoma, Blood, 119 (2012) 4215–4223. [DOI] [PubMed] [Google Scholar]
- [30].Kaiser FJ, Ansari M, Braunholz D, et al. , Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance, Hum Mol Genet, 23 (2014) 2888–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Haberland M, Mokalled MH, Montgomery RL, et al. , Epigenetic control of skull morphogenesis by histone deacetylase 8, Genes Dev, 23 (2009) 1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ramos FJ, Puisac B, Baquero-Montoya C, et al. , Clinical utility gene card for: Cornelia de Lange syndrome, Eur J Hum Genet, 23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Oehme I, Deubzer HE, Wegener D, et al. , Histone deacetylase 8 in neuroblastoma tumorigenesis, Clin Cancer Res, 15 (2009) 91–99. [DOI] [PubMed] [Google Scholar]
- [34].Rettig I, Koeneke E, Trippel F, et al. , Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation, Cell Death Dis, 6 (2015) e1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chiron D, Di Liberto M, Martin P, et al. , Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma, Cancer Discov, 4 (2014) 1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang ML, Rule S, Martin P, et al. , Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma, N Engl J Med, 369 (2013) 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Agarwal R, Chan YC, Tam CS, et al. , Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma, Nat Med, 25 (2019) 119–129. [DOI] [PubMed] [Google Scholar]
- [38].Zhao X, Lwin T, Silva A, et al. , Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma, Nat Commun, 8 (2017) 14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Romee R, Schneider SE, Leong JW, et al. , Cytokine activation induces human memory-like NK cells, Blood, 120 (2012) 4751–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.