

Abstract
The extracellular matrix (ECM) is often unaccounted for in studies that consider the stromal contribution to cancer cell signaling and response to treatment. To investigate the influence of a fibrotic microenvironment we utilize fibroblast-derived ECM scaffolds as a cell culture platform. We uncover that estrogen receptor-positive (ER+) breast cancer cells cultured within ECM-scaffolds have an increase in ER signaling that occurs via a MAPK-dependent, but estrogen-independent manner. The ECM acts as a reservoir by binding, enriching, and presenting growth factors to adjacent epithelial cells. We identified FGF2 as a specific ECM-bound factor that drives ER signaling. ER+ cells cultured on ECM matrices have reduced sensitivity to ER-targeted therapies. The sensitivity to ER-targeted therapy can be restored by inhibiting FGF2-FGFR1 binding. ECM-FGF2 complexes promote Cyclin D1 induction which prevents G1 arrest even in the presence of antiestrogens. This work reveals a previously unaccounted for role of the ECM in ER signaling and resistance to endocrine therapy and suggests that patients with ER+ breast cancer that have high mammographic breast density and an intact FGFR signaling pathway may benefit from existing FGFR-targeted therapies.
Introduction
Globally, breast cancer is the most commonly diagnosed type of cancer and the leading cause of cancer-related death among women (1). For patients with breast cancer, the presence or absence of the estrogen receptor (ER), progesterone receptor (PR), or human epidermal growth factor receptor 2 (HER2) are important factors in subtype classification as well as for determining therapeutic options. Of the subtypes, the ER+ subtype is the most commonly diagnosed type of breast cancer (2). The ER exists in both alpha and beta isoforms that are differentially expressed in various tissues throughout the body (3). ERα is the ER isoform associated with mammary gland development, the proliferation of breast cancer cells, and predicts for response to ER-targeted therapies. Upon ligand-dependent or ligand-independent activation, ERα (referred to hereafter as just ER) can dimerize with itself or cooperate with co-activators and co-repressors to regulate the transcription of many target genes that promote cell survival (4–6). Recent work suggests that ER signaling may be reprogrammed during tumorigenesis, as normal and cancerous breast cells have different transcriptional responses to estrogen stimulation (7).
ER+ breast cancers rely on estrogen stimulation for survival and proliferation. Patients with ER+ breast cancer are often treated with endocrine therapies that prevent stimulation of the ER thus preventing cell cycle progression (8). However, ER+ breast cancers commonly display de novo resistance to hormonal therapy or acquire resistance after an initial response (9). Up to one-third of patients with early-stage ER+ breast cancer treated with tamoxifen become refractory or resistant (10). Endocrine resistance is often associated with sustained, ligand-independent activation of ER (11,12). Identifying pathways that lead to the ligand-independent action of ER will open the possibility of new strategies to prevent resistance to hormone therapy.
Stromal cells can contribute to the progression and outcome of breast cancer (13,14). Multiple studies specifically implicate factors secreted by stromal cells as contributing to endocrine therapy resistance (15–20); however, in vitro studies are generally conducted on 2D surfaces in the absence of fibroblast-produced extracellular matrix (ECM), a major component of stromal tissue. The importance of using 2.5D and 3D culture to study the relationship between the ECM and breast cancer using both naturally derived and synthetic materials is emerging in the field (21). New bioinformatics approaches to study ECM are being developed and reveal additional roles for the ECM in promoting breast cancer progression (22). The ECM can also serve as a reservoir for binding growth factors and secreted proteins (23–26), but whether or not the growth factors locally concentrated in the reservoir can mediate signaling to breast cancer cells remains elusive.
Here, we investigate the crosstalk between the ECM and ER+ breast cancer cells by utilizing fibroblast-derived, decellularized ECM scaffolds to model epithelial cells in a native microenvironment. RNA sequencing reveals that ER+ MCF7 cells cultured in ECM scaffolds or co-cultured with fibroblasts have activated ER signaling. The ER pathway is activated by ECM-FGF2 complexes and results in Erk-mediated ER target gene expression including AREG and TFF1. Patients with breast cancer that have a high mammographic density also have higher expression of ER signaling in extratumoral regions. ECM-FGF2-mediated Erk phosphorylation promotes Cyclin D1 expression and reduces sensitivity to anti-estrogen therapies. Inhibiting FGFR1 reverses the effect and enhances the efficacy of endocrine therapies in vitro and in vivo. The results demonstrate a novel role for the ECM in breast cancer and suggest that inhibiting FGFR may enhance the efficacy of ER-targeted therapies particularly for patients with breast cancer that have more fibrotic breast tissue.
Materials and Methods
Cell culture
MCF7, T47D, MDA-MB-231, BJ fibroblast, and IMR90 fibroblast cells were obtained from ATCC. MCF7-GFP cells were generated as previously described (27). Breast cancer-associated fibroblasts (CAFs) were obtained from Asterand. Cell lines were authenticated by STR sequencing and confirmed to be Mycoplasma free with the Mycosensor qPCR Assay Kit (Agilent). After thaw from liquid nitrogen storage, cells were passaged at least once before usage in experiments and were discarded after 15 passages at most depending on cell line. Cancer cell lines were cultured in Dulbecco’s modified Eagle’s medium (Sigma Aldrich) (MCF7, MDA-MB-231) or RPMI medium (Sigma Aldrich) (T47D) supplemented with 10% (v/v) fetal bovine serum (Corning) and 1% penicillin-streptomycin (Invitrogen) at 37℃ and 5% CO2. Fibroblasts were cultured in DMEM supplemented with 10% (v/v) FetalClone™ III fetal bovine serum (HyClone) and 1% penicillin-streptomycin (Invitrogen). Estrogen-deprived media consisted of DMEM without L-glutamine or phenol red (Quality Biologicals) supplemented with 10% charcoal-stripped fetal bovine serum (Sigma Aldrich), 1% penicillin-streptomycin (Invitrogen), and 2.5 mM L-glutamine (Sigma Aldrich).
Fibroblast Decellularization
ECM was extracted as previously described (28) with fibroblasts plated at 60,000 cells/cm2.
Immunoblot Assays
Immunoblots were prepared as previously described (29). The following antibodies were used: ER (1:1000, Cell Signaling D6R2W), AREG (1:1000, Proteintech 16036–1-AP), TFF1 (1:500, Santa Cruz Biotechnologies A-10), phosphorylated Erk p44/42 (1:1000, Cell Signaling 9106S), Erk (1:1000, Cell Signaling 4695S), phosphorylated Akt S473 (1:1000, Cell Signaling 9271L), Akt (1:1000, Cell Signaling 9272S), Cyclin D1 (1:1000, Cell Signaling 2922S), phosphorylated ER S118 (1:1000, Cell Signaling 2511S), phosphorylated ER S167 (1:1000, Cell Signaling #5587P), phosphorylated ER S104/106 (1:500, Cell Signaling #2517), and FGFR1 (1:1000, Santa Cruz Biotechnologies M2F12).
Reverse Transcription and qPCR
Total RNA was extracted from cells using TRIzol (Invitrogen) and Direct-zol RNA Miniprep kit with DNase I treatment (Zymo). One microgram of total RNA was used for first-strand DNA synthesis with the iScript cDNA Synthesis System (Bio-Rad). qPCR was performed using human-specific primers and iTaq SYBR Green Universal Master Mix (Bio-Rad). The expression of each target mRNA relative to 18s rRNA was calculated based on the threshold cycle (Ct) as 2−Δ(ΔCt), where ΔCt = Ct,target – Ct,18s and Δ(ΔCt) = ΔCt,test – ΔCt,control. Primer sequences are shown in Supplementary Table S1.
Immunofluorescence and Microscopy
Samples were fixed and permeabilized with ice-cold acetone and blocked with 2% BSA in PBS for 30 minutes at room temperature. Samples were incubated with an antibody against collagen I (1:250, Novus NB600–408), fibronectin (1:500, Abcam ab2413), and/or FGF-2 (1:100, Santa Cruz Biotechnologies G-2) for 2 hours at room temperature. ECM was incubated with anti-mouse Alexa Fluor 488 and/or anti-rabbit Alexa Fluor 647 antibodies (1:200 each, Life Technologies) for 1 hour at room temperature. Other samples were incubated with conjugated phalloidin (1:200, Cytoskeleton PHDG1) or conjugated antibody against Ki67 (1:100, BD 558615) for 1 hour at room temperature followed by DAPI (1 μg/mL) for 10 minutes at room temperature. PBS washes were performed between steps. Fluorescent and phase images were obtained using a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek). Confocal microscopy to obtain z projections was performed by using a 10 × /0.45 PlanApo (dry, no DIC) objective in a Zeiss LSM780-FCS microscope. Z stacks spaced at 6.3 μm-intervals were processed into a 3D image via Imaris version 9.2 (Bitplane).
Automated Analysis for Percentage of Cells Positive for Ki67
A graphic representation of this process is provided in Supplementary Figure 7A. Ki67 and DAPI-stained cells were imaged with a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek). Using the Gen5 3.05 software (BioTek), the DAPI channel was used to count the number of cells and create an individual mask on the area of each nucleus. Ki67 intensity within each mask was then quantified. A Ki67 threshold intensity of greater than 10,000 was deemed positive. Percentage was determined for each well by dividing the number of positive cells over the total number of counted cells.
RNA Library Preparation
All RNA samples were isolated as described previously and had a RIN value of >9.0 when measured on an Agilent Bioanalyzer. Libraries for RNA sequencing were prepared with KAPA Stranded RNA-Seq Kit. The workflow consisted of mRNA enrichment, cDNA generation, end repair to generate blunt ends, A-tailing, adaptor ligation and 14 cycles of PCR amplification. Unique adaptors were used for each sample to multiplex samples into several lanes. Sequencing was performed on Illumina Hiseq 3000/4000 with a 150bp pair-end run by Quick Biology (Pasadena, CA).
RNA Sequencing Analysis
A data quality check was done on Illumina SAV. Demultiplexing was performed with Illumina Bcl2fastq2 v 2.17 program. The sequence reads were mapped to the latest UCSC transcript set using Howtie2 version 2.1.0 (30) and the gene expression level was estimated using RSEM v1.2.15 (31). TMM (Trimmed Mean of M-values) was used to normalize gene expression. Differentially expressed genes were identified using the edgeR program (32). Genes showing altered expression with p < 0.05 and more than 2-fold change were considered differentially expressed. Normalized (TMM) and raw data were uploaded to GEO (GSE137528).
GSEA was performed to determine the specific Hallmark gene sets from The Molecular Signatures Database collection (33) that showed statistically significant and concordant differences between ECM-cultured versus uncoated surface-cultured and fibroblast-cocultured versus monocultured MCF7 cells using the GSEA desktop application. TMM normalized data was used as the expression dataset, the Hallmark or E2-inducible gene sets were selected as the gene set database, and the number of permutations used was set to the recommended (1000). Clustergrams were generated using MATLAB R2018b (MathWorks) with TMM data for the 40 specific genes indicated in Supplementary Table S2. A Euclidean distance metric was used for both row and column clustering.
Magnetically Activated Cell Sorting (MACS)
The isolation of purified MCF7 and fibroblast cells from the MCF7/fibroblast coculture was achieved by incubating cocultured suspensions with anti-human fibroblasts microbeads (Miltenyi) for 30 minutes at room temperature. The cells were washed and resuspended in MACS buffer (2 mM EDTA and 0.5% BSA in PBS) then passed through a magnetic MACS separator MS column (Miltenyi) mounted on a MACS Multistand magnet (Miltenyi).
Flow Cytometry for MACS Validation and Cell Cycle Analysis
Flow cytometry for MACS validation was performed on a FACS Calibur (BD Biosciences). The fibroblasts and GFP-expressing MCF7 cells were first treated with anti-fibroblast microbeads and then with anti-fibroblast APC conjugated antibody (Miltenyi) for 10 minutes at 4℃ before magnetic separation according to manufacturer’s instruction. Flow cytometry for cell cycle analysis was performed on an LSR II (BD Biosciences).
For cell cycle analysis, cells were pelleted, resuspended in water, and fixed by adding 100% ethanol drop-wise to a final concentration of 70%. Fixed cells were maintained on ice for 2 hours. Cells were then washed with PBS, pelleted, and incubated in staining buffer (PBS with 100 μg/mL RNase A and 50 μg/mL propidium iodide) overnight at 4℃ in the dark. Data was analyzed with FlowJo V10 software (Tree Star Inc.). For cell cycle analysis, gates were drawn to capture the regions of G0/G1, S, and G2/M phases.
CRISPR Mediated Knock-down of ER and FGF2
LentiCRISPR v2 plasmid used for generating a CRISPR-Cas9 endonuclease was a gift from Feng Zhang (Broad Institute, Massachusetts Institute of Technology, Cambridge, MA, obtained via Addgene (Addgene plasmid #52961)). Knockout by CRISPR/Cas9 was performed as previously described with slight modifications (34). Three insert ER-specific oligonucleotides that include a guide RNA sequence (as previously described (35)) were annealed and inserted into the BsmBI cloning site of the LentiCRISPR v2 plasmid. The FGF2-specific sequence was as follows: CACCGCTTCCTTCATAGCCAGGTAA. DNA sequences of plasmid constructs were confirmed by Sanger sequencing. The LentiCRISPR v2 ER/FGF2 or a non-targeting gDNA control (NTC, sequence: CACCGCACCACGGTCCATACATACA) were transfected along with 4 μg PsPAX2 and 1 μg pMD2.G into 293T cells using PolyJet™ transfection reagent (SignaGen Laboratories, Rockville, MD) according to the manufacturer’s instructions. Media was refreshed 16 hours following transfection and filtered viral supernatant was collected 48 hours later. Viral supernatant of all three ER constructs together or the NTC construct was added to MCF7 cells and puromycin (0.5 μg/mL) was added to the medium of cells 24 hours after transduction for positive selection.
ELISA
ELISAs were performed using an FGF2 Human ELISA Kit (ThermoFisher, KHG0021) according to the manufacturer’s instruction. Conditioned media samples were collected prior to fibroblast decellularization. After decellularization, ECM-coated 96-wellplate wells were treated in the same manner as antibody-coated wells.
Cell Proliferation Assays
Cells (1–2 × 105) were seeded in 6-well plates and then treated with tamoxifen (Sigma, H7904), fulvestrant (Sigma, I4409), paclitaxel (Actavis, 45963-613-53), doxorubicin (Actavis, 45963-733-55), or vehicle. After 3–4 days, cells were harvested and counted after 0.4% Trypan Blue (Gibco) staining using a Countess II FL Automated Cell Counter (Thermo Fisher).
Alternatively, cells were treated with the aforementioned drugs for 3–4 days and then incubated with 10% PrestoBlue™ Cell Viability Reagent (ThermoFisher) for 1–2 hours. The fluorescence intensity of the reagent was measured at excitation 560 nm and emission 590 nm on a Cytation 5 Multi-Mode Reader (BioTek).
Immunohistochemistry Staining
Paraffin-embedded tissue sections were dewaxed with xylenes and hydrated with decreasing gradients of ethanol. Afterwards, slides were treated with for antigen retrieval in Tris-EDTA buffer (10 mM Tris-Cl, 1 mM EDTA, pH 9.0) at near-boiling temperature for 20 minutes. Immunohistochemistry was conducted with the Vectastain® Elite® ABC HRP Kit (Vector Laboratories PK-7200) and DAB Peroxidase (HRP) Substrate Kit (Vector Laboratories SK-4100) according to the manufacturer’s instruction. Primary antibodies against TFF1 (1:400, Santa Cruz Biotechnologies A-10) was diluted in 1% BSA in PBS and applied to slides for one hour at room temperature. Masson’s trichrome stain was performed by the Reference Histology Laboratory at Johns Hopkins Hospital. Slides were imaged in color bright field on a Cytation 5 Cell Imaging Multi-mode Reader (BioTek).
Morphology Analysis
Using ImageJ, cells were manually traced and analyzed for size and circularity with ImageJ’s built-in analysis. Distance between cells was also measured in ImageJ by drawing a line from the approximate center of one cell to the center of its closest neighboring cell.
Patient Data & Analysis
Microarray data of extratumoral regions and MBD scoring from 120 patients with breast cancer (78% had ER+ tumors) were obtained as previously described (36) and accessed through the Gene Expression Omnibus (GSE49175). Probes without ENTREZ ID were eliminated and duplicates were collapsed by averaging. GSEA was performed on the aforementioned gene set lists using the GSEA desktop application as described above. The clustergram was created by scaling the data within each gene and using the ComplexHeatmap package in R. A Euclidean distance metric was used for both row and column clustering.
Animal Studies & Tumor Decellularization
Female 5- to 7-week-old NSG mice were anesthetized, and 2 × 106 cells were injected into the mammary fat pad. Slow-release estradiol pellets (2 mg per pellet) were implanted subcutaneously three days before MCF7 injection. Tumors were measured in three dimensions (a, b, and c) and volume (V) was calculated as V = abc × 0.52. Mice were treated by intraperitoneal injection (daily, 5x weekly) with 5 mg/kg PD173074, 5 mg/kg OHT, a combination of both drugs, or vehicle only (5% ethanol in corn oil) when all tumors were palpable and reached ~25 mm3 in size.
For tumor decellularization, mice were inoculated in the same manner without drug treatment. After approximately 2 months, mice were sacrificed and tumors were collected and sectioned into 100 μm slices in a Compresstome® Tissue Slicer (Precisionary) according to the manufacturer’s protocol. To decellularize, slices were incubated at 60 RPM and 37℃ with 500 μL of a detergent solution of PBS containing 0.5% (v/v) Triton X-100 and 20 mM NH4OH for 4 days with solution being refreshed daily. On the 5th day, slices were incubated with 10 μg/mL DNase I and 10 μg/mL RNase A in PBS at the same rotating conditions. The following day, slices were washed quickly twice with 500 μL of water then 500 μL of PBS for another 24 hours rotating. Next, slices were incubated at 60℃ for 10 minutes in 250 μL of 1X RNAsecure™ RNase Inactivation Reagent (ThermoFisher AM7006). Slices were placed in an incubator for 10 minutes with no buffer to allow them to adhere to the bottom of the well. A 10 μL drop containing 100,000 MCF7 cells was dropped directly onto the slice or onto an uncoated surface as control and plates were returned to the incubator. An hour later, 500 μL of media was added. The following day RNA was collected as previously described from slices or the uncoated wells. For imaging, slices were fixed and stained as previously described. Slices were imaged with confocal microscopy to obtain z projections by using a 10 × /0.45 PlanApo (dry, no DIC) objective in a Zeiss LSM780-FCS microscope.
Statistics
All the values in text and figures are presented as mean ± SEM unless otherwise stated. Statistical significance was determined when appropriate by unpaired t-test or one/two-way ANOVA. P-values of <0.05 were considered significant.
Data Availability
The RNA sequencing data have been deposited in the GEO database under the accession code GSE137528.
Study Approval
Mouse studies were conducted according to protocols approved by the Johns Hopkins University Animal Care and Use Committee.
Results
Fibroblast-derived ECM scaffolds alter breast cancer cell morphology.
To investigate the effects of ECM signaling on breast cancer cells, we cultured MCF7 cells in a dense ECM scaffold created by decellularizing a confluent layer of fibroblasts (Figure 1A). ECM scaffolds retained both native ECM proteins and a fibrillary architecture after decellularization (Figure 1B). The ECM provided a quasi-three-dimensional scaffold often referred to as 2.5D (37) (Figure 1C). MCF7 cells cultured within the ECM scaffold displayed a disrupted cortical actin structure (Figure 1D) and did not form multicellular colonies compared to cells cultured on uncoated surfaces (Figure 1E). MCF7 cells cultured on ECM were larger, less circular, and localized farther from the nearest adjacent cell (Figure 1F).
Figure 1. Fibroblast-derived ECM scaffolds alter breast cancer cell morphology.
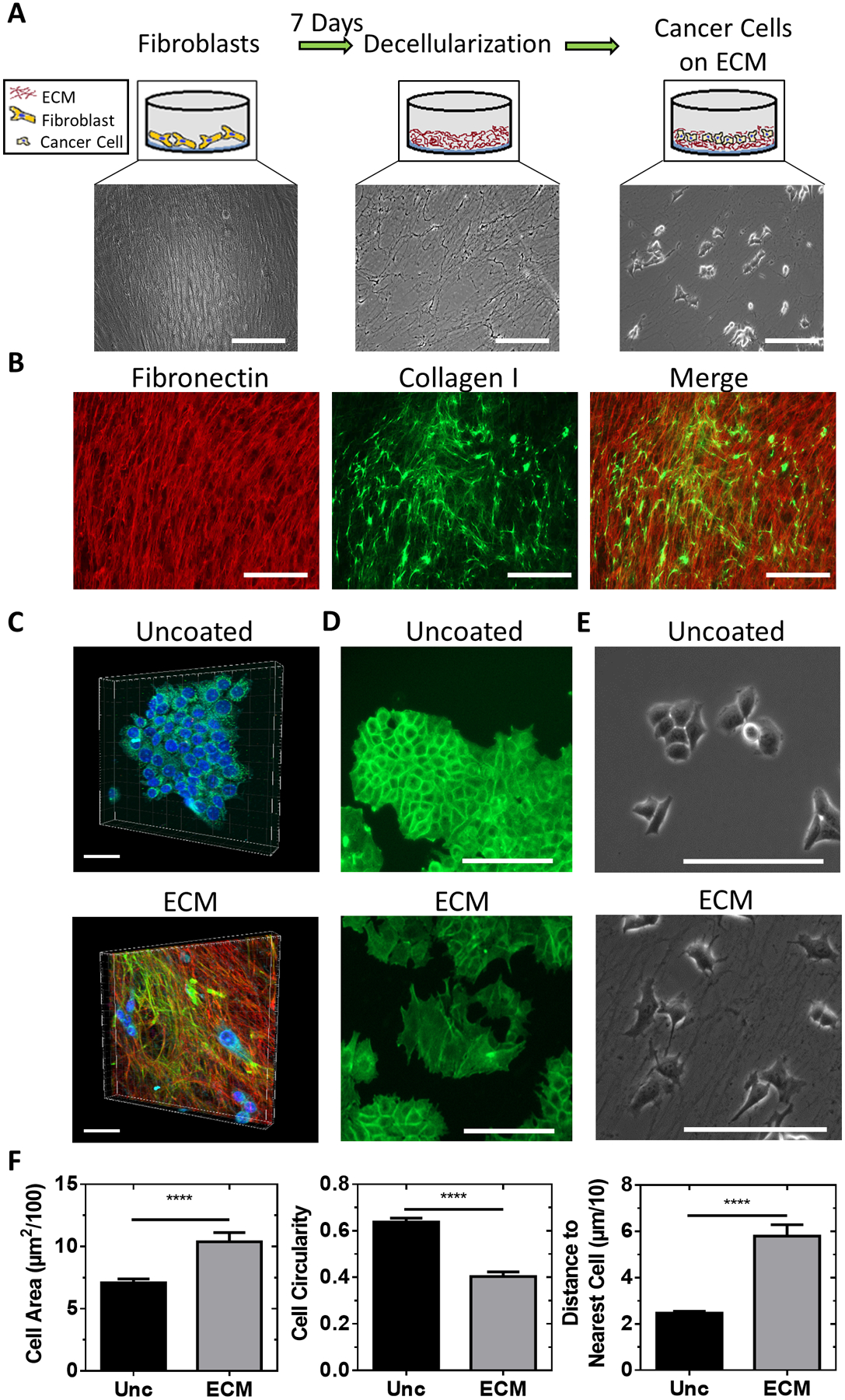
A, Schematic and phase contrast images of the fibroblast decellularization process. Scale bar = 200 μm. B, Immunofluorescent images of extracted ECM. Scale bar = 200 μm. C, 3D projection of MCF7 cells plated onto uncoated surfaces or ECM scaffolds and labeled with fibronectin (red) and collagen (green) antibodies. Scale bar = 40 μm. D, Immunofluorescent images of actin in MCF7 cells plated onto uncoated surfaces or ECM scaffolds Scale bar = 100 μm. E, Phase contrast image of MCF7 cells plated onto an uncoated surface or ECM scaffold. Scale bar = 200 μm. F, Cell area and circularity measurements and distance between MCF7 cells plated onto uncoated surfaces or ECM scaffolds. n = 55 cells (unc) and 65 cells (ECM) from three different image regions. Unpaired t-test, **** P< 0.0001.
Fibroblast-derived ECM induces estrogen signaling pathways in breast cancer cells.
To investigate changes in gene expression caused by culturing cells in a native, dense ECM environment, we isolated RNA from MCF7 cells cultured on uncoated surfaces or in ECM scaffolds generated by fibroblasts isolated from two different anatomical sites (Figure 2A). RNA sequencing revealed 280 genes that are significantly upregulated and 209 genes that were significantly downregulated, by at least 2-fold, in cells cultured on ECM scaffolds derived from either fibroblast type (Figure 2B). We reasoned that we could further enrich our gene list by identifying altered gene expression patterns that occur when MCF7 cells are co-cultured with fibroblasts and then magnetically separated for further analysis (Figure 2A & Supplementary Figure 1A). RNA sequencing and analysis showed that 84 genes were upregulated and 31 genes were downregulated by at least 2-fold in common across both experimental set-ups (Figure 2B and Supplementary Table S2). Among the top 6 altered pathways were the Hallmark early and late response to estrogen gene sets (Figure 2C & Supplementary Figures 1B–C). Gene set enrichment analysis (GSEA) confirmed that the Hallmark early and late estrogen response gene sets were enriched in MCF7 cells cultured on ECM scaffolds and in co-culture with fibroblasts. (Figures 2D and 2E). A clustergram analysis of 40 genes involved in the ER pathway response derived from these Hallmark lists (Supplementary Table S3) further demonstrated ER pathway enrichment (Figures 2F and 2G). We also found enrichment, in both setups, of a second list of genes that have previously been shown to be upregulated in response to estradiol in 7 ER+ breast cancer cell lines (38) (Supplementary Figures 1D–E).
Figure 2. ECM matrices induce estrogen signaling pathways in breast cancer cells.
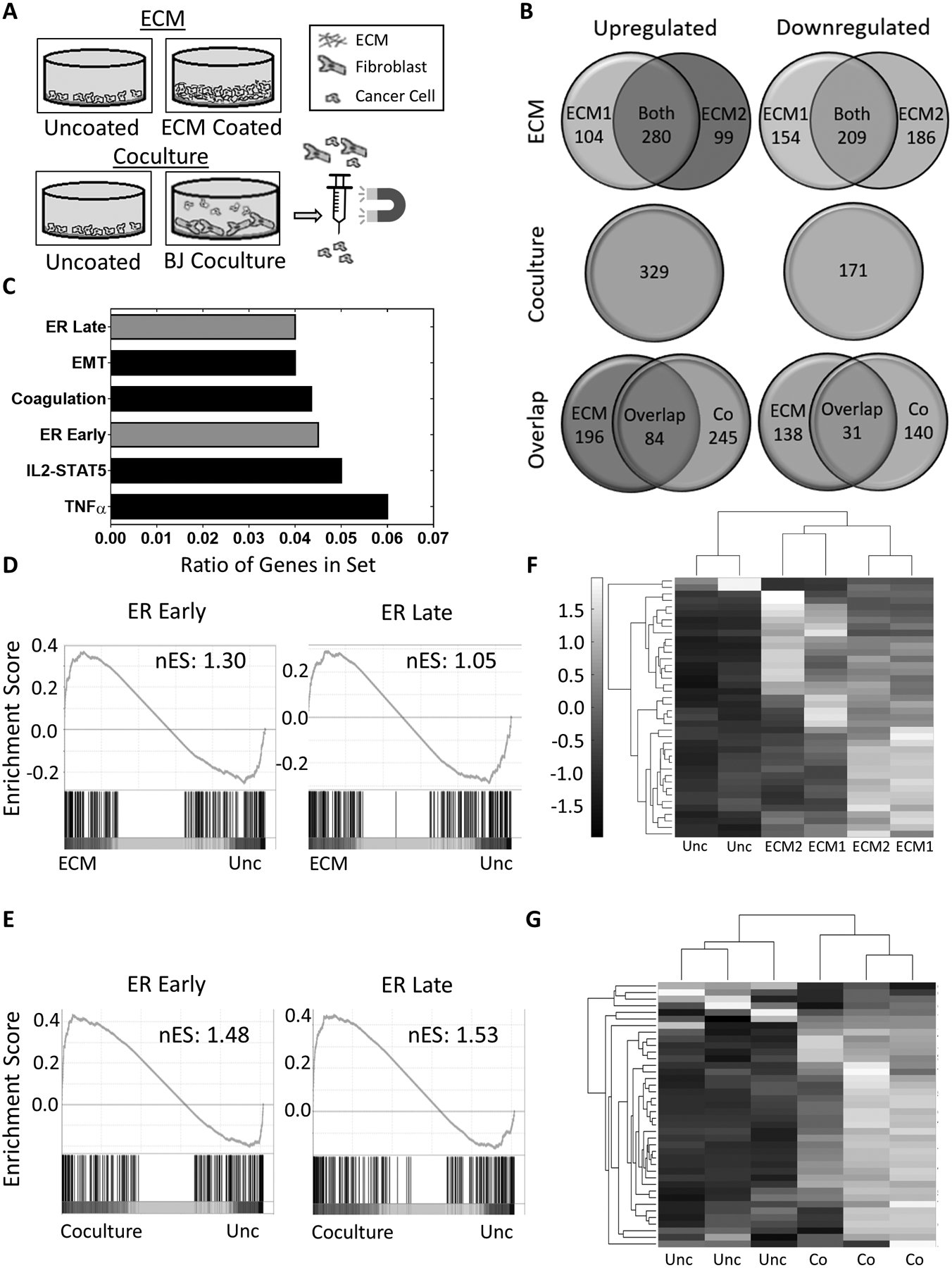
A, Schematic of the mRNA sequencing experiments. MCF7 cells were cultured in two setups: MCF7 cells cultured 1) on uncoated surfaces or ECM from BJ (ECM1) or IMR90 (ECM2) fibroblasts and 2) alone or in coculture with BJ fibroblasts before isolation by magnetically activated cell sorting. B, Venn Diagram of genes significantly (p<0.05) upregulated (left side) or downregulated (right side) by at least 2-fold in MCF7 cells cultured in either ECM scaffolds derived from BJ (ECM1) or IMR90 (ECM2) fibroblasts, or in BJ fibroblast coculture (Co) compared to cells on uncoated surfaces. C, Hallmark gene sets with highest ratios of genes (k/K) overlapping with the 84 upregulated genes shared between the ECM and coculture sets. D and E, Gene set enrichment analysis (GSEA) of Hallmark Late or Early estrogen response gene sets using mRNA sequencing data of MCF7 cells plated on ECM (D) or cocultured with BJ fibroblasts (E) with nominal enrichment score (nES). Nominal p-value <0.02 for Early (D) and <0.01 for Early (E) and Late (E). F and G, Clustergram of the gene expression of 40 ER-regulated genes in MCF7 cells from the ECM set (F) or the coculture set (G). Data is standardized by each row to a mean of zero and a standard deviation of 1. A list of the genes corresponding to each row can be found in Supplementary Table 3.
We generated cDNA from ER+ MCF7 and T47D cells as well as triple-negative MDA-MB-231 cells cultured on ECM scaffolds from normal fibroblasts (ECM1, ECM2) and breast cancer-associated fibroblasts (CAFs) or on uncoated surfaces and assessed the level of expression of 5 estrogen-responsive genes (Figures 3A–C) using quantitative polymerase chain reaction (qPCR). The transcription of estrogen-inducible genes, TFF1, AREG, and EGR1, were increased whereas estrogen-repressible genes, CDH1 and ESR1, were decreased when MCF7 cells were cultured either in the presence of estradiol (Figure 3D) or when cultured on 3 different fibroblast-derived ECM scaffolds (Figure 3A). MCF7 cells responded similarly to foreskin (ECM1), lung (ECM2) and breast cancer-associated fibroblast-derived (CAF) scaffolds. Given the large number of cells required to generate the ECM scaffolds, and the limited availability and poor growth characteristics of CAFs, foreskin fibroblasts were used to generate ECM for the majority of the experiments. Culturing cells on artificially coated collagen I or fibronectin surfaces did not recapitulate the response to cells cultured in ECM scaffolds (Supplementary Figure 2A). T47D cells had a similar response to culture in ECM scaffolds (Figure 3B), whereas MDA-MB-231 (ER-) cells did not (Figure 3C). TFF1 and AREG mRNA expression also increased when MCF7 cells were co-cultured with fibroblasts (Supplementary Figure 2B).
Figure 3. ECM signaling activates estrogen-responsive genes in an estrogen-independent manner.
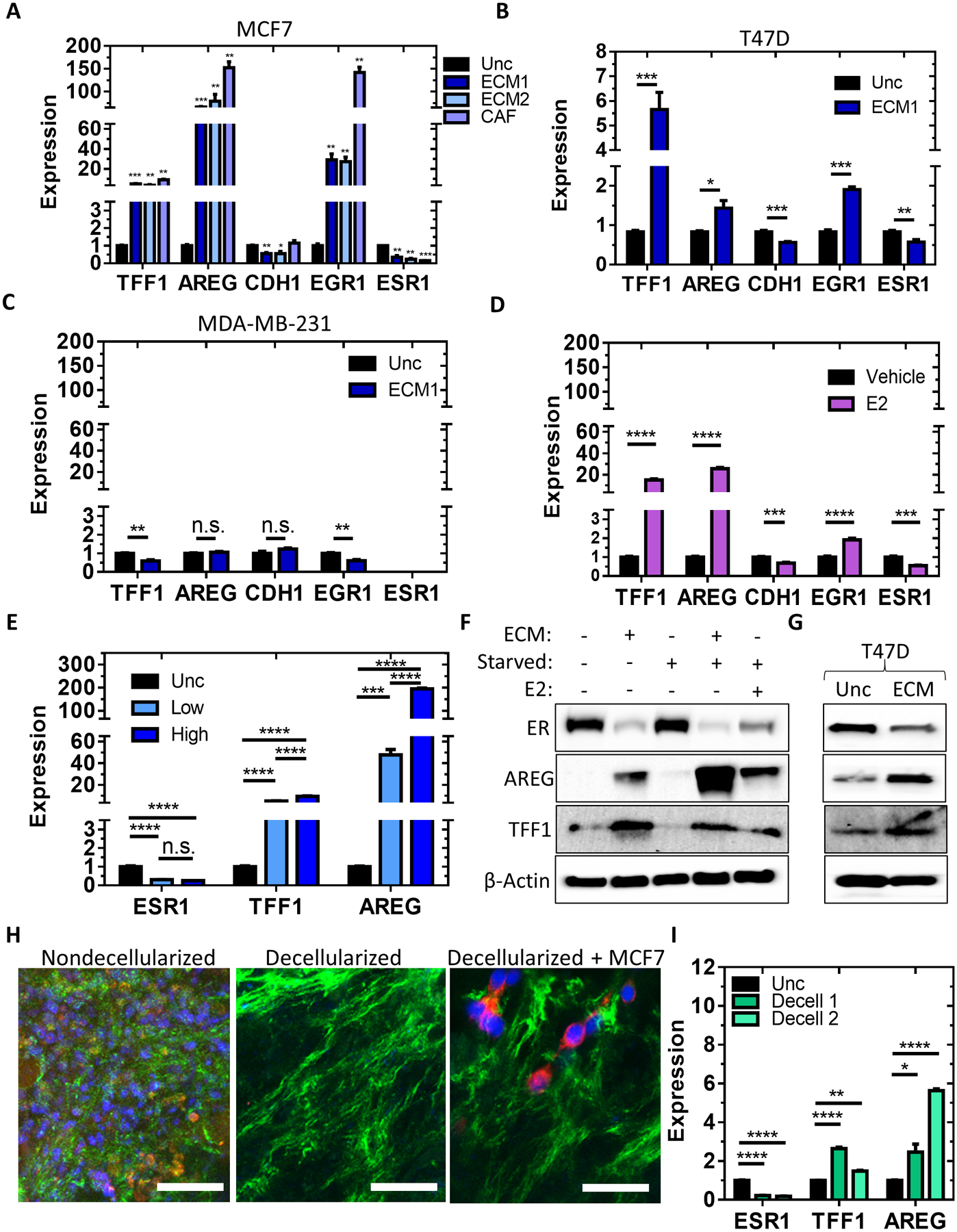
A-C, qPCR results for the quantification of mRNA expression of TFF1, AREG, CDH1, EGR1, and ESR1 in MCF7 (A), T47D (B), or MDA-MB-231 (C) cells plated onto uncoated surfaces or ECM scaffolds derived from BJ fibroblasts (ECM1), IMR90 fibroblasts (ECM2), or cancer-associated fibroblasts (CAF). n = 3–6 from N = 1–2 independent experiments. Unpaired t-test against uncoated. *** P<0.001, ** P<0.01, * P<0.05. D, qPCR results for the quantification of mRNA expression of TFF1, AREG, CDH1, EGR1, and ESR1 in MCF7 cells plated on uncoated surfaces and treated with vehicle or 10 nm of E2. n = 3. Unpaired t-test. **** P<0.0001. E, qPCR results for the quantification of mRNA expression of TFF1, AREG, and ESR1 in MCF7 cells plated onto uncoated surfaces or ECM scaffolds derived from BJ fibroblasts plated at 20,000 cells/cm2 (Low) or 60,000 cells/cm2 (High) for 24 hours. n = 3. One-way ANOVA with Bonferonni post-test for each gene. F, Immunoblot performed using protein lysates from MCF7 cells plated onto uncoated surfaces or ECM scaffolds with (+) or without (–) estrogen starvation or 100 nM E2 to detect ER, AREG, TFF1, and β-actin protein expression. G, Immunoblot performed using protein lysates from T47D cells plated onto uncoated surfaces or ECM scaffolds with complete culture media to detect ER, AREG, TFF1, and β-actin protein expression without estrogen starvation. H, Confocal of non-decellularized MCF7 xenograft tumor slices or regions of decellularized slices with or without MCF7 cells cultured in the slice. Slices were stained for actin (red), fibronectin (green), and DNA (blue). Scale bar = 50 μm. I, qPCR results for the quantification of mRNA expression of TFF1, AREG, and ESR1 in MCF7 cells plated onto uncoated surfaces or decellularized MCF7 xenograft tumor slices for 24 hours. n=3. One-way ANOVA within each gene.
To determine whether ECM-density dictated the estrogen-like response, we generated ECM from fibroblasts plated at low and high confluence to alter the abundance of the ECM (Supplementary Figure 2C). The level of TFF1 and AREG mRNA expression correlated with ECM density (Figure 3E), indicating that increasing the amount of fibroblast-derived ECM enhances the estrogen-like signaling response.
MCF7 and T47D cells had increased TFF1 and AREG protein expression, and decreased ESR1 protein expression, when cultured on fibroblast-derived ECM, and MCF7 cells displayed this regulation even when cultured in steroid-deprived media (Figure 3F and 3G, Supplementary Figure 2D–E). TFF1 and AREG levels surpassed levels induced by estradiol stimulation. To verify our findings in tumor tissue, we generated 100 μm slices from mouse tumors and decellularized to generate tissue-derived ECM scaffolds for MCF7 cell culture (Figure 3H). MCF7 cells cultured on decellularized tumor slices had increased TFF1 and AREG expression, and decreased ESR1 mRNA expression, compared to cells cultured on uncoated surfaces (Figure 3I). This confirms that the effects we observe in fibroblast-generated ECM scaffolds can be replicated in decellularized tumor tissue.
Inhibition of ER signaling or Erk phosphorylation prevents ECM-stimulated induction of AREG and TFF1.
To determine whether the ER is required for ECM-mediated induction of TFF1 and AREG, we treated MCF7 cells cultured on ECM or uncoated surfaces with selective estrogen receptor modulator (SERM), 4-hydroxytamoxifen (OHT), or a selective estrogen receptor degrader (SERD), fulvestrant (Fulv). Inhibiting ER function, significantly, reduced mRNA and protein expression of AREG and TFF1 in MCF7 and T47D cells cultured on ECM scaffolds (Figures 4A–B & 4D–E, Supplementary Figure 3A). We also observed increased Erk, phosphorylation in both MCF7 and T47D cells cultured on ECM scaffolds concomitant with the upregulation of AREG and TFF1 (Supplementary Figures 3B). To determine if the cancer cell-fibroblast interaction contributes to the production of ECM that would further potentiate ERK phosphorylation, we co-cultured MCF7 cells with BJ fibroblasts prior to decellularization to generate ECM. MCF7-fibroblast co-culture further augmented Erk phosphorylation (Supplementary Figure 3C).
Figure 4. Inhibition of ER or Erk phosphorylation prevents the ECM-mediated induction of AREG and TFF1.
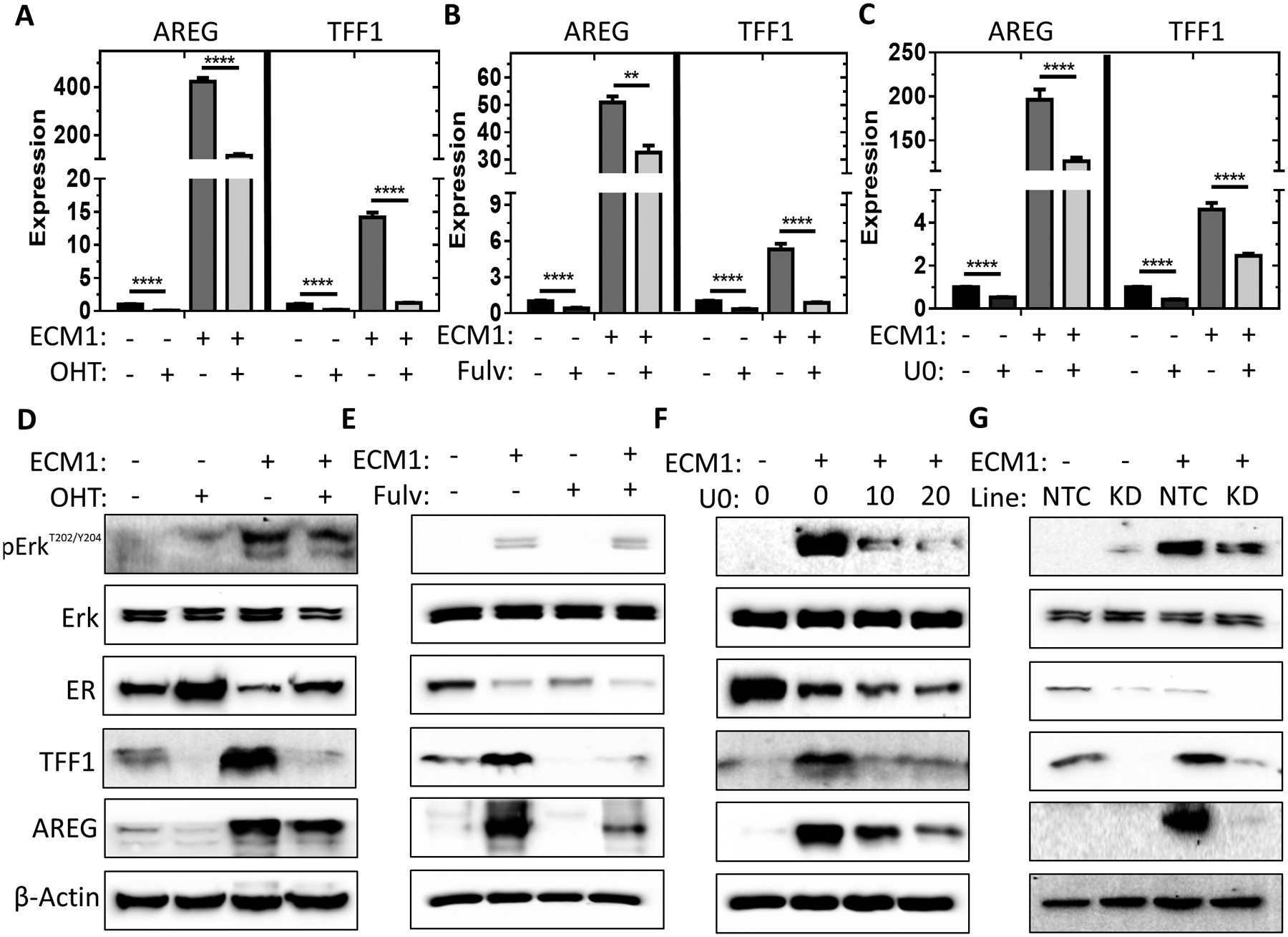
A-C, qPCR results for the quantification of mRNA expression of AREG and TFF1 in MCF7 cells plated onto ECM scaffolds or uncoated surfaces and treated with either vehicle, 1 μM of OHT (A), 1 μM of fulvestrant (B), or 10 μM of U0126 (C) for 24 hours. n = 3–6 from N = 1–2 independent experiments. Two-way ANOVA of log-transformed fold change with Bonferonni post-test within each gene. **** P<0.0001, ** P<0.01. D-F, Immunoblot performed using protein lysates of MCF7 cells plated onto ECM scaffolds or uncoated surfaces and treated with vehicle or 1 μM OHT (D), 1 μM fulvestrant (fulv) (E), or 10–20 μM U0126 (F) for 24 hours to detect Erk, Erk phosphorylation, AREG, TFF1, β-actin, and ER protein expression. G, Immunoblot performed using protein lysates from a CRISPR-mediated ER knockdown or a non-target control (NTC) sequence MCF7 cells plated onto ECM scaffolds or on uncoated surfaces to detect Erk, Erk phosphorylation, AREG, TFF1, β-actin, and ER protein expression.
To determine whether ERK phosphorylation is upstream or downstream of ER signaling, we treated MCF7 cells cultured on ECM with U0126, an inhibitor of MAPK activation, which significantly reduced the mRNA and protein levels of AREG and TFF1 (Figure 4C and 4F). On the other hand, OHT and fulvestrant treatment did not prevent Erk phosphorylation (Figure 4D and 4E). Taken together, the results suggest that MAPK pathway activation is required for ECM-mediated ER activation. In support of this notion, MCF7 cells that were depleted of ER using CRISPR technology were unable to induce AREG or TFF1 gene expression but retained increased levels of Erk phosphorylation when cultured in ECM scaffolds (Figure 4G). Although MAPK can enhance the transcriptional activity of ER through serine phosphorylation (39,40), we did not detect increased levels of serine 104/106, 118, or 167 ER phosphorylation when MCF7 cells were cultured on ECM (Supplementary Figure 3D).
The estrogen-like response elicited by ECM signaling requires a factor bound to ECM.
To assess whether the estrogen-like response caused by the ECM may be activated by factors secreted by stromal cells or cancer cells in response to contact with the ECM, we performed several conditioned media assays (Figure 5A). Conditioned complete media (CM) was collected from cultures containing MCF7 cells alone (Unc CM), ECM scaffolds alone (Blank CM), ECM scaffolds seeded with MCF7 cells (ECM CM), or BJ fibroblasts. MCF7 cells were cultured in CM or directly on ECM scaffolds. The CM did not induce AREG, TFF1, or Erk phosphorylation (Figures 5B–C). Next, we incubated MCF7 cells with a mechanically disrupted or intact ECM scaffold (Figure 5D). The disrupted ECM fully recapitulated the response of a cell interacting with intact ECM. Taken together, our results indicate that either the ECM itself or a factor bound to the ECM elicits an estrogen-like response that does not depend on the specific architecture of the scaffold.
Figure 5. Estrogen-like signaling is activated by a component bound to ECM.
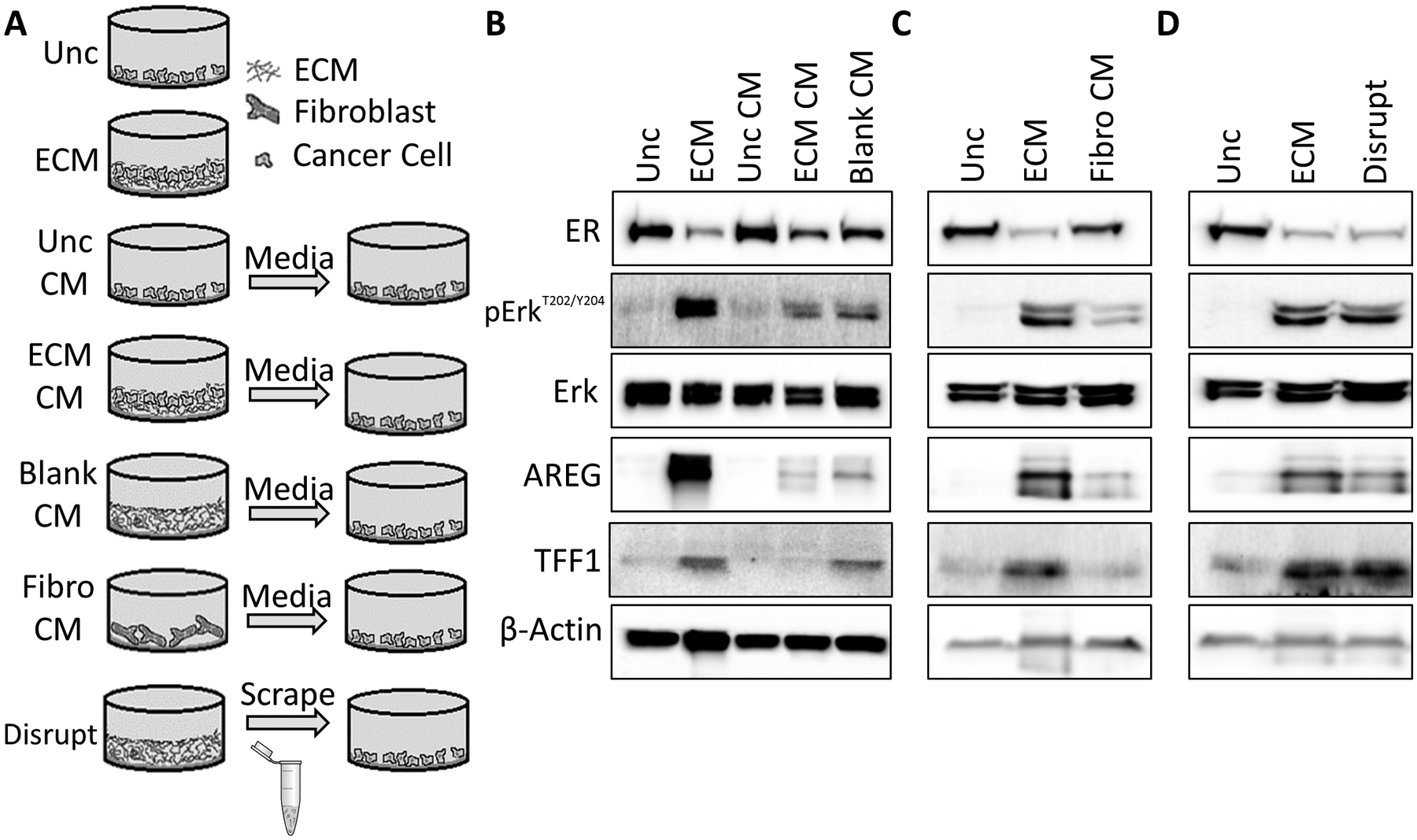
A, Schematic depicting experimental setup used for B-D. B-D, Immunoblots were performed using protein lysates from MCF7 cells plated onto ECM scaffolds (ECM) or uncoated surfaces with conditioned complete culture media from cancer cells (B), conditioned complete culture media from fibroblasts (C), or with disrupted decellularized ECM (D). Total Erk, phosphorylated Erk, AREG, TFF1, β-actin, and ER protein expression was detected.
ECM-bound FGF2 promotes estrogen-like signaling in ER+ breast cancer cells.
Erk phosphorylation is known to be stimulated by extracellular growth factors via the activation of receptor tyrosine kinase pathways (41). Growth factors bind to extracellular matrix proteins, specifically heparan sulfate, and can be presented to adjacent cells (23–25). We hypothesized that a growth factor bound to the ECM may mediate the estrogen-like response. We tested several growth factors and their inhibitors to determine whether ER activation was altered. (Supplementary Figure 4A–D). Our results demonstrate that the FGF receptor (FGFR) inhibitor, PD173074, inhibited ECM-mediated ER regulation (Figure 6A–B). The addition of FGF2 to MCF7 cells on uncoated surfaces phenocopied culture in ECM (Figure 6C and Supplementary Figure 4E). OHT, fulvestrant, or U0126 treatment prevented FGF-dependent induction of AREG and TFF1 (Figure 6C). Likewise, the addition of FGF2 or E2 to steroid-depleted medium each led to increased proliferation of MCF7 cells (Figure 6D & Supplementary Figure 4F). Taken together our results show for the first time that FGF2 stimulates ER-dependent cell proliferation in the absence of estrogen.
Figure 6. ECM-FGF2 complexes promote estrogen-like signaling in ER+ breast cancer cells.
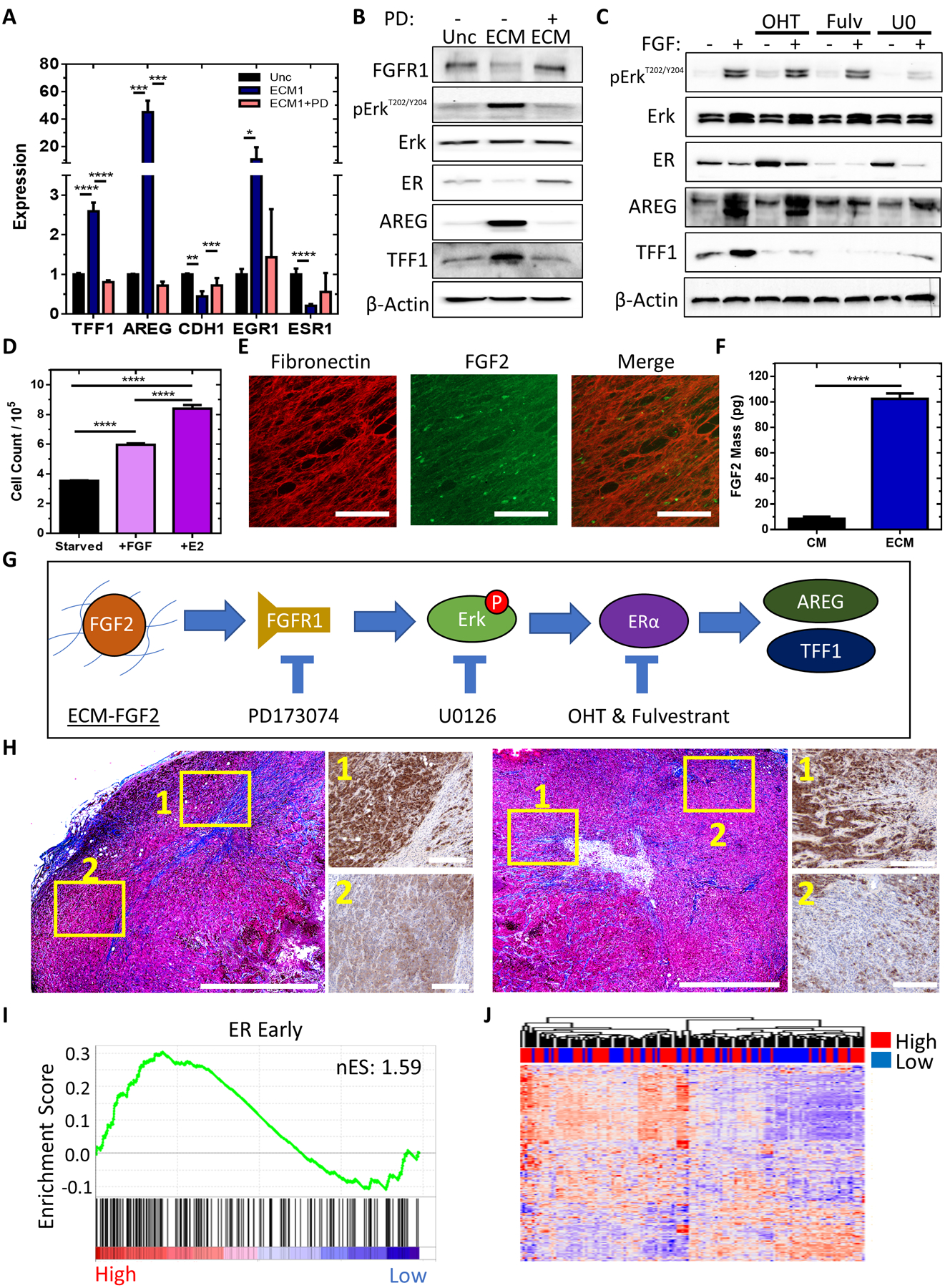
A, qPCR results for the quantification of mRNA expression of TFF1, AREG, CDH1, EGR1, and ESR1 in MCF7 cells plated onto uncoated surfaces or ECM scaffolds derived and treated with vehicle or 1 μM PD173074 for 24 hours. n = 6 from N = 2 independent experiments. One-way ANOVA with Bonferonni post-test of log-transformed fold change within each gene. **** P<0.0001, *** P<0.001, ** P<0.01, * P<0.05. B, Immunoblot performed using protein lysates from MCF7 cells cultured on uncoated surfaces or ECM and treated with vehicle or 1 μM PD173074 for 24 hours to detect Erk, Erk phosphorylation, AREG, TFF1, β-actin, FGFR1, and ER protein expression. C, Immunoblot performed using protein lysates from MCF7 cells treated with or without 40 ng/mL FGF-2 and with or without 1 μM of OHT, 1 μM of fulvestrant, or 30 μM of U0126 for 24 hours to detect Erk, Erk phosphorylation, AREG, TFF1, β-actin, and ER protein expression. D, Total cell count of MCF7 cells cultured in estrogen-deprived medium for 3 days then treated with 1 nM E2 or 40 ng/mL FGF-2 for 3 days. n = 3. One-way ANOVA with Bonferonni post-test. E, Immunofluorescent images of decellularized ECM labeled with fibronectin (red) or FGF2 (green) antibodies. Scale bar = 100 μm. F, ELISA data for the mass of FGF2 bound to ECM in a 96-wellplate versus the mass of soluble FGF2 in fibroblast-conditioned media (CM) of the fibroblasts that generated the ECM. n = 3 wells per condition. Unpaired t-test. G, Proposed mechanism. H, Masson’s trichrome stain and TFF1 immunohistochemistry stains of MCF7 xenograft tumors. Trichrome stain bars 1000 μm. IHC bars 200 μm. I, GSEA of the Hallmark estrogen response early set using extratumoral biopsies from 120 patients with breast cancer stratified by <25% mammographic breast density (MBD) (Low, N=56) or >=25% MBD (High, N=64) with nES. Nominal p-value <0.01. J,Heatmap of expression of the Hallmark estrogen response early gene set in extratumoral regions of 120 patients with breast cancer annotated by low or high MBD. Complete clustergram provided in Supplementary Figure 6.
The results demonstrated that the addition of 40 ng/mL of exogenous FGF2 to MCF-7 cells elicited a robust ER-dependent transcriptional response (Figure 6C) whereas incubating cells with the conditioned media from fibroblast cells did not (Figure 5C). Therefore, we wanted to determine the amount of FGF2 secreted into CM and compare to the amount of FGF2 adhered to the ECM or a concentration that is typically used in cell culture studies (25–50 ng/mL (16,20)) (Figure 6C). We first visually confirmed the presence of FGF2 in ECM matrices using immunofluorescence (Figure 6E). Using an enzyme-linked immunosorbent assay (ELISA), we found that ECM scaffolds contained 12 times the amount of FGF2 (102 pg/well or 320 pg per cm2 ECM) as compared to the fibroblast conditioned media (8.5 pg FGF2/well or 42.5 pg per mL of media) (Figure 6F and Supplementary Figure 4G). These results demonstrate that the ECM can bind and enrich FGF2 and can stimulate ER activation at a concentration that is 80-fold lower than adding exogenous FGF2 (8ng FGF2/well or 40ng/mL). In addition, we used CRISPR technology to generate an NTC and FGF2-knockdown BJ fibroblast cell line. FGF2 reduction was verified with ELISA of conditioned media (Supplementary Figure 4H). The CRISPR-mediated reduction of FGF2 in fibroblasts used to produce ECM decreased ECM-induced TFF1 and AREG expression and prevented ER-down regulation. (Supplementary Figure 4I). The results suggest a mechanism whereby stromal ECM-FGF2 complexes activate ER signaling (Figure 6G).
To determine if cancer cells within fibrotic tumor regions have higher ER activity in vivo, we used TFF1 protein expression as a surrogate for ER activation. Orthotopic tumors derived from MCF-7 cells implanted into NSG mice display a distinct spatial patterning of TFF1 expression. Cancer cells that are adjacent to regions of fibrosis (Figure 6H) have a marked increase in TFF1 expression.
The amount of stromal tissue in the breast varies between patients and is assessed by measuring mammographic breast density (MBD) (42). The breast tissue in women with higher MBD contains more ECM proteins and growth factors than women with lower MBD (43,44). A recent study investigating the relationship between MBD and gene expression patterns in normal tissue adjacent to breast cancer reported a correlation between high (>25%) or low (<25%) MBD and expression of an E2 gene signature (36). We performed a gene set enrichment analysis (GSEA) using the same dataset and found that extratumoral regions in women with higher MBD are enriched for the Hallmark estrogen response early and late pathways (Figure 6I and Supplementary Figure 4J), as well as a published gene set derived from ER+ cells exposed to estradiol (Supplementary Figure 4K) (38). An unsupervised clustering of each gene in the Hallmark estrogen response early gene set displays expression level by individual patient sample. (Figure 6J and Supplementary Figure 5). GSEA using the Gene Ontology: FGFR Signaling Pathway gene set did not demonstrate statistically significant enrichment between patients with low and high MBD (data not shown), suggesting that ECM-bound FGF2 may not stimulate classical FGFR1 target genes. Ultimately, the results suggest a potential role for dense stromal tissue to activate ER signaling in ER+ breast cancer cells.
ECM signaling reduces the anti-estrogen sensitivity of ER+ breast cancer cells through FGF2 via G1 arrest prevention.
Previous studies have shown that the addition of exogenous FGF2 (25 ng/mL) reduced the sensitivity of ER+ breast cancer cells to endocrine therapy in vitro (16). Likewise, breast cancer cell lines harboring an amplified FGFR1 gene are less sensitive to endocrine therapies (45). Therefore, we questioned whether ECM-FGF2 complexes altered sensitivity to ER targeting agents. To test this hypothesis, we treated GFP-expressing MCF7 cells cultured on ECM scaffolds or uncoated surfaces with OHT for four days (Figure 7A). An analysis of the area occupied by GFP+ cells served as a proxy for the number of cells remaining after treatment and revealed that cells cultured on ECM proliferated more in the presence of OHT than cells cultured on uncoated surfaces (Supplementary Figure 6A). In comparison, MCF7 cells cultured in ECM scaffolds remained sensitive to doxorubicin and paclitaxel (Figures 7B–C and Supplementary Figures 6B–H). Reduced OHT and fulvestrant sensitivity were also observed in T47D cells cultured on ECM scaffolds (Supplementary Figures 6I–K).
Figure 7. ECM signaling reduces anti-estrogen sensitivity of ER+ breast cancer cells via G1 arrest prevention.
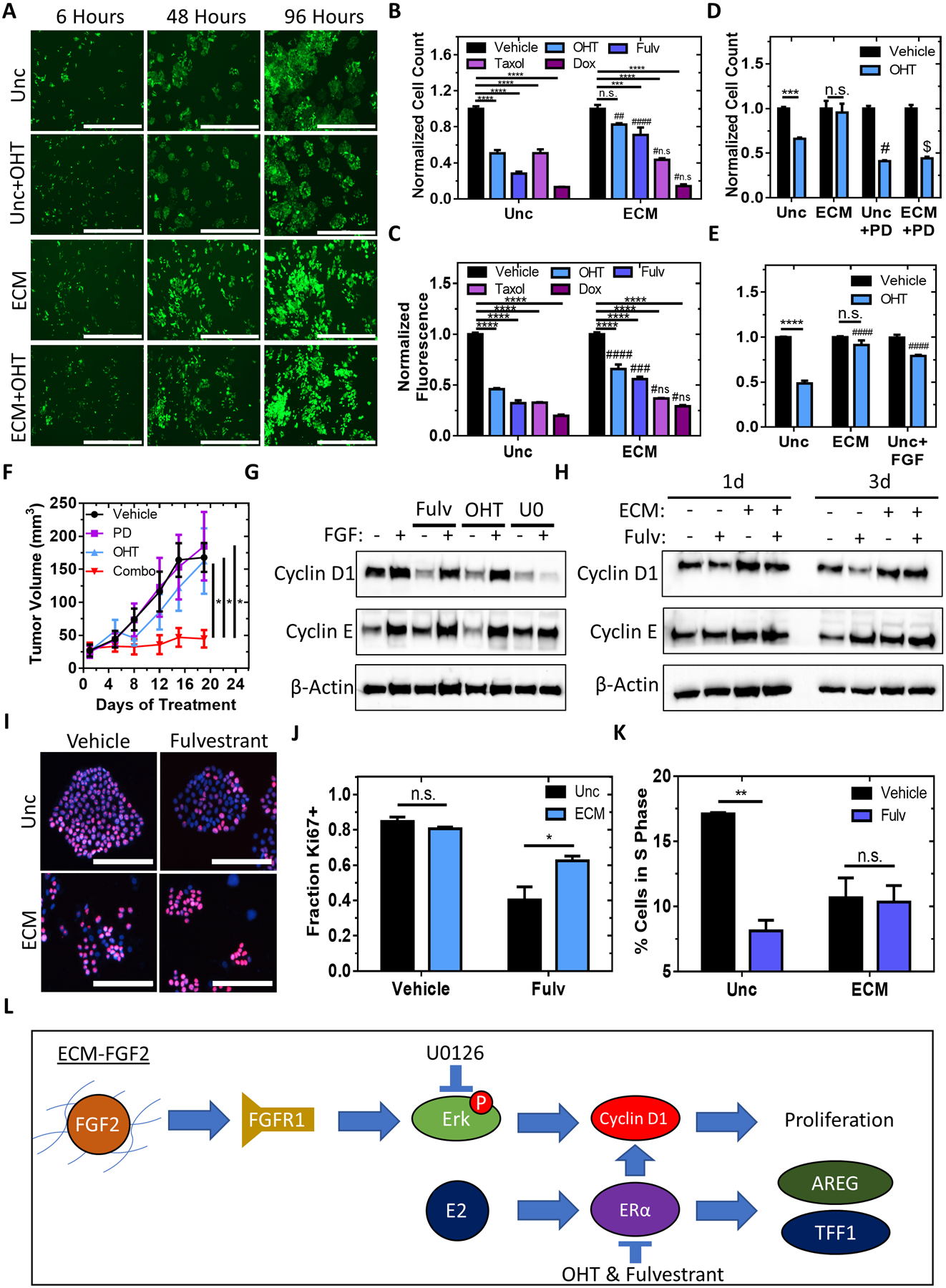
A, Representative time-lapse images of GFP-expressing MCF7 cells plated onto uncoated surfaces or ECM scaffolds with or without 1 μM OHT for 96 hours. Scale bar = 1000 μm. B, Normalized cell count of MCF7 cells treated for 4 days with 1 μM OHT, 1 μM fulvestrant, 100 nM doxorubicin, or 3 days with 5 nM taxol on uncoated surfaces or ECM scaffolds. n = 3–6 per condition from N = 1–2 independent experiments. Two-way ANOVA with Bonferonni post-test. **** P<0.0001, *** P<0.001, #### P<0.0001 compared to the same treatment with uncoated, ## P<0.01 compared to the same treatment with uncoated, #n.s. P >0.05 compared to the same treatment with uncoated. C, Normalized fluorescence of viability reagent after exposure to MCF7 cells plated on uncoated surfaces or ECM scaffolds treated for 4 days with 1 μM OHT, 1 μM fulvestrant, 2.5 nM taxol, or 100 nM doxorubicin. n=3–6. Two-way ANOVA with Bonferonni post-test. ### P <0.001 compared to the same treatment with uncoated. D and E, Normalized cell counts of MCF7 cells treated for 4 days with vehicle, 1 μM OHT, 1 μM OHT and 40 ng/mL FGF (D), or 1 μM OHT and 1 μM PD173074 (E) on uncoated surfaces or ECM scaffolds. n = 6 from N = 2 independent experiments. Two-way ANOVA with Bonferonni post-test, # P<0.05 compared to the same treatment with uncoated, $ P<0.0001 compared to the same treatment with ECM. F, Measurements of tumor volume of mice inoculated with MCF7 cells and treated with vehicle, PD173074, OHT, or a combination of both. n = 4–5 mice per condition. One-way ANOVA. * P<0.05. G, Immunoblot performed using protein lysates from MCF7 cells treated with or without 40 ng/mL FGF-2 and treated with 1 μM of OHT, 1 μM of fulvestrant, or 30 μM of U0126 for 72 hours to detect Cyclin D1, Cyclin E, and β-actin protein expression. H, Immunoblot performed using protein lysates from MCF7 cells cultured on uncoated surfaces or ECM scaffolds and treated with or without 1 μM of fulvestrant for 24 or 72 hours to detect Cyclin D1, Cyclin E, and β-actin protein expression. I, Representative images of Ki67- and DAPI-stained MCF7 cells plated onto uncoated surfaces or ECM scaffolds with or without 1 μM fulvestrant for 96 hours. Scale bar = 200 μm. J, Fraction of MCF7 cells positive for Ki67 on ECM scaffolds or uncoated surfaces after treatment with or without 1 μM fulvestrant for 96 hours. n = 2–3 wells per condition with total cell counts ranging from 905–6888 cells. Two-way ANOVA with Bonferonni post-test. K, Percentage of cells in S-phase after cell cycle analysis using propidium iodide staining of MCF7 cells on uncoated surfaces or ECM-scaffolds treated with or without 1 μM fulvestrant for 48 hours. N = 2 independent experiments with n = 8776–11674 cells per condition. Unpaired t-test. ** P < 0.01. L, Proposed mechanism.
We confirmed that the addition of exogenous FGF2 reduced OHT sensitivity in MCF7 cells cultured on uncoated surfaces, whereas PD173074 restored sensitivity to OHT for MCF7 cells cultured on ECM scaffolds (Figures 7D–E and Supplementary Figures 6L–M). To determine if PD173074 could also enhance the efficacy of hormone therapy in vivo, we treated mice bearing orthotopic tumors derived from MCF7 cells with vehicle, PD173074, OHT, or a combination of PD173074 and OHT. The combination of 5 mg/kg OHT with 5 mg/kg PD173074 reduced tumor growth (Figure 7F, Supplementary Figure 6N) indicating that FGFR inhibition enhanced the efficacy of OHT in vivo.
The addition of exogenous FGF2 to cell culture media has been shown to induce Cyclin D1 expression which reduces the sensitivity of ER+ cells to fulvestrant in vitro (16). Interestingly, we observed that both Cyclin D1 and Cyclin E are upregulated in MCF7 cells by both the addition of recombinant FGF2 (Figure 7G) or by culturing cells within ECM scaffolds (Figure 7H). FGF-induced Cyclin D1, but not Cyclin E, expression can be abrogated by blocking Erk phosphorylation (Figure 7G). Cyclin D1 expression is also maintained after fulvestrant treatment by culture within ECM scaffolds but not on uncoated surfaces (Figure 7H). To determine whether the increased level of Cyclin D1 caused reduced sensitivity to fulvestrant, we assessed the proliferative capacity of MCF7 cells plated on uncoated surfaces or ECM scaffolds (Figure 7I). The percentage of Ki-67 expressing cells decreased by 50% upon treatment with fulvestrant in cells cultured on uncoated surfaces whereas cells grown on ECM maintained their proliferative capacity and were resistant to fulvestrant-dependent G1 arrest (Figure 7J–K, Supplementary Figure 7A–B). Overall, the data demonstrated that ECM-FGF2 complexes stimulate the proliferation of ER+ breast cancer cells even in the presence of anti-estrogens (Figure 7L).
Discussion
Our goal was to determine the effects of culturing cells in a cell-derived ECM scaffold compared to standard 2D culture conditions. We identified a novel role for the ECM in FGF2-FGFR1 signaling that drives ER activity in the absence of estrogen, resulting in the induction of ER-responsive genes that promote tumor progression. ECM matrix proteins contain heparan sulfate proteoglycans that bind to and increase the local concentration of growth factors, such as FGF2 (46,47). We determined that ER+ breast cancer cells require 80-fold less ECM-bound FGF2 (Figure 6F) than exogenous FGF2 (Figure 6C) to achieve the same level of estrogen-independent ER activation. The results indicate that ECM-bound FGF2 may be more bioactive than soluble FGF2.
Recent work has demonstrated that exogenous FGF2 treatment can activate ER through ERK1/2 in a ligand-independent manner and can induce expression of TFF1, a hallmark ER-regulated gene (20). A recent study utilizing a 3D model of MCF7 cells demonstrated that ER transactivation in response to estrogen stimulation was stronger when MCF7 cells were cocultured with mammary fibroblasts (48); however, the authors did not investigate whether factors secreted from the fibroblasts were magnifying this effect. Our results show that fibroblast-produced ECM can act as a novel mediator of FGF2-induced ER signaling. We found that cells localized in fibrotic tumor regions had markedly higher levels of TFF1 (Figure 6G), a surrogate for ER-activation that has been implicated in tumor growth in xenograft models. We also show that in patients with breast cancer that have higher MBD, extratumoral tissue is enriched for the hallmark early and late estrogen response pathways (Figure 6), similar to our findings with MCF7 cells cultured on ECM scaffolds or in coculture with fibroblasts (Figure 2). Ultimately, these data demonstrate that fibrotic breast tissue may play a role in ligand-independent ER transcriptional activation.
The transcriptional activity of the ER can be regulated through several mechanisms, including dimerization and phosphorylation (4–6). The phosphorylation of the ER has been extensively studied, with nine characterized phosphorylation sites (39,40). Of these, phosphorylation at serine-118 and serine-167 occur via an Erk-dependent mechanism. We did not observe an increase in phosphorylation at serine-118 or serine-167 as a result of culturing ER+ cells on ECM (Supplementary Figure 3D). It remains possible that Erk phosphorylates a previously uncharacterized site on the ER or another protein that cooperates with ER, as both the total and phosphorylated levels of coactivator proteins that bind to ER also regulate the transcriptional activity of ER (49).
Previous work implicates factors secreted by stromal cells in the tumor microenvironment as contributing to hormone therapy resistance (15–20), but they do not consider the contribution of the ECM in this response. Our work demonstrates that ECM-FGF2 complexes reduce sensitivity to anti-estrogens via a Cyclin D1-dependent manner by preventing therapy-induced G1 arrest. The sensitivity to anti-estrogens can be restored by inhibiting FGFR1 both in vitro and in vivo. FGFR1 amplifications that lead to aberrant signaling are observed in up to 10% of breast cancers and drive endocrine therapy resistance (50) as well as act as an unfavorable, independent prognostic factor for overall survival in a cohort of 880 unselected breast tumors (51). FGFR1 amplification is also associated with resistance to inhibitors of CDK4/6 (45), of which Cyclin D1 is the regulatory subunit. A recent study found that patient-derived, ER+, FGFR1-amplified tumors are significantly enriched for the Hallmark early and late estrogen response pathways compared to non-amplified tumors when estrogen-deprived through letrozole treatment (52). As a result of these findings, clinical investigations of FGFR1 antagonists in combination with anti-estrogens are beginning to emerge. Following our in vitro data, our in vivo results revealed that the inhibition of FGFR1 with PD173074 enhances the anti-tumor effects of tamoxifen. Recent work indicates that FGFR1 amplification also promotes resistance to HER2 therapy in breast cancer (53), suggesting FGF2-ECM complex signaling may play a role in other breast cancer subtypes as well.
Our work demonstrates how ECM signaling can reduce sensitivity to endocrine therapy. Although high MBD is a risk factor for the development of breast cancer and its reduction is often used as a marker of success in preventative therapies, the biological relevance of MBD in breast cancer is still unknown (54). This suggests the possibility that the novel mechanism we uncovered may induce endocrine resistance in patients with ER+ breast cancer that have high MBD and an intact FGFR signaling pathway. Thus, patients with high MBD or high circulating levels of FGF2 may benefit from existing FGFR1-targeted therapies in combination with endocrine therapy. In addition to the ECM, the contribution of additional tumor microenvironmental factors such as adipose tissue and the immune milieu should be also considered. Additional research into the specific composition of fibrotic regions of breast tissue is warranted to lead to more effective and personalized therapies for patients with breast cancer.
Supplementary Material
Implications.
This work uncovers how the ECM may mediate signaling between growth factors and ER+ breast cancer cells to promote estrogen-independent ER signaling and resistance to endocrine therapy.
Acknowledgments
We thank I Chae Ye for assistance with RNA library preparation, Beza Woldemeskel for assistance with the preparation of RNA samples, and Dr. Elana Fertig for assistance with data visualization. Work in the Gilkes lab is supported by U54-CA210173 (NCI), R00-CA181352 (NCI), The V Scholar Foundation, Susan G. Komen Foundation (CCR17483484), The Jayne Koskinas Ted Giovanis Foundation for Health and Policy, The Emerson Collective, The Allegany Health Network, the NIH Predoctoral and Postdoctoral Training Program in Nanotechnology and Cancer (T32CA153952), and the SKCCC Core Grant (P50CA006973 (NCI)).
Footnotes
The authors declare no conflicts of interest.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 2018;68:394–424 [DOI] [PubMed] [Google Scholar]
- 2.Haque R, Ahmed SA, Inzhakova G, Shi J, Avila C, Polikoff J, et al. Impact of breast cancer subtypes and treatment on survival: an analysis spanning two decades. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2012;21:1848–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jia M, Dahlman-Wright K, Gustafsson JA. Estrogen receptor alpha and beta in health and disease. Best practice & research Clinical endocrinology & metabolism 2015;29:557–68 [DOI] [PubMed] [Google Scholar]
- 4.Sommer S, Fuqua SA. Estrogen receptor and breast cancer. Seminars in cancer biology 2001;11:339–52 [DOI] [PubMed] [Google Scholar]
- 5.Ikeda K, Horie-Inoue K, Inoue S. Identification of estrogen-responsive genes based on the DNA binding properties of estrogen receptors using high-throughput sequencing technology. Acta pharmacologica Sinica 2015;36:24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamaga R, Ikeda K, Horie-Inoue K, Ouchi Y, Suzuki Y, Inoue S. RNA sequencing of MCF-7 breast cancer cells identifies novel estrogen-responsive genes with functional estrogen receptor-binding sites in the vicinity of their transcription start sites. Hormones & cancer 2013;4:222–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chi D, Singhal H, Li L, Xiao T, Liu W, Pun M, et al. Estrogen receptor signaling is reprogrammed during breast tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America 2019;116:11437–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howell SJ, Johnston SR, Howell A. The use of selective estrogen receptor modulators and selective estrogen receptor down-regulators in breast cancer. Best practice & research Clinical endocrinology & metabolism 2004;18:47–66 [DOI] [PubMed] [Google Scholar]
- 9.AlFakeeh A, Brezden-Masley C. Overcoming endocrine resistance in hormone receptor-positive breast cancer. Curr Oncol 2018;25:S18–S27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan W, Chang J, Fu P. Endocrine therapy resistance in breast cancer: current status, possible mechanisms and overcoming strategies. Future medicinal chemistry 2015;7:1511–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeselsohn R, De Angelis C, Brown M, Schiff R. The Evolving Role of the Estrogen Receptor Mutations in Endocrine Therapy-Resistant Breast Cancer. Current oncology reports 2017;19:35. [DOI] [PubMed] [Google Scholar]
- 12.Nardone A, De Angelis C, Trivedi MV, Osborne CK, Schiff R. The changing role of ER in endocrine resistance. Breast 2015;24 Suppl 2:S60–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer metastasis reviews 2013;32:303–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast cancer research : BCR 2016;18:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nature reviews Cancer 2015;15:261–75 [DOI] [PubMed] [Google Scholar]
- 16.Shee K, Yang W, Hinds JW, Hampsch RA, Varn FS, Traphagen NA, et al. Therapeutically targeting tumor microenvironment-mediated drug resistance in estrogen receptor-positive breast cancer. The Journal of experimental medicine 2018;215:895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holton SE, Bergamaschi A, Katzenellenbogen BS, Bhargava R. Integration of molecular profiling and chemical imaging to elucidate fibroblast-microenvironment impact on cancer cell phenotype and endocrine resistance in breast cancer. PloS one 2014;9:e96878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nature reviews Clinical oncology 2018;15:366–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shee K, Jiang A, Varn FS, Liu S, Traphagen NA, Owens P, et al. Cytokine sensitivity screening highlights BMP4 pathway signaling as a therapeutic opportunity in ER(+) breast cancer. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2019;33:1644–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giulianelli S, Riggio M, Guillardoy T, Perez Pinero C, Gorostiaga MA, Sequeira G, et al. FGF2 induces breast cancer growth through ligand-independent activation and recruitment of ERalpha and PRBDelta4 isoform to MYC regulatory sequences. International journal of cancer 2019;145:1874–88 [DOI] [PubMed] [Google Scholar]
- 21.Rijal G, Li W. 3D scaffolds in breast cancer research. Biomaterials 2016;81:135–56 [DOI] [PubMed] [Google Scholar]
- 22.Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife 2014;3:e01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu J, Clark RAF. Fibronectin at select sites binds multiple growth factors and enhances their activity: expansion of the collaborative ECM-GF paradigm. The Journal of investigative dermatology 2014;134:895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009;326:1216–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macri L, Silverstein D, Clark RA. Growth factor binding to the pericellular matrix and its importance in tissue engineering. Advanced drug delivery reviews 2007;59:1366–81 [DOI] [PubMed] [Google Scholar]
- 26.Sawicka KM, Seeliger M, Musaev T, Macri LK, Clark RA. Fibronectin Interaction and Enhancement of Growth Factors: Importance for Wound Healing. Advances in wound care 2015;4:469–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhoyar S, Godet I, DiGiacomo JW, Gilkes DM. A software tool for the quantification of metastatic colony growth dynamics and size distributions in vitro and in vivo. PloS one 2018;13:e0209591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem 2013;288:10819–29 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Godet I, Shin YJ, Ju JA, Ye IC, Wang G, Gilkes DM. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nature communications 2019;10:4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods 2012;9:357–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems 2015;1:417–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 2014;11:783–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, Yakir M, et al. Structural and Molecular Mechanisms of Cytokine-Mediated Endocrine Resistance in Human Breast Cancer Cells. Molecular cell 2017;65:1122–35 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun X, Gierach GL, Sandhu R, Williams T, Midkiff BR, Lissowska J, et al. Relationship of mammographic density and gene expression: analysis of normal breast tissue surrounding breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2013;19:4972–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prestwich GD. Simplifying the extracellular matrix for 3-D cell culture and tissue engineering: a pragmatic approach. Journal of cellular biochemistry 2007;101:1370–83 [DOI] [PubMed] [Google Scholar]
- 38.Guan J, Zhou W, Hafner M, Blake RA, Chalouni C, Chen IP, et al. Therapeutic Ligands Antagonize Estrogen Receptor Function by Impairing Its Mobility. Cell 2019;178:949–63 e18 [DOI] [PubMed] [Google Scholar]
- 39.Anbalagan M, Rowan BG. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Molecular and cellular endocrinology 2015;418 Pt 3:264–72 [DOI] [PubMed] [Google Scholar]
- 40.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995;270:1491–4 [DOI] [PubMed] [Google Scholar]
- 41.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007;26:3291–310 [DOI] [PubMed] [Google Scholar]
- 42.Boyd NF, Rommens JM, Vogt K, Lee V, Hopper JL, Yaffe MJ, et al. Mammographic breast density as an intermediate phenotype for breast cancer. The Lancet Oncology 2005;6:798–808 [DOI] [PubMed] [Google Scholar]
- 43.Guo YP, Martin LJ, Hanna W, Banerjee D, Miller N, Fishell E, et al. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2001;10:243–8 [PubMed] [Google Scholar]
- 44.Sherratt MJ, McConnell JC, Streuli CH. Raised mammographic density: causative mechanisms and biological consequences. Breast cancer research : BCR 2016;18:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nature communications 2019;10:1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ornitz DM. FGFs, heparan sulfate and FGFRs: complex interactions essential for development. BioEssays : news and reviews in molecular, cellular and developmental biology 2000;22:108–12 [DOI] [PubMed] [Google Scholar]
- 47.Quarto N, Amalric F. Heparan sulfate proteoglycans as transducers of FGF-2 signalling. Journal of cell science 1994;107 (Pt 11):3201–12 [DOI] [PubMed] [Google Scholar]
- 48.Morgan MM, Livingston MK, Warrick JW, Stanek EM, Alarid ET, Beebe DJ, et al. Mammary fibroblasts reduce apoptosis and speed estrogen-induced hyperplasia in an organotypic MCF7-derived duct model. Scientific reports 2018;8:7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osborne CK, Schiff R, Fuqua SA, Shou J. Estrogen receptor: current understanding of its activation and modulation. Clinical cancer research : an official journal of the American Association for Cancer Research 2001;7:4338s–42s; discussion 411s-412s [PubMed] [Google Scholar]
- 50.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer research 2010;70:2085–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast cancer research : BCR 2007;9:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, Jansen VM, et al. Association of FGFR1 with ERalpha Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER(+) Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23:6138–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hanker AB, Garrett JT, Estrada MV, Moore PD, Ericsson PG, Koch JP, et al. HER2-Overexpressing Breast Cancers Amplify FGFR Signaling upon Acquisition of Resistance to Dual Therapeutic Blockade of HER2. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23:4323–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cuzick J, DeCensi A, Arun B, Brown PH, Castiglione M, Dunn B, et al. Preventive therapy for breast cancer: a consensus statement. The Lancet Oncology 2011;12:496–503 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data have been deposited in the GEO database under the accession code GSE137528.