

Abstract
Purpose:
A subset of pancreatic ductal adenocarcinomas (PDACs) is highly resistant to systemic chemotherapy, but no markers are available in clinical settings to identify this subset. We hypothesized that a glycan biomarker for PDAC called sTRA could be used for this purpose.
Experimental Design:
We tested for differences between PDACs classified by glycan expression in multiple systems: sets of cell lines, organoids, and isogenic cell lines; primary tumors; and blood plasma from human subjects.
Results:
The sTRA-expressing models tended to have stem-like gene expression and the capacity for mesenchymal differentiation, in contrast to the non-expressing models. The sTRA cell lines also had significantly increased resistance to seven different chemotherapeutics commonly used against pancreatic cancer. Patients with primary tumors that were positive for a gene-expression classifier for sTRA received no statistically significant benefit from adjuvant chemotherapy, in contrast to those negative for the signature. In another cohort, based on direct measurements of sTRA in tissue microarrays, the patients who were high in sTRA again had no statistically significant benefit from adjuvant chemotherapy. Furthermore, a blood-plasma test for the sTRA glycan identified the PDACs that showed rapid relapse following neoadjuvant chemotherapy.
Conclusions:
This research demonstrates that a glycan biomarker could have value to detect chemotherapy-resistant PDAC in clinical settings. This capability could aid in the development of stratified treatment plans and facilitate biomarker-guided trials targeting resistant PDAC.
Introduction
Systemic therapy is considered necessary for all patients with pancreatic ductal adenocarcinoma (PDAC), even those with localized disease, because most patients already have occult metastases at the time of diagnosis (1). Chemotherapy particularly benefits patients who have surgical resection of the tumor. In a seminal study that established adjuvant chemotherapy (chemotherapy applied after surgery) as standard of care for PDAC, the median disease-free survival after surgery improved to 13.4 months with gemcitabine from 6.9 months with observation (2). Further improvements in chemotherapy were demonstrated using the stronger FOLFIRINOX regimen (3), or gemcitabine in combination with capecitabine (4) or nab-paclitaxel (5). Systemic therapy is increasingly applied prior to surgery—called neoadjuvant therapy—in order to increase the percentage of patients who receive chemotherapy (6), since some patients have a delay or reduction in their adjuvant chemotherapy as a consequence of surgery. Neoadjuvant therapy could have the additional advantages of identifying patients with rapid progression who would not benefit from surgical intervention; treating occult metastases earlier; and downsizing the tumors to increase the chance for a margin-free (R0) resection (6).
While the combination of surgery plus systemic therapy results in significant benefit relative to surgery alone, a subset of PDACs is highly resistant to systemic therapy. Nearly 40% of patients receiving surgery plus gemcitabine monotherapy experience relapse within one year of surgery (2). Even in the subset of fit patients who are candidates for more aggressive chemotherapy regimens, over 25% relapse within one year (3). Currently, identifying this chemotherapy-resistant cohort prior to treatment remains a challenge, since conventional imaging, liquid biopsy, and molecular biomarkers are lacking.
The gene-expression subtypes defined in prior research (7–9) potentially provide some guidance to this problem. The consensus subtypes have been termed classical, basal (also referred to as quasi-mesenchymal), and exocrine, terms chosen to reflect the normal cell types that most closely correspond to the cancer cells. In retrospective evaluations of outcomes following curative resection, tumors with transcriptome profiles matching with the classical subtype had longer survival than the others (7–9). Likewise, among patients with metastatic PDAC, the classical subtype was associated with longer survival in retrospective analyses (10,11). On the other hand, patients with the classical subtype demonstrated no benefit from adjuvant chemotherapy (9,12), in contrast to patients with the basal type, and cell lines of the classical subtype are more resistant to chemotherapy than those of the basal type (8). Therefore, the predictive role of molecular subtyping in PDAC treatment remains to be established.
We recently identified a new biomarker of PDAC that is a cell-surface and secreted glycan called sTRA (sialylated tumor-related antigen) (13–15). Nothing was known about the relationship of this biomarker to subtype of cancer or drug resistance, but some clues were evident. A different subset of PDACs express sTRA, and the ones that do have outwardly different characteristics. The tumors expressing primarily sTRA tended to be sparse, poorly differentiated, or highly vacuolated, while those expressing mainly CA19-9 were part of well differentiated or moderately differentiated secretory glands (14). These facts suggested that the two groups represent distinct subtypes of tumors having differing biology and clinical behavior. Of particular interest was the possibility that the drug-resistant group observed in clinical care corresponds to a glycan-defined subtype.
Materials and Methods
Cell Culture
The PaTu-8988S and PaTu8988T cell lines were obtained from Creative Bioarray (Shirley, NY), and Colo357, L3.3, and L3.6PL lines were kindly provided by Dr. Isaiah J. Fidler (University of Texas, MD Anderson Cancer Center). The remaining cell lines were obtained from ATCC (Manassas, VA). All cell lines were cultured in RPMI-1640 supplemented with 5% fetal bovine serum, 2 mM L-Glutamine, and 100 IU/mL penicillin/streptomycin. The cells were grown at 37 °C in a humidified atmosphere supplemented with 5% (v/v) CO2. All cell lines were within ten passages of collection when used in the described experiments. The authenticities of the cell lines were confirmed by comparing their RNA expression profiles to those of previously authenticated cell lines and/or by short tandem repeat profiling (ATCC). Cross-contamination between cell lines was excluded by Infinium QC Array (see Supplemental Methods). All cell lines were mycoplasma free, as determined by RNAseq profiling and DAPI staining.
Drug Treatment Studies
The chemotherapeutic reagents cisplatin, etoposide, gemcitabine, and 5-FU were obtained from Sigma (St. Louis, MO). Irinotecan, oxaliplatin, and paclitaxel were obtained from Cayman Chemical (Ann Arbor, MI). All drugs were dissolved in dimethyl sulfoxide or dimethylformamide. For the preparation of FOLFIRINOX, 5-FU was first prepared in dimethylformamide, and the leucovorin, irinotecan, and oxaliplatin were each added in a 1:5 ratio by weight to 5-FU. The concentration of FOLFIRINOX was calculated based on the 5-FU concentration. Cells were seeded into 96 well plates at 2 × 103 cells per well and cultured for 3 d before treatment with a drug or drug mixture at six different concentrations each. After 3 d, cell viability was estimated using CellTiter-Glo (Promega, Madison, WI). The IC50 values were calculated using GraphPad Prism 6 (GraphPad Software, San Diego, CA) with 5-parameter, variable-slope fits.
Immunofluorescence
All immunofluorescent and chemical stains were performed using 5-μm-thick sections cut from formalin-fixed, paraffin-embedded blocks. Paraffin was removed from the sections using CitriSolv Hybrid (04355121, Fisher Scientific), and tissue was rehydrated through an ethanol gradient. Following rehydration, antigen retrieval was achieved through incubating slides in citrate buffer at 100 °C for 20 min. Slides were blocked in phosphate-buffered saline with 0.05% Tween-20 (PBST0.05) and 3% bovine serum albumin (BSA) for 1 h at RT. Primary antibodies (Supplemental Table 4) were labeled for immunofluorescent staining with either Sulfo-Cyanine5 NHS ester (13320, Lumiprobe) or Sulfo-Cyanine3 NHS ester (11320, Lumiprobe) so that two primary antibodies could be used simultaneously in each round of staining. Dialysis was performed following labeling to remove unreacted conjugate and the primary antibodies were then diluted into the same solution of PBST0.05 with 3% BSA to a final concentration of 10 μg/mL. Slides were incubated overnight with this solution at 4 °C in a humidified chamber.
The following day, the antibody solution was decanted, and the slides were washed twice in PBST0.05 and once in 1X PBS, each time for 3 min. The slides were dried via blotting and then incubated with DAPI at 10 μg/mL in 1X PBS for 15 min at room temperature (RT). Two 5-min washes were performed in 1X PBS, and then slides were cover-slipped and scanned using a fluorescent microscope. All slides were scanned for fluorescence using either Vectra (PerkinElmer) for the TMAs or the Axio Scan.Z1 (Zeiss) for the organoid sections. Each system collected data for each field of view at three different emission spectra. All image data were quantified using the SignalFinder-IF software (16).
Following scanning, slides were stored in a humidified chamber. Coverslips were removed for the subsequent rounds of staining by submerging the slide in deionized water at 37 °C until the coverslip floated free (between 30 and 60 min). Fluorescence was quenched between rounds by incubating the slides with 6% H2O2 in 250 mM sodium bicarbonate (pH 9.5-10) twice for 20 min at RT. Subsequent incubations and scanning steps were repeated as described above with different primary antibodies.
To perform sialidase treatment, slides were incubated with a 1:200 dilution (from a 50,000 U/mL stock) of α2-3,6,8 neuraminidase (P0720L, New England Biolabs) in 1X reaction buffer (5 mM CaCl2, 50 mM sodium acetate, pH 5.5) overnight at 37 °C. Slides were washed as described above, followed by subsequent antibody detections. Hematoxylin and eosin staining was performed following a standard protocol.
Immunoassays
The immunoassays were based on the method presented earlier (15). The capture antibodies were CA19-9, anti-MUC5AC, and anti-MUC16, and the biotinylated primary antibodies were CA19-9 or TRA-1-60 (details in Supplemental Table 4). The secondary detection agent was Cy5-conjugated streptavidin (Roche Applied Science). We diluted the samples of human plasma (8-fold and 32-fold) and cell line or organoid media (2-fold and 8-fold), into a buffer (1X PBS with 0.1% Tween-20, 0.1% Brij-35, species-specific blocking antibodies, and protease inhibitor) and incubated each sample on an antibody array overnight at 4 °C.
For sTRA detection, an extra step of enzyme treatment before antibody detection was needed. After sample incubation we prepared α2-3 neuraminidase (P0728L, New England Biolabs, Ipswich, MA) at a concentration of 250 U/mL in the supplied reaction buffer and incubated on arrays overnight at 37 °C. We incubated each array with a biotinylated antibody (3 μg/mL in 1X PBS with 0.1% Tween-20 and 0.1% BSA) and subsequently with Cy5-conjugated streptavidin (43-4316, Invitrogen, Carlsbad, CA) (2 μg/mL in the same buffer as the primary antibody). The slides were scanned for fluorescence at 635 nm using a microarray scanner (Innopsys InnoScan 1100 AL). The quantification of fluorescence was performed using SignalFinder-MA (16). All plasma and media samples were repeated in at least three independent experiments. The CA19-9 values were obtained through the clinical laboratory services at the Medical College of Wisconsin for the discovery set, and they were determined using a custom immunoassay previously validated in the Haab laboratory (15) for the test set.
Statistics
Differences between marker-positive and marker-negative cell lines in IC50 and percent viability were tested using the Mann-Whitney U test. Differential expression in the RNAseq data was tested using empirical Bayes quasi-likelihood F-tests, and the p values were adjusted using the Benjamini-Hochberg method. Differences in OS between patient groups in the survival analyses were evaluated with the log-rank test. Differences between patient groups in proportions of patients with long or short OS were analyzed with the Fisher’s Exact test and the Breslow-Day test for homogeneity of odds ratios. Differences in sensitivity and the average of sensitivity and specificity were analyzed with the Wald test based on bootstrap SE estimate. P values of less than 0.05 were considered significant.
Human Specimens
The plasma samples were collected under protocols approved by the Institutional Review Boards at the University of Pittsburgh Medical Center and the Medical College of Wisconsin. The donors consisted of patients diagnosed with pancreatic cancer who were scheduled to undergo neoadjuvant therapy. The plasma collections (EDTA plasma) took place prior to any surgical, diagnostic, or medical procedures and were performed according to the standard operating procedure from the Early Detection Research Network. The samples were frozen at −70 °C or colder within 4 h of time of collection. Aliquots were shipped on dry ice and thawed no more than three times prior to analysis.
The tissue samples for tissue microarrays were collected under approved protocols at the Medical University of South Carolina, University of Pittsburgh Medical Center, and Memorial Sloan Kettering Cancer Center. All subjects provided written, informed consent, and all methods were performed in accordance with an assurance filed with and approved by the U.S. Department of Health and Human Services and in accordance with the guidelines contained in the Belmont Report.
Results
We initially tested for differences between PDACs classified by glycan expression using a panel of 27 cell lines. We classified each cell line based on the sTRA glycan or the CA19-9 glycan (Figure 1A). Both glycans are capped with sialic acid on type-1 N-acetyl-lactosamine (LacNAc), the disaccharide of galactose linked β1,3 to N-acetyl-glucosamine (GlcNAc), and the CA19-9 epitope has a fucose attached to the N-acetyl-glucosamine, which is necessary for its recognition by selectin receptors. Type-1 LacNAc, as recognized by the TRA-1-60 antibody (17), is a marker for induced pluripotent stem cells, but the sialylated version has not been well studied due to lack of an effective antibody. We indirectly detected the sialylated structure using sialidase to uncover the TRA-1-60 epitope (Figure 1A).
Figure 1. Complementary glycan expression in model systems.
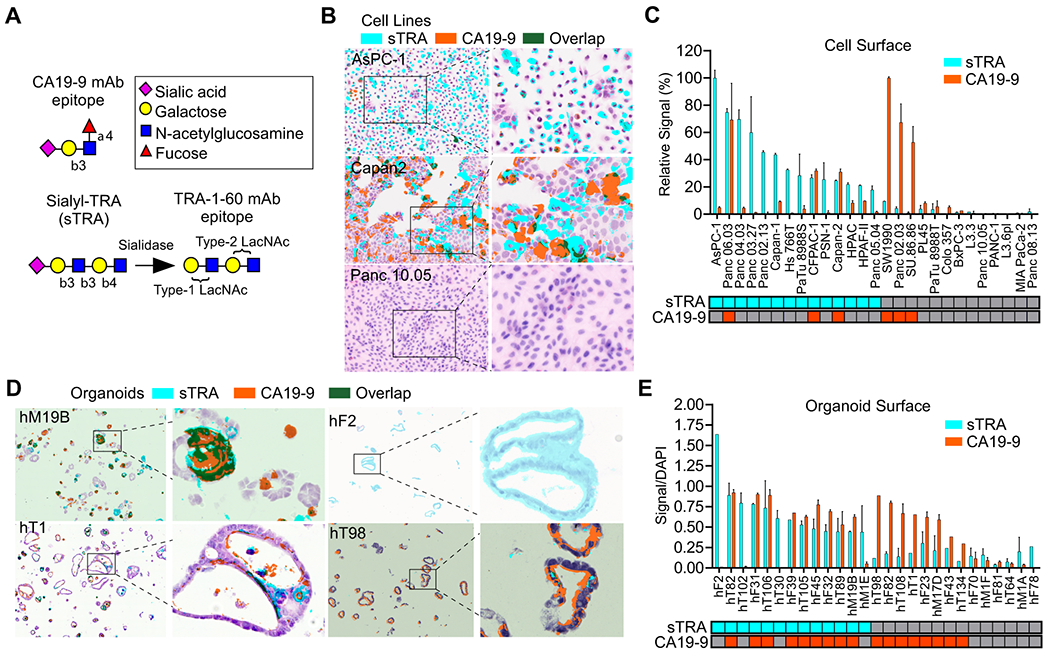
A) Glycan structures. B) Diverse sTRA and CA19-9 expression in cell culture. C) Cell-surface expression of 27 cell lines. The classification of positivity at the bottom was based on a cutoff of signal-to-noise ratio > 3. D) Diverse glycan expression in organoid models. E) Cell-surface glycan expression of 27 organoid models. The classification of positivity at the bottom used cutoffs that optimized the discrimination between background signal and true marker expression. The magnification was 20X for the non-zoomed images.
The cell-surface expression of sTRA and CA19-9 was variable among the cell lines, with some primarily expressing only one glycan and others expressing both or neither (Figs. 1B and 1C). Organoid models of pancreatic cancer(18,19) likewise showed variable expression of one, both, or neither of the glycans (Figure 1D), with slightly different proportions among them (Figure 1E).
Gene-expression programs distinguishing the glycan-defined subtypes
Using the cell lines and organoids, we then asked whether similar differences exist in gene transcription programs. A total of 267 genes were differentially expressed between the sTRA-expressing cell lines (not including the three lines also expressing CA19-9) and all others (Figure 2A and Supplemental Table 1). No individual genes were differentially expressed between the CA19-9 and sTRA groups at p < 0.05 after multiple-hypothesis correction, possibly due to the lower number of CA19-9-positive lines. The sTRA-associated genes had ontologies that were enriched in developmental, drug metabolism, and glycan-biosynthesis pathways (Figure 2A). The developmental gene BMP4 was a strong individual marker of sTRA cells, as was CYP3A5 (Figure 2A), a gene previously identified as a marker of PDACs identified as classical and exocrine (20). In addition, 9 of 14 sTRA-expressing lines were identified as classical, based on the gene classifier called PDAssigner (8), compared with 2 of 6 CA19-9-expressing and 0 of 10 glycan-negative lines (Figure 2A), suggesting that sTRA is more likely to recognize the classical subtype. Gene-set enrichment analysis showed that sets defining stem-like differentiation, stem-like metabolism, and the classical subtype were enriched in the sTRA-expressing cells (Figure 2B and Supplemental Table 1). Among individual genes that have been proposed as markers of classical, the expressions of GATA6 and CYP3A5 were higher in the sTRA cells (Figure 2C). KRT81 and HNF1A showed weak associations with sTRA.
Figure 2. Differential gene expression.
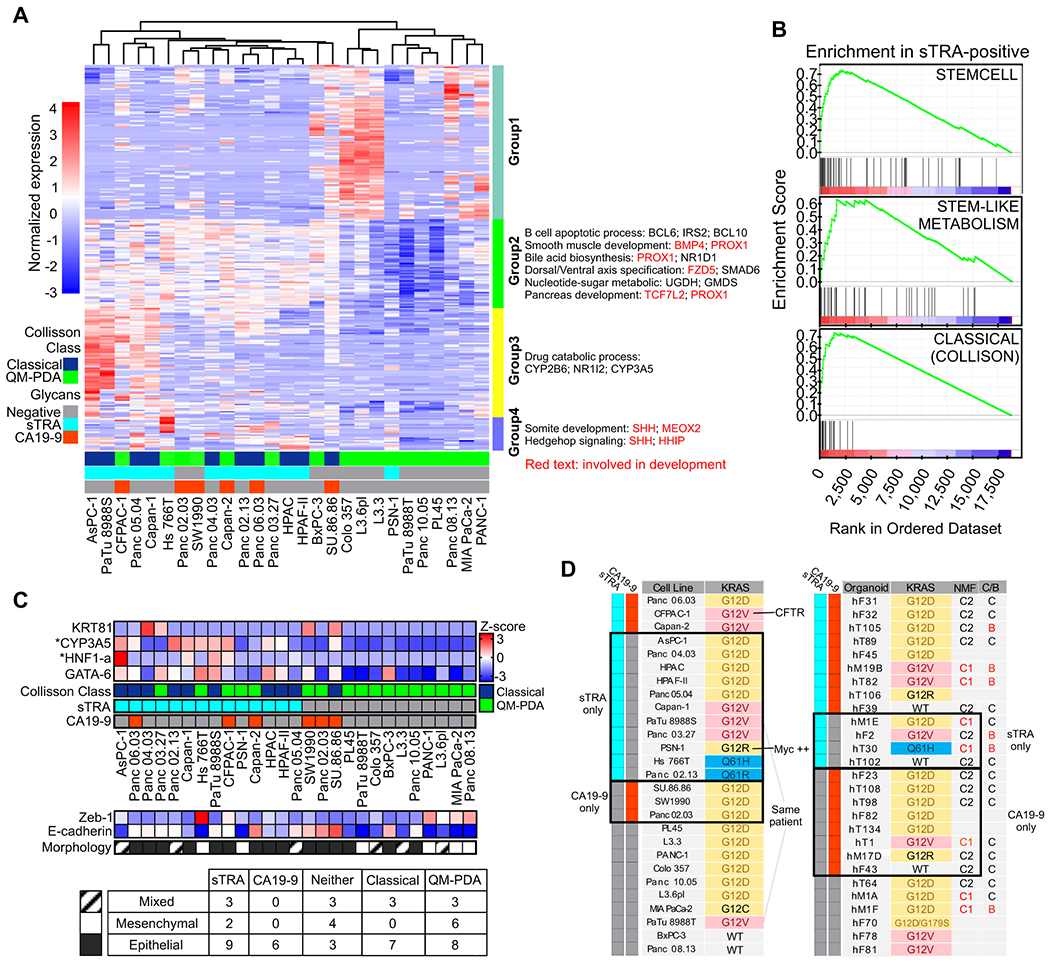
A) Differentially expressed genes and pathways. B) Enrichment of previously identified sets in the sTRA-expressing cells. C) Relationships between class markers, morphology, and glycan expression across the cell lines. CYP3A5 and HNF1-a were significantly elevated in the sTRA group compared with the non-sTRA group after multiple-testing correction D) KRAS mutation status associated with the glycan-defined groups. NMF, nonnegative matrix factorization to identify C1 and C2 clusters in the organoids. The C/B column indicates the classical or basal status based on a gene set developed for the organoids.
The epithelial/mesenchymal state of cancer cells has been widely explored as an indicator of their origin, invasiveness, or overall tumor-forming aggressiveness. All six of the CA19-9-expressing cell lines were epithelial, as determined by the gene expression of Zeb-1 and E-cadherin (Figure 2C) and by morphology (Supplemental Figure 1), but the sTRA-expressing cells and those expressing neither glycan were of various types (Figure 2C). The organoid models all had epithelial morphologies, but three of the models had mesenchymal characteristics by gene expression (not shown). Two of these produced sTRA exclusively and the third produced neither glycan. The data from both model systems suggest that some sTRA-expressing cancer cells have the potential for mesenchymal-like differentiation, in contrast to CA19-9-expressing cells.
The type of KRAS mutation in cancer potentially can drive differences in phenotype (21). The less-common Q61 alteration appeared exclusively in the cell lines and organoids that expressed only sTRA (Figure 2D). The G12V mutation was in 3 of 11 cell lines and 1 of 3 organoids that expressed only sTRA, in comparison to 0 of 3 cell lines and 1 of 8 organoids that expressed only CA19-9. While these observations are based on relatively small sample sizes, they suggest that the Q61H/R mutation fosters cancers that express sTRA in the absence of CA19-9.
Resistance to chemotherapy in sTRA-high cultures
We determined the resistance of the 27 cell lines to eight chemotherapeutics that are either front-line or alternative treatments against pancreatic cancer. Our hypothesis was that sTRA-positive cancers are more treatment-resistant than those which are sTRA-negative. In a single-dose study, the sTRA-expressing cell lines were more resistant than the sTRA-negative cell lines in each case (Figure 3A). In dose-response analyses to obtain the IC50 concentrations (Figure 3B, 3C, and Supplemental Figure 2), the sTRA cells were significantly more resistant than the non-sTRA cells (p < 0.05, Mann-Whitney U Test) to 6 of the 8 drugs. For gemcitabine, the resistance was higher in the sTRA group but with less statistical significance (p = 0.07, Mann-Whitney U Test); for oxaliplatin, resistance was similar between the groups. As a negative control, we performed the same analysis for CA19-9, since it is a glycan that is not a biomarker for resistance. CA19-9 did not define a resistant group (Supplemental Figure 2), indicating that we are not non-specifically detecting a general elevation in glycans.
Figure 3. Drug resistance differences between glycan-defined types.
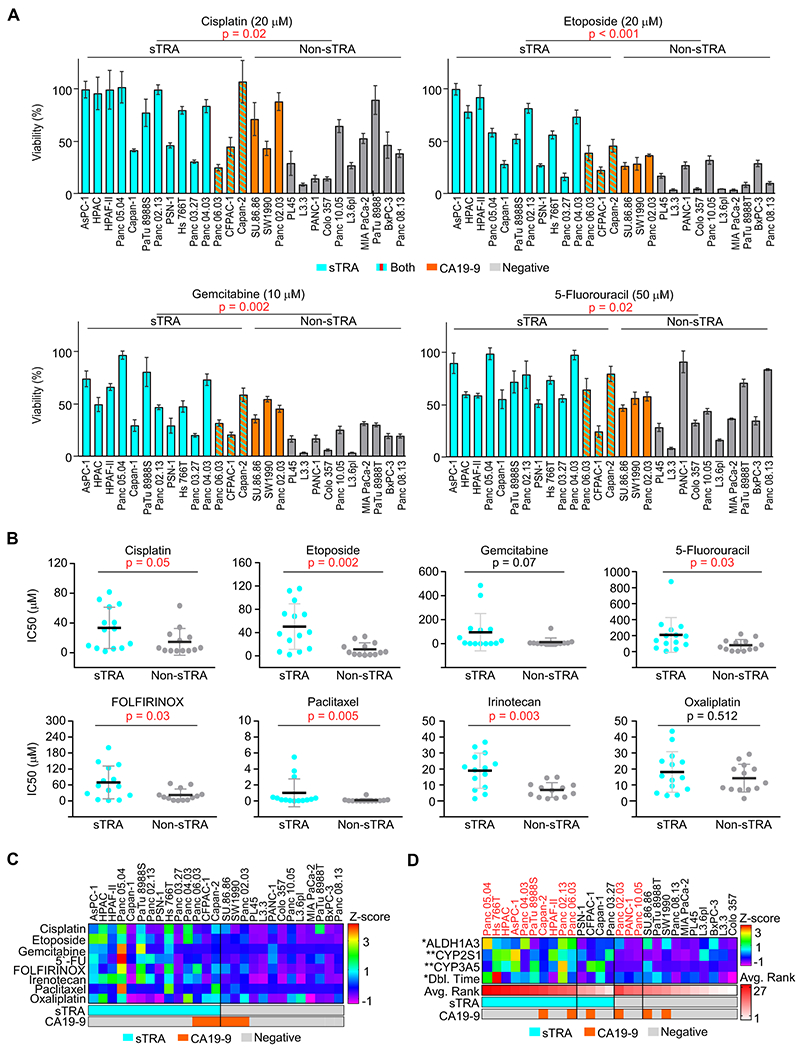
A) Single-dose viability in the panel of 27 cell lines. B) IC50 values calculated from dose-response curves, grouped by marker group. P Values were calculated by Mann–Whitney test. C) Summarized IC50 values for all drugs and cell lines. Z-scores were used to normalize the scales of the IC50 values for comparisons. D) Factors and markers associated with resistance. Z-scores were used to normalize the gene-expression data and the doubling times. Avg. Rank is the average across drugs of the ranks in IC50 of each cell line among the panel of 27.
We asked whether the high resistance corresponded to traits that have been associated with resistance. The sTRA-positive lines had higher levels of drug-metabolizing enzymes from the cytochrome P450 family (p < 0.05, Mann-Whitney U test), and they had trends toward higher levels of the stem marker ALDH1A3 and longer doubling times (Figure 3D). Thus, some sTRA lines have resistance traits, but the mechanism of resistance may differ between cell lines.
We further tested the above relationships using sets of isogenic cell lines, where all cell lines in a set are from the same individual. We repeatedly cultured the L3.3 cell line in sublethal concentrations of each of four drugs, followed by recovery and outgrowth of the surviving cells, and found that sTRA expression was increased in several of the sublines, both in the percentage of stained cells and in total staining intensity. The sublines with higher sTRA coincided with significantly increased resistance to the drugs (Supplement Figure 3).
Predictive value of the sTRA levels in primary tumors
Next, we tested whether the sTRA levels in primary tumors are associated with resistance to systemic chemotherapy. We determined sTRA levels in two ways, by a gene-expression classifier and by immunofluorescence. To develop a gene-expression classifier, we identified the significantly up-regulated or down-regulated genes (p < 0.02, Bayes quasi-likelihood F-test after multiple-testing correction) associated with sTRA expression in the panel of 27 cell lines (Supplemental Table 1) and used the algorithm from PDAssigner (8) to assign classes. We used this algorithm because of its previous robust performance and its simplicity for adoption with new gene sets. We applied the classifier to 150 cases of PDAC from The Cancer Genome Atlas (22) and to 180 cases from the International Cancer Genome Consortium (23) that had survival information. In both cohorts, distinct groups of patients showed overall differences in expression between genes associated with high sTRA and those associated with low sTRA (Figure 4A). Tests of group differences in central tendency using the classifier genes showed significance (p = 0.001, Adonis test, Vegan R package). This finding confirmed the consistency between the cell lines and both cohorts in the differential expression of the gene groups, and it supports the idea that the classifier identifies true subtypes rather than random variation in expression patterns.
Figure 4. A gene-expression classifier associated with drug resistance.
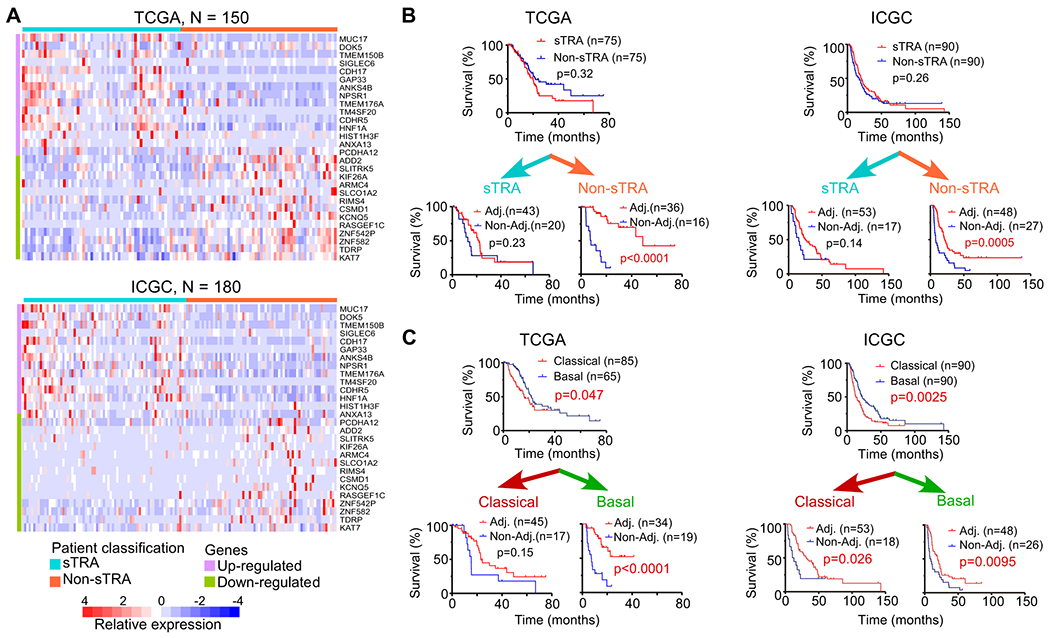
A) Classification of patients using a signature for sTRA. The 28-gene classifier applied to the TCGA (top) and ICGC (bottom) datasets produced two distinct groups of patients. The color bar at top shows the classification used in the subsequent analyses. B) Survival curves grouped by the gene-expression classifier for sTRA positive or negative. C) Survival curves grouped by classical or basal status. The classical and basal classes were from a previous publication for the TCGA dataset and were calculated from the data for the ICGC dataset. The p values are based on the log-rank test.
We assigned the patients to an sTRA or non-sTRA group based on the median of the calculated score of the classifier. No difference in overall survival (OS) was evident between the sTRA and non-sTRA groups of patients, but among the patients assigned to the non-sTRA group, those receiving adjuvant therapy had significantly longer overall survival (OS) than those who did not (p < 0.001, Log-rank test) (Figure 4B). Among the patients assigned to the sTRA group, no difference was observed. Both the TCGA and ICGC data sets showed this relationship. An analysis of progression-free survival (PFS) instead of OS showed the same results but on a shorter timescale (Supplemental Fig. 4), indicating that OS results were not biased by differences in treatments applied after progression. Other classifiers for the classical subtype gave similar results (Figure 4C), but not as consistently between data sets as the sTRA classifier.
In a parallel approach, we asked whether the directly measured amount of sTRA in primary tumors associated with a lack of response to adjuvant therapy. This experiment used tissue microarrays (TMAs) that included tumor tissue collected at the Memorial Sloan Kettering Cancer Center from patients who either did or did not receive adjuvant therapy and who had long (> 3 years) or short (< 1 year) OS following surgery (Supplemental Table 2 and ref. (24)). The adjuvant chemotherapy consisted mainly of gemcitabine or fluorouracil, based on the institutional treatment paradigm at the time.
We measured sTRA and CA19-9 in the TMAs using used multimarker immunofluorescence (14,25) in conjunction with previously developed software that enables unbiased, automated quantification of multimarker IF data (14, 16) (Supplemental Fig. 5A and Supplemental Table 2). The sTRA and CA19-9 levels showed little correlation with each other (Supplemental Fig. 5B), but the sTRA levels were higher in the short-OS group (p = 0.0077, Wilcoxon Rank-Sum test) (Supplemental Fig. 5C). Among the patients receiving adjuvant chemotherapy, those with high sTRA showed a significantly lower proportion of long OS than those with low sTRA (p = 0.003, Fisher’s Exact test) (Supplemental Fig. 5D). Among the patients with high sTRA, those receiving adjuvant chemotherapy had a significantly lower proportion of long OS than those not receiving neoadjuvant chemotherapy (p = 0.01, Fisher’s Exact test). No other comparison showed a significant difference. The Breslow-Day test for homogeneity of odds ratios (for association between survival and therapy) across biomarker-defined subgroups was highly significant for sTRA (p = 0.006) but was not significant for CA19-9 (p = 0.18). These results indicate a differential effect of adjuvant therapy on patient survival between the sTRA-high and sTRA-low groups.
Another pair of TMAs provided complementary information in that the tumors had been exposed to neoadjuvant therapy. The TMAs included tumor tissue from PDAC resections (14) at the University of Pittsburgh Medical Center. We found that the tumors that were dominated by cells producing only sTRA or only CA19-9 were in the short-OS group, with few exceptions, while tumors without such clonal outgrowth were evenly distributed in the short-OS and long-OS groups (Supplemental Figure 6 and Supplemental Table 2). This observation suggests that the persistence of sTRA-dominant cells following neoadjuvant chemotherapy portends poor outcome.
Predicting rapid relapse using a blood test
The above findings presented the possibility that high plasma sTRA identifies PDACs that do not benefit from systemic chemotherapy. We investigated this possibility using plasma samples from patient scheduled to receive neoadjuvant therapy at the Medical College of Wisconsin (MCW). The patients received treatments as determined by stage of disease, resectable cancer receiving chemoradiation and borderline-resectable cancer receiving chemotherapy followed by chemoradiation. The chemotherapy consisted mainly of FOLFIRINOX or gemcitabine/nab-paclitaxel, as determined by age and performance status. No association with outcome was detected for any particular regimen. Many of the patients received additional adjuvant chemotherapy after surgery, but the use of additional treatment was not associated with outcome.
We measured the plasma levels of sTRA glycan using sandwich immunoassays, in which we detect sTRA on the proteins captured by one of three different capture antibodies (Figure 5A). The use of this assay as a surrogate for tumor sTRA was supported by separate analyses. First, the agreement of the cell-surface expression of the glycan and the amount in the conditioned media was high for both the cell lines and organoids (Supplemental Figure 7). Furthermore, in a previous study we found that the peripheral blood glycans correlated with tumor glycans for cell-line xenograft mouse models, patient-derived xenograft mouse models, and human PDAC patients (14).
Figure 5. A blood test to predict rapid relapse following chemotherapy.
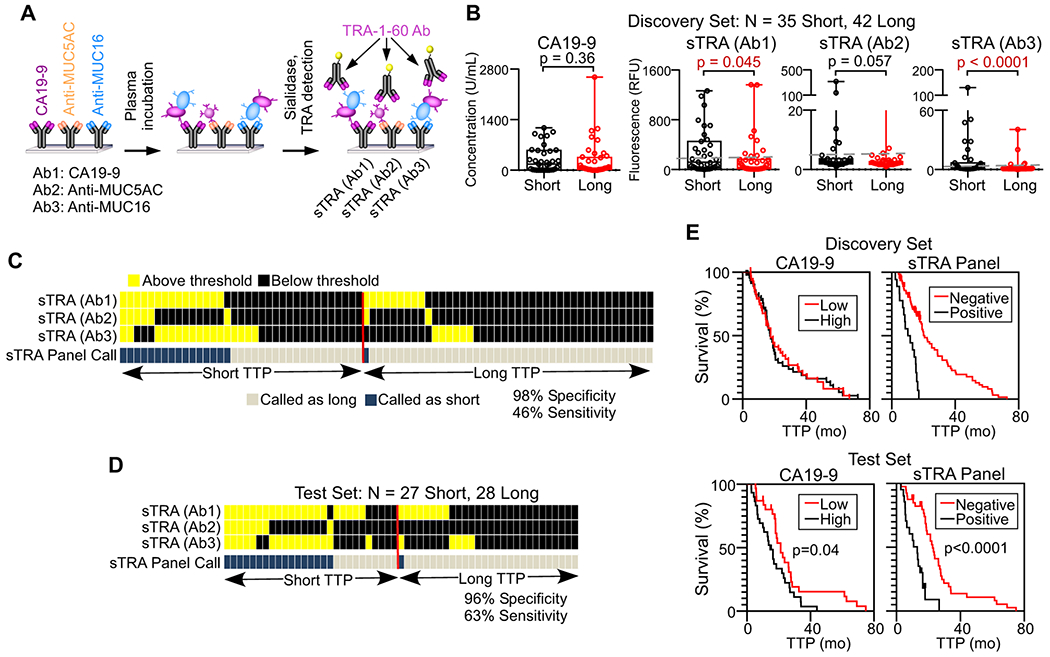
A) Immunoassays used for measuring sTRA in patient plasma. B) Comparisons between short and long TTP, based on 18 months, of the indicated immunoassays. The values are the averages of three independent experiments. C) Patterns of high and low values across the three sTRA immunoassays in the discovery set. Each column represents a patient sample. Samples with elevation in two or three markers were classified as cases. D) Patterns of high and low values across the three sTRA immunoassays in the test set, using optimized thresholds. E) Survival curves in the discovery and test sets. For CA19-9, patients above the median were classified as high. For the panel, patients with elevations in two or three markers were classified as positive.
We asked whether any of the biomarkers could serve as an indicator of short time-to-progression (TTP), with disease progression diagnosed based on CT scans at 3-4 months intervals for the first two years and at six months intervals thereafter. We dichotomized the patients using a cutoff of 18 months from the time of diagnosis (Supplemental Table 3), based on the approximate rate of 50% recurrence within one year after the completion of treatments and on the clinical observation that such recurrence strongly suggests treatment resistance. We did not dichotomize by pathological response (based on surgical pathology from resection), as it is a subjective assessment that does not correlate with TTP or OS, or by radiographic response prior to surgery, since it is weakly associated with TTP and OS.
In a discovery set, two of the sTRA immunoassays were significantly higher in subjects with short TTP than in subjects with long TTP (Figure 5B). Patients elevated in two or more of the sTRA assays (using thresholds optimized for each marker, Figure 5B) were especially likely to have short TTP, as 16 of 17 had short TTP (94% PPV) (Figure 5C and Table 1). To minimize the effect of overfitting on the estimate of panel performance, we further assessed the panel performance using five-fold cross-validation with 200-fold bootstrapping (re-samplings of the cohort). The improvements in cross-validated sensitivity and the average of sensitivity and specificity were statistically significant (p < 0.0001, Wald test based on bootstrap SE estimate) (Table 1).
Table 1. Performance of the sTRA Panel and CA19-9 in the discovery and test sets.
CI, confidence interval. Bolded text of numbers indicates statistical significance.
| Sample set/performance | Marker | Sensitivity (95% CI) | Specificity (95% CI) | (Sens + Spec)/2 (95% CI) |
|---|---|---|---|---|
| Discovery Set | ||||
| Naïve Performance | sTRA Panel | 45.7 | 97.6 | 71.7 |
| CA19-9 | 2.9 | 95.2 | 49 | |
| Cross-Validated Performance | sTRA Panel | 45.4 (28.2, 63.7) | 92.5 (80.7, 97.3) | 68.9 (57.7, 78.3) |
| CA19-9 | 3.0 (0.1, 48.9) | 96.2 (85.9, 99.1) | 49.6 (44.8, 54.4) | |
| Difference | **42.3 (21.7, 62.9) | −3.7 (−11.8, 4.4) | **19.3 (8.1, 30.6) | |
| Test Set | ||||
| Blinded Performance | sTRA Panel | 55.6 (37.2, 72.5) | 96.4 (78.1, 99.5) | 76.0 (64.8, 84.5) |
| CA19-9 | 11.1 (3.6, 29.2) | 96.4 (78.8, 99.5) | 53.8 (46.9, 84.5) | |
| Difference | **44.4 (25.8, 63.1) | 0 (−10.1, 10.1) | **22.2 (11.6, 32.9) | |
| Naïve Performance | sTRA Panel | 63 | 96.4 | 79.7 |
| CA19-9 | 11.1 | 96.4 | 53.8 | |
| Cross-Validated Performance | sTRA Panel | 66.2 (44.8, 82.5) | 90.6 (75.4, 96.8) | 78.4 (64.6, 87.8) |
| CA19-9 | 13.3 (1.4, 61.6) | 96.0 (82.4, 99.2) | 54.6 (41.7, 67.0) | |
| Difference | **52.9 (24.4, 81.4) | −5.4 (−16.1, 5.3) | *23.8 (8.5, 39.0) |
p < 0.0001
p < 0.01 (Wald test with bootstrap standard error estimate).
We then applied the panel to a blinded test set. We applied the thresholds that were derived from the discovery set to the test set and made case/control calls on the blinded samples. The calls were sent to the collaborators who collected the samples, and upon comparison with the true outcomes data, the result was 96% specificity (27/28 with long TTP were high in 1 or less) and 56% sensitivity (15/27 with short TTP were high in 2 or more) (Table 1). The improvements in sensitivity and the average of sensitivity and specificity were statistically significant (p < 0.0001, Wald test based on 1000-fold bootstrap SE estimate, Table1).
Adjustments to the individual marker thresholds (naïve performance) gave 94% PPV (17/18 with two or more marker elevations had short TTP), 96% specificity (27/28 with long TTP), and 63% sensitivity (17/27 with short TTP) (Figure 5D and Table 1). The three individual sTRA assays showed strong associations with short TTP (p = 0.008 to 0.00008, Mann-Whitney U test), as did CA19-9 (p = 0.007) (Supplemental Figure 8). The sTRA panel also identified differences in TTP in Kaplan-Meier analysis (Figure 5E). Kaplan-Meier analysis is appropriate here because the cohort is a random selection of the patients seen in the clinic. In both sets, patients positive in the panel had shorter TTP than the rest of the patients. In the test set, the difference was highly significant (p < 0.0001, log-rank test) for sTRA and moderately associated (p = 0.04) for CA19-9.
To further test these associations, we performed additional analysis on data from a previous study of these markers (15) that used plasma collected at the University of Pittsburgh Medical Center. We obtained outcomes for a subset of patients who were scheduled to receive neoadjuvant therapy (data in Supplemental Table 3). The study was not designed for this question but nevertheless could provide insights. In two separate cohorts, two of the sTRA assays trended with short overall survival (p = 0.05 and 0.06, Supplemental Figure 8). CA19-9 showed no such trend in either cohort. These findings substantiate the use of a blood test for sTRA to identify a subtype of pancreatic cancers that is resistant to chemotherapy.
Discussion
This research demonstrates the use of the sTRA glycan to identify the PDAC cases that are highly resistant to chemotherapy. The precise performance of the biomarker for treatment prediction will need to be determined and validated in larger, prospective studies, but the work here establishes the relationship and the validity of the finding in clinical samples as well as model systems. The immediate implication of this result relates to the development of treatment plans for patients with resistant PDAC. For patients with resectable PDAC, potentially morbid operations could be avoided if rapid relapse following surgery could be predicted a priori. For patients with metastatic disease and patients undergoing neoadjuvant therapy, a practical biomarker could guide the choices and comparisons of the treatment options. For example, FOLFIRINOX is suggested to be slightly better than gemcitabine for the classical subtype (10,26), possibly indicating a difference between the sTRA-positive and sTRA-negative types. Patient stratification also could improve the selection of patients that receive nab-paclitaxel (5). Furthermore, the sTRA assay could be used in subgroup analyses of the many human trials currently underway, given that many trials do not meet primary objectives but show evidence of efficacy in subgroups. Trials could involve targeted therapies suggested from studies on the cell-culture and organoid models that are available for sTRA-positive and sTRA-negative PDACs. Thus, the biomarker could have value for patient stratification using current options, as well as for research using model systems and in biomarker-guided drug trials.
A blood test has particular value in the clinical setting because physicians could stratify patients prior to any treatments, without a biopsy. Furthermore, a blood test would capture secretions from the whole tumor rather than just the cells that are sampled by a biopsy, which may not reflect the heterogeneity of the tumor. Various blood tests have potential value for detecting or diagnosing PDAC, including mutated DNA in the circulation (27,28), tumor exosomes, and metabolites (29–31), but they do not predict therapeutic responses. Highly elevated CA19-9 in the blood and the failure to drop to normal levels following neoadjuvant therapy or surgery (32,33) are unfavorable prognostic factors (34), but its value is as a tumor-volume indicator rather than as a subtype indicator or a means to predict resistance to chemotherapy (32). The sTRA marker, in contrast, differentiates biological subtypes. As with any new test, blinded, prospective studies using a clinical assay will be required to fully assess the value of the sTRA test to patients and physicians.
The sTRA and CA19-9 tests could be used in combination, however, in the surveillance setting (15). For example, if a patient were high in either sTRA or CA19-9 (or both), the patient would be further evaluated in order to confirm or negate a diagnosis of cancer. In the event of a diagnosis of pancreatic cancer, the sTRA-elevated cancers would be predicted to be more resistant to chemotherapy than the sTRA-non-elevated cancers. The cutoff used for sTRA likely would be different in the surveillance setting. The previous study of sTRA in the surveillance setting (15) used cutoffs that captured about 65% of the patients, whereas cutoffs optimized for treatment-response prediction captured a smaller percentage, suggesting that both resistant and sensitive PDACs would be detected by the combined sTRA and CA19-9 panel.
In the use of sTRA for treatment-response prediction, a current limitation is that some of the resistant PDACs would be missed because they do not make the sTRA antigens. In our study of patients receiving neoadjuvant therapy (Fig. 5), 47% of the combined cohorts had short TTP, but 25% of the patients were high in plasma sTRA, working out to 53% of the subjects with short TTP. Consistent with this observation, one of the very-resistant cell lines, AsPC-1, makes cell-surface sTRA but does not secrete it well (Supplemental Figure 7), suggesting that a subset of the resistant tumors do not secrete the antigen in enough quantity to be detectable in blood. Nevertheless, the test picks up a substantial proportion of the resistant cancers and could represent a valuable step forward that can be built upon. Additional research could test for biomarkers to detect the remainder of the resistant PDACs.
This work extends previous findings relating to the prognostic and predictive value of the molecular subtypes. The classical subtype in previous research indicated a better prognosis than the basal subtype, but, on the other hand, it tended to benefit less from chemotherapy (9,12). This result is also consistent with an in vitro study in cultured cells, which indicated that classical-subtype cells were more resistant to gemcitabine (8). But the relative value for prognosis versus treatment prediction was not clear, and a recent study suggested that the classical subtype is more sensitive to chemotherapy than the basal subtype (11). The differences between the studies may result from several sources. The latter study included non-resectable, advanced PDAC (the COMPASS Trial), while the former studies involved resectable PDAC. Advanced cancer could be less responsive in general than localized disease. The COMPASS study also used a different classifier that was more stringent than the original, and the study was not designed to distinguish prognostic from predictive value, since it did not include a non-treatment control group. Overall, the results indicate that native prognosis and sensitivity to chemotherapy are not necessarily linked, and that the classical subtype is suboptimal for decoupling these traits.
The sTRA subtype seems to distinguish the traits better: it was indicative of chemotherapy resistance, but not of a poor prognosis. It encompassed resistant cancers of both subtypes and was more consistent than the classical/basal system in identifying resistance in the primary tumors. A valid model is that the sTRA subtype more precisely identifies resistant tumors, but the classical subtype could be more effective for stratifying by native prognosis. Ultimately, the typing of cancers and prediction of drug responses could involve both glycans and other types of markers. Further studies should focus on clarifying additional markers that are suggestive of other subtypes. The genes HNF1A, CDH17, LGALS4, and CYP3A5 have been variously assigned as markers of non-basal subtypes including exocrine and classical but without good agreement between studies (35). The elevation of these genes in most sTRA cancers could indicate that a third subtype is at least partially encompassed by sTRA.
Building on these findings, our next steps will involve the validation of clinical assays for prospective studies and the analyses of model systems in order to understand the susceptibilities of sTRA-positive cancers. Based on the gene-expression results, a successful path may involve metabolic approaches (36). Alternatively, probing the sTRA-positive subtype for dependencies on particular nutrient sources may be feasible. These directions in research are made possible because we now have a practical assay to detect chemoresistant PDAC using either tissue or plasma. The use of such an assay in model systems, and then in clinical specimens to detect and follow the resistant subtype, should help both the development and the application of new treatments against PDAC.
Supplementary Material
Translational Relevance.
Patients afflicted with pancreatic ductal adenocarcinoma (PDAC) face a dismal prognosis, but headway could be made if physicians could identify subgroups with differing responses to therapy. In previous work, we identified a new biomarker of pancreatic cancer—a glycan called sTRA—but nothing was known about its relationship to drug resistance. We found that assays for sTRA using either tumor tissue or blood plasma identified patients who had no benefit from adjuvant chemotherapy or rapid relapse following neoadjuvant therapy. The translational relevance of this work is that physicians could be provided a practical assay to stratify PDAC patients according to predicted response to chemotherapy. A blood-plasma assay is important because biopsy specimens from the pancreas can be difficult and risky to obtain, with sometimes uncertain results. In addition, researchers could use the biomarker in research to develop new therapies targeting treatment-resistant PDAC and to test the therapies in biomarker-guided drug trials.
Acknowledgements
We thank the core services at the Van Andel Institute for expert support of this research, particularly the Optical Imaging, Bioinformatics and Biostatistics, Genomics, and Pathology and Biorepository cores. We thank Zachary Klamer, MS, at the Van Andel Institute for support in the processing and analysis of the plasma biomarker data; Toshinori Hinoue, PhD, at the Van Andel Institute for advising on the analysis of TCGA and ICGC datasets, and Christine Decapite at the University of Pittsburgh Medical Center for assistance with data coordination.
Funding
U01CA152653, Early Detection Research Network, National Cancer Institute, to R.E.B. and B.B.H.; U01CA226158, Alliance of Glycobiologists for Cancer Detection, National Cancer Institute, B.B.H.; D. A. Tuveson: Distinguished Scholar and Director of the Lustgarten Foundation–Designated Laboratory of Pancreatic Cancer Research. D. Plenker is supported by the German Research Foundation (DFG) (PL 894/1-1).
Footnotes
Conflict of Interest Statement: The authors declare no potential conflicts of interest.
References
- 1.Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol 2018;15:333–48. [DOI] [PubMed] [Google Scholar]
- 2.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med 2018;379:2395–406. [DOI] [PubMed] [Google Scholar]
- 4.Neoptolemos JP, Palmer DH, Ghaneh P, Psarelli EE, Valle JW, Halloran CM, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011–24. [DOI] [PubMed] [Google Scholar]
- 5.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asare EA, Evans DB, Erickson BA, Aburajab M, Tolat P, Tsai S. Neoadjuvant treatment sequencing adds value to the care of patients with operable pancreatic cancer. J Surg Oncol 2016;114:291–5. [DOI] [PubMed] [Google Scholar]
- 7.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 8.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;4:500–03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin Cancer Res 2018;24:1344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Kane GM, Grunwald BT, Jang GH, Masoomian M, Picardo S, Grant RC, et al. GATA6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin Cancer Res 2020;epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 12.Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2019;16:207–20. [DOI] [PubMed] [Google Scholar]
- 13.Tang H, Partyka K, Hsueh P, Sinha JY, Kletter D, Zeh H, et al. Glycans related to the CA19-9 antigen are elevated in distinct subsets of pancreatic cancers and improve diagnostic accuracy over CA19-9. Cell Mol Gastroenterol Hepatol 2016;2:201–21 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnett D, Liu Y, Partyka K, Huang Y, Tang H, Hostetter G, et al. The CA19-9 and Sialyl-TRA Antigens Define Separate Subpopulations of Pancreatic Cancer Cells. Sci Rep 2017;7:4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staal B, Liu Y, Barnett D, Hsueh P, He Z, Gao CF, et al. The sTRA Plasma Biomarker: Blinded Validation of Improved Accuracy over CA19-9 in Pancreatic Cancer Diagnosis. Clin Cancer Res 2019;29:2745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barnett D, Hall J, Haab B. Automated Identification and Quantification of Signals in Multichannel Immunofluorescence Images: The SignalFinder Platform. Am J Pathol 2019;189:1402–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews PW, Banting G, Damjanov I, Arnaud D, Avner P. Three monoclonal antibodies defining distinct differentiation antigens associated with different high molecular weight polypeptides on the surface of human embryonal carcinoma cells. Hybridoma 1984;3:347–61. [DOI] [PubMed] [Google Scholar]
- 18.Boj SF, Hwang C, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2014;160:324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov 2018;8:1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noll EM, Eisen C, Stenzinger A, Espinet E, Muckenhuber A, Klein C, et al. CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma. Nat Med 2016;22:278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamidi H, Lu M, Chau K, Anderson L, Fejzo M, Ginther C, et al. KRAS mutational subtype and copy number predict in vitro response of human pancreatic cancer cell lines to MEK inhibition. Br J Cancer 2014;111:1788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biankin AV. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32:185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connor AA, Denroche RE, Jang GH, Timms L, Kalimuthu SN, Selander I, et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol 2017;3:774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winter JM, Tang LH, Klimstra DS, Brennan MF, Brody JR, Rocha FG, et al. A novel survival-based tissue microarray of pancreatic cancer validates MUC1 and mesothelin as biomarkers. PLoS ONE 2012;7:e40157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A 2013;110:11982–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muckenhuber A, Berger AK, Schlitter AM, Steiger K, Konukiewitz B, Trumpp A, et al. Pancreatic Ductal Adenocarcinoma Subtyping Using the Biomarkers Hepatocyte Nuclear Factor-1A and Cytokeratin-81 Correlates with Outcome and Treatment Response. Clin Cancer Res 2018;24:351–59. [DOI] [PubMed] [Google Scholar]
- 27.Cohen JD, Javed AA, Thoburn C, Wong F, Tie J, Gibbs P, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci U S A 2017;114:10202–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359:926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lennon AM, Wolfgang CL, Canto MI, Klein AP, Herman JM, Goggins M, et al. The Early Detection of Pancreatic Cancer: What Will It Take to Diagnose and Treat Curable Pancreatic Neoplasia? Cancer Res 2014;74:3381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly KA, Hollingsworth MA, Brand RE, Liu CH, Singh VK, Srivastava S, et al. Advances in Biomedical Imaging, Bioengineering, and Related Technologies for the Development of Biomarkers of Pancreatic Disease: Summary of a National Institute of Diabetes and Digestive and Kidney Diseases and National Institute of Biomedical Imaging and Bioengineering Workshop. Pancreas 2015;44:1185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young MR, Wagner PD, Ghosh S, Rinaudo JA, Baker SG, Zaret KS, et al. Validation of Biomarkers for Early Detection of Pancreatic Cancer: Summary of The Alliance of Pancreatic Cancer Consortia for Biomarkers for Early Detection Workshop. Pancreas 2018;47:135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai S, George B, Wittmann D, Ritch PS, Krepline AN, Aldakkak M, et al. Importance of Normalization of CA19-9 Levels Following Neoadjuvant Therapy in Patients With Localized Pancreatic Cancer. Ann Surg 2018;271:740–47. [DOI] [PubMed] [Google Scholar]
- 33.Boone BA, Steve J, Zenati MS, Hogg ME, Singhi AD, Bartlett DL, et al. Serum CA 19-9 response to neoadjuvant therapy is associated with outcome in pancreatic adenocarcinoma. Ann Surg Oncol 2014;21:4351–8. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa K, Akahori T, Nishiwada S, Nagai M, Nakamura K, Tanaka T, et al. Prognostic factors for actual long-term survival in the era of multidisciplinary treatment for pancreatic ductal adenocarcinoma. Langenbecks Arch Surg 2018;403:693–700. [DOI] [PubMed] [Google Scholar]
- 35.Martens S, Lefesvre P, Nicolle R, Biankin AV, Puleo F, Van Laethem JL, et al. Different shades of pancreatic ductal adenocarcinoma, different paths towards precision therapeutic applications. Ann Oncol 2019;30:1428–36. [DOI] [PubMed] [Google Scholar]
- 36.Halbrook CJ, Lyssiotis CA. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017;31:5–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.