

Abstract
Signaling from multiple receptor tyrosine kinases (RTKs) contributes to therapeutic resistance in glioblastoma (GBM). Heparan sulfate (HS), present on cell surfaces and in the extracellular matrix, regulates cell signaling via several mechanisms. To investigate the role for HS in promoting RTK signaling in GBM, we generated neural progenitor cells deficient for heparan sulfate (HS) by knockout of the essential HS-biosynthetic enzyme Ext1, and studied tumor initiation and progression. HS-null cells had decreased proliferation, invasion, and reduced activation of multiple RTKs compared to control. In vivo tumor establishment was significantly decreased and rate of tumor growth reduced with HS deficient cells implanted in an HS-poor microenvironment. To investigate if HS regulates RTK activation through platelet-derived growth factor receptor α (PDGFRα) signaling, we removed cell surface HS in patient-derived GBM lines and identified reduced cell surface PDGF-BB ligand. Reduced ligand levels were associated with decreased phosphorylation of PDGFRα suggesting HS promotes ligand-receptor interaction. Using human GBM tumorspheres and a murine GBM model, we show that ligand-mediated signaling can partially rescue cells from targeted RTK inhibition and that this effect is regulated by HS. Indeed, tumor cells deficient for HS had increased sensitivity to EGFR inhibition in vitro and in vivo.
INTRODUCTION
The malignant behavior of glioblastoma (GBM) is dependent, in part, on signaling from multiple receptor tyrosine kinases (RTKs). Among the most commonly altered RTKs is epidermal growth factor receptor (EGFR), which is amplified and/or mutated in over 50% of these tumors (1,2). Despite its importance in the malignant biology of GBM, attempts to inhibit EGFR signaling have failed to improve outcomes for GBM patients (3). One explanation for this failure is RTK functional redundancy, which has shown that alternative RTKs, such as platelet-derived growth factor receptor α (PDGFRA), insulin-like growth factor receptor (IGFR1), fibroblast growth factor receptors (FGFRs), and c-Met, can compensate for reduced RTK signaling when EGFR is being therapeutically targeted (4,5).
An aspect of RTK signaling that has experienced only limited investigation in GBM, as well as in the response to RTK targeted therapy, is growth factor bioavailability (6,7). Heparan sulfate proteoglycans (HSPGs), present on cell surfaces and in the extracellular matrix (ECM), bind to diverse partners, including soluble growth factors, membrane proteins and components of the ECM (8,9). Thus, HSPGs play an important role in regulating ligand bioavailability and RTK signaling. While HSPG function depends on both its core protein and covalently attached heparan sulfate (HS) glycosaminoglycan chains, the HS chains are primarily responsible for determining ligand specificity and binding. A critical step essential for HS biosynthesis is performed by the HS-polymerizing enzymes EXT1 (exostosin 1) and EXT2 (exostosin 2) that encode glycosyltransferases that elongate heparan sulfate chains (10,11). Conditional disruption of EXT1 has established its essential role in mammalian brain development (12), and EXT1–/– and EXT2–/– mice have profound developmental defects resulting in embryonic lethality (13,14). In the embryonic spinal cord HS 6-O-sulfation levels, regulated by the extracellular sulfatases (Sulfs), create a morphogen gradient of Shh necessary for the normal motor neuron to oligodendrocyte progenitor cell switch (15,16). In the adult brain, HS continues to regulate cell signaling. Alterations in HS structure promote glial scar formation after central nervous system injury (17) and mediate tau localization in neurodegenerative disease (18).
In GBM, HS is an important component of the cellular microenvironment and promotes PDGFRA signaling, angiogenesis, and tumor cell invasion (19–22). Data from in vitro studies and model organisms have indicated several potential mechanisms by which HS can regulate ligand-mediated signaling, including: direct binding to and sequestration of soluble ligands; localization of ligand to specific cell surface regions; and acting as a co-receptor on the cell surface (8,23–25).
Given the importance of HS in regulating growth factor-mediated signaling in the brain, we investigated HS regulation of multiple RTKs in GBM and its contribution to innate resistance to RTK inhibitor therapy. We have pursued this in three ways: 1) By generation of tumor-prone murine neural progenitor cells with Ext1knockout, therefore preventing biosynthesis of HS; 2) use of explant cultures derived from patient-derived xenografts enzymatically cleared of cell surface HS; and 3) investigation of in vivo tumor growth in different tumor microenvironments. Loss of cell surface HS decreased the activity of multiple RTKs, reduced cell surface bound PDGF-BB, and decreased tumor cell growth in vitro and in vivo. Our studies highlight the importance of HS-mediated regulation of growth factor signaling in the biology of GBM and its contribution to RTK inhibitor resistance.
MATERIALS AND METHODS
Cell culture conditions & reagents.
Murine neural progenitor cells (mNPCs), including both untransduced cells and tumor-prone cells containing EGFRvIII (mNPC-E*), and patient-derived GBM tumorspheres were cultured as described previously using minimal essential media under non-adherent conditions (20,22,26). Patient-derived GBM tumorsphere lines GBM43 (from C.D.J.), GBM5 (from C.D.J.), and GBM1 (SF9487 from UCSF) established and demonstrated to have tumor-generating capacity, were maintained under non-adherent conditions as tumorspheres (20,26–28). The three human lines were selected based on their molecular and transcriptional properties as follows: GBM43 has moderate expression of cell surface HS (20), expression of EGFR with response to erlotinib, and expression of PDGFRA with a robust signaling response to PDGF-BB (20); GBM1 also has moderate expression of cell surface HS and expression of EGFR with response to erlotinib (20); and GBM5 has high levels of cell surface HS expression (20). All cell lines were analyzed before using and each subsequent year by short tandem repeat (STR) analysis to ensure the identity and validity of cells and confirm by PCR that they are mycoplasma negative. Cells were passaged no more than 15–20 times and then a new frozen aliquot of cells was used. Unless otherwise specified media and supplements were purchased from Thermo Fisher Scientific (Waltham, MA). Invasion assays use Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix (Corning Inc., Corning, NY). The EGFR tyrosine kinase inhibitor, erlotinib (#OSI-744), was purchased from Selleckchem (Houston, TX). Cell viability was assessed using CellTiter-Glo® 3D Reagent (Promega, Madison, WI) or DyLight800 (Thermo Fisher Scientific). To measure self-renewal (sphere-forming ability) and to assess sphere size, neurospheres were dissociated by trituration, then replated at clonal density in round bottom 96 well ultra-low attachment plates (Corning). Wells with more than one cell per well were eliminated by visual inspection and secondary neurospheres were counted and imaged on days 4 and 6 (DFC3000G, Leica, Wetzlar, Germany). Sphere size was quantified using ImageJ (RRID:SCR_003070).
Generation of HS-null murine neural progenitor cells transduced with EGFRvIII (mNPC-E*).
Mice harboring loxP-flanked Ext1 (Ext1f/f) (strain B6;129-Ext1<tm1Vcs>/J, #009326) were purchased from The Jackson Laboratory (Bar Harbor, ME) and backcrossed a minimum of 10 generations into FVB/N Ink4a/Arf-+/−. All experiments were executed in compliance with institutional guidelines and regulations and after approval from the appropriate institutional review board (UCSF Office of Research Institutional Animal Care and Use Program, #AN169893). Murine neural progenitor cells were isolated from the subventricular zone (SVZ) of 4-week-old male Ink4a/Arf−/−;Ext1fl/f mice as described previously (22,26,29). Neural progenitor cells maintained as neurospheres were transduced with EGFRvIII by retrovirus and Cre recombinase by adenovirus (Ad-GFP-2A-iCre, #1772, Vector Biolabs, Malvern, PA) sequentially. The pBabe vector based on the Moloney Murine Leukemia Virus (MoMuLV) containing EGFRvIII was used to generate retrovirus (21, 25, 28). After expanding, heparan sulfate-negative cells (HS-null) were enriched by cell sorting (SH800, Sony Biotechnology Inc., San Jose, CA) following immunostaining with anti-heparan sulfate antibody (10E4 epitope) and Alexa488-labeled anti-mouse IgM antibody. Isogenic control cells were treated in parallel and were transduced with GFP (Ad-CMV-GFP, #1768, Vector Biolabs, Malvern, PA), expanded, and sorted for GFP-positive cells (HS-wt) by cell sorting. Cell surface HS levels were re-evaluated every six months and in tumor cells cultured from in vivo tumors and remained stable.
Antibodies.
Antibodies used for flow cytometry, Western blotting, and immunostaining included: mouse anti-heparan sulfate (clone 10E4, US Biological Cat #H1890, RRID:AB_10013601); mouse anti-heparan sulfate (clone F69–3G10, AMSBIO Cat# 370260–1, RRID:AB_10892311); mouse anti-chondroitin sulfate (clone CS-56, Sigma-Aldrich Cat #C8035, RRID:AB_476879); rabbit anti-Ki-67 (clone 30–9, Ventana Medical Systems Cat #790–4286, RRID:AB_2631262); mouse anti-phosphotyrosine (Millipore Cat #05–321, RRID:AB_309678), rabbit anti-phosphorylated PDGFRα (Y742) (R&D Systems Cat# AF2114, RRID:AB_416551); rabbit anti-PDGFRα (Millipore Cat #07–276, RRID:AB_310485); mouse anti-GAPDH antibody (Millipore Cat #MAB374, RRID:AB_2107445); and from Cell Signaling Technology rabbit anti-phosphorylated AKT (S473) (Cat #4060, RRID:AB_2315049), rabbit anti-AKT (Cat #9272, RRID:AB_329827), rabbit anti-phosphorylated ERK1/2 (T202/T204) (Cat #9101, RRID:AB_331646), mouse anti-ERK1/2 Cat #9107, RRID:AB_10695739), and rabbit anti-β-actin antibody (Cat #4967, RRID:AB_330288). Secondary antibodies included Alexa488-labeled goat anti-rabbit IgG antibody (Cat #A11008, RRID:AB_143165), Alexa488-labeled anti-mouse IgM (Cat #A-11001, RRID:AB_2534069), and Alexa568-labeled goat anti-mouse IgG antibody (Cat #A11004, RRID:AB_2534072) from Thermo Fisher Scientific. For immunostaining of formalin-fixed paraffin-embedded sections the 3G10 neo-epitope was generated by digestion of HS by heparitinase I and II for four hours at 37°C following proteinase K antigen retrieval. The negative control is staining in the absence of enzyme.
Removal of cell surface heparan sulfate and chondroitin sulfate.
Cells were dissociated by treatment with Accutase™ (Sigma) and incubated with or without heparitinase I, II, III from Flavobacterium heparinum as described previously (20) (kind gift from Dr. Kuberan Balagurunathan, University of Utah) (Sigma) for 1 h at 37°C. Cells (5 × 105) were washed with PBS, blocked in PBS plus 5% goat serum on ice for 15 min, washed twice with ice-cold PBS plus 1% BSA, incubated on ice for 1 h with an anti-heparan sulfate antibody diluted in PBS plus 1% BSA, and detected with Alexa488-labeled secondary antibodies. Cells were analyzed by FACS Canto II Flow Cytometer (BD Bioscience, CA).
Assessment of receptor tyrosine kinase signaling pathway activity.
Protein lysates from mNPC-E* were analyzed by Mouse Phospho-RTK Array Kit (R&D Systems) per the manufacture’s protocol (300 μg) or by Western blotting (20μg total protein) using an Odyssey detection system (LI-COR, Lincoln, NE). The murine RTK array contains 39 RTK capture and 4 control antibodies spotted in duplicate on a membrane. The human RTK array contains 49 RTK capture antibodies spotted in duplicate in addition to the control antibodies.
In vivo experiments.
All experiments were executed in compliance with institutional guidelines and regulations and after approval from the appropriate institutional review board (UCSF Office of Research Institutional Animal Care and Use Program, #AN169893). Cell suspensions of HS-null and HS-wt mNPC-E* (1 × 107 cells/ml in PBS) were diluted with equal volume of matrigel (8.4 mg/ml) (BD). Under inhalation anesthesia with isoflurane, 1 × 106 cells were injected subcutaneously in 4-weeks-aged male athymic nude mice (Charles River Laboratories, RGD Cat #2312499, RRID:RGD_2312499) (2 μl per injection site). Tumor volumes were calculated as major axis × minor axis × height. For inhibition of EGFR, when tumor volume reached 200 mm3 (designated Day 0 of treatment), erlotinib (150 mg/kg) was administered intraperitoneal in 15% Captisol® (Ligand Pharmaceuticals, Inc., San Diego, CA) every other day for a total of three doses. Mice were weighed and subcutaneous tumor volumes were measured daily. For intracranial allografts 1 × 105 cells diluted in sterile saline were injected using a stereotactic device over 5 minutes as described previously (21). Mice were weighed daily and monitored for signs of tumor development. Numbers of mice used in each experiment are listed in corresponding figure legend.
Cell surface tagged PDGF-BB ligand.
Cells were washed twice, incubated on ice for 1h with biotinylated-human PDGF-BB (Fluorokine® Biotinylated-human PDGF-BB Kit) (R&D Systems), and bound ligand was detected using a FITC-labeled avidin probe and a FACS Canto II Flow Cytometer (BD). Cell treatment with heparitinase I, II and III, Chondroitinase ABC, or heparin was performed prior to incubation with biotinylated-human PDGF-BB.
Statistics.
All statistics were performed using Graph Pad Prism 6.0 software (Graph Pad Software, San Diego, CA). All data are presented as mean ± standard deviation (SD). One-way ANOVA, the student’s t-test analysis, and Mann-Whitney test were performed at confidence interval levels of 95%. Log-rank (Mantel-Cox) test was used for survival analysis.
RESULTS
Decreased tumor cell growth and invasion from cells deficient in heparan sulfate.
Carbohydrates on the cell surface and in the extracellular matrix, including glycosaminoglycans, O-glycans, and glycolipids, influence cell signaling by modulating RTK function (21,22,30,31). To investigate the role for HS glycosaminoglycans in ligand-mediated signaling in GBM, we developed a murine model for GBM in which HS biosynthesis could be knocked out. Exostosin-1 (Ext1) encodes an enzyme that elongates HS chains and is essential for HS biosynthesis and for embryonic viability (13,14). Ext1f/f/Ink4a/Arf−/− mice were generated from mice harboring loxP-flanked Ext1 (Ext1f/f) and Inka4a/Arf-null (Ink4a/Arf−/−) alleles. Neural progenitor cells were isolated from the subventricular zone (SVZ) and transduced with EGFRvIII, a constitutively active and oncogenic form of EGFR. These cells were then transduced with Cre-expressing adenovirus, then flow sorted to identify cells lacking surface HS (HS-null) (Fig 1A and B). Biosynthetic functions of EXT1 could then be investigated using these HS-null tumor-prone progenitor cells. Control cells were transduced with GFP-expressing adenovirus and sorted for GFP expression (HS-wt).
Fig. 1. Heparan sulfate deficient cells exhibit attenuated tumor phenotypes.
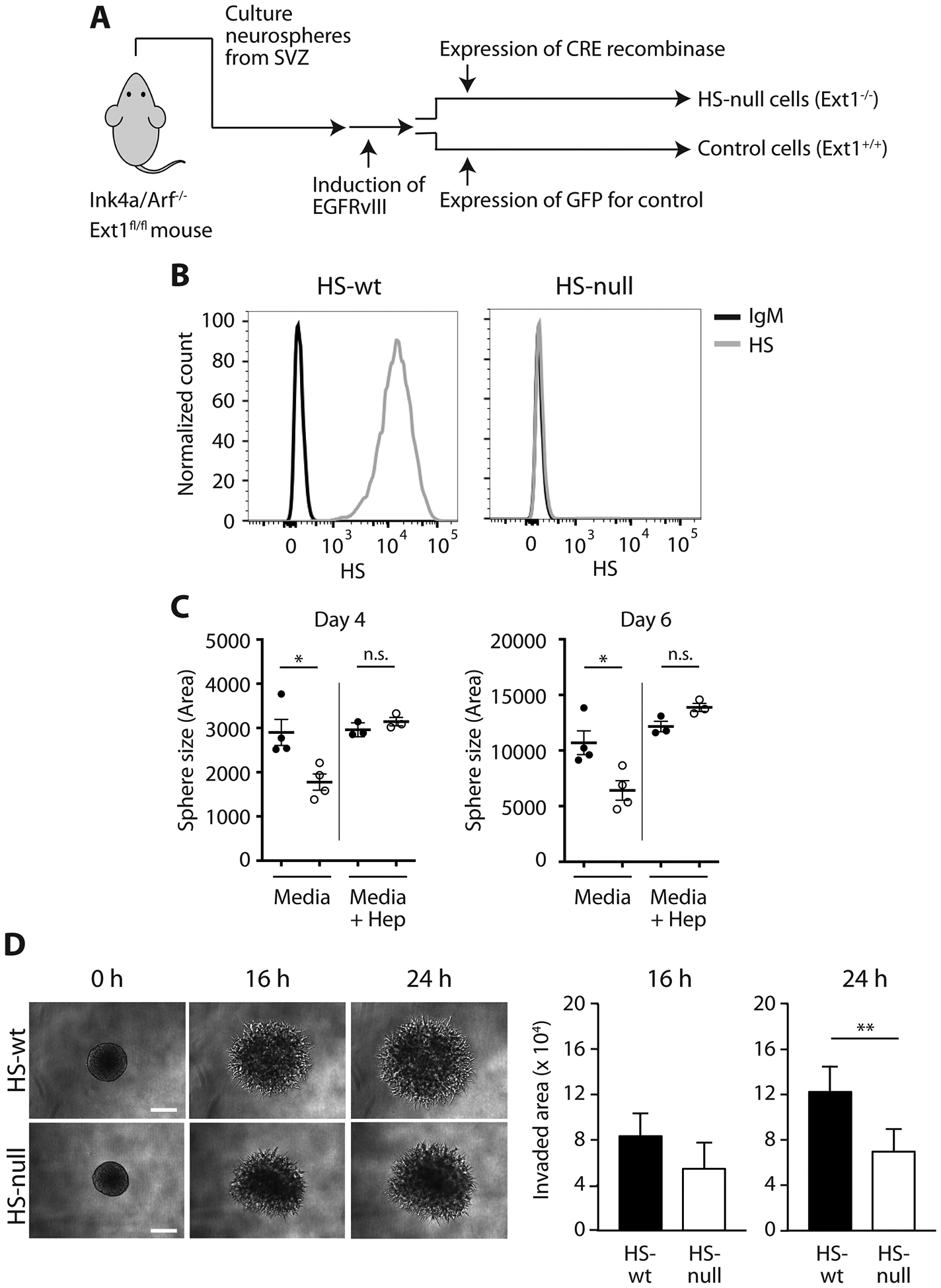
(A) Scheme for establishment of Ext1-knockout mNPC-E* cells, heparan sulfate (HS)-null and control cells (HS-wt). (B) Expression of cell surface HS by flow cytometry. Gray line anti-HS and black line control IgM. (C) Sphere size at 4 days and 6 days following plating as single cells in HS-null (open circles, n=4) and HS-wt (black circles, n=4). Addition of exogenous heparin (right of gray line in each panel) fully rescued the HS-null growth defect (open circles, n=3) but had no effect on HS-wt (black circles, n=3). Mean of biologic replicates (n=16–53 spheres counted per condition per replicate) ± SD, *p < 0.05, **p < 0.01. (D) Representative images and quantification of 3D matrigel invasion assay in HS-null and HS-wt cells at 0 h, 16 h and 24 h. Bars, 200 μm. Mean ± SD for HS-null (n=8) and HS-wt spheres (n=7). **p < 0.01. Experiments were performed in biologic triplicate.
HS-null cells demonstrated a significant reduction in cell growth as determined by assessment of sphere size at days 4 and 6 post-plating (Fig 1C; p < 0.05) and cell growth in bulk cultures at day 3 (Supplementary Fig S1C; p<0.01). Secondary neurosphere assays demonstrated a similar reduction in sphere size in HS-null cells (HS-null sphere area normalized to HS-wt 0.75 ± 0.0, p<0.01, n=5 per condition). The overall ability of HS-null and HS-wt cells to form spheres from single cells (self-renewal), however, was not altered in primary or secondary neurosphere assays (Supplementary Fig S1A–B). Exogenous heparin rescued cell growth in HS-null cells (Fig 1C; p<0.01 day 4 and p<0.005 day 6). HS-null cells also demonstrated decreased invasion in 3D matrigel invasion assays relative to HS-wt at 24 h (Fig 1D, p<0.01).
Absence of HS reduces receptor tyrosine kinase activity.
Ext1−/− (HS-null) cells showed decreased total tyrosine phosphorylation levels and a corresponding decrease in activation of downstream signaling pathways, as indicated by reduced levels of phosphorylated Akt and Erk1/2 (Fig 2A and B). To identify specific signaling pathways affected by loss of cell surface HS we performed a comprehensive analysis of RTK phosphorylation in HS-null and control cells. HS-null cells showed reduced phosphorylation of several RTKs. RTKs with the greatest reduction included fibroblast growth factor receptors (FGFR3, FGFR4), insulin receptor family (InsulinR, IGF1R), platelet-derived growth factor receptors (PDGFRα, PDGFRβ), receptor tyrosine-protein kinase Erb-B2 (ErbB2), and receptor tyrosine-protein kinase UFO (Axl) (Fig 2C, Supplementary Fig S2).
Fig. 2. Reduced phosphorylation of receptor tyrosine kinases and decreased in vivo tumor growth in the absence of HS.
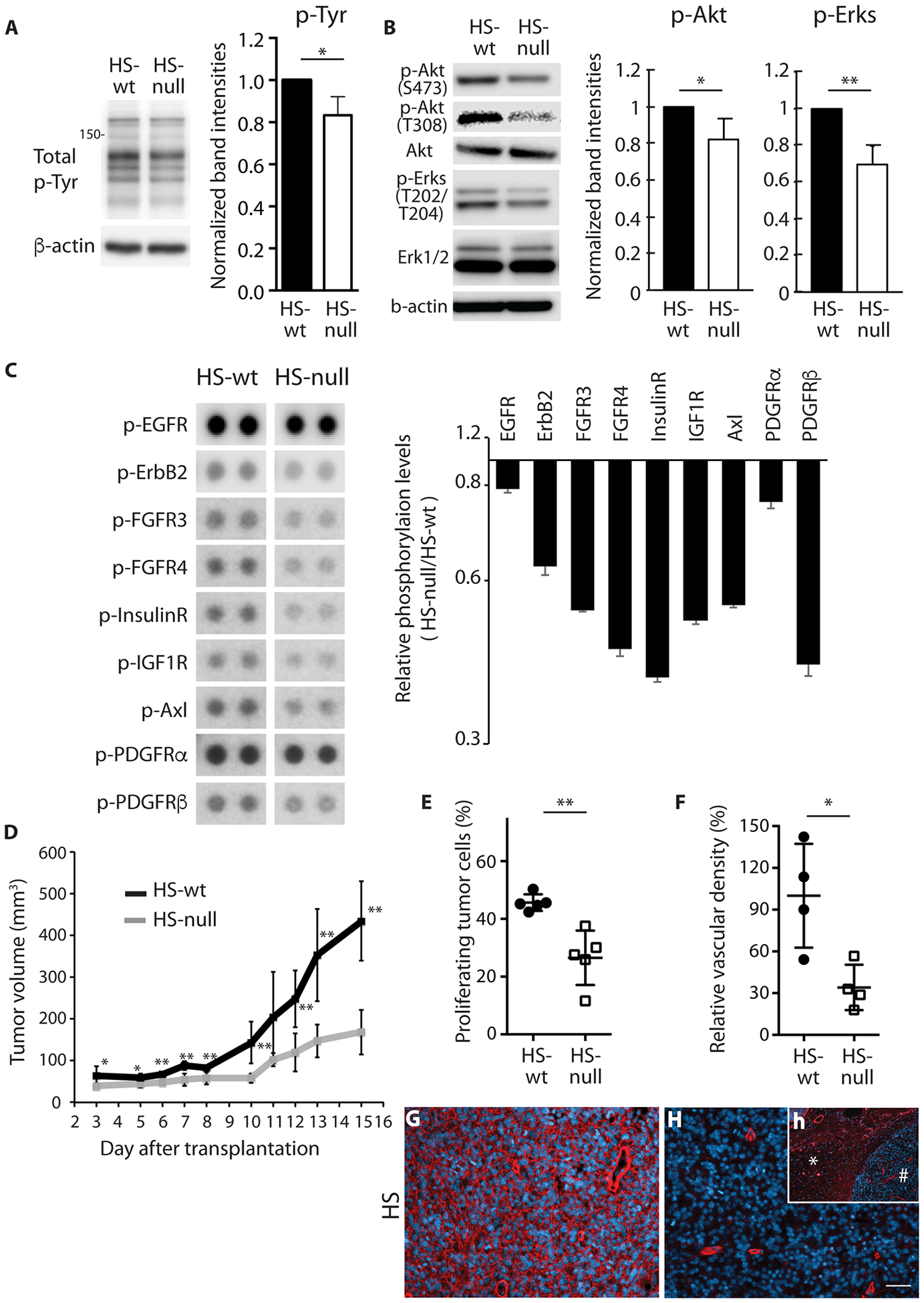
(A) Representative images and relative quantification of total phosphotyrosines in HS-null and HS-wt cells (n=3 biologic replicates, mean ± SD). β-actin loading control. (B) Representative images of phosphorylation of Akt (S473), Akt (T308), and Erk1/2 (T202/T204) and total Akt and Erk1/2. Relative quantification of phosphorylation of Akt (S473) and Erk1/2 in HS-null and HS-wt cells (n=3 biologic replicates, mean ± SD). β-actin loading control. (C) Representative images and quantification of RTK phosphorylation (EGFR, ErbB2, FGFR3, FGFR4, InsulinR, IGF1R, Axl, PDGFRα and PDGFRβ) in HS-null and HS-wt cells as determined by phospho-RTK array. For all RTKs shown HS-null versus HS-wt was different by one-way ANOVA, p<0.001. (D) Tumor volume over time in mice harboring HS-null and HS-wt cells (n=5). (E) Proliferation, as determined by Ki-67, in tumor tissue (n=5 per group). (F) Relative vascular density in representative regions of HS-null and HS-wt tumors normalized to HS-wt (n=4 per group) (G,H) Representative images of HS expression (red) in HS-wt (G) and HS-null (H) tumors. Inset (h) demonstrates circumscribed tumor (#) in subcutaneous space (*) with modest HS expression (red). Nuclei are stained with DAPI. Scale bar 30μm. All data representative of at least two experiments, mean ± SD. *p < 0.05, **p < 0.01.
HS deficient cells exhibit reduced tumor growth in vivo.
HS-wt cells readily formed tumors in athymic nude mice. In contrast, cells deficient for HS resulted in a delay in allograft establishment, and reduced rate of tumor growth upon establishment (Fig 2D). This decreased growth was associated with a decrease in proliferating tumor cells and reduced vascular density in HS-null tumors relative to HS-wt tumors (Fig 2E and F). HS-null tumors had a near complete absence of HS in tumors (Fig 2G and H). Thus, HS deficiency in a murine model for GBM resulted in decreased RTK activation, decreased tumor cell proliferation, and decreased tumor growth in vivo.
Non-cell autonomous HS, in the form of heparin, rescued cell growth in HS-null cells (Fig 1C) and promoted growth of HS-wt cells largely via FGF2-mediated signaling (Fig 3A). Thus, both cell autonomous and non-cell autonomous HS can promote ligand-mediated signaling in GBM. In the hypodermis, HS-null allografts grow in the absence of tumor cell invasion and maintain a relative HS-null phenotype (Fig 2H). In contrast, HS is abundant in the brain (Fig 3B and C) and intracerebral allografts exhibit single cell tumor invasion (Fig 3D and E). Thus, HS-null cells are surrounded by an HS-rich microenvironment (Fig 3F and G). To investigate the role for both cell autonomous and non-cell autonomous HS in GBM, HS-null and HS-wt cells were implanted orthotopically in HS-wt mice. Intracerebral allografts of HS-null cells trended towards slowed growth relative to HS-wt cells (Fig 3J, p<0.08). At the time of signs or symptoms of tumor, however, the HS-null and HS-wt tumors had similar tumor vascular density (relative vascular density 78 ± 22 vs. 100 ± 13, respectively). Comparison of tumor size, extent of invasion, extent of necrosis, and proliferation (Ki-67 immunostaining) revealed similar patterns between HS-null and HS-wt tumors. While prolonged growth of HS-null tumor cells in the brain may select for cells with HS expression, immunostaining (Fig 3H and I) failed to identify robust expression of HS on HS-null tumor cells as compared to HS-wt cells. In addition, flow cytometry on cells isolated from tumors at 42.5 days post implantation revealed robust HS expression on HS-wt cells and no detectable HS expression on HS-null cells (Supplementary Fig S3). These data suggest in the brain, both cell autonomous and non-cell autonomous HS contribute to GBM growth.
Fig. 3. Non-cell autonomous HS-mediated growth stimulation and intracerebral growth of HS-null cells.
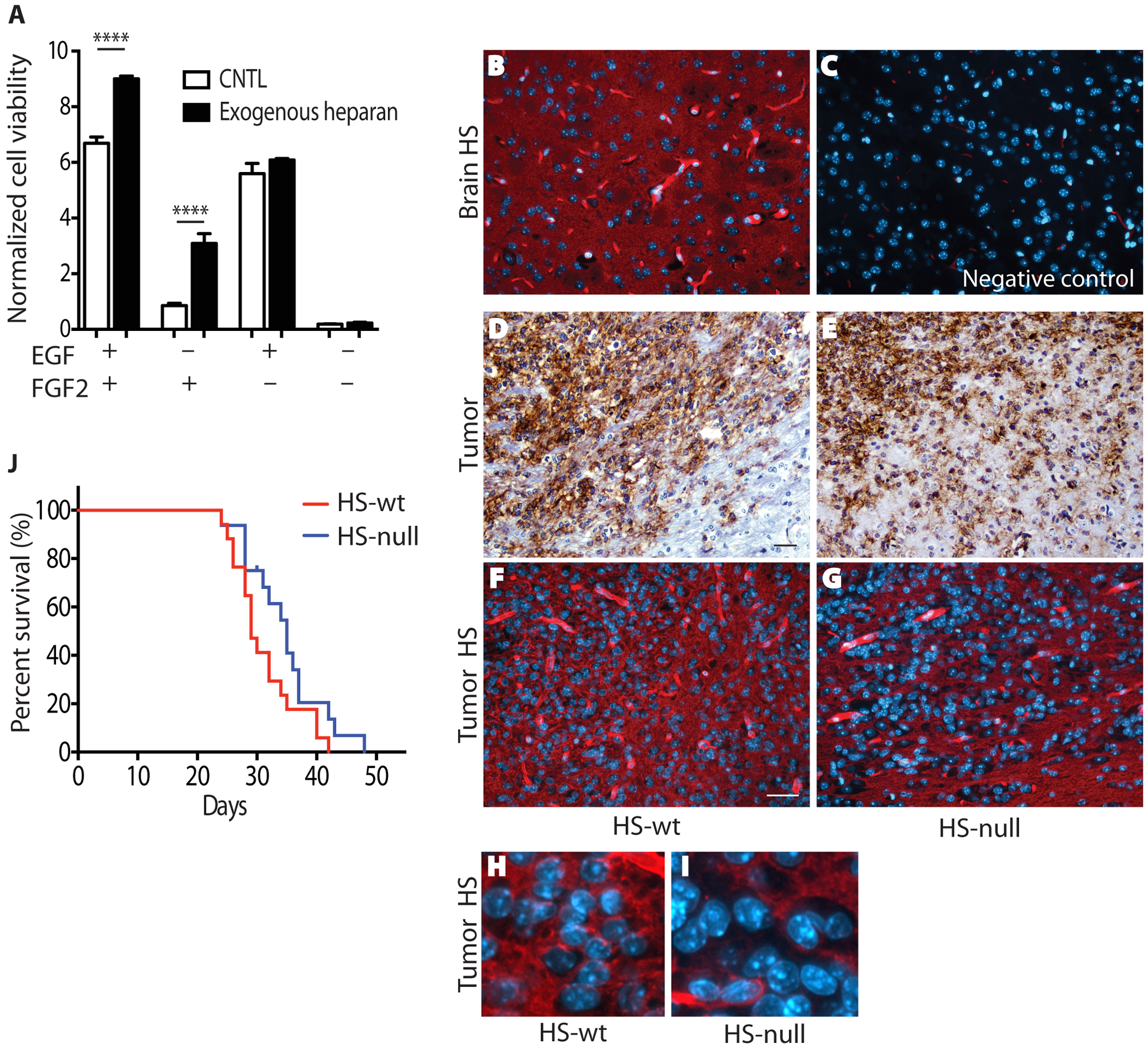
(A) Exogenous heparin, a highly sulfated type of HS, promotes growth of Ext-null cells largely via FGF2. Cell viability was determined in the presence of 100μg/mL heparin. Quantification is normalized cell viability ± SD (n=3) and data are representative of 4 independent experiments. ****, p<0.0001. (B,C) Representative images of HS expression (red) in striatum of normal brain (B) and negative staining control (C). (D,E) Representative images of HS-wt (D) and HS-null (E) tumors demonstrating single cell invasion. Tumor cells are immunostained using an antibody against human EGFR (brown). (F,G) Representative images of HS expression (red) in HS-wt (F) and HS-null (G) tumors. (H,I) Enlarged image from panels F and G highlighting lack of HS staining in many HS-null tumor cells. Note nuclei appear larger in HS-null tumors due to lack of HS (red) expression in individual tumor cells. Nuclei are stained with DAPI (B,C,F-I) and hematoxylin (D,E). Scale bar 30μm. (J) Overall survival of mice with intracranial allograft of HS-null and HS-wt cells (n=16 and n=17, respectively; p=0.08).
HS-mediated growth factor signaling in patient-derived GBM lines.
We next investigated the role for HS in RTK regulation in GBM43, a patient-derived GBM tumorsphere line with moderate cell surface HS content and expression of both EGFR and PDGFRA (20). Enzymatic removal of cell surface HS, via enzymatic clearing with heparitinase I, II, III (ΔHS) (Fig 4A, Supplementary Fig S4A), resulted in reduced phosphorylation of several RTKs. RTKs with the greatest reduction included insulin receptor family (InsulinR, IGF1R) and platelet-derived growth factor receptor (PDGFRα) (Fig 4B). PDGFRα signaling is important in GBM and we have previously identified a role for HS structure in PDGFRα signaling in GBM (22,32). To investigate potential mechanisms by which HS may influence RTK signaling, we examined effects of HS deficiency with respect to cell surface PDGF-BB ligand and PDGFRα signaling. Enzymatic removal of cell surface HS resulted in a 70% reduction of cell surface bound PDGF-BB (Fig 4C and D; p < 0.01). Removal of the glycosaminoglycan chondroitin sulfate had no effect on cell surface bound PDGF-BB (Supplementary Fig S4B–C). Enzymatic removal of cell surface HS was also associated with reduced phosphorylation of PDGFRα and ERK1/2 as compared to control-treated cells (Fig 4E and F; p < 0.01 and p<0.05, respectively). To investigate long-term effects of HS on cells, exogenous heparin, a highly sulfated form of the glycosaminoglycan heparan sulfate, was added to GBM43. Heparin promoted cell viability in a dose-dependent manner (Fig 4G). In contrast, the glycosaminoglycan chondroitin sulfate A had no effect on cell growth. Heparin-induced growth of GBM43 was associated with increased phosphorylation of PDGFRα and ERK1/2 (Fig 4H). GBM5 is a patient-derived GBM tumorsphere line with high cell surface levels of HS (20). Similar to GBM43, enzymatic removal of cell surface HS from GBM5 resulted in reduced phosphorylation of several RTKs with reduced phosphorylation of Axl being most striking (46% of control treated cells, Supplementary Fig S4E). In PDGF-BB binding assays, enzymatic removal of cell surface HS in GBM5 resulted in a 60% reduction of cell surface bound PDGF-BB while removal of the positively charged glycosaminoglycan chondroitin sulfate had no effect (Supplementary Fig S4A–D).
Fig. 4. Influence of cell surface HS on receptor tyrosine kinase signaling in patient-derived GBM lines.
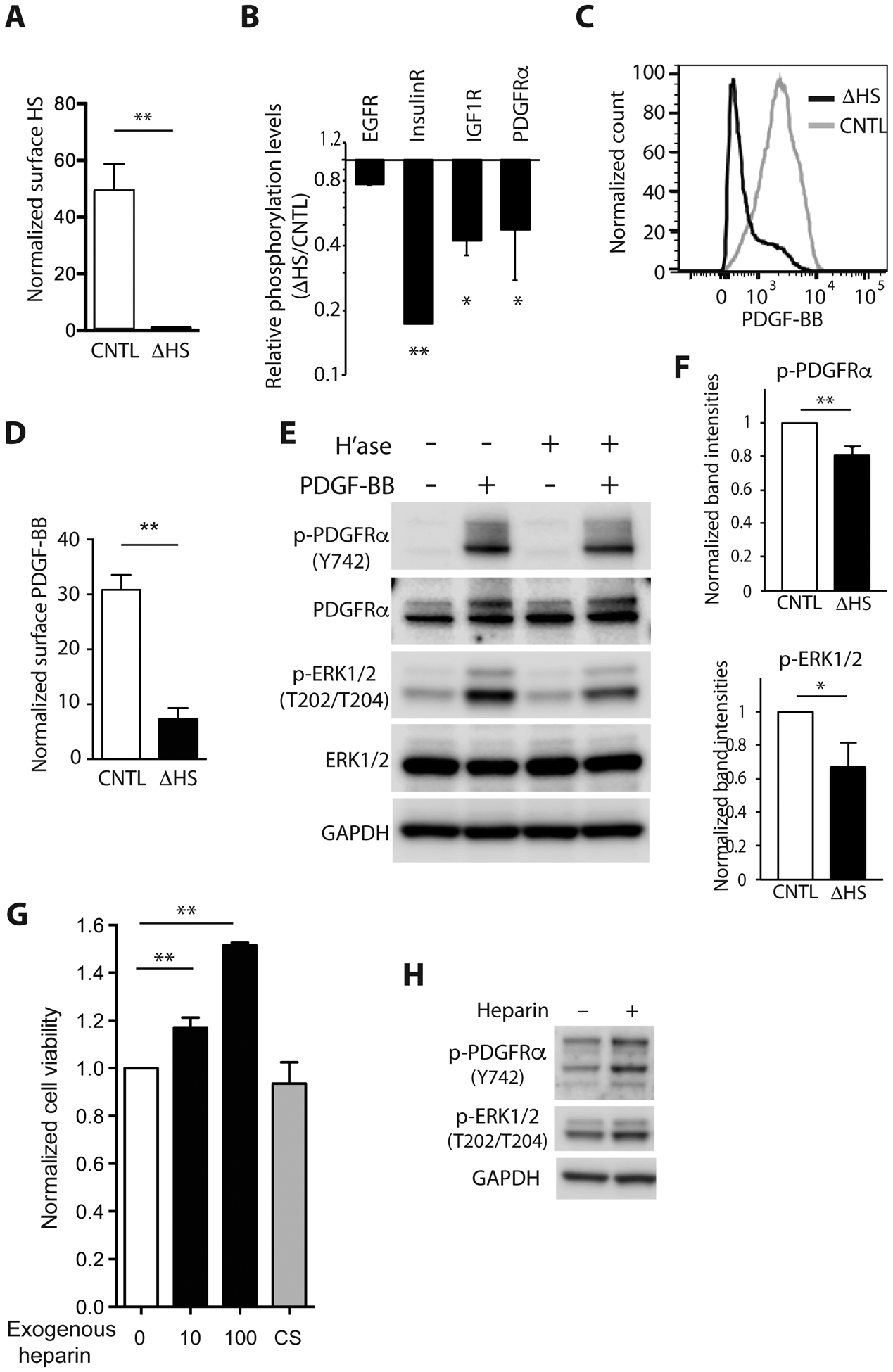
(A) Quantification of cell surface HS in GBM43 with (ΔHS, black bar) and without (CNTL, clear bar) removal of cell surface HS by heparitinase I, II and III. Normalized median fluorescence intensity ± SD for three independent experiments. (B) Quantification of RTK phosphorylation (EGFR, InsulinR, IGF1R, and PDGFRα) in GBM43 with enzymatic HS removal (ΔHS) relative to control GBM43 (CNTL) as determined by phospho-RTK array. Representative of two independent experiments. (C,D) Representative flow cytometry for GBM43 and quantification of cell surface bound tagged PDGF-BB with (ΔHS, black line and black bars) or without (CNTL, grey line and white bars) removal of HS. Quantification is normalized median fluorescence intensity ± SD for three independent experiments. Data normalized to non-relevant IgM. (E) Representative Western blot demonstrating phosphorylation of PDGFRα (Y742), AKT (S473) and ERK1/2 (T202/T204) after PDGF-BB (100 ng) stimulation for 15 min with and without enzymatic removal of HS (H’ase). (F) Relative band intensities of p-PDGFRα (Y742), p-AKT (S473) and p-ERK1/2 (T202/T204) after normalization to total-PDGFRα, total-AKT and total-ERK1/2, respectively. *p < 0.05, **p < 0.01. GAPDH, loading control. (G) Exogenous heparin, a highly sulfated type of HS, promotes growth of patient-derived GBM43 in a dose-dependent manner. Cell viability determined in the presence of 10μg/mL or 100μg/mL heparin or 100μg/mL chondroitin sulfate A, or control (0, CNTL). Quantification is normalized cell viability ± SD for three independent experiments. (H) Representative Western blot demonstrating increased phosphorylation of PDGFRα (Y742) and ERK1/2 (T202/T204) after heparin treatment in GBM43.
Influence of heparan sulfate structure on cell surface PDGF-BB binding.
To investigate how HS sulfation influences cell surface PDGF-BB binding, GBM43 cells were acutely co-incubated with a series of structurally defined polymeric heparin mimetics or fully sulfated heparin (NS2S6S) (Supplementary Fig S5A–B and S6). Relative to control treated cells all mimetics competed with cell surface HS and decreased PDGF-BB binding, with the most potent being the trisulfated form, NS2S6S.
The extracellular enzyme SULF2 removes 6O-sulfation on HSPGs and is known to affect HS binding to multiple growth factors such as stromal cell-derived factor 1 (SDF1) and vascular endothelial growth factor (VEGF)(33). We asked whether exogenous SULF2 could alter cell surface bound PDGF-BB in an experiment in which conditioned media containing SULF2 or inactive SULF2 (S2ΔCC) (34) was harvested from 293T cells that had been modified to express wild-type or mutant SULF2. Production of active SULF2 enzyme was confirmed using a fluorogenic substrate for SULF2 (Supplementary Fig S6B), and by mass-spectrometry analysis of conditioned media that revealed decreased 6-O-sulfation from trisulfated heparin (Supplementary Fig S6C). Addition of SULF2-conditioned media resulted in a small but significant reduction in cell surface bound PDGF-BB relative to S2ΔCC-conditioned media (Supplementary Fig S6D; p<0.01). Taken together these data suggest alterations in HS sulfation, including those mediated by the extracellular enzyme SULF2, alter ligand availability at the cell surface and thereby alter PDGFRA signaling.
HSPGs consist of a core protein and covalently attached glycosaminoglycan chains. Several protein cores exhibit increased expression in GBM (19). In GBM43 we identified several potential HS-containing proteins using the 3G10 antibody. Flow cytometry for GPC1 and flow cytometry and immunofluorescence for SDC1 demonstrated robust expression of both proteoglycans on the cell surface of GBM43 (Supplementary Fig S7).
Growth factor-mediated resistance to EGFR inhibition.
In diverse cancers, ligand-mediated activation of alternative receptor tyrosine kinases (RTKs) with functional redundancy to the therapeutically targeted RTK is a mechanism of innate and acquired RTK inhibitor resistance (6,7). That is RTK ligands present in the tumor microenvironment can reduce the effectiveness of targeted kinase inhibitors. Using two distinct patient-derived GBM tumorsphere lines that express EGFR and respond to EGFR inhibition (20), we investigated the ability of four different growth factors relevant in GBM to confer resistance to EGFR inhibition. In GBM43, exogenous FGF2 and PDGF-BB could partially rescue cell viability upon EGFR inhibition (Fig 5A–C). In GBM1, partial rescue was conferred by addition of FGF2 or IGF-1 and not by PDGF-BB (Fig 5D).
Fig. 5. Growth factor mediated resistance to EGFR inhibition in patient-derived GBM lines.
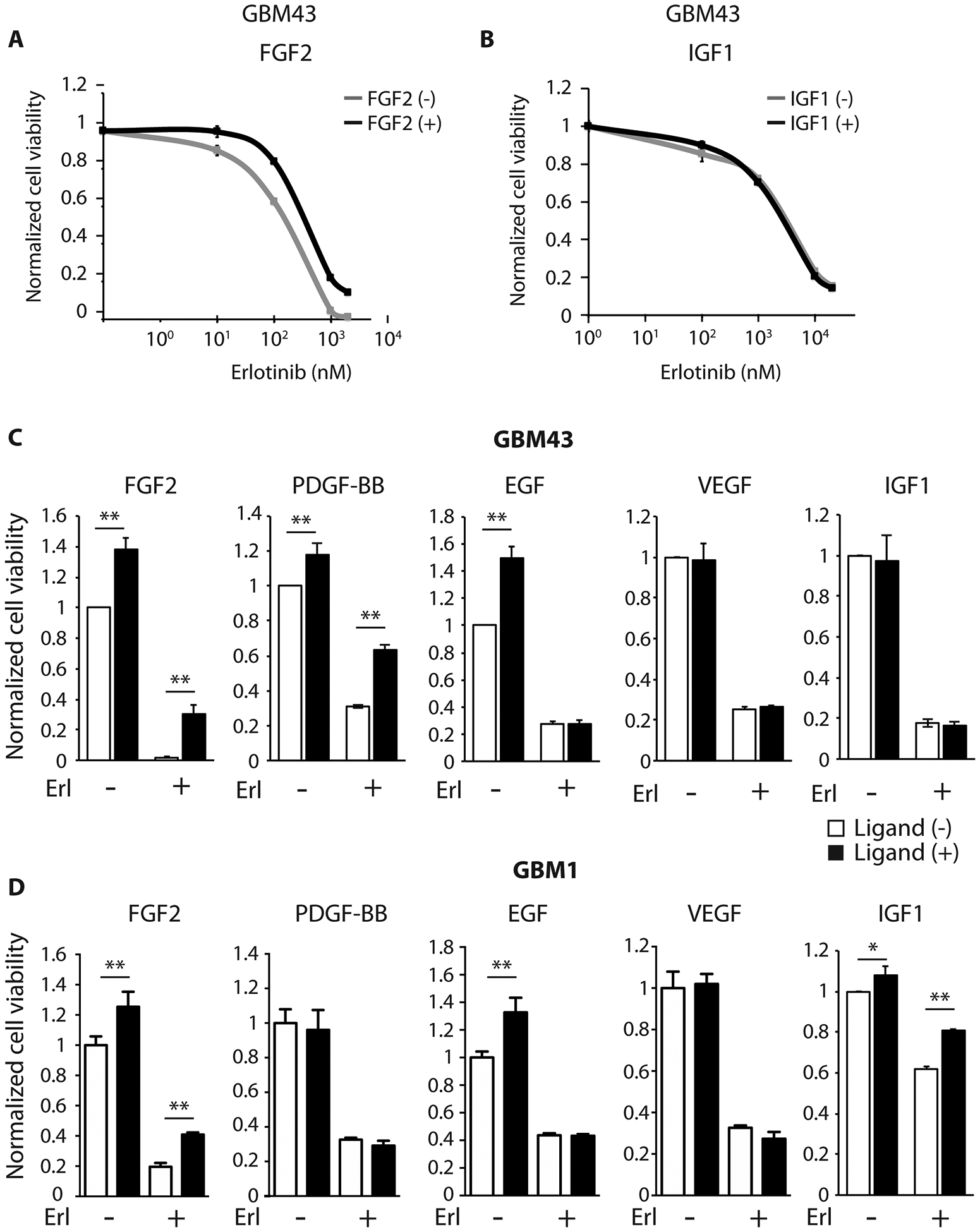
Dose-response curve of GBM43 to erlotinib (μM) in the presence (black line) or absence (gray line) of (A) FGF2 (20 ng/ml) and (B) IGF1 (50 ng/ml) after 3 days. Cell viabilities normalized to untreated cells. (C, D) GBM43 viability (C) and GBM1 viability (D) with and without exogenous growth factor in the presence and absence of EGFR inhibition (erlotinib 5 μM). White bars denote no ligand and black bars denote ligand as noted: FGF2 (20 ng/ml), PDGF-BB (50 ng/ml), EGF (20 ng/ml), VEGF (50 ng/ml), and IGF1 (50 ng/ml). Cell viabilities normalized to untreated cells. All data represent mean ± SD for three biologic replicates. *p < 0.05, **p < 0.01.
Heparan sulfate promotes EGFR inhibitor resistance in vitro and in vivo.
We next investigated whether HS contributes to ligand-dependent tumor resistance to RTK inhibition. To do this we focused on the role for HS in FGF2 mediated resistance to EGFR inhibition. In both patient-derived GBM43 and murine GBM, FGF2 partially rescued cell viability in HS deficient versus control cells upon EGFR inhibition (Fig 6A and B). As early as 24 hours after drug treatment HS-null cells exhibited increased cell death at both 500nM and 5μM erlotinib relative to HS-wt (Fig 6C). To determine whether HS mediates resistance to EGFR inhibition in vivo, HS-null and HS-wt cells were engrafted into mice and EGFR inhibitor therapy was initiated when tumors reached an equivalent size. Following completion of treatment, HS-null tumors were significantly smaller than HS-wt tumors (Fig 6D; day 5 post-treatment, p<0.05).
Fig. 6. Heparan sulfate deficient cells have reduced resistance to EGFR inhibitor in vitro and in vivo.
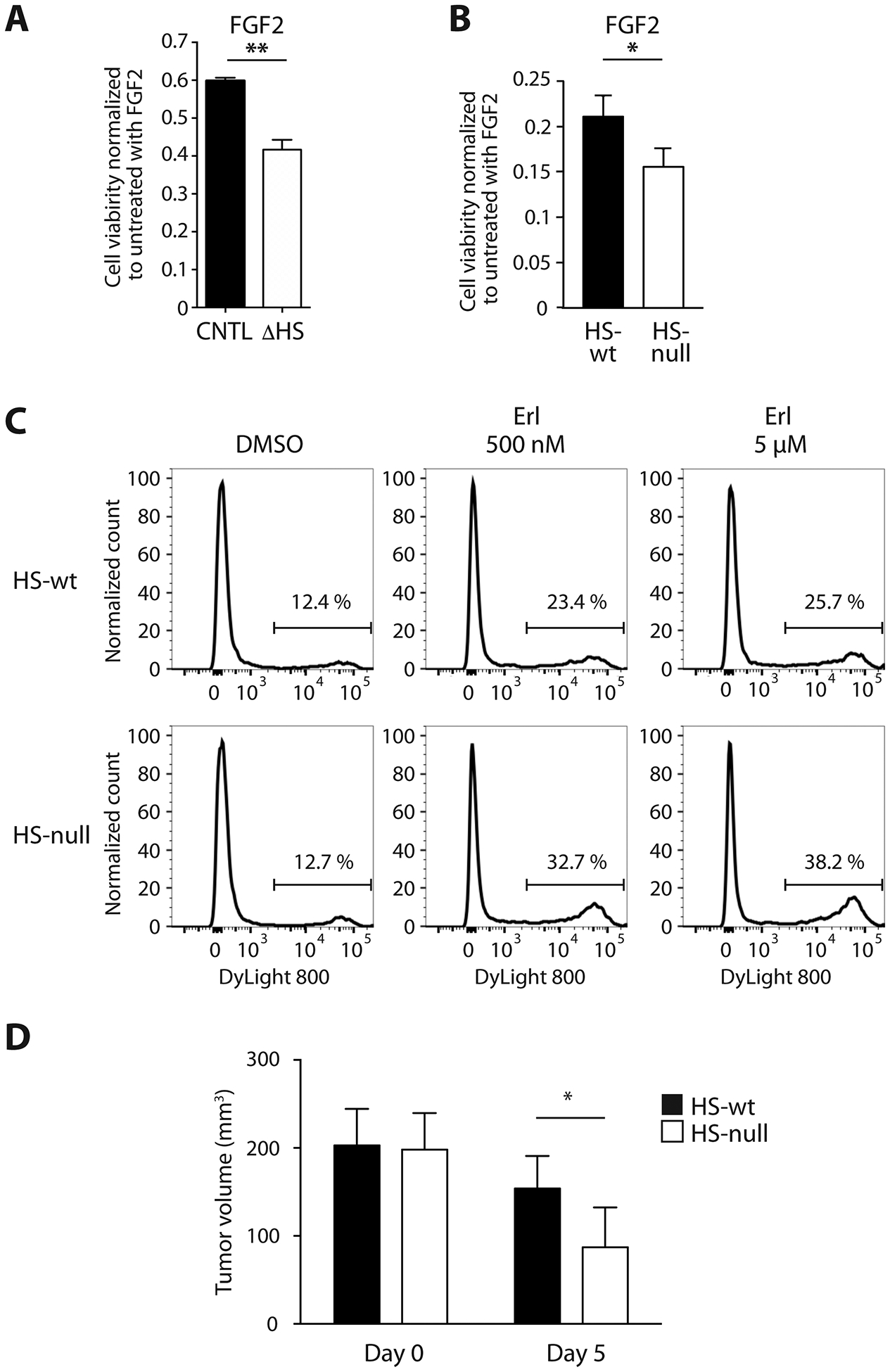
(A) FGF2-mediated partial rescue of GBM43 cell viability when treated with erlotinib (5 μM) is reduced in the absence of cell surface HS (ΔHS, white bars) as compared to control (CNTL, black bars). Data normalized to without EGFR inhibitor. (B) FGF2-mediated partial rescue of murine GBM is reduced in HS-null cells (white bar) as compared to HS-wt cells (black bar) at 3 days. Data normalized to without erlotinib (500 nM). (C) Increased cell death at 24 hours in HS-null versus HS-wt cells in the presence FGF2 (5 ng/ml) and erlotinib (500 nM and 5 μM) as determined by flow cytometry. (D) Tumor volumes following transplant of HS-null (white bars) and HS-wt (black bars) cells at the start of treatment (designated day 0) and after x3 treatments with erlotinib (150 mg/kg) on day 5 post treatment. HS-null (white bars; n=9) and HS-wt (black bars; n=10). Mean ± SD. *p < 0.05.
DISCUSSION
Glycosylation of cell surface proteins is an important post-translational modification that impacts cell-cell communication, cell signaling pathway activity, and cell adhesion (35). In GBM, alterations in heparan sulfate (HS) proteoglycans, present on the cell surface and in the extracellular matrix, promote oncogenic signaling and invasion (19,20,22). By genetically preventing HS chain biosynthesis in a murine model for GBM and enzymatically clearing cell surface HS from patient-derived cell lines, we demonstrate the importance of HS in growth factor driven glioma progression as well as response to targeted therapies. Tumor cells lacking HS had decreased proliferation, increased length of time for establishment as an allograft and decreased allograft growth rate subsequent to establishment, and decreased activation of multiple receptor tyrosine kinases (RTKs). We have shown that HS regulates PDGFRA signaling pathway activity and that decreased pathway activity is associated with reduced cell surface bound PDGF-BB ligand. HS influence of ligand-mediated signaling may be particularly important in contributing to tumor adaptive response to receptor tyrosine kinase (RTK) inhibition.
Ext1 is an essential enzyme for HS biosynthesis. Using neural progenitors isolated from Ext1fl/fl Ink4a/Arf−/− mice, we generated isogenic cell pairs (22,26) that differed only in their ability to synthesize HS glycosaminoglycan chains. Causing tumor cell HS deficiency in this manner mitigated tumor malignancy both in vitro and in vivo. HS on the cell surface and in the tumor microenvironment contributes to ligand-mediated signaling via several mechanisms (reviewed in (19,36)), including the sequestration of growth factors (37). Growth factors demonstrated to bind to HS include FGF2 (17,38,39), platelet-derived growth factor (40,41), hepatocyte growth factor (scatter factor) (42), and vascular endothelial growth factor (21,33). HS can also serve as a co-receptor for ligand-mediated signaling. Indeed, HSPGs stabilize fibroblast growth factor (FGF) ligand-receptor complex to promote FGF2 signaling (23,43,44).
In GBM, oncogenic activation of PDGFRA is common and regulated in part via HSPGs (22,32,45). Investigating how HS alters PDGF-BB-mediated signaling using patient derived GBM lines, we found that enzymatic clearing of cell surface HS reduced cell surface bound PDGF-BB ligand, reduced activation of PDGFRα, and reduced downstream signaling pathway activity. Thus, HS may regulate local PDGF-BB bioavailability.
The sulfation pattern of the HS chains is a major determinant of the specificity and the affinity of HS-ligand interactions, particularly the 6O-sulfate (6OS) of glucosamine (24,33,37,46). Regulation of 6OS levels occurs during biosynthesis and post-synthetically by the extracellular sulfatases (SULFs). Using polymeric heparin mimetics differing in their sulfation status combined with exogenous SULF2 enzyme, we demonstrated cell surface PDGF-BB binding is dependent on the sulfation status of the HS chains. Binding also has specificity for HS as reducing cell surface chondroitin sulfate glycosaminoglycans had no impact on cell surface bound ligand.
Ligand-mediated activation of RTKs with functional redundancy to a specific RTK being therapeutically targeted is one mechanism of resistance to RTK inhibitors in diverse cancers (6,7). In patient-derived GBM lines and a murine model for GBM we demonstrate HS is a major factor regulating growth factor bioavailability and promotes resistance to targeted RTK inhibition. In the absence of cell surface HS, FGF2-mediated resistance to EGFR inhibition was diminished. Importantly, inhibition of EGFR in vivo was more efficacious in HS-null than HS-wt tumors (Figure 6D).
HS acts in both a cell autonomous and a non-cell autonomous manner. In the HS-low microenvironment of the hypodermis, HS-null allografts grow as relatively circumscribed masses and exhibit profound growth defects. This is likely due to a combination of decreased growth factor signaling in tumor cells, as evidenced by decreased RTK signaling and tumor cell proliferation, and decreased tumor angiogenesis, as evidenced by decreased vascular density, in HS-null tumors. HS binds vascular endothelial growth factor and promotes signaling in normal tissues and in GBM (21). Decreased tumor angiogenesis is likely one factor in the apparent growth plateau of the HS-null cells in vivo (Fig 2). In the brain, however, HS is abundant in the microenvironment and tumor cells exhibit diffuse single cell invasion. Thus, HS-null tumor cells invade and proliferate within an HS-rich microenvironment. In this context HS-null cells exhibited a less severe growth defect. Comparing orthotopic HS-null and HS-wt tumors at late time points, tumor growth and angiogenesis were similar suggesting HS in the tumor microenvironment rescues the profound growth defects observed in the hypodermis. While HS-null tumor cells appeared to lack robust HS expression (Fig 3I), it is possible that even low-level HS expression may contribute to tumor growth in the brain. As HS-null mice are not viable we could not directly test the role for non-cell autonomous HS in tumor growth and invasion. While this is a limitation of our study, our in vitro and in vivo data, comparing two different tumor compartments, suggest HS on cell surfaces and in the brain tumor microenvironment contribute to tumor growth in GBM.
Our data with FGF2, IGF-1, and PDGF-BB suggest HS promotes ligand-mediated signaling in GBM via multiple mechanisms that collectively contribute to tumor malignancy and progression. As HS regulates multiple ligand-mediated signaling pathways, inhibitors to HS biosynthesis and function are currently being investigated and in clinical trials (47–50). In combination with diverse RTK inhibitors, the modulation of HS function may be a promising therapeutic strategy in GBM and in other therapy resistant neoplasms. This includes diffuse lower grade glioma, a tumor that may be particularly amenable to strategies that broadly target oncogenic signaling pathways and angiogenesis.
Supplementary Material
Implications:
Our study shows that HS expressed on tumor cells and in the tumor microenvironment regulates ligand-mediated signaling, promoting tumor cell proliferation and invasion, and these factors contribute to decreased tumor cell response to targeted RTK inhibition.
Acknowledgments:
This study was supported in part by NIH/NCI U01 CA229345 to J. Phillips, NIH/NINDS R01 NS081117 to J. Phillips, NIH/NCI U01 CA168878 to J. Phillips, and NIH/NCI P50CA221747 to C.D. James. We acknowledge and thank the T.J. Martell Foundation, Gerson and Barbara Bakar Philanthropic Fund, and the Sence Foundation for their support of this work. We are grateful and thank Dr. Kuberan Balagurunathan for sharing his expertise, providing us with heparitinase I, II, III, and providing us with polymeric heparin mimetics. We also acknowledge the UCSF Brain Tumor SPORE Biorepository (NIH/NCI P50CA097257) for providing histology services, NIH Grant PO1-HL107152 to Balagurunathan Kuberan for providing essential resources, the UCSF Helen Diller Family Comprehensive Cancer Center Laboratory for Cell Analysis (P30CA082103) for microscopy services, and the UCSF Mass Spec Facility (NIH NCRR P41RR001614) for use of their mass spectrometer. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of interest:
The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- 1.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. 2015;15:302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reardon DA, Wen PY, Mellinghoff IK. Targeted molecular therapies against epidermal growth factor receptor: past experiences and challenges. Neuro-oncology. 2014;16 Suppl 8:viii7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–90. [DOI] [PubMed] [Google Scholar]
- 5.Liu F, Mischel PS. Targeting epidermal growth factor receptor co-dependent signaling pathways in glioblastoma. Wiley Interdiscip Rev Syst Biol Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma Y, Tang N, Thompson RC, Mobley BC, Clark SW, Sarkaria JN, et al. InsR/IGF1R Pathway Mediates Resistance to EGFR Inhibitors in Glioblastoma. Clin Cancer Res. 2016;22:1767–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–7. [DOI] [PubMed] [Google Scholar]
- 9.Qiu H, Jiang J-L, Liu M, Huang X, Ding S-J, Wang L. Quantitative Phosphoproteomics Analysis Reveals Broad Regulatory Role of Heparan Sulfate on Endothelial Signaling. Mol Cell Proteomics. 2013;12:2160–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lind T, Tufaro F, McCormick C, Lindahl U, Lidholt K. The Putative Tumor Suppressors EXT1 and EXT2 Are Glycosyltransferases Required for the Biosynthesis of Heparan Sulfate. J Biol Chem. 1998;273:26265–8. [DOI] [PubMed] [Google Scholar]
- 11.Osterholm C, Barczyk MM, Busse M, Grønning M, Reed RK, Kusche-Gullberg M. Mutation in the heparan sulfate biosynthesis enzyme EXT1 influences growth factor signaling and fibroblast interactions with the extracellular matrix. J Biol Chem. 2009;284:34935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302:1044–6. [DOI] [PubMed] [Google Scholar]
- 13.Stickens D, Zak BM, Rougier N, Esko JD, Werb Z. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development. 2005;132:5055–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin X, Wei G, Shi Z, Dryer L, Esko JD, Wells DE, et al. Disruption of Gastrulation and Heparan Sulfate Biosynthesis in EXT1-Deficient Mice. Developmental Biology. 2000;224:299–311. [DOI] [PubMed] [Google Scholar]
- 15.Danesin C, Agius E, Escalas N, Ai X, Emerson C, Cochard P, et al. Ventral Neural Progenitors Switch toward an Oligodendroglial Fate in Response to Increased Sonic Hedgehog (Shh) Activity: Involvement of Sulfatase 1 in Modulating Shh Signaling in the Ventral Spinal Cord. J Neurosci. 2006;26:5037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braquart-Varnier C, Danesin C, Clouscard-Martinato C, Agius E, Escalas N, Benazeraf B, et al. A subtractive approach to characterize genes with regionalized expression in the gliogenic ventral neuroepithelium: identification of chick Sulfatase 1 as a new oligodendrocyte lineage gene. Molecular and Cellular Neuroscience. 2004;25:612–28. [DOI] [PubMed] [Google Scholar]
- 17.Higginson JR, Thompson SM, Santos-Silva A, Guimond SE, Turnbull JE, Barnett SC. Differential Sulfation Remodelling of Heparan Sulfate by Extracellular 6-O-Sulfatases Regulates Fibroblast Growth Factor-Induced Boundary Formation by Glial Cells: Implications for Glial Cell Transplantation. J Neurosci. 2012;32:15902–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rauch JN, Chen JJ, Sorum AW, Miller GM, Sharf T, See SK, et al. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci Rep. 2018;8:6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wade A, Robinson AE, Engler JR, Petritsch C, James CD, Phillips JJ. Proteoglycans and their roles in brain cancer. FEBS J. 2013;280:2399–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran VM, Wade A, McKinney A, Chen K, Lindberg OR, Engler JR, et al. Heparan Sulfate Glycosaminoglycans in Glioblastoma Promote Tumor Invasion. Mol Cancer Res. 2017;15:1623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cecchi F, Pajalunga D, Fowler CA, Üren A, Rabe DC, Peruzzi B, et al. Targeted Disruption of Heparan Sulfate Interaction with Hepatocyte and Vascular Endothelial Growth Factors Blocks Normal and Oncogenic Signaling. Cancer Cell. 2012;22:250–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips JJ, Huillard E, Robinson AE, Ward A, Lum DH, Polley M-Y, et al. Heparan sulfate sulfatase SULF2 regulates PDGFRα signaling and growth in human and mouse malignant glioma. J Clin Invest. 2012;122:911–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–8. [DOI] [PubMed] [Google Scholar]
- 24.Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM, Emerson CP. Regulation of Wnt Signaling and Embryo Patterning by an Extracellular Sulfatase. Science. 2001;293:1663–6. [DOI] [PubMed] [Google Scholar]
- 25.Yamada K, Miyamoto Y, Tsujii A, Moriyama T, Ikuno Y, Shiromizu T, et al. Cell surface localization of importin α1/KPNA2 affects cancer cell proliferation by regulating FGF1 signalling. Sci Rep. 2016;6:21410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindberg OR, McKinney A, Engler JR, Koshkakaryan G, Gong H, Robinson AE, et al. GBM heterogeneity as a function of variable epidermal growth factor receptor variant III activity. Oncotarget. 2016;7:79101–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7:164–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264–71. [DOI] [PubMed] [Google Scholar]
- 29.Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimitroff CJ. Galectin-Binding O-Glycosylations as Regulators of Malignancy. Cancer Res. 2015;75:3195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furukawa K, Ohmi Y, Ji S, Zhang P, Bhuiyan RH, Ohkawa Y, et al. Glycolipids: Essential regulator of neuro-inflammation, metabolism and gliomagenesis. Biochim Biophys Acta. 2017;1861:2479–84. [DOI] [PubMed] [Google Scholar]
- 32.Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M, et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010;24:2205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uchimura K, Morimoto-Tomita M, Bistrup A, Li J, Lyon M, Gallagher J, et al. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morimoto-Tomita M, Uchimura K, Bistrup A, Lum DH, Egeblad M, Boudreau N, et al. Sulf-2, a proangiogenic heparan sulfate endosulfatase, is upregulated in breast cancer. Neoplasia. 2005;7:1001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540–55. [DOI] [PubMed] [Google Scholar]
- 36.Sarrazin S, Lamanna WC, Esko JD. Heparan Sulfate Proteoglycans. Cold Spring Harb Perspect Biol [Internet]. 2011. [cited 2016 Jul 20];3 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3119907/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shipp EL, Hsieh-Wilson LC. Profiling the Sulfation Specificities of Glycosaminoglycan Interactions with Growth Factors and Chemotactic Proteins Using Microarrays. Chemistry & Biology. 2007;14:195–208. [DOI] [PubMed] [Google Scholar]
- 38.Guimond S, Maccarana M, Olwin BB, Lindahl U, Rapraeger AC. Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J Biol Chem. 1993;268:23906–14. [PubMed] [Google Scholar]
- 39.Maccarana M, Casu B, Lindahl U. Minimal sequence in heparin/heparan sulfate required for binding of basic fibroblast growth factor. J Biol Chem. 1993;268:23898–905. [PubMed] [Google Scholar]
- 40.Feyzi E, Lustig F, Fager G, Spillmann D, Lindahl U, Salmivirta M. Characterization of Heparin and Heparan Sulfate Domains Binding to the Long Splice Variant of Platelet-derived Growth Factor A Chain. J Biol Chem. 1997;272:5518–24. [DOI] [PubMed] [Google Scholar]
- 41.Rolny C, Spillmann D, Lindahl U, Claesson-Welsh L. Heparin amplifies platelet-derived growth factor (PDGF)- BB-induced PDGF α-receptor but not PDGF β-receptor tyrosine phosphorylation in heparan sulfate-deficient cells effects on signal transduction and biological responses. J Biol Chem. 2002;277:19315–21. [DOI] [PubMed] [Google Scholar]
- 42.Ashikari S, Habuchi H, Kimata K. Characterization of heparan sulfate oligosaccharides that bind to hepatocyte growth factor. J Biol Chem. 1995;270:29586–93. [DOI] [PubMed] [Google Scholar]
- 43.Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–50. [DOI] [PubMed] [Google Scholar]
- 44.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–8. [DOI] [PubMed] [Google Scholar]
- 45.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. Journal of Clinical Investigation. 2001;108:169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hossain MM, Hosono-Fukao T, Tang R, Sugaya N, Kuppevelt TH van, Jenniskens GJ, et al. Direct detection of HSulf-1 and HSulf-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology. 2010;20:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu C-J, Lee P-H, Lin D-Y, Wu C-C, Jeng L-B, Lin P-W, et al. Heparanase inhibitor PI-88 as adjuvant therapy for hepatocellular carcinoma after curative resection: A randomized phase II trial for safety and optimal dosage. Journal of Hepatology. 2009;50:958–68. [DOI] [PubMed] [Google Scholar]
- 49.Joyce JA, Freeman C, Meyer-Morse N, Parish CR, Hanahan D. A functional heparan sulfate mimetic implicates both heparanase and heparan sulfate in tumor angiogenesis and invasion in a mouse model of multistage cancer. Oncogene. 2005;24:4037–51. [DOI] [PubMed] [Google Scholar]
- 50.Boothello RS, Patel NJ, Sharon C, Abdelfadiel EI, Morla S, Brophy DF, et al. A Unique Nonsaccharide Mimetic of Heparin Hexasaccharide Inhibits Colon Cancer Stem Cells via p38 MAP Kinase Activation. Mol Cancer Ther. 2019;18:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.