

Abstract
Androgen deprivation therapy has led to elevated cases of androgen receptor (AR) pathway-independent prostate cancer with dys-regulated fatty acid metabolism. However, it is unclear how prostate cancer cells sustain dys-regulated fatty acid metabolism to drive AR-independent prostate cancer. Long-chain acyl-CoA synthetases (ACSLs) catalyze the conversion of fatty acids into fatty acyl-CoAs that are required for fatty acid metabolism. In this study, we demonstrate that expression levels of ACSL3 and 4 were oppositely regulated by androgen-AR signaling in prostate cancer cells. AR served as a transcription suppressor to bind at the ACSL4 promoter region and inhibited its transcription. Inhibition of androgen-AR signaling significantly downregulated ACSL3 and PSA but elevated ACSL4 levels. ACSL4 regulated a broad spectrum of fatty acyl-CoA levels, and its catalytic efficiency in fatty acyl-CoAs biosynthesis was about 1.9-4.3-fold higher than ACSL3. Additionally, in contrast to ACSL3, ACSL4 significantly regulated global protein myristoylation or myristoylation of Src kinase in prostate cancer cells. Knockdown of ACSL4 inhibited the proliferation, migration, invasion and xenograft growth of AR-independent prostate cancer cells. Our results suggest that the surge of ACSL4 levels by targeting AR signaling increases fatty acyl-CoAs biosynthesis and protein myristoylation, indicating the opposite yet complementary or Yin-Yang regulation of ACSL3 and 4 levels in sustaining fatty acid metabolism when targeting androgen-AR signaling. This study reveals a mechanistic understanding of ACSL4 as a potential therapeutic target for treatment of AR-independent prostate cancer.
Keywords: ACSLs, fatty acid metabolism, protein myristoylation, prostate cancer
Introduction
Prostate cancer is the second leading cause of cancer-related deaths for men in North America (1). The majority of cancer patients undergo androgen deprivation therapy (ADT) by targeting androgen receptor (AR) signaling surgically or pharmacologically as the first line of treatment (2, 3). However, some patients will eventually develop castration-resistant prostate cancer (CRPC). Recent studies have reported that among patients undergoing ADT, more than one third carry AR-independent tumors. In particular, the number of AR-independent tumor patients has increased three-fold since a new generation of effective AR-pathway treatments have been in clinical use (4).
Fatty acid (FA) metabolism is highly dys-regulated in prostate cancer (5, 6). Elevated FA metabolism results in ATP synthesis through beta-oxidation, biosynthesis of membranes, and transcriptional regulation, all of which contribute to sustaining a high proliferation rate in prostate cancer cells and tumor growth (7). Additionally, metabolism of exogenous saturated FAs such as dietary myristic and palmitic acid has been found to fuel prostate tumor progression (8, 9) and targeting FA synthesis can inhibit the growth of prostate cancer cells (5). However, it is unknown how FA metabolism is sustained in the progression of AR-independent prostate tumors.
FAs must first be converted into acyl-CoAs to participate in FA metabolism. Long-chain acyl-CoA synthetases (ACSLs) including ACSL1, 3, 4, 5, and 6 catalyze the conversion of 12-20 carbon fatty acids into the corresponding acyl-CoAs (10). The synthesis of fatty acyl-CoAs is necessary for numerous cellular activities, including posttranslational modification of proteins (10, 11). Therefore, ACSLs are potentially integral in promoting cancer cell proliferation. Among the ACSL family members, ACSL3 and ACSL4 play an important role in acyl-CoAs biosynthesis and lipid biosynthesis (12, 13). While ACSL3 is up-regulated by the addition of androgen in androgen sensitive prostate cancer cells (14–16), over-expression of ACSL4 promotes cell proliferation, migration and invasion of prostate cancer cells (17). Additionally, ACSL4 mediates the biosynthesis of phosphatidylinositol (18), and causes enrichment of long unsaturated FAs in the cytoplasmic membrane and promotes ferroptosis-related disease (19). The ACSL4 isoform mediates germline sex determination by regulation of protein myristoylation in C. elegans (20). While it is known that functions of the ACSL family members overlap, it remains to be explored if this holds true in prostate cancer progression.
Androgen deprivation is the first line of chemotherapy in prostate cancer. We studied how FA metabolism mediated by ACSL members is regulated by androgen-AR signaling. We demonstrate herein that inhibition of androgen-AR signaling suppressed ACSL3 levels, but simultaneously up-regulated ACSL4 levels in prostate cancer cells. ACSL4 had about 1.9-4.3-fold higher catalytic efficiency to regulate a broad spectrum of fatty acyl-CoAs biosynthesis in comparison with ACSL3. It regulated global protein myristoylation or myristoylation of Src kinase in prostate cancer cells. Inhibition of ACSL4 suppressed proliferation, migration, and invasion of AR-null cancer cells and growth of prostate xenograft tumors. These results suggest that ADT treatment leads to de-repression of ACSL4, and confers more efficiency in the biosynthesis of fatty acyl-CoAs and protein myristoylation in prostate cancer cells. Our study provides a mechanistic understanding of an opposite yet complementary or Yin-Yang regulation of ACSL3 and 4 expression to sustain fatty acid metabolism in prostate cancer cells.
Materials and Methods
Cell culture
Human prostate cell lines LNCaP (CRL-1740), C4-2B (CRL-3315), 22Rv1 (CRL-2505), PC-3 (CRL-1435) and DU145(HTB-81) and hepatic cell lines, HepG2 (HB-8065) and Hep3B(HB-8064) were obtained from American Type Culture Collection (ATCC). PNT2 was purchased from Sigma (Cat#, 95012613). Cell lines from ATCC and Sigma had a certificate of the mycoplasma-free and authentication when purchased. Cells were maintained in a humidified incubator under 5% CO2 at 37 °C and grown in the recommended media supplemented with 10% fetal bovine serum (FBS) or charcoal stripped FBS and 1% penicillin/streptomycin. To maintain the cell line authentication, all cell lines were passaged for no more than 20 passages after resuscitation in this study. The potential of mycoplasma contamination was periodically examined by using ATCC detection kit.
Plasmid construction for RNA interference and lentivirus production
shRNAs for ACSL3, ACSL4, AR, and NMT were generated using primers listed in Supplementary Table S2. Primers were annealed and inserted into psiRNA-W[H1.4] vector at the BbsI site. H1 promoter-driven shRNAs were further reconstructed into FUCRW or FUCGW lentiviral vectors at the PacI site. The new plasmid with insertion was confirmed by DNA sequencing. Lentivirus production and infection of prostate cell lines were performed as described previously(48). Lentivirus procedures followed the guidelines and regulations at the University of Georgia.
Quantitative RT-PCR analysis
Total RNA was isolated from cell lines by TRIzol following the manufacturers’ instruction. One microgram of the total RNA was used for first-strand cDNA synthesis using the High-Capacity cDNA Kit (Applied BioSystems, Foster City, CA). Real-time PCR was performed using SYBR Green as an indicator on the ABI StepOne Plus Real-Time PCR system. The final reaction mixture contained 10 ng of cDNA, 100 nM of each primer, 5 μL of 2× SYBR Green PCR Master Mix (Quanta Biosciences, Beverly, MA), and RNase-free water to a final volume of 12.5 μL. PCR was carried out for 40 cycles at 95 °C for 15 s and 60 °C for 1 min. The data were analyzed using the ΔΔCt method and normalized to the internal control of GAPDH. Primers were synthesized by Sigma (St. Louis, MO). Melting curve analysis of all real-time PCR products was conducted and showed a single DNA duplex.
Analysis of ACSL3 and ACSL4 in TCGA database and Gene Expression Omnibus (GEO)
Expression values were extracted from the cBioPortal for Cancer Genomics (http://www.cbioportal.org/) for ACSL3, ACSL4, KLK2, and KLK3 using the Gene Set Query. The expression values were then cross-referenced with the data sets from the TCGA Data Portal. The data was then downloaded and aligned to the respective TCGA Sample IDs. The data from total 496 patient samples were download (two outlies were excluded from the analysis). Given the fact that some samples were derived from patients under androgen deprivation therapy (ADT), AR expression levels might be still high in those samples. Therefore, the KLK2/3 levels might have been blocked due to the ADT treatment. This means that mRNA levels of AR downstream genes might not genuinely reflect AR mRNA levels. However, since ACSL3, ACSL4, KLK2 and KLK3 are all-regulated by the AR, the correlation of ACSL3 and 4 with KLK2/3 could reflect how AR regulates these genes differently under any ADT treatment circumstances.
Another data set was also downloaded from cBioPortal for Cancer Genomics (http://www.cbioportal.org/) for ACSL4, KLK2, and KLK3 in the study titled with “Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories” (21). The correlation of ACSL4 with KLK2 and KLK3 is presented in the Supplementary Figure S1E–F.
Additionally, the ACSL3 and ACSL4 mRNA levels in human prostate cancer cells was analyzed in three public databases in the GEO (GSE4016, GSE7868, GSE8702). ACSL3 and ACSL4 expression levels in androgen insensitive cells was compared with the level in androgen sensitive cells.
Chromatin immunoprecipitation (ChIP)-qPCR
LNCaP cells in 10 cm dishes were cultured in RPMI-1640 medium with 10% charcoal stripped FBS and treated with 10 nM R1881 or DMSO overnight. Cells were cross-linked using 1% formaldehyde at room temperature for 10 min and the reaction was stopped by adding 0.125 M glycine for 5 min. Cells were harvested and the pellets were suspended in 500 μL ChIP- Lysis Buffer (1 M HEPES-KOH, 4 M NaCl, 0.5 M EDTA, 10% Triton-X100, 10% DOC, 0.1% protease inhibitor, 0.1% PMSF). The cross-linked DNA was sheared to approximately 100–500 bp using a BIORUPTOR sonicator for 45 min at high power followed by centrifugation at 12,000 g for 10 min at 4 °C. The supernatant was then immunoprecipitated with anti-AR (Millipore Sigma, Cat# 06-680) or IgG antibody (Cell signaling, Cat# 7074) overnight at 4 °C, after saving 4% of the chromatin as input. Protein G beads (40 μl; Cell Signaling Technology) were added and incubated for 2 h at 4 °C. The beads were washed with lysis buffer twice and DNA-protein complexes were eluted from beads using 62.5 μL TES Buffer (41.5 mL sterile water, 2.5 mL Tris-HCl, pH 8.0, 1.0 mL 0.5 M EDTA, 5 mL 10% SDS in total 50 mL) in a 65 °C incubator for 10 min. The reversal of DNA-protein crosslinks was performed by incubation in chromatin elution buffer containing proteinase K at 65 °C overnight. The remaining RNAs were digested by adding 6.25 μL of 10 mg/mL RNase A and incubation at 50 °C for 3 h, followed by the addition of 6.25 μL 20 mg/m Proteinase-K at 50 °C for 1 h to digest proteins. DNA was isolated by phenol:chloroform:IAA and chloroform extraction. To the aqueous layer was added 1 μL glycogen, 25 μL Na-Acetate and 865 μL 100% EtOH to precipitate DNA overnight at −20 °C. DNA was re-suspended in 50 μL ultra-pure water and the target genes were amplified by real time PCR. Quantification of target sequences relative to the input chromatin, which is derived from a portion (2%) of the lysate before immunoprecipitation, was computed using the percent (%) input method, which is indicated by the following formula: the form of % input = 2Ct (Input) % (InpChIP). The primers used for ChIP-PCR are detailed in Supplementary Table S1.
ACSL4 promoter analysis
To clone two copies of ACSL4 primers 9 flanking region into the parental pGL4 vector, primers listed in Supplementary Table S2 were used for PCR amplification. The Primer 9 F (SacI) and Primer 9 R (XhoI) amplified primers 9 flanking region containing a PCR product with SacI at the 5′-end and the Xho I at the 3′-end. Similarly, the Primer 9 F (XhoI) and Primer 9 R (HindIII) amplified the same sequence containing a PCR product with Xho I at the 5′-end and the Hind III at the 3′-end. These two PCR products were inserted into the vector pGL4 containing a minimal promoter (from Promega) at the restriction sites of SacI and HindIII. ARR2PB is a gift from Dr. Yun Qiu (49), which contains a probasin promoter in front of the luciferase gene. The vector was used as a positive control. C4-2B cells were seeded into 12-well plates. When cell density reached 70-90%, cells were transfected by using a lipofectamine 3000 transfection reagent according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA). A synthetic renilla luciferase reporter, phRG-TK (Promega), was used as a luciferase internal standard. One microgram of luciferase reporter gene, and 100 ng of phRG-TK vector (internal standard) were co-transfected. Twenty-four hours following the transfection, R1881 (10 nM) or DMSO were added and incubated for another 24 h. Luciferase activities were determined using the Dual-Glo Luciferase Assay system according to the manufacturer’s protocol (Promega). The amount of luciferase activity was measured using a FlexStation 3 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA) and normalized to the amount of phRG-TK luciferase activity. The experiment was repeated four times.
Western blot and antibodies
Cells were lysed in RIPA buffer [137 mM NaCl, 20 mM Tris-HCl (pH 7.4), 10% glycerol, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM EDTA] including protease inhibitor cocktail (Millipore, #539137) on ice followed by short sonication. Sample supernatants were collected after centrifugation at 14,000 rpm for 10 min. Forty micrograms of protein were separated on a 10% SDS-polyacrylamide gel before being transferred to nitrocellulose using a Bio-Rad Mini-Blot transfer apparatus (Richmond, CA). Membranes were blocked in Tris-buffered saline (TBS) containing 5% non-fat milk powder (Lab Scientific, Livingston, NJ) for 1 h.
Immunoblotting was performed at 4 °C overnight using antibodies against ACSL3 (ab151959), ACSL4 (ab205197), CD34 (ab81289) from Abcam (Cambridge, MA), AR (N-20) from Santa Cruz Biotechnology (Dallas, TX), CDK4 (Cat# 12790T), cyclin D1 (Cat# 2978), cyclin D3 (Cat# 2936T), GAPDH (Cat# 2118) from Cell Signaling (Boston, MA), or NMT1 (HPA022949) from Sigma (St. Louis, MO). The membranes were incubated with a secondary antibody at room temperature for 1 h after washing with TBS containing 0.5% Tween-20. Protein bands were visualized using the Pierce ECL Western Blotting substrate (Rockford, IL) and quantified by Image J software.
Fatty acyl-CoA analysis by LC-MS/MS
Long chain fatty acyl-CoAs were analyzed as described previously (23, 50). Briefly, cells were treated with/without 400 μM myristic acid in serum-free media containing 2% fatty acid-free BSA for 24 h. Cells were incubated with 2 mL methanol including 15 μL pentadecenoyl-CoA (10 μg/mL, internal standard) at −80 °C for 15 min after washing with PBS twice. The cell lysate was collected and centrifuged at 14,000 rpm at 4 °C for 10 min and the supernatant was transferred to a glass tube, mixed with 1 mL acetonitrile and evaporated in a vacuum concentrator at 55 °C for 2 h. The sample was reconstituted with 120 μL methanol, briefly vortexed, centrifuged, and 100 μL supernatant was transferred to an auto-sampler vial for LC-MS/MS analysis. The results were normalized to total cellular protein and the results are expressed as pmol/mg protein.
Detecting protein myristoylation by Click chemistry
To evaluate myristoylation of proteins, PC-3, and DU145 cancer cells transduced with shRNA-control, shACSL4 or shNMT1 were grown in DMEM with 2% fatty acid free BSA containing 100 μM myristic acid-azide for 24 h. The protein lysates were extracted using M-PER buffer (Thermo Scientific) containing protease inhibitors. Forty micrograms of protein underwent the Click chemistry reaction for 1 h at room temperature mixed with one volume of Click reaction buffer (2 mM CuSO4, 1 mM bis(tert-butyl)-tris(triazolylmethyl)amine-propanol, 10 mM sodium L-ascorbate, 40 μM biotin-alkyne). The mixture was then heated at 98 °C for 10 min and separated on 10% Tris-glycine SDS-polyacrylamide gels. Myristoylated protein was detected by probing the membrane with streptavidin-horseradish peroxidase (Thermo Fisher Scientific).
Assay for cell proliferation, migration and cell cycle analysis
For cell proliferation assays, the cells were seeded into 96-well plates at a density of 1500 cells per well and incubated in 100 μL of fresh culture medium without phenol red. Ten microliters of 12 mM MTT (3-(4, 5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) reagent (Thermo Fisher Scientific, Somerset, NJ) was added to each well at different time points and incubated at 37 °C for 4 h. The reaction was stopped by addition of 100 μL of 10% SDS and incubated for 4 h. The absorbance was measured at 570 nm by an iMark Microplate Reader (Bio-Rad, Richmond, CA).
Cell migration assays were performed using the BD FluoroBlok Insert System (Becton-Dickinson, Mansfield MA). Cells were grown to ~80% confluence and incubated in serum-free medium overnight. Cells were harvested and seeded in the upper wells of the chamber at a density of 2-4×104 cells in serum-free medium while lower wells contained medium with 10% FBS. After 48 h incubation, the cells that migrated through the polycarbonate filter were fixed with 4% formaldehyde for 5 min and washed with PBS twice followed by 0.1% crystal violet staining. Stained cell images were taken by a microscope and counted in five fields.
For wound-healing assays, 8×105 cells were seeded in a 6-well plate and a wound space was created by scraping with a 1 mL pipet tip after the cells reached confluence. The floating cells were washed out and regular media was added. The scratched space was monitored and imaged by a microscope daily.
To analyze the cell cycle, cells were trypsinized and suspended in PBS at a concentration of 1 × 106 cells/mL when cells reached ~70% confluence within 4 days. Then cells were fixed with ice-cold 70% ethanol for 30 min at 4 °C and stained by FxCycleTM PI/RNase solution (Thermo Fisher Scientific) for 30 min in the dark at room temperature before analysis using flow cytometry (CyAn ADP Analyzer, 488-nm excitation and 585-nm emission). Data were analyzed by using Flowjo software (TreeStar, San Marcos, CA).
In vivo xenograft animal model and immunohistochemistry
CB.17SCID/SCID (SCID) mice were purchased from Taconic (Hudson, NY) and housed under standard conditions with a 12 h light–dark cycle. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Georgia. Prostate cancer cell lines with ACSL4-shRNA or control vector were grown in recommended media. PC-3 cells (3×105) or DU145 cells (1×106) were mixed with 30 μL of collagen type I (pH 7.0) (BD Biosciences) and inoculated subcutaneously in both lateral flank sides of male SCID mice (ACSL1 knockdown cells on the right flank and control on the left counterpart, n=6). The grafts were randomized to implanted into the mice. Sex as a biological variable is not applicable to this study. The host mice were sacrificed, and xenograft tumors were harvested after 8 weeks of incubation. Tumor volume and weight were measured and then fixed in formaldehyde or stored at −80 °C. Tumor volume based on external caliper measurements was calculated by the modified ellipsoidal formula: Tumor volume = 1/2(length × width2).
For immunohistochemistry (IHC), mice xenograft tumor tissues were fixed in 10% formalin and embedded in paraffin followed by tissue sectioning at a thickness of 4 μm. IHC was performed to measure expression levels of Ki67 (1:400; Novus Biologicals, #NB500-170B) and CD34 (1:2000; Abcam, # ab81289).
Statistical analysis
Prism software was used to carry out statistical analysis. The data are presented as mean ± SEM and analyzed using the Student’s t test. “*”: P<0.05; “**”: P<0.01; “N.S.”: not significant. The linear regression model was used to study the association between CoA levels and ACSL4 gene expression. A regression coefficient was considered significant if its P-value was less than 0.05.
Results
Expression levels of ACSL3 and 4 are oppositely regulated in prostate cancer cells.
ACSLs catalyze the conversion of 12-20 carbon fatty acids (FAs) into the corresponding acyl-CoAs. This is essential for the activation of fatty acids to participate in metabolic pathways in normal and cancer cells. Among the ACSL family members examined, ACSL3 and 4 mRNA levels were the dominant isoforms identified in prostate cancer cells (Fig. S1A–C). The relative ACSL3 mRNA levels were significantly higher in AR+ prostate cancer cells (LNCaP and 22Rv1), while ACSL4 levels were significantly higher in AR− prostate cancer cells (PC-3 and DU145 cells) (Fig. 1A–B and Fig. S1D). We further analyzed expression levels of ACSL3 and 4 mRNA in three independent microarray datasets of prostate cancer cells from the Gene Expression Omnibus (GEO). A trend of opposite regulation of ACSL3 and 4 depending on AR responsiveness was found (Fig. 1C–D). Further analysis of the TCGA database showed that ACSL3 expression levels positively correlated with AR and the AR-regulated gene KLK2 or KLK3 (Fig. 1E–F). Conversely, ACSL4 expression levels negatively correlated to the AR-regulated genes KLK2 and KLK3 (Fig. 1G–H). The negative correlation of ACSL4 with KLK2 and KLK3 was also observed in another study in the cBioPortal for Cancer Genomics database (Fig. S1E–F)(21). Collectively, the results suggest that androgen-AR responsiveness differentially regulates expression of ACSL3 and ACSL4.
Figure 1. Correlation of ACSL3 and 4 levels with androgen/AR levels or AR activity in prostate cancer.
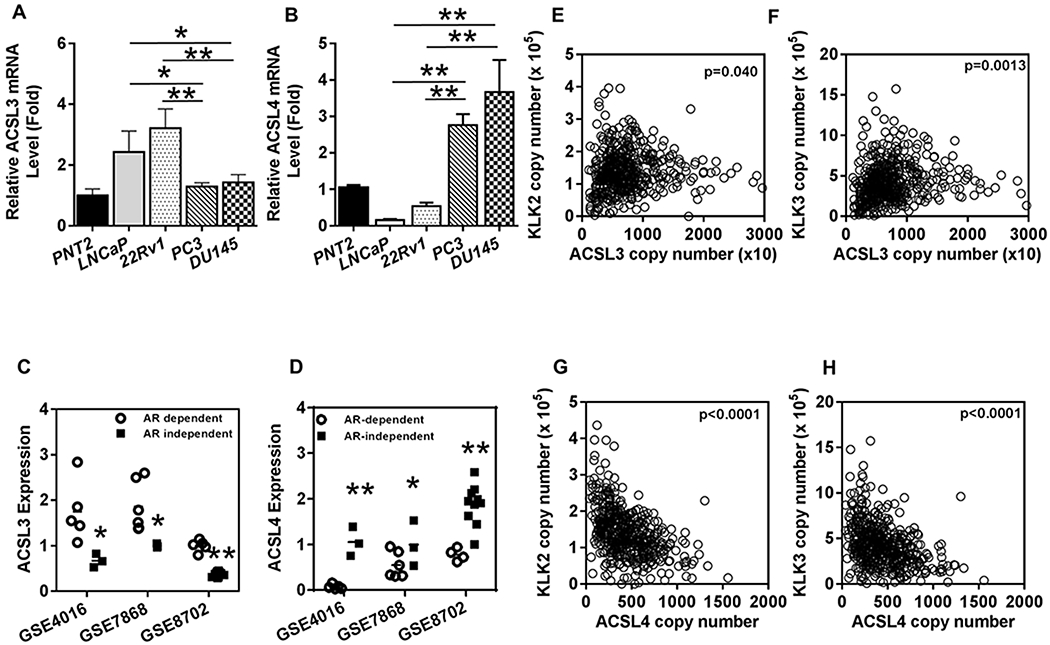
(A-B) The relative mRNA levels of ACSL3 and ACSL4 were measured in normal prostate cells (PNT2), AR+ (LNCaP and 22Rv1), and AR− (PC-3 and DU145) prostate cancer cells by Real-time RT-PCR. The relative mRNA levels of ACSL3/GAPDH (A) and ACSL4/GAPDH (B) in PNT2 cells were set at 1. The fold change of ACSL3 and ACSL4 in other cell lines relative to those in PNT cells were calculated. (C-D) The relative ACSL3 and ACSL4 mRNA levels from AR-dependent and independent prostate cancer cells were extracted from three public databases (GSE4016, GSE7868 and GSE8702) in Gene Expression Omnibus (GEO). *: P<0.05. (E-H) The mRNA copy number of ACSL3, ACSL4, and the AR-regulated downstream genes KLK2 and KLK3 were extracted from the TCGA database (n=496). ACSL3 levels were positively correlated with KLK2 (E) and KLK3 levels (F), while ACSL4 levels were inversely correlated with KLK2 (G) and KLK3 (H) levels in prostate tumors.
Activation or suppression of androgen-AR signaling oppositely regulates expression of ACSL3 and 4 in prostate cancer cells.
Androgen signaling was manipulated by androgen removal, addition of an AR agonist or antagonist, or knockdown of AR. ACSL3 levels were down-regulated (Fig. 2A) while ACSL4 levels were up-regulated (Fig. 2B) in LNCaP or C4-2B cells growing in androgen depleted media in comparison with those growing in a serum complete medium. Similarly, the AR agonist R1881 up-regulated ACSL3 levels, but down-regulated ACSL4 levels in LNCaP cells (Fig. 2C). Conversely, the addition of the AR antagonist casodex or MDV3100 significantly suppressed ACSL3 levels, but up-regulated ACSL4 levels (Fig. 2D–E). We further examined the response of ACSL3 and 4 expression under the regulation of AR levels in C4-2B cells. Knockdown of AR was confirmed by Western blotting and decreased expression of its downstream gene PSA by RT-PCR (Fig. 2F). While ACSL3 levels were suppressed, ACSL4 levels were significantly elevated in cells expressing shRNA-AR (Fig. 2G–H). Collectively, suppression of androgen-AR signaling inhibited ACSL3 expression levels but up-regulated ACSL4 expression levels and vice versa for activation of androgen-AR signaling.
Figure 2. Expression levels of ACSL3 and ACSL4 were inversely regulated by androgen-AR signaling or AR levels in prostate cancer cells.
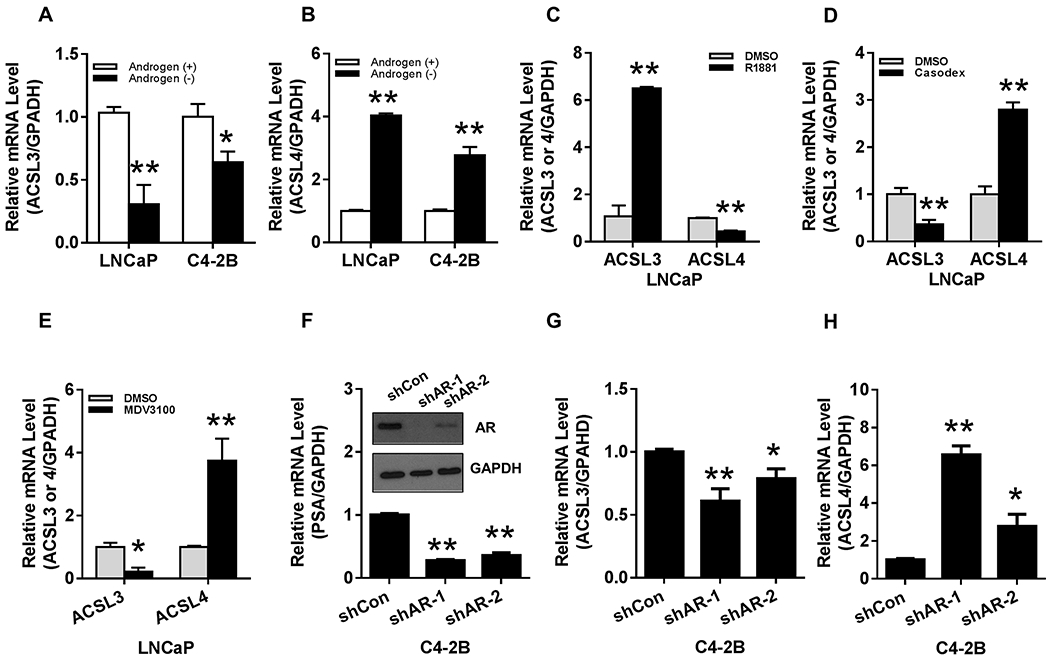
(A-B) Expression levels of ACSL3 and ACSL4 were inversely regulated by androgen levels. LNCaP and C4-2B cells were cultured in androgen depleted [charcoal stripped FBS, black bar, Androgen (−)] or androgen complete media [10% FBS, white bar, Androgen (+)]. mRNA levels of ACSL3, ACSL4, and GAPDH were measured by Real-time RT-PCR. The ratio of ACSL3 or ACSL4 to GAPDH is presented. The relative mRNA levels of ACSL3(A) and ACSL4 (B) in androgen complete media were set as 1. (C-E) LNCaP cells were cultured in medium containing 10% FBS and treated with 10 nM R1881 (AR agonist) (C), 10 μM casodex (AR antagonist) (D), or 10 μM MDV3100 (AR antagonist) (E) for 24 hours. ACSL3, ACSL4, and GAPDH mRNA levels were measured by Real-time RT-PCR. The relative ACSL3 or ACSL4 mRNA levels in the DMSO treatment group were set as 1. (F-H) C4-2B cells were transfected with shRNA-AR-1, 2, or control (shCon). AR protein levels were measured by Western blot, and relative mRNA levels of PSA, ACSL3, and ACSL4 were measured by Real-time RT-PCR. Expression levels of PSA (F), ACSL3 (G), and ACSL4 (H) in shRNA-Control were set as 1. The data represent the mean ± SEM (n=3). *: P<0.05; **: P<0.01.
AR binds to the androgen response element (ARE) in the promoter region of the ACSL4.
We explored whether AR directly regulated ACSL3 and 4 resulting in the observed opposite regulation of these genes. It has been reported that the ACSL3 promoter region contains AR-binding sites and androgen regulates its transcription (22). The promoter region of ACSL4 contained numerous potential androgen response element (ARE) sites (Fig. 3A). We examined if AR bound to these sites to regulate ACSL4 transcriptionally by designing 14 potential ARE binding site primers covering a 5 kb upstream region of the ACSL4 promoter and (Fig. 3A, Fig. S3, and Table S1). As expected, PSA and ACSL4 mRNA levels were up-regulated nearly 20-fold or down-regulated by 50%, respectively, after treatment of LNCaP cells with R1881 (Fig. 3B), demonstrating that androgen-AR signaling regulates PSA and ACSL4 mRNA levels inversely. This is supported by a 32-fold enrichment of AR binding at the PSA enhancer region in the presence of R1881 (Fig. 3C). Compared with cells treated with DMSO, a 3-fold AR enrichment at the ACSL4 promoter region was detected using primer 9, while no enrichment was observed using any of the other primers (Fig 3D–F, and Fig. S2). The data suggest that AR binds at the primers 9 flanked region of the ACSL4 promoter and suppresses its expression.
Figure 3. Identification of AR binding sites in the promoter region of the ACSL4 gene.
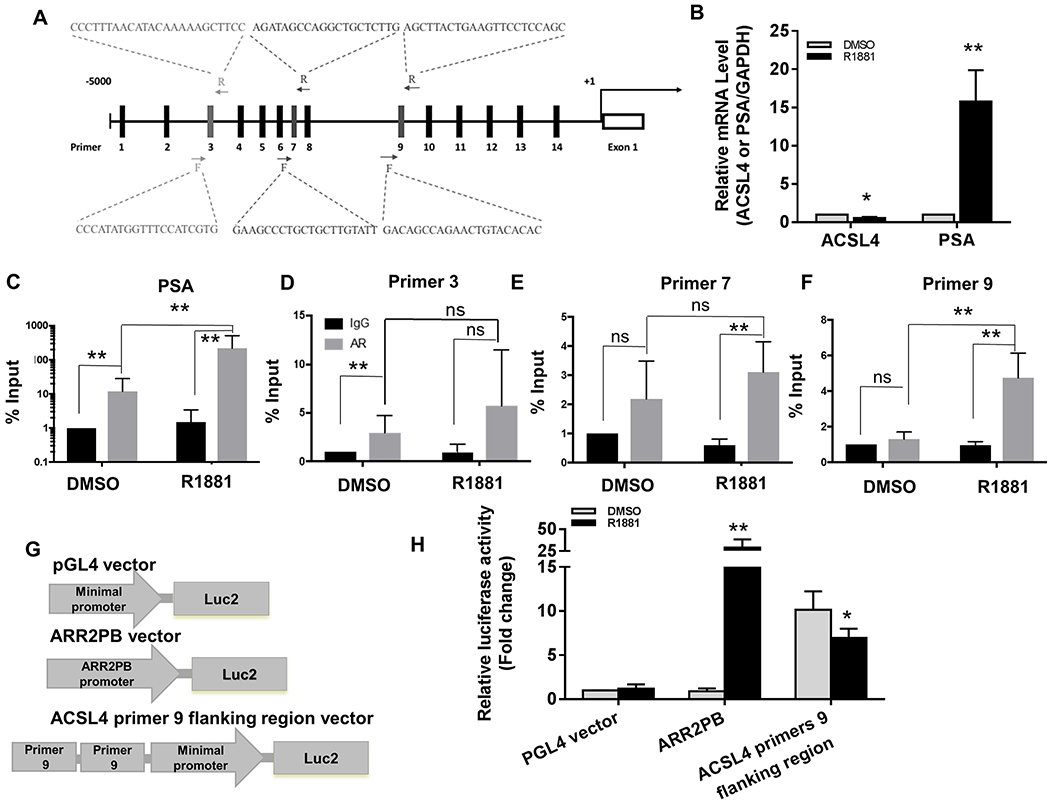
(A) The genomic DNA sequence 5 kb upstream from the first exon of ACSL4 in chromosome X was downloaded from the NCBI Gene website (https://www.ncbi.nlm.nih.gov/gene). Based on the AR binding consensus sequence, fourteen potential AR binding sites were selected in the 5 kb upstream region of ACSL4. The forward and reverse primer sequences at the locations 3, 7, and 9 are shown. The enrichment of DNA fragments at these sites were analyzed by ChIP-qPCR (C-F). (B) LNCaP cells were grown with/without 10 nM R1881 overnight. The relative expression levels of ACSL4 or PSA (control) and GAPDH were analyzed by Real-time RT-PCR. Expression levels of ACSL4 or PSA in the control (DMSO) were set as 1. (C-F) The genomic DNA derived from LNCaP cells treated with DMSO or R1881 was extracted for ChIP-qPCR as described in the Material and Methods. The presence of AR-binding sites at the promoter region of PSA was used as a positive control (C). The primers 3 (D), 7 (E), and 9 (F) sites at the ACSL4 locus region are shown. Results from the regions covered by other primer pairs are presented in Supplemental Figure 3. The data were normalized using the % input method. The IgG control in the DMSO treatment group was normalized to 1%. (G) The scheme of three vectors used for the promoter activity analysis. The pGL4 and ARR2PB vectors contain a minimal promoter (from Promega), and two copies of probasin promoter in front of the luciferase gene (positive control), respectively. In the ACSL4 primer 9 flanking region vector, two copies of ACSL4 primers 9 flanking sequence were cloned in front of minimal promoter of the pGL4 vector. (H) C4-2B cells were co-transfected with the vector expressing renilla luciferase together with the pGL4, ARR2PB, or ACSL4 primers 9 flanking region vector, respectively. After 24 h transfection, the cells were grown in medium treated with DMSO or R1881 (10 nM) for another 24 h. Luciferase activities were determined. The data represent the mean ± SEM (n=3). *: P<0.05; **: P<0.01.
To further verify if the primers 9 flanking region is regulated by AR signaling, two copies of primers 9 flanking sequence were sub-cloned into the vector containing the mini-promoter of the luciferase gene (Fig. 3G). As expected, pGL4 basal vector did not respond to stimulation by R1881, but the luciferase activity increased about 29-fold in the ARR2PB promoter (Fig. 3H). The luciferase activity was enhanced about 10-fold in the presence of the primer 9 flanking sequence, suggesting that the region might potentially recruit transcriptional machinery. Importantly, the activity was reduced by 31% under the stimulation of R1881 (Fig. 3G). Collectively, the results suggest that the primers 9 flanking region serves as a suppression site to regulate ACSL4 expression.
ACSL4 has higher catalytic efficiency than ACSL3 in regulating the biosynthesis of a broad spectrum of fatty acyl-CoAs.
ACSL4 is a member of the ACSL family that catalyzes the conversion of fatty acids into fatty acyl-CoAs (10). We have previously developed experimental approaches to examine fatty acyl-CoAs levels by LC-MS/MS (23). Knockdown of ACSL4 significantly reduced fatty acyl-CoAs levels including C12:0-, C14:0-, C16:0-, C18:0-, C18:1-, C18:2-CoA, C20:4-CoA in PC-3 and DU145 cells (Fig. 4A–N). Similarly, knockdown of ACSL3 led to a reduction of the above tested fatty acyl-CoAs levels in PC-3 cells (Fig. S3A). Considering shRNA knockdown efficiency, the reduction of fatty acyl-CoA, and abundancy of the relative ACSL3 and ACSL4 mRNA levels, we further compared the catalytic efficiency of ACSL3 versus ACSL4. ACSL4 was 4.2, 2.2, 1.9, 2.4, 1.9, 2.0, 4.3 fold higher in biosynthesis of the C12:0-, C14:0-, C16:0-, C18:0-, C18:1-, C18:2-CoA, C20:4-CoA compared to ACSL3 (Fig. S3B). These results suggest overlap of ACSL3 and ACSL4 in the biosynthesis of fatty acyl-CoAs. Although ACSL3 is more abundant compared to ACSL4 in prostate cancer cells (Fig. S1), ACSL4 has a higher catalytic efficiency in the biosynthesis of fatty acyl-CoAs.
Figure 4. ACSL4 regulates the biosynthesis of fatty acyl-CoAs and global protein myristoylation in PC-3 and DU145 cells.
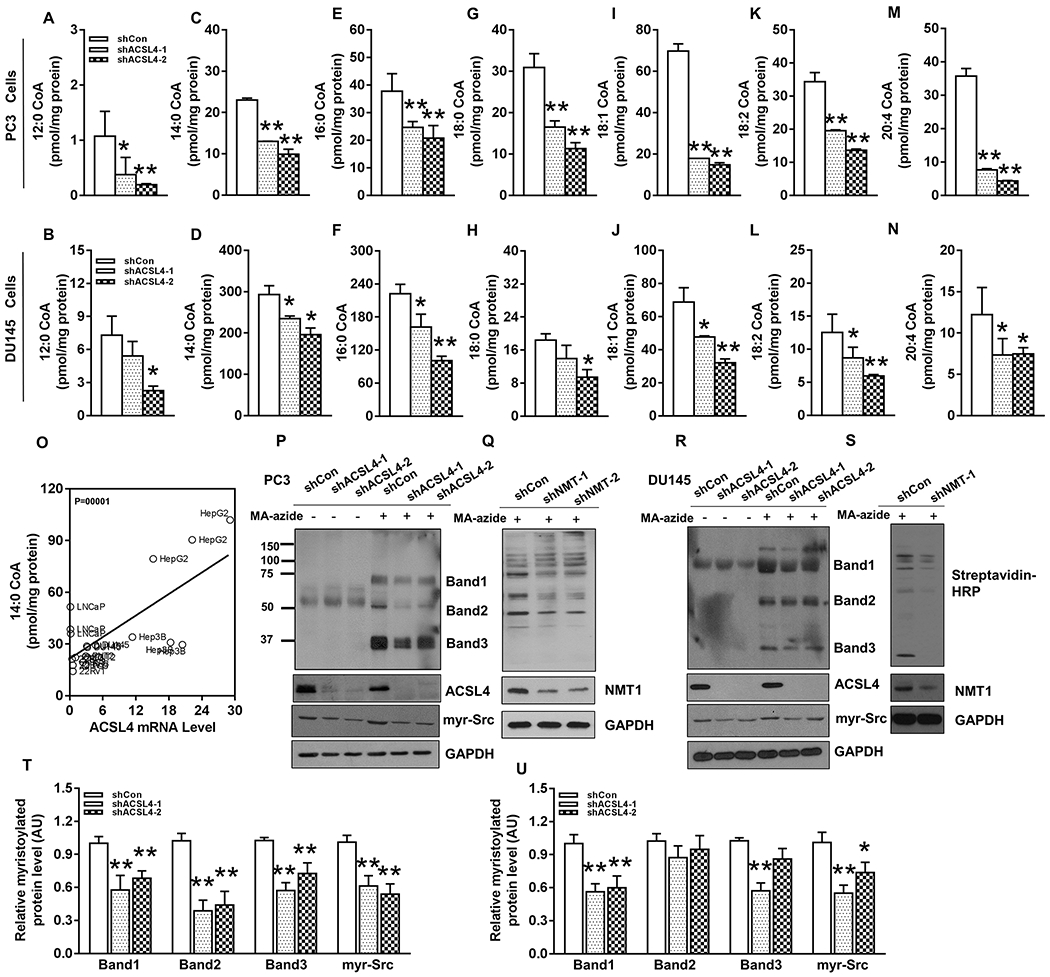
(A-N) PC-3 and DU145 cells were transduced with shRNA-control or two shRNA-ACSL4 (shACSL4-1 or 2) by lentiviral infection. The transduced cells were grown with/without myristic acid (MA, 400 μM) in DU145 and PC-3 cells, and the cell lysate was analyzed for C12:0-CoA (A-B), C14:0-CoA (C-D), C16:0-CoA (E-F), C18:0-CoA (G-H), C18:1-CoA (I-J), C18:2-CoA (K-L) and C20:4-CoA (M-N) by LC-MS/MS. Data are represented as mean ± SD (n=3). *: P<0.05; **: P<0.01. (O) Correlation of ACSL4 expression with myristoyl-CoA levels. ACSL4 mRNA levels were examined in normal prostate cells (PNT2), prostate cancer cells (LNCaP, 22Rv1, PC-3 and DU145) and hepatic cancer cells (HepG2 and Hep3B) by RT-PCR. Myristoyl-CoA levels were analyzed by LC-MS/MS. (P-S) PC-3 and DU145 cells transduced with shRNA-control (shCon), shRNA-ACSL4, or shRNA-NMT1 by lentiviral infection were cultured with/without 100 μM myristic acid-azide overnight. Global protein myristoylation in PC-3 (P-Q) or DU145 (R-S) expressing shRNA-control (shCon), shRNA-ACSL4, or shRNA-NMT1 were analyzed by Click Chemistry, and detected by streptavidin-HRP. ACSL4, NMT, Myr-Src, and GAPDH were detected by Western blot. (T-U) Three major bands of global protein myristoylation and Myr-Src in PC-3 (T) and DU145 (U) were quantified by Image J.
ACSL4 regulates global protein myristoylation in AR-independent prostate cancer cells.
Protein myristoylation is essential for the activity of a variety proteins (8, 24, 25). Myristoyl-CoA is the substrate for N-myristoyltransferases (NMT) that catalyze the attachment of the myristoyl group to proteins (26). We studied whether ACSL4 and ACSL3 regulated protein myristoylation. A linear regression analysis showed that ACSL4 mRNA levels were positively correlated with myristoyl-CoA levels (Fig. 4O). Additionally, Click Chemistry was used to examine the effect of ACSL4 on protein myristoylation. Knockdown of ACSL4 significantly suppressed global protein myristoylation in PC-3 (Fig. 4P and 4T) and DU145 (Fig. 4R and 4U), which is similar to the effect of NMT knockdown (Fig. 4Q and 4S). We have previously reported that myristoylation regulates the activity of Src kinase and its transformation potential (11). We have developed an antibody against myristoylated Src (Fig. S4). Knockdown of ACSL4 inhibited the levels of myristoylated Src kinase in PC-3 (Fig. 4P and 4T) and DU145 cells (Fig. 4R and 4U).
However, knockdown of ACSL3 did not change either myristoylation of Src kinase or the majority of myristoylated protein bands in LNCaP (Fig. S5A and C) and PC-3 cells (Fig. S5B and D). Some bands of myristoyl-proteins were elevated maybe due to a compensation of the loss of ACSL3. The data suggest that in contrast to ACSL4, loss of ACSL3 does not inhibit global protein myristoylation. Collectively, these results suggest a differential roles of ACSL3 and ACSL4 in protein myristoylation.
ACSL4 regulates the proliferation, migration, invasion, and cell cycle of AR-independent prostate cancer cells.
Suppression of androgen-AR signaling leads to up-regulation of ACSL4 levels, suggesting that ACSL4 might play an important role in AR-independent prostate cancer. We further examined whether knockdown of ACSL4 inhibited proliferation, migration, and invasion of the AR-independent prostate cancer cell lines PC-3, DU145, and C4-2B. While knockdown of ACSL4 did not affect the proliferation of C4-2B cells (of note, the endogenous ACSL4 levels were very low likely due to the suppression of active AR signaling) (Fig. 5A), it significantly inhibited proliferation (Fig. 5B–C) as well as migration and invasion of PC-3 and DU145 cells (Fig. 5D–G). Down-regulation of ACSL4 expression in PC-3 and DU145 cells with shRNA was determined by RT-PCR (Fig. S6A–B) and Western blotting (Fig. 5B–C). Knockdown of ACSL4 arrested PC-3 and DU145 cancer cells at the G0/G1 phase with a decrease of cells entering S phase or G2/M phase (Fig. 5H–I and Fig. S6C–H). The cell cycle arrest was further confirmed by the decrease of cell cycle regulators such as CDK4, cyclin D1 and cyclin D3 (Fig. 5J). The data suggest that ACSL4 facilitates proliferation of androgen-independent prostate cancer cells by regulating proliferation, migration, and invasion of prostate cancer cells and modulating the cell cycle.
Figure 5. ACSL4 regulates proliferation, migration, and cell cycle of prostate cancer cells.
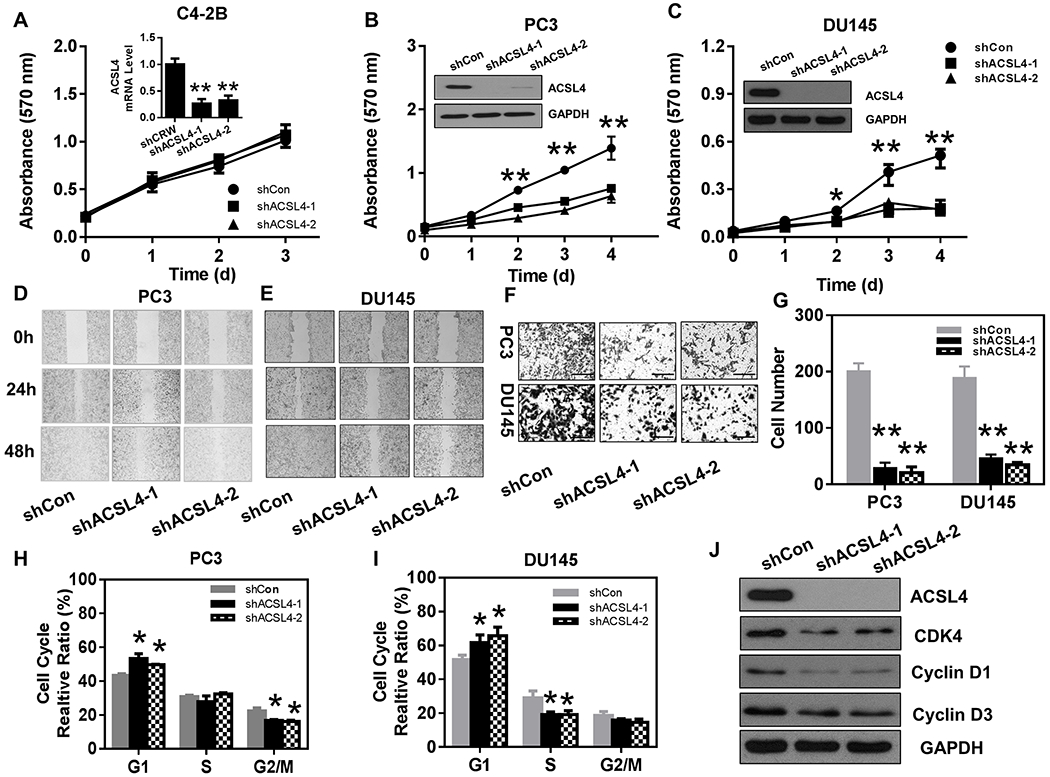
(A-C) The prostate cancer cell lines C4-2B, PC-3 and DU145 were transduced with control (shCon) or two shRNA-ACSL4 by lentiviral infection. Knockdown of ACSL4 in C4-2B cells was measured by RT-PCR due to low expression levels of endogenous ACSL4 protein likely due to AR activity (A). Down regulation of ACSL4 expression levels in PC-3 (B) and DU145 cells (C) were analyzed by Western blot. The proliferation of the transduced cancer cells was measured by the MTT assay. Data are represented as mean ± SEM (n=6). (D-E) The transduced PC-3 and DU145 cells were subjected to the wound healing assay. The healing process was observed at 24 and 48 hours. (F-G) The transduced PC-3 and DU145 cells were subjected to the transwell migration assay. Representative images were taken (Scale bars: 100 μm) (F), and the average number of migrated cells per field was calculated (G) (mean ± SEM, n=6). (H-I) The transduced PC-3 and DU145 cells were subjected to cell cycle analysis. The percentage of the cell population in the G1, S, and G2/M phases was calculated (mean ± SEM, n=3). *: P<0.05; **: P<0.01. (J) Expression levels of ACSL4 and the cell cycle regulators CDK4, cyclin D1, and cyclin D3 were examined by Western blot in PC-3 cells expressing shRNA-control or shRNA-ACSL4-1 or 2.
Knockdown of ACSL4 inhibits the growth of prostate cancer xenografts.
We examined if suppression of ACSL4 inhibited the growth of prostate tumors. PC-3 and DU145 cells transduced with shRNA-control or ACSL4 were implanted into SCID mice subcutaneously. The size and weight of tumors were significantly reduced in xenografts derived from PC-3 (Fig. 6A–C) and DU145 cells (Fig. 6D–F) expressing shRNA-ACSL4 in comparison with the control group. Expression levels of Ki67 and CD34 were decreased in tumors expressing shRNA-ACSL4 (Figure 6G–H), indicating that ACSL4 regulates growth and the formation of vasculature in these prostate tumors.
Figure 6. Knockdown of ACSL4 inhibits growth of prostate tumors in vivo.
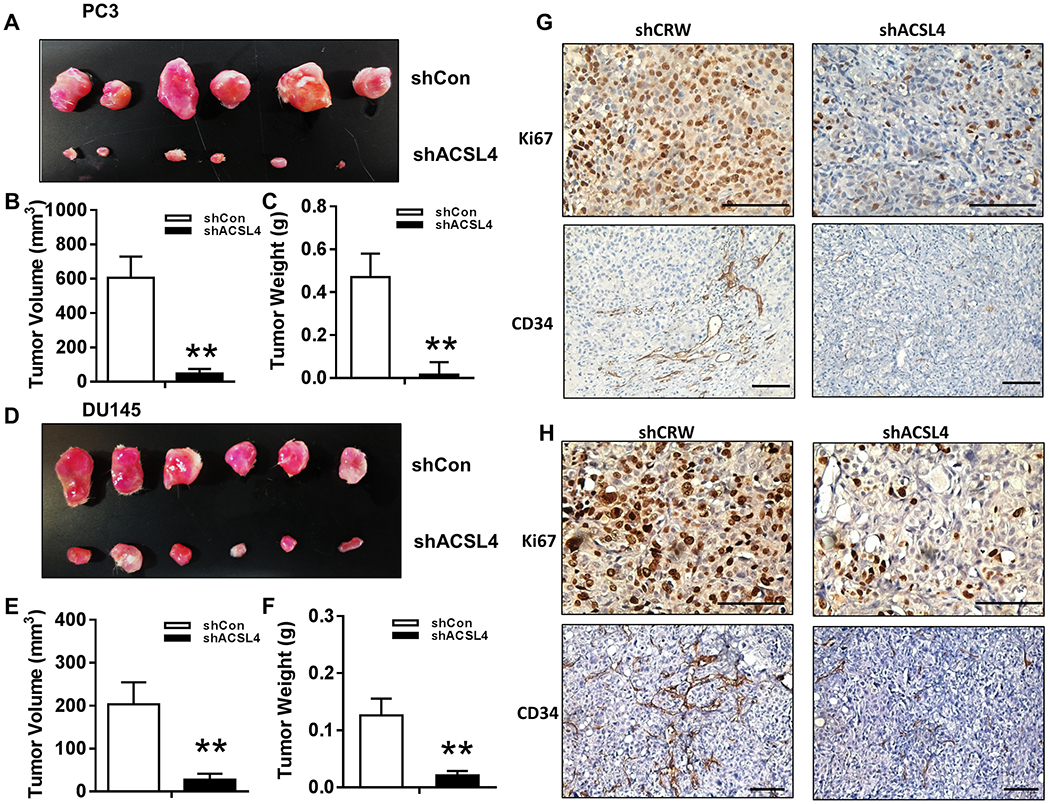
(A-H) PC-3 and DU145 cells transduced with control (shCon) or shRNA-ACSL4 were implanted in SCID male mice subcutaneously. After 8 weeks, tumors were harvested. The representative images (A and E), size (B and F), and weight (C and G) of the xenografts are presented. The data are represented as mean ± SEM (n=6). Expression levels of Ki67 and CD34 in xenograft tumors were analyzed by IHC staining (scale bars: 100 μm) (G-H). *: P<0.05; **: P<0.01.
Discussion
Our study has illustrated that long-chain acyl-CoA synthetase (ACSL) 3 and ACSL4 are differentially regulated by AR signaling in prostate cancer cells. Activation of androgen-AR signaling elevated ACSL3 expression, but down-regulated ACSL4. Conversely, suppression of androgen-AR signaling down-regulated ACSL3 expression levels, but up-regulated ACSL4 levels. Androgen deprivation treatment will lead to an elevation of ASCL4 levels. This is consistent with previous reports that mRNA expression levels of ACSL3 and ACSL4 have an inverse relationship in prostate cancer cells and tissues (27), and androgen receptor signaling is inversely correlated with ACSL4 expression (13, 17). In this study, we further demonstrated that AR plays a role as a transcriptional repressor in regulating ACSL4 expression, and identified the location (primers 9 flanking region) where AR bound to the ACSL4 promoter region. Additionally, our study demonstrated that ACSL3 and ACSL4 have overlapping functions with respect to fatty acyl-CoAs biosynthesis. However, ACSL4 had ~1.9-4.3-fold higher catalytic efficiency than ACSL3. Furthermore, ACSL4 and ACSL3 differentially regulated protein myristoylation. ACSL4, but not ACSL3 regulated global protein myristylation and specifically the myristoylation of Src kinase. Our study provides a mechanistic understanding of how prostate cancer cells might regulate ACSL levels in an opposite yet complementary way or in a Yin-Yang fashion to sustain fatty acid metabolism in response to androgen deprivation treatment (Fig. 7).
Figure 7. Expression levels of long-chain acyl-CoA synthetase 3 (ACSL3) and ACSL4 are oppositely regulated by androgen-AR signaling to maintain fatty acid metabolism in AR-dependent and AR-independent prostate cancer cells.
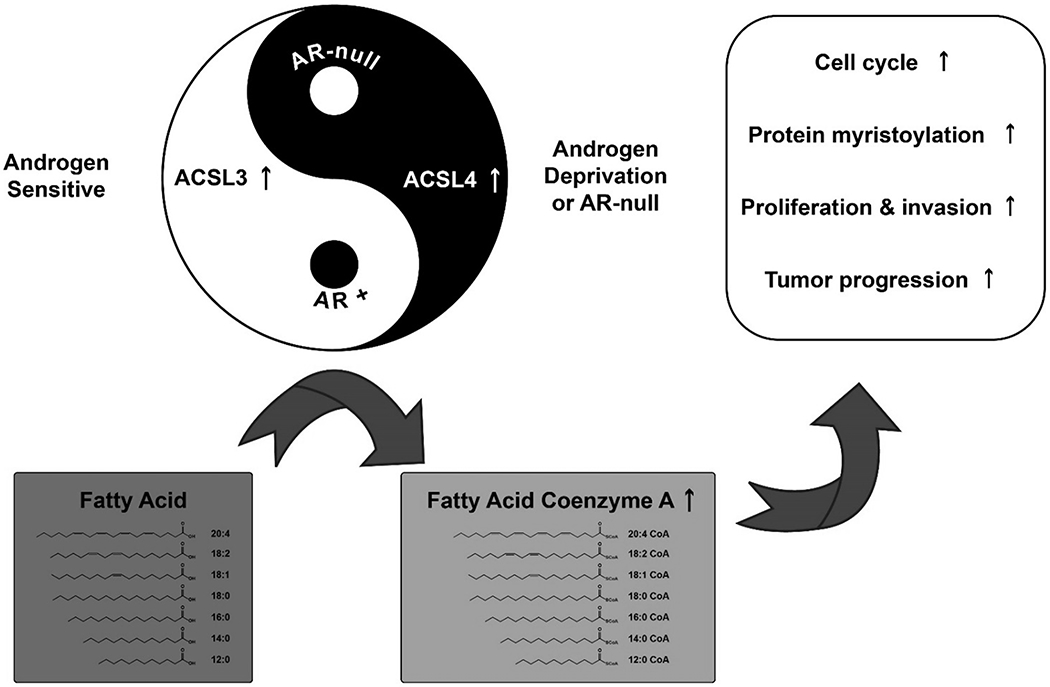
Fatty acid metabolism is dys-regulated in prostate cancer cells. ACSLs catalyze the conversion of fatty acids into fatty acyl-CoAs, which is essential for fatty acids to participate in metabolism to promote cancer cell proliferation, migration, invasion, and tumor progression. Two major ACSLs members, ACSL3 and ACSL4 are oppositely regulated by androgen-AR signaling. Suppression of androgen-AR signaling inhibits ACSL3 levels, but enhances ACSL4 levels. While ACSL3 is abundant in prostate cancer cells, it has lower efficiency to catalyze the biosynthesis of fatty acyl-CoA. Conversely, ACSL4 is less abundant, but has higher catalytic efficiency. In addition to involvement in fatty acyl-CoAs biosynthesis, ACSL4 regulates the biosynthesis of protein myristoylation. The opposite regulation occurs in AR-dependent cancer cells. This opposite complementary or Yin-Yang regulation could sustain fatty acid metabolism and survival of AR-independent prostate cancer cells or prostate cancer cells under androgen deprivation treatment.
Our study illustrates that knockdown of ACSL4 inhibits proliferation, migration, invasion and tumor growth in AR-independent cancer cells in vitro, and growth of xenograft tumors in vivo. Among castration resistant prostate cancer, more than one-third of patients have developed AR-independent tumors (4). Those tumors which have undergone androgen deprivation treatment and become AR-independent might potentially shift toward ACSL4-dependent fatty acid metabolism and cell growth. Over-expression of ACSL4 leads to an increase of proliferation and invasion in breast cancer cells (28). Elevated levels of ACSL4 have also been reported in numerous other cancers including hepatocellular carcinoma, cholangiocarcinoma, colon cancer, breast cancer, and others (29–31). For example, ACSL4 was up-regulated up to 65.0-fold at the protein level in colon adenocarcinoma (32). It serves as a biomarker for aggressive breast cancer or predictive biomarker for hepatocellular carcinoma in responding to sorafenib treatment (28, 33). Mechanistically, ACSL4 can integrate fatty acid metabolism together with oncogenic signaling to regulate proliferation of cancer cells. Our study shows that ACSL4 regulates the biosynthesis of a broad spectrum of fatty acyl-CoAs, and has a particularly high catalytic efficiency in the C20:4-CoA biosynthesis compared to ACSL3. Our results are consistent with the reported substrate preference of polyunsaturated fatty acid (PUFAs) such as arachidonic acid and eicosapentaenoic acid, which are the precursors for the formation of prostaglandin and lipid biosynthesis (30, 34). In addition, over-expression of ACSL4 also leads to activation of mTOR signaling (35), and upregulation of a set of other proteins such as pAkt, LSD1, and beta-catenin (17). Given the frequent occurrence of up-regulated ACSL4 levels in various cancer types, ACSL4 might potentially be a universal target for treatment of aggressive cancers.
Our study has demonstrated that AR plays a dual role as both a transcriptional activator and suppressor, leading to opposite regulation of ACSL3 and 4 levels simultaneously. AR is known to activate genes including KLK2, KLK3, TMPRSS2, and many others through its binding to the androgen response element (ARE). Recent studies have also recognized that AR directly suppresses an array of genes through genomic landscape analysis (36). For example, activation of AR signaling suppresses a number of genes including NOV by the synergy of AR with EZH2. NOV expression levels are de-repressed by hormone-deprivation therapy (37). While the role of the majority of AR suppressed genes is still functionally unknown in castration resistant prostate cancer, our data reveal that AR binds to an ARE site in the ACSL4 promoter region, and suppresses its transcription in the presence of the AR agonist R1881. Of note, compared with the AR-regulation of PSA, AR might only weakly regulate ACSL4 transcription based on our ChIP-qPCR results. However, biosynthesis of fatty acyl-CoAs could be well-sustained due to its higher catalytic efficiency in comparison with ACSL3. Additionally, similar to over-expression of ACSL4 in AR-null negative cells, over-expression of ACSL4 occurs in ER-null breast tumors. ER and ACSL4 are inversely correlated in breast cancer (13). It remains to be determined mechanistically how collaboration of AR/ER regulates ACSL4 and its synthesis of fatty acyl-CoAs in normal or cancer cells or tissues.
Recent studies have focused on exploring effective therapeutic strategies for targeting androgen-AR signaling to treat castration resistant prostate cancer. However, given the fact that AR could partially be a transcriptional repressor, de-repression of an array of AR down-stream genes may potentially contribute to development of resistance to androgen deprivation. For example, numerous AR degradation inhibitors have been developed based on the PROTAC principle (38, 39) and some have undergone clinic trials (40). As shown in this study, degradation of AR led to de-repression of genes such as ACSL4 in order to sustain fatty acid metabolism and thereby prostate cancer survival. Suppression of ACSL4 inhibits proliferation, invasion, and migration of AR-independent prostate cancer cells. It has also been reported that ACSL4 can serve as a biomarker and potential therapeutic target for castration-resistant prostate cancer (17). Therefore, targeting ACSL4 will be potentially beneficial by synergizing with AR degradation therapy to inhibit tumor growth.
Our study has revealed that ACSL4 levels regulate global protein myristoylation. Protein myristoylation is essential for the oncogenic signaling of numerous proteins such as Src kinase and FRS2 (8, 11, 24). Protein myristoylation requires myristoyl-CoA, which is the substrate for N-myristoyltransferases (NMTs) (41). ACSL4 plays as an important role in converting myristic acid to myristoyl-CoA, thus providing the substrate for the biosynthesis of myristoylated proteins (8). Additionally, we have previously shown that a high fat diet (HFD) accelerates Src kinase-mediated prostate cancer progression through protein myristoylation. It is speculated that myristic acid (MA) in a HFD will promote tumor progression by increasing protein myristoylation. Numerous epidemiological studies suggest that obesity, higher body mass index, or adult weight gain significantly increases the risk of aggressiveness, mortality, and recurrence of prostate cancer (42–45). Dietary MA is one of the major saturated FAs found in the Western diet. It accounts for about 9.5% of total fatty acids in retail milk or milk labeled as organic in the U.S. (46, 47). Therefore, the role of ACSL4 in HFD-accelerated tumor progression by exogenous MA metabolism needs to be explored as a potential intervention to prevent prostate cancer progressing to advanced stages.
Supplementary Material
Implication.
Androgen receptor coordinately regulates the expression of ACSL3 and ACSL4, such that AR pathway-independent prostate tumors become dependent on ACSL4-mediated fatty acid metabolism.
Acknowledgments
This work was supported by NIH (R01CA172495), DOD (W81XWH-15-1-0507), and NIH (U01CA225784) to H. Cai
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. [DOI] [PubMed] [Google Scholar]
- 2.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. 2015;4:365–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell. 2017;32:474–89 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baron A, Migita T, Tang D, Loda M. Fatty acid synthase: a metabolic oncogene in prostate cancer? Journal of cellular biochemistry. 2004;91:47–53. [DOI] [PubMed] [Google Scholar]
- 6.Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nature reviews Cancer. 2016;16:732–49. [DOI] [PubMed] [Google Scholar]
- 8.Kim S, Yang X, Li Q, Wu M, Costyn L, Beharry Z, et al. Myristoylation of Src kinase mediates Src-induced and high-fat diet-accelerated prostate tumor progression in mice. J Biol Chem. 2017;292:18422–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim S, Yang X, Yin A, Zha J, Beharry Z, Bai A, et al. Dietary palmitate cooperates with Src kinase to promote prostate tumor progression. Prostate. 2019;79:896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grevengoed TJ, Klett EL, Coleman RA. Acyl-CoA metabolism and partitioning. Annual review of nutrition. 2014;34:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim S, Alsaidan OA, Goodwin O, Li Q, Sulejmani E, Han Z, et al. Blocking Myristoylation of Src Inhibits Its Kinase Activity and Suppresses Prostate Cancer Progression. Cancer Res. 2017;77:6950–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Migita T, Takayama KI, Urano T, Obinata D, Ikeda K, Soga T, et al. ACSL3 promotes intratumoral steroidogenesis in prostate cancer cells. Cancer Sci. 2017;108:2011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monaco ME, Creighton CJ, Lee P, Zou X, Topham MK, Stafforini DM. Expression of Long-chain Fatty Acyl-CoA Synthetase 4 in Breast and Prostate Cancers Is Associated with Sex Steroid Hormone Receptor Negativity. Transl Oncol. 2010;3:91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendriksen PJ, Dits NF, Kokame K, Veldhoven A, van Weerden WM, Bangma CH, et al. Evolution of the androgen receptor pathway during progression of prostate cancer. Cancer Res. 2006;66:5012–20. [DOI] [PubMed] [Google Scholar]
- 15.Wang G, Jones SJ, Marra MA, Sadar MD. Identification of genes targeted by the androgen and PKA signaling pathways in prostate cancer cells. Oncogene. 2006;25:7311–23. [DOI] [PubMed] [Google Scholar]
- 16.Zhao H, Kim Y, Wang P, Lapointe J, Tibshirani R, Pollack JR, et al. Genome-wide characterization of gene expression variations and DNA copy number changes in prostate cancer cell lines. Prostate. 2005;63:187–97. [DOI] [PubMed] [Google Scholar]
- 17.Wu X, Deng F, Li Y, Daniels G, Du X, Ren Q, et al. ACSL4 promotes prostate cancer growth, invasion and hormonal resistance. Oncotarget. 2015;6:44849–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuch EM, Vellaramkalayil R, Zhang I, Lehnen D, Brugger B, Sreemmel W, et al. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim Biophys Acta. 2014;1841:227–39. [DOI] [PubMed] [Google Scholar]
- 19.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature chemical biology. 2017;13:91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang H, Han M. Fatty Acids Regulate Germline Sex Determination through ACS-4-Dependent Myristoylation. Cell. 2017;169:457–69 e13. [DOI] [PubMed] [Google Scholar]
- 21.Gerhauser C, Favero F, Risch T, Simon R, Feuerbach L, Assenov Y, et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell. 2018;34:996–1011 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han W, Gao S, Barrett D, Ahmed M, Han D, Macoska JA, et al. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene. 2018;37:710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X, Ma Y, Li N, Cai H, Bartlett MG. Development of a Method for the Determination of Acyl-CoA Compounds by Liquid Chromatography Mass Spectrometry to Probe the Metabolism of Fatty Acids. Anal Chem. 2017;89:813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q, Alsaidan OA, Ma Y, Kim S, Liu J, Albers T, et al. Pharmacologically targeting the myristoylation of the scaffold protein FRS2alpha inhibits FGF/FGFR-mediated oncogenic signaling and tumor progression. J Biol Chem. 2018;293:6434–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin DD, Beauchamp E, Berthiaume LG. Post-translational myristoylation: Fat matters in cellular life and death. Biochimie. 2011;93:18–31. [DOI] [PubMed] [Google Scholar]
- 26.French KJ, Zhuang Y, Schrecengost RS, Copper JE, Xia ZP, Smith CD. Cyclohexyl-octahydro-pyrrolo[1,2-a] pyrazine-based inhibitors of human N-myristoyltransferase-1. J Pharmacol Exp Ther. 2004;309:340–7. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. Am J Clin Exp Urol. 2014;2:111–20. [PMC free article] [PubMed] [Google Scholar]
- 28.Wu X, Li Y, Wang J, Wen X, Marcus MT, Daniels G, et al. Long chain fatty Acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PloS one. 2013;8:e77060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ndiaye H, Liu J, Hall A, Minogue S, Morgan M, Waugh MG. Immunohistochemical staining reveals differential expression of ACSL3 and ACSL4 in hepatocellular carcinoma and hepatic gastrointestinal metastases. Biosci Rep. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuwata H, Hara S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 2019;144:106363. [DOI] [PubMed] [Google Scholar]
- 31.Rossi Sebastiano M, Konstantinidou G. Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao Y, Dave KB, Doan TP, Prescott SM. Fatty acid CoA ligase 4 is up-regulated in colon adenocarcinoma. Cancer Res. 2001;61:8429–34. [PubMed] [Google Scholar]
- 33.Feng J, Lu PZ, Zhu GZ, Hooi SC, Wu Y, Huang XW, et al. ACSL4 is a predictive biomarker of sorafenib sensitivity in hepatocellular carcinoma. Acta Pharmacol Sin. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang MJ, Fujino T, Sasano H, Minekura H, Yabuki N, Nagura H, et al. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc Natl Acad Sci U S A. 1997;94:2880–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orlando UD, Castillo AF, Dattilo MA, Solano AR, Maloberti PM, Podesta EJ. Acyl-CoA synthetase-4, a new regulator of mTOR and a potential therapeutic target for enhanced estrogen receptor function in receptor-positive and -negative breast cancer. Oncotarget. 2015;6:42632–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao JC, Yu J, Runkle C, Wu L, Hu M, Wu D, et al. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012;22:322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu L, Runkle C, Jin HJ, Yu J, Li J, Yang X, et al. CCN3/NOV gene expression in human prostate cancer is directly suppressed by the androgen receptor. Oncogene. 2014;33:504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salami J, Alabi S, Willard RR, Vitale NJ, Wang J, Dong H, et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol. 2018;1:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang CY, et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J Med Chem. 2019;62:941–64. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Ma J, Liu Y, Xia J, Li Y, Wang ZP, et al. PROTACs: A novel strategy for cancer therapy. Semin Cancer Biol. 2020. [DOI] [PubMed] [Google Scholar]
- 41.Sulejmani E, Cai H. Targeting protein myristoylation for the treatment of prostate cancer. Oncoscience. 2018;5:3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38. [DOI] [PubMed] [Google Scholar]
- 43.Wright ME, Chang SC, Schatzkin A, Albanes D, Kipnis V, Mouw T, et al. Prospective study of adiposity and weight change in relation to prostate cancer incidence and mortality. Cancer. 2007;109:675–84. [DOI] [PubMed] [Google Scholar]
- 44.Hu MB, Xu H, Bai PD, Jiang HW, Ding Q. Obesity has multifaceted impact on biochemical recurrence of prostate cancer: a dose-response meta-analysis of 36,927 patients. Med Oncol. 2014;31:829. [DOI] [PubMed] [Google Scholar]
- 45.Joshu CE, Mondul AM, Menke A, Meinhold C, Han M, Humphreys EB, et al. Weight gain is associated with an increased risk of prostate cancer recurrence after prostatectomy in the PSA era. Cancer Prev Res (Phila). 2011;4:544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Donnell-Megaro AM, Barbano DM, Bauman DE. Survey of the fatty acid composition of retail milk in the United States including regional and seasonal variations. J Dairy Sci. 2011;94:59–65. [DOI] [PubMed] [Google Scholar]
- 47.O’Donnell AM, Spatny KP, Vicini JL, Bauman DE. Survey of the fatty acid composition of retail milk differing in label claims based on production management practices. J Dairy Sci. 2010;93:1918–25. [DOI] [PubMed] [Google Scholar]
- 48.Xin L, Ide H, Kim Y, Dubey P, Witte ON. In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc Natl Acad Sci U S A. 2003;100 Suppl 1:11896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309–19. [DOI] [PubMed] [Google Scholar]
- 50.Brennen WN, Isaacs JT. Cellular Origin of Androgen Receptor Pathway-Independent Prostate Cancer and Implications for Therapy. Cancer Cell. 2017;32:399–401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.