

Abstract
The Ehlers-Danlos syndromes (EDS) are a group of heritable, connective tissue disorders characterized by joint hypermobility, skin hyperextensibility, and tissue fragility. There is phenotypic and genetic variation among the 13 subtypes. The initial genetic findings on EDS were related to alterations in fibrillar collagen, but the elucidation of the molecular basis of many of the subtypes revealed several genes not involved in collagen biosynthesis or structure. However, the genetic basis of the hypermobile type of EDS (hEDS) is still unknown. hEDS is the most common type of EDS and involves generalized joint hypermobility, musculoskeletal manifestations, and mild skin involvement along with the presence of several comorbid conditions. Variability in the spectrum and severity of symptoms and progression of patient phenotype likely depend on age, gender, lifestyle, and expression domains of the EDS genes during development and postnatal life. In this review, we summarize the current molecular, genetic, epidemiologic, and pathogenetic findings related to EDS with a focus on the hypermobile type.
Keywords: Ehlers-Danlos syndrome, hypermobility, musculoskeletal
1 |. HISTORY
Edvard Ehlers and Henri Danlos described variances in cutis laxa in 1908 and 1904, respectively. Ehlers-Danlos syndrome (EDS) was first named in 1946 and categorization of the EDSs followed in the late 1960’s. The EDS subtypes were then formalized in the Berlin nosology, which recognized 11 numbered subtypes of EDS.1 As the underlying molecular basis of various subtypes emerged, the Villefranche nosology came out in 1998, consisting of six subtypes with descriptive names.2 The Villefranche nosology for hEDS was originally defined as joint hypermobility with or without skin manifestations, family history, joint pain, dislocations, and systemic manifestations. The Villefranche criteria poorly defined hEDS and broadly labeled individuals with generalized joint hypermobility as having hEDS. Thus, in 2017, the EDS International Consortium proposed the 2017 International Classification for the EDSs to replace the Villefranche nosology, recognizing 13 subtypes of EDS (Table 1) with major and minor diagnostic criteria outlined for each subtype.74 The new criteria for hypermobile EDS were more strict than previous criteria, in an attempt to better define the disease subtypes and reduce future misdiagnoses.
TABLE 1.
Description of subtypes of EDS comparing 2017 nomenclature with the Villefranche and Berlin nosology
| Molecular biology of Ehlers-Danlos syndromes | |||||||
|---|---|---|---|---|---|---|---|
| Gene | Mutation(s) | Inheritance | Molecular biology | 2017 EDS nomenclature | Villefranche nosology | Berlin nosology | References |
| ADAMSTS2 | Homozygous nonsense mutations (p.(Gln225) and p.(Trp795*)), skipping in frame and out of frame of various exons, homozygous loss of function mutations (c.2927_2928delCT, p. (Pro976Argfs*42); c.669–670dupG, p. (Pro224Argfs*41); c.2751–2A>T) and compound heterozygous (p. (Met295Thrfs25*)) | AR | Enzymatic component of the ECM | Dermatosparaxis EDS (dEDS) | Dermatosparaxis type | Type VIIC | 3–5 |
| AEBP1 | Compound heterozygous (c,1470delC [p. Asn490_Met495delins(40)] and c,1743C>A [p.Cys581*]), homozygous (c,1320_1326del [p.Arg440Serfs*3]) homozygous splice site variant (c.1630J)1G>A), homozygous c,1925T>C p.(Leu642Pro) | AR | Regulates collagen fibrillogenesis | Classic-like EDS (clEDS) | N/A | N/A | 6,7 |
| B3GALT6 | Missense and frameshift mutations, in-frame deletions, start codon mutations, splice site mutations and in-frame duplication. Homozygous and compound heterozygous mutations. The following are the most commonly reported: p.(Pro67Leu), p. (Thr79Ala), p.(Arg232Cys), p.(Asp207His), p. (Phel86Leu), p.(Arg6Trp) p.(Glu265Asp), p. (Ser309Thr), p.(Glul74Alafs*266), p.(Metl?) | AR | Post-translational modification of proteins including proteogylcans | Spondylodysplastic EDS (spEDS) | EDS progeroid type | N/A | 8–13 |
| B4GALT7 | c.808C>T, p.(Arg270Cys); c,122T>C, p. (Leu41Pro); c.421C>T, p.(Argl41Trp); c.557C>A, p.(Alal86Asp); c.617T>G, p. (Leu206Pro); c.641G>A, p.(Cys214Tyr); c.277dup,p.(His93Profs*73); c.970T>A, p. (Cys324Ser) | AR | Post-translational modification of proteins including proteoglycans | Spondylodysplastic EDS (spEDS) | EDS progeroid type | N/A | 14–19 |
| C1R | Heterozygous missense and in-frame insertion/ deletion variants; p.Asp290Gly, p.Gly297Asp, p.Leu300Pro, p.Arg301Pro, p.Tyr302Cys, p. Ile306_Cys309delinsArgArg p.Cys309Trp, p. Cys309Trp, p.Cys338Arg, p.Cys358Phe, p. Trp364Cys, p.Cys371Trp, p. Arg401_Tyr405delinsHisValIle, p.Trp435Arg | AD | Complement immune system | Periodontal EDS (pEDS) | EDS periodontitis | Type VIII | 20 |
| CIS | Heterozygous missense and in-frame insertion/deletion variants p.Cys294Arg, p.Val316del | AD | Complement immune system | Periodontal EDS (pEDS) | EDS periodontitis | Type VIII | 20 |
| CHST14 | Loss of function, missense, frameshift and nonsense mutations p.(Pro281Leu), p. (Val49*), p.(Arg213Pro), p.(Tryr293Cys), p. (Arg29Gfs*113), p.(Lys69*), p. (Glnl 13Argfs*14), p.(Argl35Gly), p. (Leul37Gln), p.(Cysl52Leufs*10), p. (Phe209Ser), p.(Arg218Ser), p. (Gly228Leufs*13, p.(Glu262Lys), p. (Arg274Pro), p.(Met280Leu), p.(Cys289Ser), p.(Trp327Cfs*29), p.(Glu334Glyfs*107) | AR | Post-translational modification— conversion of sulfate to dermatan sulfate | Musculocontractural EDS (mcEDS) | N/A | N/A | 21–23 |
| C0L1A1 | c.934C>T, p.(Arg312Cys); c,1720C>T, p. (Arg574Cys); and c.3277C>T, p. (Argl093Cys) | AD | Structural ECM component | cEDS with vascular fragility, vEDS | Classical type, vascular type | Typel/II/IV | 24–26 |
| C0L1A1 | Splice site mutations that lead deletions of exon 6 (intron 5–2A>G/T; intron 5-lG>A/C/ T; exon 6-lG>A/C) | AD | Structural ECM component | Arthrochalasia EDS (aEDS) | Athroscorasia type | Type VIIA/B | 27–29 |
| C0L1A2 | c.213dupC,p.(Arg99*) homozygous, six splice site mutations (two homozygous (c.3105+ 2T>C and c.3601G>T)) and two compound heterozygous (c.70+717A>G; c.1404+1G>A and c.540+5G>A; c,1404G>C) | AR | Structural ECM component | Cardiac-valvular (cvEDS) | N/A | N/A | 30–33 |
| Splice site mutations that lead deletions of exon 6 | 34 | ||||||
| C0L1A2 | (intron 5–2A>G; intron 5-lG>A/C; exon 6-lG>A; intron 6+lG>A/T/C; intron 6+2T>C/G) | AD | Structural ECM component | Arthrochalasia EDS (aEDS) | Athroscorasia type | Type VIIA/B | 29,35–39 |
| C0L3A1 | p.(Gly637Ser) | AD | Structural ECM component | Hypermobile EDS (hEDS)? One Family | Hypermobility type (EDS-HT) | Type III | 40 |
| C0L3A1 | Glycine substitutions, splice-site insertions/ deletions, in-frame insertions/deletions, haploinsufficiency, nonglycine missense variants in the triple helix, nonglycine missense variants and in-frame insertions/ deletions, in the N- or C-terminal | AD | Structural ECM component | Vascular EDS (vEDS) | Vascular type | Type IV | 41,42 |
| C0L5A1 | Mutations leading to nonsense mediated mRNA decay, haploinsufficiency and structural mutations | AD | Structural ECM component | Classical EDS (cEDS) | Classical type | Type I/II | 26,43 |
| COL5A2 | Structural mutations and splice site mutations | AD | Structural ECM component | Classical EDS (cEDS) | Classical type | Type I/II | 26,43 |
| C0L12A1 | Heterozygous missense mutations that are autosomal dominant (c.7167 T>C, p. (Ile2334Thr), C.C5893T, p.(Argl965Cys), 8329G>C, p.(Gly2777Arg), C.G8357A, p. (Gly2786Asp)) and a homozygous frameshift mutation that is autosomal recessive (c.8006 +1 G>A, p.(2567Asp>Phefs*2) | AR or AD | ECM component | Myopathic EDS (mEDS) | N/A | N/A | 44–46 |
| DSE | Homozygous loss of function, missense mutations (p.(Arg267Gly), p.(Ser268Leu)) | AR | Biosynthesis of dermatan sulfate | Musculocontractural EDS (mcEDS) | N/A | N/A | 23,47 |
| FKPB14 | Duplication (c.362dup, p.(Glul22Argfs*7), homozygous deletion (c,197+5_197+ 8delGTAA), compound heterozygous mutations | AR or compound heterozygosity | ER folding/transport of collagen | Kyphoscoliotic EDS (kEDS) | Kyphoscoliosis type | Type VI, Type VIA | 48–51 |
| LZTS1 | p.(His211Gln) | AD | Tumor suppressor | Hypermobile EDS (hEDS)? One Family | Hypermobility type (EDS-HT) | Type III | 52 |
| PRDM5 | c. 1517_1527dell 1, p.(Val506Glufs*5); c.974delG, p.(Cys325Leufs*2); c.711_714delTGTT, p.(Val238Alafs*35); c,1768C>T, p.(Arg590*); c.320A>G, p. (Tyrl07Cys); c,17T>G, p.(ValôGly); C.93+ 1G>A | AR | Collagen synthesis | Brittle cornea syndrome (BCS) | N/A | N/A | 53–55 |
| PL0D1 | p.Ile454IlefsX2, p.Ala667Thr, and p.His706Arg, homozygous for exons 10–16 duplication, p. Ile454IlefsX2, homozygous for p.Arg319X | AR | ER folding/transport of collagen | Kyphoscoliotic EDS (kEDS) | Kyphoscoliosis type | Type VI, Type VIA | 56–64 |
| SLC39A13 | Homozygous 9-bp in-frame deletion in exon 4 | AR | Zinc transporter | Spondylodysplastic EDS (spEDS) | EDS progeroid type | N/A | 65–67 |
| ZNF469 | 5294delA, 9527delG, Cys 3339Tyr, Glul392Ter | AR | Collagen synthesis | Brittle cornea syndrome (BCS) | N/A | N/A | 68–73 |
Note: Gene mutation, inheritance pattern, molecular biology, nomenclature, and references are listed.
Similar to hEDS, hypermobility spectrum disorders (HSD) are a poorly recognized group of connective tissue disorders that involve a spectrum ranging from asymptomatic hypermobility or hypermobility affecting only one joint, to generalized joint hypermobility, subluxations, and dislocations. Patients with symptomatic joint hypermobility that do not meet the criteria for hEDS are often labeled as HSD. Prior to the 2017 criteria, patients may have been categorized as having joint hypermobility syndrome (JHS) or benign joint hypermobility syndrome (BJHS), both of which overlap phenotypically and may be indistinguishable from hEDS.75 Due to the lack of a genetic marker for hEDS or HSD/JHS and similarity in clinical and molecular phenotype, the two diagnostic labels are often grouped together. Unless further genetic discoveries are able to accurately separate patients into different subgroups, it has been recommended that JHS and hEDS should be viewed as one.76 Due to reclassifications of EDS subtypes, hEDS may also be referred to as EDS III or grouped with HSD or JHS in the literature reflected in this review.
2 |. EPIDEMIOLOGY
Generalized joint hypermobility, impacting four or more joints, has been reported to be present in anywhere from 12% to 28% of children, adolescents and young adults.77–83 This has been shown to be both age and gender specific, where females and children tend to be more hypermobile.79–82,84–86 It is important to note that joint hypermobility alone is somewhat common in the general population and may be present with no complications, especially in children.78,79,82,85 Furthermore, there are forms of acquired hypermobility that may be present in ballet dancers, gymnasts, wrestlers, cheerleaders, and other athletes where hypermobility is trained and is not indicative of a connective tissue disorder.87
There is an unexpected female predominance in hEDS despite suspected autosomal dominant inheritance.88 It is possible that women may be more frequently diagnosed due to a higher severity of symptoms than men. This may be explained by muscle mass and ligament stiffness being controlled by sex and leading to greater joint stability in men, as it is already known that women are more flexible than men.89 Additionally, women are also known to engage the medical system earlier than males.89 Understanding the protective factors that lead to reduced penetrance in men may be a key component of treating hEDS.
The exact prevalence of hEDS has proved challenging to determine due to changes in categorization of subtypes and lack of a clear diagnostic test for hEDS. EDS as a whole has been estimated to impact 1 in 5,000 people worldwide, however, there is limited evidence to support this statistic.90 Demmler et al recently reported the combined prevalence of hEDS and HSD to be 1 in 500 people in Wales with 70% of the diagnosed being women, suggesting that hEDS/HSD prevalence has been grossly underestimated.91 With the spectrum of severity, variation in symptom presentation and the lack of an effective clinical diagnostic test, it is not uncommon for patients to go years undiagnosed with hEDS. Because of this, it can be assumed that the actual prevalence of hEDS is higher than reported. This highlights the need for a better understanding of the genetics and biology of hEDS to develop accurate diagnostic tools that will allow for accurate detection and epidemiological reports.
3 |. DIAGNOSTIC CRITERIA
Current diagnostic guidelines indicate major and minor criteria for each EDS subtype. Due to the heterogeneity and overlap of phenotype among subtypes, a genetic diagnosis is preferred for all subtypes, except hEDS. The presence of criteria 1, 2, and 3 (Table 2) must all be met for a clinical diagnosis of hEDS. Criterion 1 includes generalized joint hypermobility, to be assessed by the Beighton score with ≥6 out of 9 for children and adolescents, 5 out of 9 for adults up to 50 yr of age and 4 out of 9 in adults over 50. Limitations such as prior surgery or joint injury should be taken into consideration, as well as history of hypermobility; a five-point questionnaire was developed for this purpose (Table 3).92 A “yes” answer to two or more of the questions would suggest joint hypermobility and add one additional point to the Beighton score.74 For criterion 2, patients must meet two or more of the following; must have systemic manifestations of a more generalized connective tissue disorder, positive family history and/or musculoskeletal complications. Criterion 3 requires all of the following: absence of unusual skin fragility that would prompt consideration for another type of EDS, exclusion of other heritable and acquired connective tissue disorders, exclusion of alternate diagnosis that may also include joint hypermobility.74
TABLE 2.
EDS diagnostic checklist listing the three main criterion
| Criterion 1: Generalized joint hypermobility |
| Beighton score: |
| ≥6 in prepubertal children and adolescents |
| ≥5 in pubertal men and women up to age 50 |
| ≥4 men and women over the age of 50 |
| If one point below cut off two or more “yes” answers to the five point questionnaire should be considered (Table 3). |
| Criterion 2: Two or more of the following features (A, B, or C) must be present |
| Feature A (five must be present): |
| • Unusually soft or velvety skin |
| • Mild skin hyperextensibility |
| • Unexplained striae distensae or rubae at the back, groin, thighs, breasts and/or abdomen in adolescents, men or prepubertal women without a history of significant changes in weight |
| • Bilateral piezogenic papules of the heel |
| • Recurrent or multiple abdominal hernias |
| • Atrophic scarring involving at least two sites without the formation of papyraceous and/or hemosideric scars |
| • Pelvic floor, rectal and/or uterine prolapse in children, men or nulliparous women without history of morbid obesity or predisposing medical condition |
| • Dental crowing and high or narrow palate |
| • Arachnodactyly (positive Walker sign or Steinberg sign on both sides) |
| • Arm span to height ratio ≥1.05 |
| • Mitral valve prolapse |
| • Aortic root dilatation with Z-score >+2 |
| Feature B: |
| • Positive family history (one or more first degree relatives meeting current criteria for hEDS) |
| Feature C (must have at least one): |
| • Musculoskeletal pain in two or more limbs, recurring daily for at least 3 months |
| • Chronic, widespread pain for ≥3 months |
| • Recurrent joint dislocations or joint instability in the absence of trauma |
| Criterion 3: All of the following must be met |
| • Absence of unusual skin fragility, which should prompt consideration of other types of EDS |
| • Exclusion of other heritable and acquired connective tissue disorders. In patients with an acquired connective tissue disorder, additional diagnosis of hEDS requires meeting both features A and B of criterion 2. Feature of criterion C cannot be counted in this situation |
| • Exclusion of alternative diagnoses that may also include joint hypermobility by means of hypotonia and/or connective tissue laxity. Alternative diagnoses may include: neuromuscular disorders (e.g., Bethlem myopathy), other hereditary connective tissue disorders (e.g., Loeys-Dietz syndrome, Marfan syndrome, other types of EDS) and skeletal dysplasias (e.g., osteogenesis imperfecta) |
TABLE 3.
The five-point questionnaire91
| 1. Can you now (or could you ever) place your hands flat on thefloor without bending your knees? |
| 2. Can you now (or could you ever) bend your thumb to touchyour forearm? |
| 3. As a child, did you amuse your friends by contorting yourbody into strange shapes or could you do the splits? |
| 4. As a child or teenager did your shoulder or kneecapdislocated on more than one occasion? |
| 5. Do you consider yourself double-jointed? |
Although a Marfanoid habitus is known to be associated with Marfan syndrome, it is commonly seen in other heritable disorders of connective tissue. Arm span to height ratio, upper segment to lower segment ratio, high or narrow palate, dental crowding, ectopia lentis, and pes planus are commonly evaluated in hEDS. Arachnodactyly, or spider fingers, is also common in Marfanoid habitus and a sign of connective tissue disorders.93 The Steinberg sign, also known as the thumb sign, in which the thumb extends past the palm of the hand in a closed fist and Walker-Murdoch, or wrist sign, in which the thumb and fifth finger overlap around the wrist are typically used to asses arachnodactyly. The Beighton criteria was designed to be a nonsubjective measurement of joint hypermobility but is often criticized for not including other relevant joints where hypermobility may be present. It is recommended that other joints that are commonly affected in patients with hEDS, such as the hips and shoulders, are not excluded during clinical assessment.74
De Wandele et al emphasized that nonmusculoskeletal complaints are extremely common in hEDS and greater awareness of the heterogeneity of symptoms is necessary for diagnosis and treatment.94 It is essential that physicians recognize signs of hypermobility and ligament laxity early on and make necessary recommendations and referrals to prevent chronic joint damage and complications of the disease. Patients who do not meet enough criteria for hEDS are often diagnosed with HSD or may lose a previous diagnosis after the new 2017 criteria, despite hEDS/HSD being indistinguishable in regard to severity of symptoms.95,96 Because of this, the 2017 diagnostic criteria have received much criticism.95
4 |. PHENOTYPE
Hypermobile EDS presents heterogeneously and varies in degree of severity. Multiple body systems may be impacted, and the presence of several comorbidities is common, including widespread chronic pain, autonomic dysfunction, mast cell activation syndrome (MCAS), psychological disorders and gastrointestinal dysfunction. The natural history of hEDS progression has been described by Castori et al, in a continuum of three overlapping phases.88 The first includes ligament laxity in children without complaining of pain, despite sometimes frequent dislocations and subluxations, occasionally, problems persist and lead to complaints of pain. The second phase is termed the pain phase which begins in the 20s, where joint hypermobility may decrease but pain worsens. The third phase, stiffness phase, is later in life and results in limited joint motion and reduction of vertebral curves with chronic pain.88
4.1 |. Musculoskeletal manifestations
The main features of hEDS consist of musculoskeletal complications resulting from hypermobility and joint instability. Hypermobility of the joints is due to laxity of ligaments, joint capsules, and tendons.90 Instability can occur in any joint and can lead to early complications such as subluxations, dislocations, sprains, soft tissue lesions and later complications such as tendonitis, tendon ruptures, muscle and ligament tears, muscle tension and spams, osteoarthritis and chronic joint pain in both children and adults.74,88,97–100 It is not uncommon for patients to have multiple joint complications occurring simultaneously or for patients to experience a domino effect of existing instability impacting surrounding joints.99 Recurrent dislocations are not uncommon, but reduction may occur spontaneously and without difficultly.101 Ligament laxity has been indicated in soft tissue lesions such as ganglion cysts.102 Other soft tissue lesions may include molluscoid pseudotumors, spheroids, and piezogenic papules.74,103
There have been conflicting studies suggesting that hEDS may lead to an increased incidence of fractures or lower bone mineral density (BMD). Infants with EDS were found to have no increased fracture risk, while ambulatory older children with EDS had an increased risk.104 Unlike other connective tissue diseases such as osteogenesis imperfecta (OI), there is little evidence to suggest that hEDS alone should be viewed as a brittle bone disease or as an explanation for multiple fractures presenting in infants or children.105,106 Focusing on adults, it was found that vertebral fractures have been reported at a higher incidence in hEDS and classical EDS (cEDS) patients.107 In further studies, when evaluating a patient population with hEDS or cEDS, reduced bone mineral density and a higher prevalence of general bone fractures have been reported,108,109 but normal BMD and increase in vertebral fractures was also reported. 107 EDS has been indicated to have no influence on bone fragility or fracture healing as well.101 However, in a Chilean population with JHS, 26% of patients had osteoporosis, but interpretation of this data is confounded by the absence of a reported control population for comparison.110 Additionally, altered proprioception and gait mechanics may lead to an increased susceptibility of falls, accounting for the reported fracture incidence. It is also possible that patients with hEDS have lower bone mineral density due to deconditioning, rather than a direct mechanism influencing bone integrity.
4.2 |. Cutaneous/dermatological
Skin hyperextensibility is seen in many types of EDS but typically to a lesser degree than is observed in hEDS or cEDS.111,112 Soft, velvety skin is a common feature of hEDS, and reduced dermal thickness and increased skin fragility has been reported in hEDS and cEDS patients.74,112,113 Wound healing defects may be present and may cause atrophic scarring.74,111 Unexplained striae without significant changes in weight may occur.74,114 Capillary fragility causes frequent bruising with delayed resolution and deep vessel fragility can result in subcutaneous and intramuscular hematomas.111 Patients also present with peizogenic papules (small subcutaneous fat herniations through the dermis) in the heels and wrists.74,111 Additionally, keratosis pilaris has been reported in the hEDS population and can present in other EDS types as well.111 Alterations in sweating, such as hypohidrosis or hyperhidrosis, are seen in some patients and may be due to an underlying autonomic dysfunction.111
4.3 |. Gynecological manifestations
Gynecological manifestation of hEDS can range from pelvic organ prolapse (POP), to pregnancy and menstrual cycle complications. In a small sample of patients with unspecified EDS subtypes, patients experienced both urinary incontinence and history of POP.115 POP has also been found to be more common in patients with benign joint hypermobility syndrome.115,116 In a patient-reported survey, infertility issues have been reported in 44% of patients with EDS, hEDS was the most affected type of EDS compared to 10% of general population.117 Spontaneous abortions have been reported in 28% of hEDS patients and 57% of EDS patients while impacting only 15% of the general population.117,118 Despite some evidence of pregnancy complications, including prelabor membrane rupture, preterm labor and failure to progress in labor,119 other published data indicates that hEDS/JHS are associated with a normal risk of serious adverse pregnancy outcomes.120
An increase in dislocations and symptoms at puberty, during pregnancy, postpartum and during the perimenstrual period have both been reported along with an improvement after menopause.117,118 In the general population, ligament laxity has been shown to be influenced by estrogen, progesterone, relaxin and testosterone and has been best evaluated in the context of anterior cruciate ligament (ACL) injury in females. Knee ligament laxity and risk of ACL injury occurs more frequently during preovulatory phase and ovulatory phase of the menstrual cycle, when estrogen exceeds progesterone.121–123 Hormonal contraceptives have been found to have a possible protective role in ACL tears.122,124,125 The influence of hormones on ligament laxity, combined with patient-reported fluctuations in symptoms that coincide with hormonal shifts, indicate that more research is needed to establish the role of hormones in hEDS.
4.4 |. Ocular manifestations
Present in many heritable disorders of connective tissue, ocular involvement may occur in hypermobile EDS as well. Ocular involvement is typically milder in hEDS than other heritable connective tissue disorders. Thin, blue sclerae, angioid streaks, retinal detachment, ketatoglobus, keratoconus, and lens subluxation or dislocation may occur in hEDS but are more frequently reported in other connective tissue disorders.110,126,127 Myopia is more common in the hEDS population and can be severe.127 Stromal keratocytes are also more prevalent in hEDS patients than the general population, along with xerophthalmia (dry eye).127 It has been suggested that an underlying autonomic nervous system dysfunction could be impacting tear production or, alternatively, tear production may be reduced due to alterations in the extracellular matrix of the lacrimal gland.110,127 Increased eyelid laxity and prominent folds of the upper eyelid skin are also common in patients with hEDS and JHS.128 Patients with EDS may also have an increased risk of surgical complications in ophthalmological surgeries.129 The presence of ectopia lentis (ocular lens dislocation) should raise suspicion for other conditions, such as Marfan syndrome.
4.5 |. Oral and mandibular manifestations
Oral and mandibular manifestations, resulting from compromised oral soft tissue and orofacial structures, have been identified among hEDS patients.96,130 Dental crowding and high or narrow palate are common features of hEDS included in the current diagnostic criteria.74 Compromised soft tissue and orofacial structures among hEDS patients may be attributed to the altered production and organization of collagen, subsequently affecting oral mucosa and periodontium.111,131 The increased fragility of mucosal tissue, capillaries, and perivascular connective tissues132 should be anticipated during dental procedures for hEDS patients, due to the high incidence of injury from oral appliances.133 Compromised oral soft tissue may also present as periodontal recession, which may be exacerbated by accelerated tooth mobility among hEDS populations.134,135
Early onset periodontitis is prevalent among a variety of EDS subtypes, inclusive of the periodontal form (type VIII).136 Periodontal abnormalities may be considered a nonspecific consequence of heritable connective tissue disorders of various subtypes.136 Early onset periodontitis among hEDS patients may be attributed to compromised oxygen and nutrient diffusion, resulting from abnormal composition of the extracellular matrix, therefore increasing susceptibility to bacterial pathogens.131
Alterations to the composition of the extracellular matrix subsequently compromise the ability of the extracellular matrix (ECM) to maintain tissue homeostasis and diffuse oxygen, nutrients, and other small molecules.137 This may consequentially effect overall tissue health, and may play a role in general and regional anesthesia complications.138 Insufficient effects of local anesthetics are not uncommon and can range from no analgesic affect to shortened affect, mostly reported at the dentist.139,140 The precise mechanism of partial or complete failure of local anesthesia among hEDS patients is not fully understood; however, some patients experienced analgesic effects with the use of intradermal lidocaine,139 although effects lasted for shorter durations than control groups.137
Anatomical abnormalities in orofacial structures have been identified in hEDS populations, such as the absence of lingual and inferior labial frenula.141,142 Subluxations and dislocation of the temporomandibular joint, along with temporomandibular disorders (TMD) are a significant occurrence in hEDS patients,143–146 which may be attributed to vertebral posture and cervical functions of the head and neck.147 As craniocervical instability has been noted among hEDS populations, muscle spasms bordering the anterior, posterior, and suboccipital triangle muscles may result in the overuse and spasm of mastication muscles, sequentially resulting in subluxation of the temporomandibular joint.131
4.6 |. Cardiovascular and autonomic dysfunction
Cardiovascular dysfunction among hEDS patients is often attributed to autonomic dysfunction, typically presenting with a constellation of both cardiac and noncardiac symptoms including tachycardia, positional hypotension, gastrointestinal dysmotility, disturbed bladder function and sweating regulation.148 Highly debilitating symptoms in hEDS patients may be attributed to compromised autonomic function, presenting as postural orthostatic tachycardia syndrome (POTS), vasovagal syncope or neurally mediated hypotension (NMH), orthostatic hypotension (OH), and orthostatic intolerance.148,149 Recent estimates on the prevalence of both hEDS and POTS in a small study with 91 patients revealed 24% of POTS patients had generalized joint hypermobility without fully meeting clinical criteria for hEDS, while 31% of POTS patients did meet hEDS criteria.150 Prior to this, the estimated prevalence of hEDS in POTS patients was between 15% and 22%,151–153 while hypermobility spectrum disorder in POTS patients was seen at 35%.153
The association between cardiovascular autonomic dysfunction and hEDS is not well understood, although several plausible mechanisms have been suggested in accordance with clinical experience. Possible mechanisms for cardiovascular autonomic dysfunction among hEDS patients include: low blood pressure, increased peripheral venous dilation and blood pooling, elevated circulating catecholamines, excess systemic levels of histamine, and brainstem or cervical cord impingement attributed to Chiari malformations or craniocervical instability.148 Rowe et al also proposed that connective tissue laxity in hEDS increases vascular and venous compliance, impacting vasoconstriction and venoconstriction when upright.154 More recently, work by Miller et al,150 has supported increased arterial elasticity in these patients as a plausible reason for lower blood pressure and cardiovascular complications, although more work is still needed in this area.
Cardiac valve abnormalities, including mitral valve prolapse (MVP), may occur in patients with hEDS and is considered in the diagnostic criteria (Table 2).74,88,155 Aortic Root dilation (ARD) is also included in hEDS diagnostic criteria and has been reported to occur in some individuals with hEDS (Table 2).74,155–157 In a study of 15 patients evaluated with joint hypermobility syndrome, 13 were found to have increased aortic compliance that correlated with an increase in age and 10 were identified as having MVP.158 Despite the inclusion of MVP and ARD also in hEDS diagnostic criteria, they are not seen as frequently as in connective tissue disorders with vascular complications such as vascular Ehlers-Danlos syndrome (vEDS), cEDS, Marfan syndrome, and Loeys-Dietz syndrome.159
In retrospective chart review of adults age 15 and older with hEDS, BJHS and cEDS, 6.4% and 1.6% were found to have MVP and ARD (Z-score ≥2), respectively.160 Inclusion criteria for the study were based on a diagnosis of EDS, cEDS or BJHS and at least one echocardiogram. This study had several limitations including a change in diagnostic criteria during the study period, variability in clinician interpretation of echocardiograms and the timing of echocardiogram evaluation. Cardiac abnormalities like MVP and ARD typically develop later in life and echocardiography at an early time point in some of these patients may not reflect their current cardiac pathology.
Pediatric patient data suggests that ARD and MVP are less common in hEDS than previously reported with MVP occurring in less than 1% of patients and no ARD identified in the hEDS cohort.161 In the same study, there were no vascular abnormalities reported in vEDS patients. Routine echocardiography may only be necessary in patients with cardiac symptoms or family history of aortic disease or MVP.161 Another vascular pathology, Raynaud’s phenomenon, is correlated with joint hypermobility and EDS, as well as other connective tissue disorders.88,96,101,110,162 Raynaud’s phenomenon is a recurrent vasospasm that occurs due to stress or cold temperatures, leading to decreased blood flow to the fingers or toes. Raynaud’s phenomenon is also reported in more than half of POTS patients.163 However, acrocyanosis may also be a plausible explanation for similar symptoms in this patient population.88,101,164
4.7 |. Immunological manifestations
Non-IGE mediated hypersensitivity, such as histamine intolerance and mast cell activation disorders, often referred to as mast cell activation syndrome (MCAS) are comorbid clinical manifestations among hEDS patients.165–167 Mast cells that reside in the connective tissue produce tryptase, while mast cells in the gut secrete both tryptase and chymase.165 Mast cell activation disorders are described as an increased number of mast cells, increased mast cell activity, or both. This leads to abnormal degranulation in the presence of inappropriate stimuli and can potentially impact all organ systems.167 Histamine, tryptase and chymase are the main chemical mediators released by mast cells that may lead to allergy-like symptoms in patients. Elevated serum tryptase is often used in the diagnosis of MCAS.165 However, symptoms may occur alongside normal plasma histamine and serum tryptase levels; for this reason, diagnostic criteria is initially suspected on clinical grounds, in the presence of symptoms including flushing, cholinergic urticaria, angioedema, hypotension, diarrhea, and rhinitis165 Patients often experience fluctuations in the frequency, duration, and intensity of symptoms, with trends of increasing intensity.
Mast cells are known to modulate connective tissue metabolism. Both chymase and tryptase positive cells have been identified in the papillary dermis of patients with signs of connective tissue dysplasia, mimicking hEDS symptoms.168 Although tryptase positive cells were similar in numerical density in the patient and control groups, chymase positive cells had significant increased density in the patient group.168 Increased chymase positive mast cells in the skin of those with connective tissue dysplasia may be a compensatory mechanism to increased collagen.168 Along with MCAS, asthma is prevalent among hEDS/HSD patients.169
4.8 |. Gastrointestinal
Hypermobile EDS diagnostic criteria is consistently limited to skin fragility or elasticity and hypermobile joints.2 Due to the emphasis on specific joint and skin elasticity, diagnostic criteria for hEDS frequently neglects gastrointestinal manifestations, despite their high prevalence.170 The frequency of gastrointestinal symptoms is higher than previously assessed among hEDS patients. While GI symptoms experienced by affected individuals are primarily functional and nonlife threatening in nature, their impact upon the patient’s quality of life is significant. Clinical assessment of gastrointestinal symptoms associated with hEDS should be constructed to address diagnosed and under-treated gastrointestinal complaints among hEDS patients.170 Gastrointestinal complaints are common in EDS and generalized joint hypermobility.88,171–173 Abdominal pain, bloating, nausea, reflux symptoms, vomiting, constipation and diarrhea are commonly experienced GI symptoms.171 In a widespread survey inquiring about GI symptoms among hEDS populations, 79.3% of participants reported gastroesophageal disease (GERD), 48% reported symptoms congruent with irritable bowel syndrome, and 36% reported motility issues, specifically functional constipation.170 Dysmotility and delayed gastric emptying (gastroparesis) was highly reported, which may be attributed to the high prevalence of dysautonomia among hEDS patients.171,174
Gastrointestinal physiological studies by Mayo Clinic surveyed 36 EDS patients of various subtypes, a majority of whom presented with type III (hypermobile type): 28% of patients who underwent colonic transit studies had abnormal results, with either slow or fast transit.172 There is currently no standardized clinical assessment nor care guidelines for the management of hEDS-related gastrointestinal symptoms.174 Anatomical abnormalities among hEDS patients may be attributed to structural changes in collagen located in the smooth muscle of gastrointestinal pathology, presenting as diverticulosis, rectoceles, and prolapse. Celiac disease is also reported to be more prevalent in hEDS.175 Recurrent abdominal pain, chronic gastritis and constipation/diarrhea was reported by hEDS patients.176
4.9 |. Neurological manifestations
Neurological manifestations can be serious in hEDS and may require surgical intervention. Chiari malformation Type I may occur in hEDS patients and can be associated with recurrent cerebrospinal fluid leak.177,178 Spinal instability is also prevalent and may present in various ways. Atlantoaxial instability (AAI) and craniocervical instability (CCI) are spinal manifestations directly due to ligament laxity.179 Additionally, spinal instability in the form of spondylolisthesis was reported in 10% of patients.100 Cervical and thoracic instability and discopathy in EDS can lead to a loss of the normal cervical lordosis and myelopathy.179 Scoliosis, neck and back pain are also expected in hEDS.100 Tethered cord syndrome (TCS) may also be present in EDS due to abnormalities in filum terminale, although the exact prevalence of TCS in the EDS population is unknown.179
It has been suggested that neuropathic pain in EDS is associated with axonal polyneuropathy and compression neuropathy.180 Nerural conditions such as small fiber neuropathy181 and peripheral nerve entrapments may develop.182 Brachial plexopathies and sciatic neuropathies have been reported in EDS and may require surgical decompression.99,183–185 It is possible that dislocations and subluxations due to ligament laxity may stretch or apply pressure to peripheral nerves resulting in neuropathy or plexopathy.186 Headache and migraine were shown to be commonplace hEDS patients.187–189 Headache may be a result of ligament laxity and AAI and CCI, or a separate entity. Potential associations between Idiopathic intracranial hypertension and EDS may also be related and can contribute to headaches.179
4.10 |. Sleep, fatigue, pain, and psychological impact
Chronic fatigue and chronic pain are prevalent in hEDS.173,189–193 Poor sleep quality, pain, orthostatic intolerance, physical deconditioning and muscle weakness may be possible causes for chronic fatigue.154,194,195 Joint pain is often associated with subluxations, degenerative joint disease and hypermobility but can be both acute and chronic.189,196–198 About one third of children with hEDS reported chronic back pain, arthralgias and myalgias, which increased to two thirds of patients by age 20 and almost all patients over age 40, indicating that pain progresses over time.176,189 A significant number of patients with fibromyalgia have joint hypermobility.199,200 The relationship between joint hypermobility and fibromyalgia may be relevant in widespread pain. An increased sensitivity to pain, generalized hyperalgesia, has also been reported in hEDS and HSD.201 When investigating pain in hEDS patients, Leone et al recently revealed that hEDS patients may have a deficit in endogenous pain inhibitory control.202
Sleep problems such as insomnia and poor sleep quality are common in children and adults with hEDS.203–205 In 26% to 42% of children and 32% of adults, obstructive sleep apnea has been reported.203,206,207 Psychological manifestations of hEDS have been focused mainly on anxiety and depression, which have both been described in hEDS/JHS patient population.208–210 It is unknown whether these are a result of living with a chronic disorder and pain, or if there is another explanation for the relationship. Chronic pain, mood disorders, anxiety, disordered sleep, and comorbid conditions can have significant impacts on social life, and overall quality of life. Poor quality of life has been reported by children and adults with hEDS/JHS.190,205,211,212 A proposed association between joint hypermobility and neurodevelopment disorders including autism spectrum disorders (ASD), has been suggested.213,214 Immune mediated and endocrine mediated symptoms were also reported to occur at a higher rate in patients with ASD and hEDS than ASD alone.215
4.11 |. Development and proprioception
Often, EDS is first noticeable during childhood. Although all babies are hypermobile, there may be some signs of EDS during infancy. Children may delay in walking and have coordination issues and clumsiness.216,217 Clumsiness was reported by almost half of hEDS patients regardless of age.176 Because many patients have muscular hypotonicity due to ligament laxity, babies might be diagnosed as “floppy infant,” where EDS as a differential diagnosis should be considered.101 Hypotonia is often accompanied by poor postural control and impaired proprioception. Poor postural control has been described in EDS and joint hypermobility due to excess joint movement as stretched or lax ligaments are unable to align the body properly.218–220 This can lead to abnormal stress on the joints, pain and long-term musculoskeletal complications.221
Proprioception is necessary for maintaining joint stability. Proprioceptive impairment is very prevalent in hEDS/JHS and has been shown to be correlated with patients’ Beighton score in some cases, but not in others.222–226 Abnormal, nonphysiological gait pattern has also been observed in hEDS/JHS due to biomechanical consequences of hypermobility and is associated with an increased frequency of falls.220,227
5 |. GENETICS
The genetic basis for most of the EDS subtypes are well characterized and were initially discovered to be involved in production and processing of the collagen extracellular matrix (ECM). More recently, the discovery of the genetic etiology of several subtypes has revealed that not all types are directly involved in collagen biosynthesis (Table 1). However, for the majority of EDS genes, their involvement in the synthesis, folding, transport and post-translational modification of ECM components, namely collagen, tenascin, proteoglycans and dermatan sulfate overwhelmingly points to ECM alterations as a generalized theme in EDS pathogenesis. Hypermobile EDS, on the other hand, has challenged researchers and very little is known about the underlying biology. An autosomal dominant pattern of inheritance has been observed, however, at this time there have been no definitive gene candidates identified.
The genetic etiology of hEDS is largely unknown. A few reports have been published identifying gene variants involved in hEDS, but the majority of hEDS cases are of unknown genetic origin. A mutation in COL3A1 in a single family with autosomal dominant hEDS phenotype was identified that led to reduced collagen secretion and over modification of collagen.40 The family’s affected individuals had a glycine 637 to serine substitution in COL3A1, similar to what has been observed in vEDS; however, patients lacked vascular phenotype. There have been no other reports of this COL3A1 variant in hEDS. As compared to those with a clinical diagnosis of vEDS, those with a pathogenic variant in COL3A1 were less likely to have hypermobility.
In a multigenerational Belgian family with hEDS phenotype, genome wide linkage analysis identified a chromosomal locus, 8p22–8p21.1 A heterozygous missense variant in the LZTS1 gene was confirmed in all affected individuals and none of the unaffected. Screening of hEDS patients revealed 3 additional LZTS1 variants.52 LZTS1 encodes leucine zipper tumor suppressor 1, and pathogenic variants in this gene are associated with several types of cancer. Other potentially more plausible candidate genes in this chromosomal locus were tested, but excluded when analyzed more closely. To date, data supporting the role of LZTS1 in hEDS is very limited and lacks conclusive evidence for its involvement in connective tissue biology. Additional research is needed to determine the pathogenicity of these variants and care should be taken to extrapolate causation between this gene and disease phenotype.
More recently, Lyons et al revealed that copy number alterations in TPSAB1, encoding alpha-tryptase was found to be associated with increased basal serum tryptase levels in 35 families with symptoms related to autonomic dysfunction, gastrointestinal disorders, allergic and cutaneous symptoms, and connective tissue abnormalities.228 A gene dose effect relating to copy number and basal serum tryptase levels was also reported. However, not all affected individuals met diagnostic criteria for dysautonomia, MCAS or hEDS and elevated basal serum tryptase is considered relatively common in the general population.
Tenascin X is an extracellular matrix protein that is important for collagen organization. It is encoded by the TNXB gene. TNXB is part of the complex RCCX module, composed of RP1, RP2, C4A, C4B, CYP21A2 and pseudogene CYP21AP, TNXB and pseudogene TNXA. The RCCX module contains pseudogenes, tandem copy number variation and promotes gene rearrangements and deletions. Tenascin X deficiency has been indicated in a recessive form of EDS in which patients meet major and minor diagnostic criteria of classical EDS.229 The Tenascin X deficient patients were found to have truncating mutations or deletions in TNXB.229,230 This has since been reclassified as classic-like EDS, a rare, autosomal recessive type of EDS.231 Some other point mutations in TNXB have also been suggested to cause hEDS or classic-like EDS (clEDS).231–233 TNXB haploinsufficiency has been identified in patients with an autosomal dominant form of hEDS who did not have the easy bruising and skin hyperextensibility as seen in TNX deficient EDS.234 Patients with TNX haploinsufficiency and point mutations have distinct abnormalities in elastic fibers and normal collagen appearance.233 It is likely that this only represents a small group of patients with hEDS.234 Interestingly, in a small study, there was evidence to indicate that some hEDS/JHS patients may have reduced serum TNX concentration compared to healthy controls, without any discernible mutations in TNXB.235 Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder causing 21-hydroxylase deficiency. Some patients with CAH will have a mutation or deletion in CYP21A2 that also affects TNXB, leading to contiguous deletion syndrome (CAH-X).236 TNXA/TNXB chimeras have been identified in which TNXB exons are replaced with TNXA.236–238 These patients have phenotypic characteristics of both hEDS and CAH. The tenascin x protein has been proposed to be essential for collagen I deposition by dermal fibroblasts and important for maintaining the distance between collagen fibrils by forming bridges through direct interactions with collagen fibrils.239–242
6 |. MOLECULAR BIOLOGY
6.1 |. Collagen fiber analysis
Collagens are among the most abundant proteins in the body and a major component of skin, bone, tendons and ligaments. Collagens are an integral part of the ECM and provides structure and support throughout the body. Fibrillar collagens are transcribed as three alpha chains which are then modified and assembled into a triple helical conformation known as procollagen. After further modification by enzymes like lysyl hydroxylase and procollagen peptidase, the collagen fiber is then incorporated into a cross-linked fibril that is part of the ECM. Collagen fibril analysis has been performed on patient dermal biopsies to investigate possible changes in collagen structure and appearance but has provided limited information in regard to hEDS.
Dermal biopsies have shown a wide variety of findings and many of the studies were performed prior to recent reclassification of EDS subtypes. In one study, biopsies were examined for collagen structure among people who clinically appeared to have EDS I, II or III and were then grouped based on collagen appearance and patient phenotype. It is important to note that this was prior to the discovery of variants related to classical and classic-like EDS so patients were characterized based on clinical phenotype alone. Some patients presented with “flower-like” collagen cross sections and “rope-like” longitudinal sections. In other patients who were more mildly affected with joint hypermobility and lacked hypertrophic scars, the dermis appeared normal.243
Flower-like collagen fibrils and collagen fibril abnormalities, including reduced collagen fibril thickness and disarray of fibrils, were found in some patients with hEDS and patients with low Beighton scores.244–248 There was evidence that abnormal collagen appearance did not always correlate with the degree of hypermobility.246,248 Yet, many patients do exhibit decreased collagen I to collagen III ratios.248 Conflicting evidence on elastic fiber changes has been reported, indicating both changed and unchanged elastic fibers.246,247,249 Angwin et al, recently published transmission electron microscopy findings that revealed no specific collagen abnormalities associated with any subtype of EDS, except collagen flowers in most patients with cEDS.250 Eighty-six of ninety patients diagnosed with hEDS had what were considered normal biopsy appearance by transmission electron microscopy (TEM), despite some variations in collagen and elastin, possibly due to age variation of the patients evaluated.250 To date, collagen structure analysis is mostly outdated and provides limited information on the pathophysiology of hEDS. Additionally, most of the studies investigating extracellular matrix structure have focused only on collagen and elastin structure, specifically by TEM. There is a strong need for further investigation of ECM components and analysis of fibroblasts of hEDS patients.
6.2 |. The ECM and integrins
Integrins are vital components of ECM adhesion. They link the ECM proteins to the cytoskeleton and serve as mechanotransducers for various cellular process, including development, cell proliferation, cell movement and tissue homeostasis. Various types of EDS have shown changes in αvβ3, α5β1, and α2β1 integrin expression in dermal fibroblasts isolated from patients.231,251–254 The αvβ3 integrin is widely expressed in endothelial cells, which interacts with several proteins including vitronectin, laminin, fibronectin, fibrillin as well as collagen. Recruitment of αvβ3 integrin is also an indicator of fibroblast activation. The α5β1 integrin and α2β1 integrin are involved in ECM organization of fibronectin and fibrillar collagens, respectively.
In a small study of four hEDS and six HSD patients, dermal biopsies from both were reported to have a “myofibroblast-like” phenotype, exhibiting organized α-smooth muscle actin, increased cadherin-11 expression, αvβ3 integrin expression and enhanced cell migration. Additionally, the fibroblasts showed a decreased in CCN1/CYR61 and increased CCN2/CTGF in both hEDS and HSD compared to controls, along with an increase in MMP-9 that could help explain ECM abnormalities. ILK was found to be localized mainly in focal adhesions in hEDS and HSD cells and coimmunoprecipitated with αvβ3 integrin, suggesting they may form a complex that promotes α-smooth muscle actin assembly in hEDS/HSD. Snail1/Slug was found to be increased in expression in hEDS/HSD and localized in the cytoplasm and nucleus. Coimmunoprecipitation with αvβ3 integrin and ILK indicated Snail1/Slug enables the fibroblast to myofibroblast transition in hEDS/HSD cells through an αvβ3 integrin—ILK mediated interaction.255
6.3 |. Gene expression analysis
Transcriptome-wide expression analyses from five female patients’ fibroblasts with hEDS/JHS has identified numerous differentially expressed genes in pathways involved in composition and homeostasis of connective tissue (e.g., FNDC1, GPC4, MMP16, SPON2, SULF1, TGM2), inflammation (e.g., CFD, COLEC12, IGSF10, IL11, IL6, NFKBIA), cell adhesion (e.g., CLDN11, DSP, FLG, ITGA4, ITGA2, CDH10, CDH2, PCDH9, PCDHB16, PCDHB8), signal transduction (e.g., AQP9, CHRM2, CLIC2, KCNQ5, OPCML, PRLR, SLCO2A1, NPR3), and redox homeostasis (e.g., ADH1B, ADH1C, AKR1C3, GSTM5).252 This study was limited in its small sample size but can guide future research to understand the pathogenicity of hEDS.
Gene expression was also investigated in women with generalized joint hypermobility. The authors found that serum levels of zinc, strontium and lithium were altered in generalized joint hypermobility (GJH) patients, along with several differentially expressed genes. GJH patients had lower COL1A1 and COL1A2. Contrary to other studies, GJH patients had higher TNXB than controls. Additionally, B3GALT6, encoding for galactosyltransferase II was elevated, while B4GALT7, also involved in galactosyltransferase was reduced compared to health controls. Recessive mutations in both B3GALT6 and B4GALT7 have been implicated in Spondylodysplastic EDS. Dermatan sulfate epimersase-1 (DSE) was found to be reduced in GJH patients and is important for biosynthesis of iduronic acid blocks which is indicated in Musculocontractural EDS. Mutations in FKBP14 are causative for kyphoscoliotic EDS and expression of FKBP14 was reduced in GJH patients. SLC39A13 which encodes a Zn exporter ZIP13, which plays a role in connective tissue development, was increased in GJH patients. Spondylodysplastic EDS is also associated with mutations in SLC39A13.65 Further research on gene expression and serum levels may help to identify biomarkers for hEDS.
An overall view of EDS gene expression is presented in Figure 1 and detailed gene mutational analyses for each EDS subtype are presented in Table 1. While many of the collagen genes are abundantly expressed, some of the EDS genes display more specific spatial distribution. For example, Fkbp14 and Plod1, which cause kyphoscoliotic EDS are most abundantly expressed in the developing spinal column. Additionally, C1S, as part of the complement system, is expressed primarily in the developing craniofacial structures and may help explain some of the phenotypes related to oral-mandibular conditions in patients with periodontal EDS (pEDS). Interestingly, collagen 5α2 and to a lesser extent collagen 3α1, which cause classical and vascular EDS, respectively display unique neurological patterns not observed with the other genes, and may play a role in pain accentuation, pain management and/or migraines in this subset of patients.
FIGURE 1.
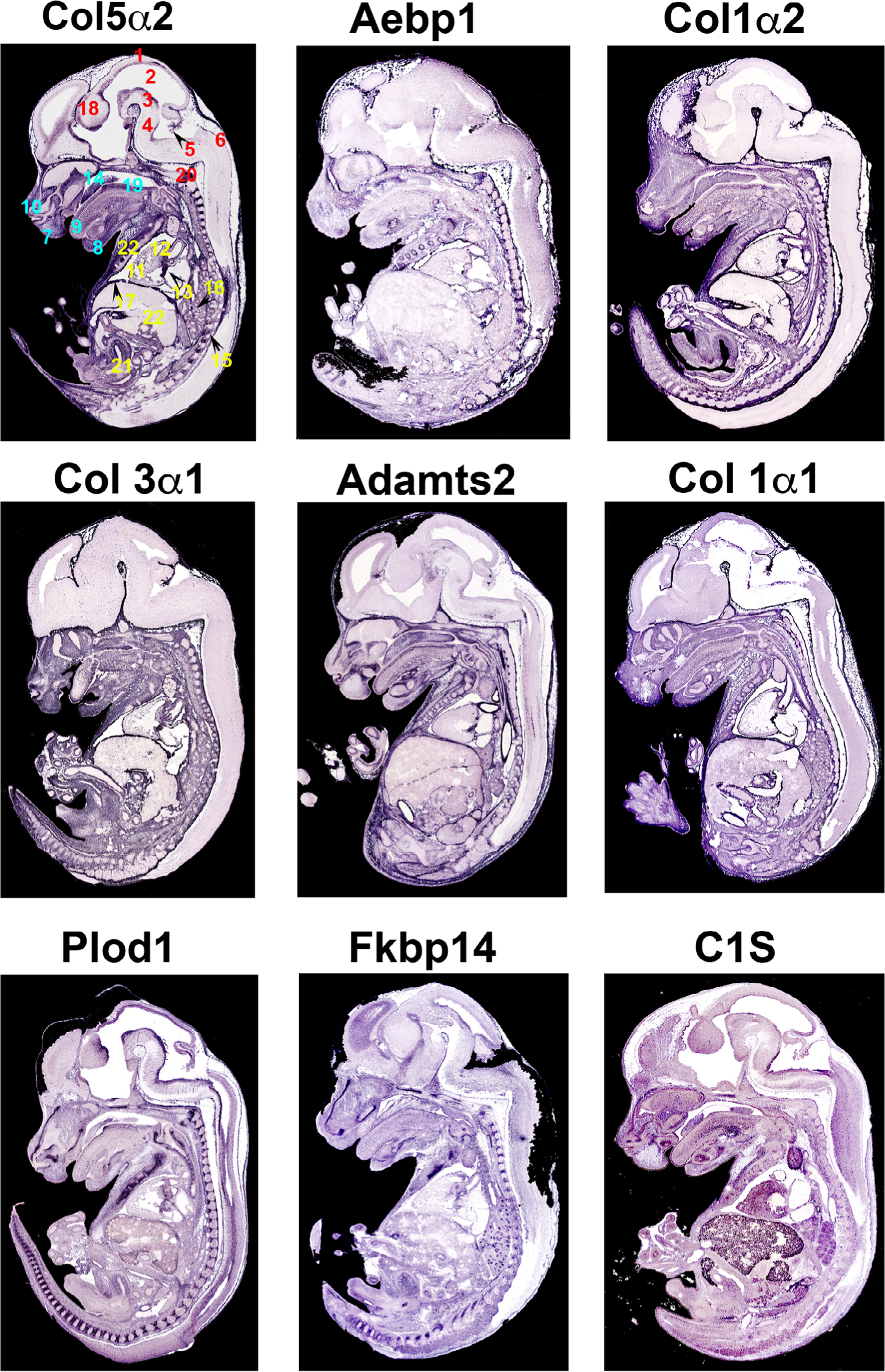
Section in situ hybridization of EDS genes. Messenger RNA expression of EDS genes on sagittal mouse sections at E14.5. Purple staining reveals expression of mRNAs showing widespread expression of EDS genes in regions of connective tissue development. Numbers correspond to specific anatomical locations (red: neural tissues, teal: craniofacial tissues, yellow: thoracic/abdomen tissues). 1, Roof of midbrain; 2, mesencephalic vesicle; 3, ventral part of midbrain; 4, pons; 5, choroid plexus within central part of lumen of fourth ventricle; 6, dorsal part of medulla oblangata; 7, upper lip; 8, Meckel’s cartilage; 9, dorsal surface of tongue; 10, cartilage primordium of nasal bone; 11, right ventricle of the heart; 12, aortic valve and ascending aorta; 13, mitral valve; 14, cartilage primordium of the palatal shelf; 15, lower thoracic dorsal root ganglion; 16, apical part of caudal lobe of right lung; 17, right dome of diaphragm; 18, diencephalon; 19, median circumvallate papilla/cartilage primordium of basisphenoid bone; 20, evidence of ossification within cartilage primordium of basioccipital bone (cilvus); 21, lumen of urogenital sinus (future bladder); 22, liver; 23, costal cartilage of ribs
6.4 |. Model systems
Mouse models have provided insight into the underlying molecular biology of some EDS subtypes revealing abnormal ECM synthesis, deposition, and/or organization.256,257 For example, a conditional knockout of Col5a1 in tendon and ligaments resulted in joint laxity, abnormal gate and early onset osteoarthritis associated with abnormal collagen structure and organization.258 Additionally, knocking out a known gene for mcEDS in mice, CHST14, provided functional evidence for the glycosaminoglycan, dermatan sulfate, in connective tissues.259 More recently, knock in mice with heterozygous mutations in Col3a1 mimicking patient genetics, revealed abnormal signaling through the PLC/IP3/PKC/ERK pathway and that inhibition of these targets may prevent death due to aortic rupture.260 Despite TNXB not encoding for collagen or any enzyme involved in collagen modification, it was revealed to play a role in collagen deposition though studies involving inactivation of Tenascin-X in a murine model of EDS.240 Tenascin-X knockout mice also exhibit mild muscular features and evidence of increased ECM turnover.261 Using an in vitro model, there was evidence of wound healing defects involving up-regulated TGF-β1 and increased MMP activity in mouse embryonic fibroblasts lacking Tenascin-X.262 Although, some models for studying a few EDS subtypes exists, there are currently no established model system for studying hEDS. It is intriguing that nearly all of the genes that have thus far been identified for the various subtypes of EDS are related to the extracellular matrix. These genes are primarily related to collagen isoforms themselves or collagen synthesis (ZNF469, PRDM5), intracellular folding or transport of collagens (PLOD1, FKBP14), post-translational modifications of glycosaminoglycans such as proteoglycans or dermatan sulfate (B4GALT7, B3GALT6, CHST14) or the synthesis of dermatan sulfate (DSE), or other noncollagen, ECM genes (TNXB, ADAMTS1). The correlation of gene-phenotype relationship to particular subtypes of EDS indicates that each of these genes likely have unique cell-specific expression domains and/or levels that are required for normal development and homeostasis within affected tissues in each of the EDS subtypes as discussed above and shown in Figure 1.
The lack of hEDS models is likely based on a lack of genetic knowledge around the causes of hEDS in the human population, further underscoring the necessity for large-scale genetic analyses in this EDS patient group. Only through a combination of this genetic knowledge with animal models will the field be able to advance understanding for the etiology and pathogenesis of the disease.
7 |. MANAGEMENT
Early diagnosis of hEDS allows for disease monitoring and management. Although there are no therapies approved to treat hEDS, symptoms of hEDS and comorbidities can be managed through physical therapy, exercise, lifestyle modification, medication and when needed, surgical intervention. Physical therapy is a crucial part of hEDS management. Treatment is aimed at treating problems due to joint laxity and instability, often seen with muscle weakness from underuse of key muscles and proprioception.263 Therapy is used both preventatively to prevent subluxations and as a treatment for acute injury.264 It is not uncommon for patients to seek treatment for multiple issues at once, for extended periods of time. The use of stretching exercises should be limited due to risk of subluxation and dislocation. Physical therapy should include manual therapy for overactive muscles and an emphasis on core and trunk stabilization.265 Posture reeducation and joint awareness using biofeedback are important to improve proprioception and poor posture due to ligament laxity.265 Adaptive lifelong activity and fitness is recommended at every stage of life for patients EDS. Daily exercise consistent with mild strength training, proprioception, dynamic joint stability, flexibility, and cardiovascular exercise is beneficial in most cases. Aquatic therapy has been noted to be very helpful in treating the more severely affected patients. There are various pharmacologic therapies that may ease the suffering of certain aspects of the EDS patient. Other anecdotal treatments such as meditation, massage, kinseotaping, CBD oil, mindfulness, acupuncture, dry needling, and so forth, need more evidence-based research to ascertain their effectiveness. Early recognition and appropriate treatment of issues with nutrition, sleep function, mobility, chronic pain, and psychologic conditions is important for successful holistic treatment of EDS patients. Additionally, bracing of unstable joints including finger splints, knee and ankle stabilizing braces and cervical collars is sometimes recommended.99,265,266 Instability, weakness and pain may result in the need for part time or full-time mobility aids. Frequent chiropractic care and spinal manipulation are generally not recommended for patients with joint hypermobility, ligament laxity, connective tissue disease, or spinal instability.267–270 Surgical intervention for orthopedic issues involving hEDS should always be carefully considered and failure rate of surgical repair and postoperative complications are higher in EDS patients.99,271 The risks and benefits of surgery should always be taken into careful consideration. Slow wound healing may require careful surgical closure with extra precautions.272 Medications for pain, inflammation, and management of comorbid conditions including MCAS and POTS are frequently prescribed.3,98,99,148
8 |. CONCLUDING REMARKS AND PERSPECTIVES
Modification of the ECM likely play a major role in impairing the mechanical stability of the affected tissues in EDS patients. These ECM alterations likely feedback to the cells, resulting in altered mechanosensing and cell phenotype, probably through an integrin-dependent mechanism. As a consequence, propagation of altered mechanical, cellular and physiological result in a chronic, feed-forward disease responses with profound effects on tissue damage and instability. Stabilizing the ECM environment may set in motion positive mechanical cues that can revert cell and physiological phenotypes with long-term benefits for patients. This highlights the necessity for careful physiotherapy while taking into consideration already compromised connective tissues. While this concept may apply broadly for EDS, its application to hEDS is not yet known. The clinical spectrum of hEDS has been described for decades, with significant advancements made in recent years in regard to diagnostic guidelines, knowledge of comorbidities, treatment options, and awareness among healthcare practitioners. However, most of the available data on the underlying biology of hEDS is broad in scope and lacks clear information regarding genetic pathways that contribute to disease etiology and pathogenesis. Establishing this information will not only provide molecular pathways for hEDS but also other diseases that affect musculoskeletal, ocular, neurological, and cardiovascular tissues. To date, basic sciences discoveries on EDS diseases are woefully lacking. Collective research networks should continue identifying genetic factors that contribute to disease. But, we as a scientific community most move these genetic discoveries into appropriate, genetically accurate animal models to understand the molecular, biochemical, mechanical, and physiological mechanisms that contribute to disease origin and its progression. Without this fundamental knowledge, diagnostic tools and more effective or curative therapeutics for hypermobile EDS and EDS disease subtypes will remain enigmatic.
EDS patients may endure years without proper diagnoses and/or treatments. Increased awareness in the medical community is needed to ensure proper care for this group of affected individuals. This medical deficiency may originate during training as medical students are taught “when you hear hoofbeats, look for horses not zebras.” This means to look for the more common diagnosis and not the rare one. As such, the zebra has become the symbol for EDS. Just like zebras in the wild, no two EDS zebras have the same stripes. All patients’ symptoms and experiences are different, but the community comes together as a group of zebras, called a dazzle. A mission of this review is to inform the scientific and lay community of the necessity for increased awareness and involvement in connective tissue disease societies like the EDS society. This, in turn, will lead to a brightening, or dazzling of research and provide new hope to patients with these life altering diseases.273
ACKNOWLEDGMENTS
The work at MUSC was performed in a facility constructed with support from the National Institutes of Health grant number C06 RR018823 from the Extramural Research Facilities Program of the National Center for Research Resources. Other funding sources include: National Institutes of Health: HL131546 (R. A. N.), GM103444 (R. A. N.), and HL149696 (R. A. N.); T32GM132055 and T32HL007260 (C. G.); Grant Sponsor: American Heart Association; Grant Numbers: 19TPA34850095 (R. A. N.), 17CSA33590067 (R. A. N.). We would also like to thank the reviewers for their insightful comments as well as the Ehlers Danlos Society (https://www.ehlers-danlos.com), especially Nancy Feracco, RN and Susan Chalela, PT, who work tirelessly for EDS awareness and outreach. They are constant beacons of hope and light for the many that are so adversely affected by EDS and related connective tissue diseases.
Funding information
American Heart Association, Grant/Award Numbers: 33590067, 34850095; National Center for Research Resources, Grant/Award Number: rr018823; National Heart, Lung, and Blood Institute, Grant/Award Numbers: 127692, 131546, 007260; National Institute of General Medical Sciences, Grant/Award Numbers: 103444, 132055
REFERENCES
- 1.Beighton P, De Paepe A, Finidori G, et al. International nosology of heritable disorders of connective tissue Berlin. Am J Med Genet. 1986;29:581–594. [DOI] [PubMed] [Google Scholar]
- 2.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-danlos syndromes: revised nosology, Villefranche, 1997. Am J Med Genet. 1998;77(1):31–37. [DOI] [PubMed] [Google Scholar]
- 3.Colige A, Sieron AL, Li SW, et al. Human ehlers-danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet. 1999;65(2):308–317. 10.1086/302504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colige A, Nuytinck L, Hausser I, et al. Novel types of mutation responsible for the dermatosparactic type of Ehlers-Danlos syndrome (type VIIC) and common polymorphisms in the ADAMTS2 gene. J Invest Dermatol. 2004;123(4):656–663. 10.1111/j.0022-202X.2004.23406.x. [DOI] [PubMed] [Google Scholar]
- 5.Van Damme T, Colige A, Syx D, et al. Expanding the clinical and mutational spectrum of the Ehlers-Danlos syndrome, dermatosparaxis type. Genet Med. 2016;18:882–891. 10.1038/gim.2015.188. [DOI] [PubMed] [Google Scholar]
- 6.Blackburn PR, Xu Z, Tumelty KE, et al. Bi-allelic alterations in AEBP1 lead to defective collagen assembly and connective tissue structure resulting in a variant of Ehlers-Danlos syndrome. Am J Hum Genet. 2018;102:696–705. 10.1016/j.ajhg.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritelli M, Cinquina V, Venturini M, et al. Expanding the clinical and mutational spectrum of recessive AEBP1-related classical-like Ehlers-Danlos syndrome. Genes (Basel). 2019; 10(135):1–14. 10.3390/genes10020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakajima M, Mizumoto S, Miyake N, et al. Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am J Hum Genet. 2013;92(6):927–934. 10.1016/j.ajhg.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malfait F, Kariminejad A, Van Damme T, et al. Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder. Am J Hum Genet. 2013;92(6):935–945. 10.1016/j.ajhg.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vorster AA, Beighton P, Ramesar RS. Spondyloepimetaphyseal dysplasia with joint laxity (Beighton type); mutation analysis in eight affected South African families. Clin Genet. 2015;87(5):492–495. 10.1111/cge.12413. [DOI] [PubMed] [Google Scholar]
- 11.Ritelli M, Chiarelli N, Zoppi N, et al. Insights in the etiopathology of galactosyltransferase II (GalT-II) deficiency from transcriptome-wide expression profiling of skin fibroblasts of two sisters with compound heterozygosity for two novel B3GALT6 mutations. Mol Genet Metab Rep. 2015;2:1–15. 10.1016/j.ymgmr.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sellars EA, Bosanko KA, Lepard T, Garnica A, Schaefer GB. A newborn with complex skeletal abnormalities, joint contractures, and bilateral corneal clouding with sclerocornea. Semin Pediatr Neurol. 2014;21(2):84–87. 10.1016/j.spen.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Honey EM. Spondyloepimetaphyseal dysplasia with joint laxity (Beighton type): a unique south African disorder. South Afr Med J. 2016;106(6):S54–S56. 10.7196/SAMJ.2016.v106i6.10994. [DOI] [PubMed] [Google Scholar]
- 14.Salter CG, Davies JH, Moon RJ, Fairhurst J, Bunyan D, Foulds N. Further defining the phenotypic spectrum of B4GALT7 mutations. Am J Med Genet Part A. 2016;170(6): 1556–1563. 10.1002/ajmg.a.37604. [DOI] [PubMed] [Google Scholar]
- 15.Guo MH, Stoler J, Lui J, et al. Redefining the progeroid form of ehlers-danlos syndrome: report of the fourth patient with B4GALT7 deficiency and review of the literature. Am J Med Genet Part A. 2013;161(10):2519–2527. 10.1002/ajmg.a.36128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faiyaz-Ul-Haque M, Zaidi SHE, Al-Ali M, et al. A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danlos syndrome resembling the progeroid type. Am J Med Genet. 2004;128A(1):39–45. 10.1002/ajmg.a.30005. [DOI] [PubMed] [Google Scholar]
- 17.Kresse H, Rosthoj S, Quentin E, et al. Glycosaminoglycan-free small proteoglycan core protein is secreted by fibroblasts from a patient with a syndrome resembling progeroid. Am J Hum Genet. 1987;41(3):436–453. [PMC free article] [PubMed] [Google Scholar]
- 18.Okajima T, Fukumoto S, Furukawat K, Urano T, Furukawa K. Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J Biol Chem. 1999;274(41):28841–28844. 10.1074/jbc.274.41.28841. [DOI] [PubMed] [Google Scholar]
- 19.Quentin E, Gladen A, Rodén L, Kresse H. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc Natl Acad Sci U S A. 1990; 87(4):1342–1346. 10.1073/pnas.87.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kapferer-Seebacher I, Heiss-Kisielewsky I, Pepin M, et al. Periodontal Ehlers-Danlos syndrome is caused by mutations in C1R and C1S, which encode subcomponents C1r and C1s of complement. Am J Hum Genet. 2016;99(5):1005–1014. 10.1016/j.ajhg.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyake N, Kosho T, Mizumoto S, et al. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum Mutat. 2010;31(8):966–974. 10.1002/humu.21300. [DOI] [PubMed] [Google Scholar]
- 22.Malfait F, Syx D, Vlummens P, et al. Musculocontractural Ehlers-Danlos syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum Mutat. 2010;31(11):1233–1239. 10.1002/humu.21355. [DOI] [PubMed] [Google Scholar]
- 23.Syx D, Van Damme T, Symoens S, et al. Genetic heterogeneity and clinical variability in musculocontractural ehlers-danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum Mutat. 2015;36(5):535–547. 10.1002/humu.22774. [DOI] [PubMed] [Google Scholar]
- 24.Nuytinck L, Freund M, Lagae L, Pierard GE, Hermanns-Le T, De Paepe A. Classical Ehlers-Danlos syndrome caused by a mutation in type I collagen. Am J Hum Genet. 2000;66(4): 1398–1402. 10.1086/302859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malfait F, Symoens S, De Backer J, et al. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat. 2007;28(4):387–395. 10.1002/humu.20455. [DOI] [PubMed] [Google Scholar]
- 26.Ritelli M, Dordoni C, Venturini M, et al. Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: Identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet J Rare Dis. 2013;8(58):1–19. 10.1186/1750-1172-8-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole WG, Chan D, Chambers GW, Walker ID, Bateman JF. Deletion of 24 Amino acids from the pro-alpha 1(I) chain of type I Procollagen in a patient with the Ehlers-Danlos syndrome type VII - PubMed. J Biol Chem. 1986;261(12):5496–5503. [PubMed] [Google Scholar]
- 28.D’Alessio M, Ramirez F, Blumberg BD, et al. Characterization of a COL1A1 splicing defect in a case of Ehlers-Danlos syndrome type VII: further evidence of molecular homogeneity. Am J Hum Genet. 1991;49(2):400–406. [PMC free article] [PubMed] [Google Scholar]
- 29.Byers PH, Duvic M, Atkinson M, et al. Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am J Med Genet. 1997;72(1): 94–105. 10.1002/(SICI)1096-8628(19971003)72:1<94::AID-AJMG20>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 30.Hata R-I, Kurata S-I, Shinkai H. Existence of malfunctioning proα2(I) collagen genes in a patient with a proα2(I)-chain-defective variant of Ehlers-Danlos syndrome. Eur J Biochem. 1988;174(2):231–237. 10.1111/j.1432-1033.1988.tb14087.x. [DOI] [PubMed] [Google Scholar]
- 31.Nicholls AC, Valler D, Wallis S, Pope FM. Homozygosity for a splice site mutation of the COL1A2 gene yields a nonfunctional proα2 (I) chain and an EDS/OI clinical phenotype [10]. J Med Genet. 2001;38(2):132–135. 10.1136/jmg.38.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwarze U, Hata RI, McKusick VA, et al. Rare autosomal recessive cardiac Valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am J Hum Genet. 2004;74(5):917–930. 10.1086/420794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malfait F, Symoens S, Coucke P, Nunes L, De Almeida S, De Paepe A. Total absence of the alpha2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems. J Med Genet. 2006;43 (e36):1–5. 10.1136/jmg.2005.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinmann B, Tuderman L, Peltonen L, Martin GR, McKusick VA, Prockop DJ. Evidence for a structural mutation of procollagen type I in a patient with the Ehlers-Danlos syndrome type VII. J Biol Chem. 1980;255(18):8887–8893. [PubMed] [Google Scholar]
- 35.A Heterozygous Collagen Defect in a Variant of the Ehlers-Danlos Syndrome Type VII. Evidence for a Deleted Amino-Telopeptide Domain in the Pro-Alpha 2(I) Chain. https://pubmed.ncbi.nlm.nih.gov/2993307/?dopt=Abstract. Accessed May 20, 2020. [PubMed]
- 36.Nicholls AC, Oliver J, Renouf DV, McPheat J, Palan A, Pope FM. Ehlers-Danlos syndrome type VII: a single base change that causes exon skipping in the type I collagen α2 (I) chain. Hum Genet. 1991;87(2):193–198. 10.1007/BF00204180. [DOI] [PubMed] [Google Scholar]
- 37.Vasan NS, Kuivaniemi H, Vogel BE, et al. A mutation in the pro alpha 2(I) gene (COL1A2) for type I procollagen in Ehlers-Danlos syndrome type VII: evidence suggesting that skipping of exon 6 in RNA splicing may be a common cause of the phenotype. Am J Hum Genet. 1991;48(2):305–317. [PMC free article] [PubMed] [Google Scholar]
- 38.Klaassens M, Reinstein E, Hilhorst-Hofstee Y, et al. Ehlers-Danlos arthrochalasia type (VIIA-B)—expanding the phenotype: from prenatal life through adulthood. Clin Genet. 2012;82(2):121–130. 10.1111/j.1399-0004.2011.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatamochi A, Hamada T, Yoshino M, Hashimoto T. The first Japanese case of the arthrochalasia type of Ehlers-Danlos syndrome with COL1A2 gene mutation. Gene. 2014;538(1):199–203. 10.1016/j.gene.2014.01.033. [DOI] [PubMed] [Google Scholar]
- 40.Narcisi P, Richards A, Ferguson S, Pope MF. A family with Ehlers-Danlos syndrome type III/articular hypermobility syndrome has a glycine 637 to serine substitution in type III collagen. Hum Mol Genet. 1994;3(3):1617–1620. [DOI] [PubMed] [Google Scholar]
- 41.Frank M, Albuisson J, Ranque B, et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur J Hum Genet. 2015; 23(12):1657–1664. 10.1038/ejhg.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 2014;16(12):881–888. 10.1038/gim.2014.72. [DOI] [PubMed] [Google Scholar]
- 43.Symoens S, Syx D, Malfait F, et al. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum Mutat. 2012;33(10):1485–1493. 10.1002/humu.22137. [DOI] [PubMed] [Google Scholar]
- 44.Mutations in the Collagen XII Gene Define a New Form of Extracellular Matrix-Related Myopathy. https://pubmed.ncbi.nlm.nih.gov/24334769/?dopt=Abstract. Accessed May 21, 2020. [DOI] [PubMed]
- 45.Zou Y, Zwolanek D, Izu Y, et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum Mol Genet. 2014;23(9): 2339–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Punetha J, Kesari A, Hoffman EP, et al. Novel Col12A1 variant expands the clinical picture of congenital myopathies with extracellular matrix defects. Muscle Nerve. 2017;55(2):277–281. 10.1002/mus.25232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Müller T, Mizumoto S, Suresh I, et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum Mol Genet. 2013;22(18): 3761–3772. [DOI] [PubMed] [Google Scholar]
- 48.Baumann M, Giunta C, Krabichler B, et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am J Hum Genet. 2012;90:201–216. 10.1016/j.ajhg.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aldeeri AA, Alazami AM, Hijazi H, Alzahrani F, Alkuraya FS. Excessively redundant umbilical skin as a potential early clinical feature of Morquio syndrome and FKBP14-related EhlersDanlos syndrome. Clin Genet. 2014;86(5):469–472. 10.1111/cge.12414. [DOI] [PubMed] [Google Scholar]
- 50.Murray ML, Yang M, Fauth C, Byers PH. FKBP14-related Ehlers-Danlos syndrome: expansion of the phenotype to include vascular complications. Am J Med Genet Part A. 2014; 164(7):1750–1755. 10.1002/ajmg.a.36492. [DOI] [PubMed] [Google Scholar]
- 51.Dordoni C, Ciaccio C, Venturini M, Calzavara-Pinton P, Ritelli M, Colombi M. Further delineation of FKBP14 -related Ehlers-Danlos syndrome: a patient with early vascular complications and non-progressive kyphoscoliosis, and literature review. Am J Med Genet Part A. 2016;170(8):2031–2038. 10.1002/ajmg.a.37728. [DOI] [PubMed] [Google Scholar]
- 52.Syx D, Symoens S, Steyaert W, De Paepe A, Coucke PJ, Malfait F. Ehlers-Danlos Syndrome, Hypermobility Type, Is Linked to Chromosome 8p22–8p21.1 in an Extended Belgian Family. Disease Markers. London, United Kingdom: Hindawi Publishing Corporation; 2015. 10.1155/2015/828970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burkitt Wright EMM, Spencer HL, Daly SB, et al. Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance. Am J Hum Genet. 2011;88(6):767–777. 10.1016/j.ajhg.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Avgitidou G, Siebelmann S, Bachmann B, Kohlhase J, Heindl LM, Cursiefen C. Brittle cornea syndrome: case report with novel mutation in the PRDM5 gene and review of the literature. Case Rep Ophthalmol Med. 2015;2015:637084. 10.1155/2015/637084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aldahmesh MA, Mohamed JY, Alkuraya FS. A novel mutation in PRDM5 in brittle cornea syndrome. Clin Genet. 2012; 81(2):198–199. 10.1111/j.1399-0004.2011.01808.x. [DOI] [PubMed] [Google Scholar]
- 56.Giunta C, Randolph A, Steinmann B. Mutation analysis of the PLOD1 gene: an efficient multistep approach to the molecular diagnosis of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA). Mol Genet Metab. 2005;86(1–2):269–276. 10.1016/j.ymgme.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 57.Hyland J, Ala-Kokko L, Royce P, Steinmann B, Kivirikko KI, Myllylä R. A homozygous stop codon in the lysyl hydroxylase gene in two siblings with Ehlers-Danlos syndrome type VI. Nat Genet. 1992;2(3):228–231. 10.1038/ng1192-228. [DOI] [PubMed] [Google Scholar]
- 58.Ha VT, Marshall MK, Elsas LJ, Pinnell SR, Yeowell HN. A patient with Ehlers-Danlos syndrome type VI is a compound heterozygote for mutations in the lysyl hydroxylase gene. J Clin Invest. 1994;93(4):1716–1721. 10.1172/JCI117155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brinckmann J, Açil Y, Feshchenko S, et al. Ehlers-Danlos syndrome type VI: Lysyl hydroxylase deficiency due to a novel point mutation (W612C). Arch Dermatol Res. 1998;290(4):181–186. 10.1007/s004030050287. [DOI] [PubMed] [Google Scholar]
- 60.Walker LC, Marini JC, Grange DK, Filie J, Yeowell HN. A patient with Ehlers-Danlos syndrome type VI is homozygous for a premature termination codon in exon 14 of the lysyl hydroxylase 1 gene. Mol Genet Metab. 1999;67(1):74–82. 10.1006/mgme.1999.2824. [DOI] [PubMed] [Google Scholar]
- 61.Walker LC, Overstreet MA, Siddiqui A, et al. A novel mutation in the lysyl hydroxylase 1 gene causes decreased lysyl hydroxylase activity in an ehlers-danlos VIA patient. J Invest Dermatol. 2005;124(5):914–918. 10.1111/j.0022-202X.2005.23727.x. [DOI] [PubMed] [Google Scholar]
- 62.Yeowell HN, Allen JD, Walker LC, Overstreet MA, Murad S, Thai SF. Deletion of cysteine 369 in lysyl hydroxylase 1 eliminates enzyme activity and causes Ehlers-Danlos syndrome type VI. Matrix Biol. 2000;19(1):37–46. 10.1016/S0945-053X(99)00055-4. [DOI] [PubMed] [Google Scholar]
- 63.Yeowell HN, Walker LC, Farmer B, Heikkinen J, Myllyla R. Mutational analysis of the lysyl hydroxylase 1 gene (PLOD) in six unrelated patients with Ehlers-Danlos syndrome type VI: prenatal exclusion of this disorder in one family. Hum Mutat. 2000;16(1). 10.1002/1098-1004(200007)16:1<90::AID-HUMU19>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 64.Abdalla EM, Rohrbach M, Bürer C, et al. Kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA) in six Egyptian patients presenting with a homogeneous clinical phenotype. Eur J Pediatr. 2015;174(1):105–112. 10.1007/s00431-014-2429-9. [DOI] [PubMed] [Google Scholar]
- 65.Tuna F, Doganlar ZB, Özdemir H, Kabayel DD. Ehlers-Danlos syndrome-related genes and serum strontium, zinc, and lithium levels in generalized joint hypermobility: a case-control study. Connect Tissue Res. 2019;00(00):1–11. 10.1080/03008207.2019.1675648. [DOI] [PubMed] [Google Scholar]
- 66.Giunta C, Elçioglu NH, Albrecht B, et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome-an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am J Hum Genet. 2008;82(6):1290–1305. 10.1016/j.ajhg.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fukada T, Civic N, Furuichi T, et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-β signaling pathways. PLoS One. 2008;3(11). 10.1371/journal.pone.0003642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abu A, Frydman M, Marek D, et al. Deleterious mutations in the zinc-finger 469 gene cause brittle cornea syndrome. Am J Hum Genet. 2008;82(5):1217–1222. 10.1016/j.ajhg.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Christensen AE, Knappskog PM, Midtbø M, et al. Brittle cornea syndrome associated with a missense mutation in the zinc-finger 469 gene. Investig Ophthalmol Vis Sci. 2010;51(1): 47–52. 10.1167/iovs.09-4251. [DOI] [PubMed] [Google Scholar]
- 70.Khan AO, Aldahmesh MA, Alkuraya FS. Brittle cornea without clinically-evident extraocular findings in an adult harboring a novel homozygous ZNF469 mutation. Ophthalmic Genet. 2012;33 (4):257–259. 10.3109/13816810.2012.670362. [DOI] [PubMed] [Google Scholar]
- 71.Al-Owain M, Al-Dosari MS, Sunker A, Shuaib T, Alkuraya FS. Identification of a novel ZNF469 mutation in a large family with Ehlers-Danlos phenotype. Gene. 2012;511 (2):447–450. 10.1016/j.gene.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 72.Rohrbach M, Spencer HL, Porter LF, et al. ZNF469 frequently mutated in the brittle cornea syndrome (BCS) is a single exon gene possibly regulating the expression of several extracellular matrix components. Mol Genet Metab. 2013;109(3):289–295. 10.1016/j.ymgme.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ramappa M, Wilson ME, Rogers RC, Trivedi RH. Brittle cornea syndrome: a case report and comparison with Ehlers Danlos syndrome. J AAPOS. 2014;18(5):509–511. 10.1016/j.jaapos.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 74.Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet Part C Semin Med Genet. 2017;175(1):8–26. 10.1002/ajmg.c.31552. [DOI] [PubMed] [Google Scholar]
- 75.Castori M, Tinkle B, Levy H, Grahame R, Malfait F, Hakim A. A framework for the classification of joint hypermobility and related conditions. Am J Med Genet Part C Semin Med Genet. 2017;175:148–157. 10.1002/ajmg.c.31539. [DOI] [PubMed] [Google Scholar]
- 76.Tinkle BT, Bird HA, Grahame R, Lavallee M, Levy HP, Sillence D. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet Part A. 2009;149A(11):2368–2370. 10.1002/ajmg.a.33070. [DOI] [PubMed] [Google Scholar]
- 77.Al-Jarallah K, Shehab D, Al-Jaser MT, et al. Prevalence of joint hypermobility in Kuwait. Int J Rheum Dis. 2017;20(8): 935–940. 10.1111/1756-185X.12556. [DOI] [PubMed] [Google Scholar]
- 78.Gocentas A, Jascaniniene N, Pasek M, et al. Prevalence of generalised joint hypermobility in school-aged children from east-central European region. Folia Morphol. 2016;75(1):48–52. 10.5603/FM.a2015.0065. [DOI] [PubMed] [Google Scholar]
- 79.Rikken-Bultman DG, Wellink L, van Dongen PW. Hypermobility in two Dutch school populations. Eur J Obstet Gynecol Reprod Biol. 1997;73(2):189–192. 10.1016/s0301-2115(97)02745-0. [DOI] [PubMed] [Google Scholar]
- 80.Jansson A, Saartok T, Werner S, Renström P. General joint laxity in 1845 Swedish school children of different ages: age- and gender-specific distributions. Acta Paediatr Int J Paediatr. 2004; 93(9):1202–1206. 10.1080/08035250410023971. [DOI] [PubMed] [Google Scholar]
- 81.El-Garf AK, Mahmoud GA, Mahgoub EH. Hypermobility among Egyptian children: prevalence and features. J Rheumatol. 1998;25(5):1003–1005. [PubMed] [Google Scholar]
- 82.Clinch J, Deere K, Sayers A, et al. Epidemiology of generalized joint laxity (hypermobility) in fourteen-year-old children from the UK: a population-based evaluation. Arthritis Rheum. 2011;63(9):2819–2827. 10.1002/art.30435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Subramanyan V, Janaki KY. Joint hypermobility in south Indian children. Indian Pediatr. 1996;33(9):771–772. [PubMed] [Google Scholar]
- 84.Gedalia A, Person DA, Brewer EJ, Giannini EH. Hypermobility of the joints in juvenile episodic arthritis/arthralgia. J Pediatr. 1985;107(6):873–876. 10.1016/S0022-3476(85)80178-5. [DOI] [PubMed] [Google Scholar]
- 85.Larsson LG, Baum J, Mudholkar GS, Srivastava DK. Hypermobility: prevalence and features in a swedish population. Rheumatology. 1993;32(2):116–119. 10.1093/rheumatology/32.2.116. [DOI] [PubMed] [Google Scholar]
- 86.Birrell FN, Adebajo AO, Hazleman BL, Silman AJ. High prevalence of joint laxity in west africans. Rheumatology. 1994; 33(1):56–59. 10.1093/rheumatology/33.1.56. [DOI] [PubMed] [Google Scholar]
- 87.Grahame R. Joint hypermobility and genetic collagen disorders: are they related? Arch Dis Child. 1999;80(2):188–191. 10.1136/adc.80.2.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Castori M, Camerota F, Celletti C, et al. Natural history and manifestations of the hypermobility type Ehlers-Danlos syndrome: a pilot study on 21 patients. Am J Med Genet Part A. 2010;152A(3):556–564. 10.1002/ajmg.a.33231. [DOI] [PubMed] [Google Scholar]
- 89.Castori M, Camerota F, Celletti C, Grammatico P, Padua L. Ehlers-Danlos syndrome hypermobility type and the excess of affected females: possible mechanisms and perspectives. Am J Med Genet Part A. 2010;152A(9):2406–2408. 10.1002/ajmg.a.33585. [DOI] [PubMed] [Google Scholar]
- 90.Steinmann B, Royce PM, Superti-Furga A. The Ehlers-Danlos syndrome. Connective Tissue and Its Heritable Disorders. Hoboken, New Jersey: Wiley-Liss, Inc; 2003:431–523. [Google Scholar]
- 91.Demmler JC, Atkinson MD, Reinhold EJ, Choy E, Lyons RA, Brophy ST. Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK: a national electronic cohort study and case-control comparison. BMJ Open. 2019;9(11):e031365. 10.1136/bmjopen-2019-031365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. IntJClinPract. 2003;57(3):163–166. [PubMed] [Google Scholar]
- 93.Grahame R, Hakim AJ. Arachnodactyly—a key to diagnosing heritable disorders of connective tissue. Nat Rev Rheumatol. 2013;9(6):358–364. 10.1038/nrrheum.2013.24. [DOI] [PubMed] [Google Scholar]
- 94.De Wandele I, Rombaut L, Malfait F, De Backer T, De Paepe A, Calders P. Clinical heterogeneity in patients with the hypermobility type of Ehlers-Danlos syndrome. Res Dev Disabil. 2013;34(3):873–881. 10.1016/j.ridd.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 95.Mcgillis L, Mina DS, Soever L, et al. Utilization of the 2017 diagnostic criteria for hEDS by the Toronto GoodHope Ehlers-Danlos syndrome clinic: a retrospective review. Am J Med Genet Part A. 2020;182:484–492. 10.1002/ajmg.a.61459. [DOI] [PubMed] [Google Scholar]
- 96.Copetti M, Morlino S, Colombi M, Grammatico P, Fontana A, Castori M. Severity classes in adults with hypermobile Ehlers-Danlos syndrome/hypermobility spectrum disorders: a pilot study of 105 Italian patients. Rheumatol (United Kingdom). 2019;58(10):1722–1730. 10.1093/rheumatology/kez029. [DOI] [PubMed] [Google Scholar]
- 97.Stern CM, Pepin MJ, Stoler JM, Kramer DE, Spencer SA, Stein CJ. Musculoskeletal conditions in a pediatric population with Ehlers-Danlos syndrome. J Pediatr. 2017;181:261–266. 10.1016/j.jpeds.2016.10.078. [DOI] [PubMed] [Google Scholar]
- 98.Chopra P, Tinkle B, Hamonet C, et al. Pain management in the Ehlers-Danlos syndromes. Am J Med Genet Part C Semin Med Genet. 2017;175(1):212–219. 10.1002/ajmg.c.31554. [DOI] [PubMed] [Google Scholar]
- 99.Ericson WB, Wolman R. Orthopaedic management of the Ehlers-Danlos syndromes. Am J Med Genet Part C Semin Med Genet. 2017;175(1):188–194. 10.1002/ajmg.c.31551. [DOI] [PubMed] [Google Scholar]
- 100.Stanitski DF, Nadjarian R, Startitski CL, Bawle E, Tsipouras P. Orthopaedic Manifestations of Ehlers-Danlos Syndrome. Vol 376. Philadelphia, Pennsylvania: Lippincott Williams & Wilkins; 2000. [DOI] [PubMed] [Google Scholar]
- 101.Beighton P. Articular manifestations of the Ehlers-Danlos syndrome. Semin Arhtritis Rheum. 1972;3:246–261. [DOI] [PubMed] [Google Scholar]
- 102.McKeon KE, London DA, Osei DA, et al. Ligamentous hyperlaxity and dorsal wrist ganglions. J Hand Surg Am. 2013; 38(11):2138–2143. 10.1016/j.jhsa.2013.08.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Inamadar AC, Palit A. Cutaneous signs in heritable disorders of the connective tissue. Indian J Dermatol Venereol Leprol. 2004;70(4):253–255. [PubMed] [Google Scholar]
- 104.Rolfes MC, Deyle DR, King KS, Hand JL, Graff AH, Derauf C. Fracture incidence in Ehlers-Danlos syndrome—a population-based case-control study. Child Abuse Negl. 2019; 91:95–101. 10.1016/j.chiabu.2019.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Castori M. Ehlers-Danlos syndrome(s) mimicking child abuse: is there an impact on clinical practice? Am J Med Genet Part C Semin Med Genet. 2015;169(4):289–292. 10.1002/ajmg.c.31460. [DOI] [PubMed] [Google Scholar]
- 106.Tinkle B, Castori M, Berglund B, et al. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome type III and Ehlers-Danlos syndrome hypermobility type): clinical description and natural history. Am J Med Genet Part C Semin Med Genet. 2017;175:48–69. 10.1002/ajmg.c.31538. [DOI] [PubMed] [Google Scholar]
- 107.Mazziotti G, Dordoni C, Doga M, et al. High prevalence of radiological vertebral fractures in adult patients with Ehlers-Danlos syndrome. Bone. 2016;84:88–92. 10.1016/j.bone.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 108.Dolan AL, Arden NK, Grahame R, Spector TD. Assessment of bone in Ehlers Danlos syndrome by ultrasound and densitometry. Ann Rheum Dis. 1998;57:630–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eller-Vainicher C, Bassotti A, Imeraj A, et al. Bone involvement in adult patients affected with Ehlers-Danlos syndrome. Osteoporos Int. 2016;27:2525–2531. 10.1007/s00198-016-3562-2. [DOI] [PubMed] [Google Scholar]
- 110.Bravo JF, Wolff C. Clinical study of hereditary disorders of connective tissues in a Chilean population. Arthritis Rheum. 2006;54(2):515–523. 10.1002/art.21557. [DOI] [PubMed] [Google Scholar]
- 111.Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with Mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:1–22. 10.5402/2012/751768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Catala-Pétavy C, Machet L, Georgesco G, Pétavy F, Maruani A, Vaillant L. Contribution of skin biometrology to the diagnosis of the Ehlers-Danlos syndrome in a prospective series of 41 patients. Skin Res Technol. 2009;15(4):412–417. 10.1111/j.1600-0846.2009.00379.x. [DOI] [PubMed] [Google Scholar]
- 113.Eisenbeiss C, Martinez A, Hagedorn-Greiwe M, Reinhardt DP, Bätge B, Brinckmann J. Reduced skin thickness: a new minor diagnostic criterion for the classical and hypermobility types of Ehlers-Danlos syndrome. Br J Dermatol. 2003;149(4):850–852. 10.1046/j.1365-2133.2003.05439.x. [DOI] [PubMed] [Google Scholar]
- 114.Hakim AJ, Sahota A. Joint hypermobility and skin elasticity: the hereditary disorders of connective tissue. Clin Dermatol. 2006; 24:521–533. 10.1016/j.clindermatol.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 115.Carley ME, Schaffer J. Urinary incontinence and pelvic organ prolapse in women with Marfan or Ehlers-Danlos syndrome. Am J Obstet Gynecol. 2000;182(5):1021–1023. 10.1067/mob.2000.105410. [DOI] [PubMed] [Google Scholar]
- 116.Mastoroudes H, Giarenis I, Cardozo L, et al. Prolapse and sexual function in women with benign joint hypermobility syndrome. BJOG Int J Obstet Gynaecol. 2013;120(2):187–192. 10.1111/1471-0528.12082. [DOI] [PubMed] [Google Scholar]
- 117.Hurst BS, Lange SS, Kullstam SM, et al. Obstetric and gynecologic challenges in women with ehlers-danlos syndrome. Obstet Gynecol. 2014;123(3):506–513. 10.1097/AOG.0000000000000123. [DOI] [PubMed] [Google Scholar]
- 118.Hugon-Rodin J, Lebègue G, Becourt S, Hamonet C, Gompel A. Gynecologic symptoms and the influence on reproductive life in 386 women with hypermobility type Ehlers-Danlos syndrome: a cohort study. Orphanet J Rare Dis. 2016;11:124. 10.1186/s13023-016-0511-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Khalil H, Dfsrh R, Hla TT. A case report of obstetrical management of a pregnancy with hypermobile Ehlers-Danlos syndrome and literature review. Obstet Med. 2013;6(2):80–82. 10.1177/1753495X13482894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sundelin HEK, Stephansson O, Johansson K, Ludvigsson JF. Pregnancy outcome in joint hypermobility syndrome and Ehlers-Danlos syndrome. Acta Obstet Gynecol Scand. 2017; 96(1):114–119. 10.1111/aogs.13043. [DOI] [PubMed] [Google Scholar]
- 121.Beynnon BD, Johnson RJ, Braun S, et al. The relationship between menstrual cycle phase and anterior cruciate ligament injury: a case-control study of recreational alpine skiers. Am J Sports Med. 2006;34(5):757–764. 10.1177/0363546505282624. [DOI] [PubMed] [Google Scholar]
- 122.Wojtys EM, Huston LJ, Boynton MD, Spindler KP, Lindenfeld TN. The effect of the menstrual cycle on anterior cruciate ligament injuries in women as determined by hormone levels. Am J Sports Med. 2002;30(2):182–188. 10.1177/03635465020300020601. [DOI] [PubMed] [Google Scholar]
- 123.Adachi N, Nawata K, Maeta M, Kurozawa Y. Relationship of the menstrual cycle phase to anterior cruciate ligament injuries in teenaged female athletes. Arch Orthop Trauma Surg. 2008;128(5):473–478. 10.1007/s00402-007-0461-1. [DOI] [PubMed] [Google Scholar]
- 124.Rahr-Wagner L, Thillemann M, Mehnert F, Pedersen AB, Lind M. Is the use of oral contraceptives associated with operatively treated anterior cruciate ligament injury? A case-control study from the Danish Knee Ligament Reconstruction Registry. Am J Sports Med. 2014;42:2897–2905. 10.1177/0363546514557240. [DOI] [PubMed] [Google Scholar]
- 125.Gray AM, Gugala Z, Baillargeon JG. Effects of oral contraceptive use on anterior cruciate ligament injury epidemiology. Med Sci Sports Exerc. 2016;48(4):648–654. 10.1249/MSS.0000000000000806. [DOI] [PubMed] [Google Scholar]
- 126.Jen M, Nallasamy S. Ocular manifestations of genetic skin disorders. Clin Dermatol. 2016;34(2):242–275. 10.1016/j.clindermatol.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 127.Gharbiya M, Moramarco A, Castori M, Parisi F. Ocular features in joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type: a clinical and in vivo confocal microscopy study. AJOPHT. 2012;154:593–600.e1. 10.1016/j.ajo.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 128.Mishra MB, Ryan P, Atkinson P, et al. Extra-articular features of benign joint hypermobility syndrome. Br J Rheumatol. 1996;35:861–866. [DOI] [PubMed] [Google Scholar]
- 129.Louie A, Meyerle C, Francomano C, et al. Survey of Ehlers-Danlos patients’ ophthalmic surgery experiences. Mol Genet Genomic Med. 2020;8:e1155. 10.1002/mgg3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Honoré MB, Lauridsen EF, Sonnesen L. Oro-dental characteristics in patients with hypermobile Ehlers-Danlos syndrome compared to a healthy control group. J Oral Rehabil. 2019;46 (11):1055–1064. 10.1111/joor.12838. [DOI] [PubMed] [Google Scholar]
- 131.Mitakides J, Tinkle BT. Oral and mandibular manifestations in the Ehlers-Danlos syndromes; Oral and mandibular manifestations in the Ehlers-Danlos syndromes. Am J Med Genet Part C Semin Med Genet. 2017;175:220–225. 10.1002/ajmg.c.31541. [DOI] [PubMed] [Google Scholar]
- 132.De Paepe A, Malfait F. Bleeding and bruising in patients with Ehlers-Danlos syndrome and other collagen vascular disorders. Br J Haematol. 2004;127(5):491–500. 10.1111/j.1365-2141.2004.05220.x. [DOI] [PubMed] [Google Scholar]
- 133.Létourneau Y, Pérusse R, Buithieu H. Oral manifestations of Ehlers-Danlos syndrome. J Can Dent Assoc. 2001;67(6): 330–334. [PubMed] [Google Scholar]
- 134.Norton LA, Assael LA. Orthodontic and temporomandibular joint considerations in treatment of patients with Ehlers-Danlos syndrome. Am J Orthod Dentofacial Orthop. 1997; 111(1):75–84. 10.1016/s0889-5406(97)70305-6. [DOI] [PubMed] [Google Scholar]
- 135.Abel MD, Carrasco LR. Ehlers-Danlos syndrome: classifications, oral manifestations, and dental considerations. Oral Surg Oral Med Oral Pathol Oral Radiol Endodontol. 2006;102 (5):582–590. 10.1016/j.tripleo.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 136.Reinstein E, Delozier CD, Simon Z, Bannykh S, Rimoin DL, Curry CJ. Ehlers-Danlos syndrome type VIII is clinically heterogeneous disorder associated primarily with periodontal disease, and variable connective tissue features. Eur J Hum Genet. 2013; 21(2):233–236. 10.1038/ejhg.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hakim AJ, Grahame R, Norris P, Hopper C. Local anaesthetic failure in joint hypermobility syndrome. J R Soc Med. 2005; 98(2):84–85. 10.1258/jrsm.98.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wiesmann T, Castori M, Malfait F, Wulf H. Recommendations for anesthesia and perioperative management in patients with Ehlers-Danlos syndrome(s). Orphanet J Rare Dis. 2014; 9(1):109. 10.1186/s13023-014-0109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Arendt-Nielsen L, Kaalund SS, Bjerring P, Hogsaa B. Insufficient effect of local analgesics in Ehlers Danlos type III patients (connective tissue disorder). Acta Anaesthesiol Scand. 1990;34(5):358–361. [DOI] [PubMed] [Google Scholar]
- 140.Schubart JR, Schaefer E, Janicki P, et al. Resistance to local anesthesia in people with the Ehlers-Danlos syndromes presenting for dental surgery. J Dent Anesth Pain Med. 2019; 19(5):261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.De Felice C, Toti P, Di Maggio G, Parrini S, Bagnoli F. Absence of the inferior labial and lingual frenula in EhlersDanlos syndrome. Lancet. 2001;357(9267):1500–1502. 10.1016/S0140-6736(00)04661-4. [DOI] [PubMed] [Google Scholar]
- 142.Savasta S, Bassanese F, Hruby C, et al. Absence of lingual frenulum in children with Ehlers-Danlos syndrome: a retrospective study of forty cases and literature review of a twenty years long debate. Minerva Pediatr. 2019. 10.23736/S0026-4946.19.05530-0. [DOI] [PubMed] [Google Scholar]
- 143.Mitakides JE. The effect of Ehlers-Danlos syndromes on TMJ function and craniofacial pain. Cranio J Craniomandib Pract. 2018;36(2):71–72. 10.1080/08869634.2018.1435092. [DOI] [PubMed] [Google Scholar]
- 144.De Coster PJ, Van Den Berghe LI, Martens LC. Generalized joint hypermobility and temperomandibular disorders: inherited connective tissue disease as a model with maximum expression. J Orofac Pain. 2005;19(1):47–57. [PubMed] [Google Scholar]
- 145.Di Giacomo P, Celli M, Ierardo G, Polimeni A, Di Paolo C. Evaluation of temporomandibular disorders and comorbidities in patients with Ehler-Danlos: clinical and digital findings. J Int Soc Prev Community Dent. 2018;8(4):333–338. 10.4103/jispcd.JISPCD_103_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Coster PJ, Martens LC, Paepe A. Oral health in prevalent types of Ehlers-Danlos syndromes. J Oral Pathol Med. 2005;34(5): 298–307. 10.1111/j.1600-0714.2004.00300.x. [DOI] [PubMed] [Google Scholar]
- 147.Walczynska-Dragon K, Baron S, Nitecka-Buchta A, Tkacz E. Correlation between TMD and cervical spine pain and mobility: is the whole body balance TMJ related? Biomed Res Int. 2014;2014:1–7. 10.1155/2014/582414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hakim A, O’Callaghan C, De Wandele I, Stiles L, Pocinki A, Rowe P. Cardiovascular autonomic dysfunction in Ehlers-Danlos syndrome—hypermobile type. Am J Med Genet Part C Semin Med Genet. 2017;175(1):168–174. 10.1002/ajmg.c.31543. [DOI] [PubMed] [Google Scholar]
- 149.Roma M, Marden CL, De Wandele I, Francomano CA, Rowe PC. Postural tachycardia syndrome and other forms of orthostatic intolerance in Ehlers-Danlos syndrome. Auton Neurosci Basic Clin. 2018;215(January):89–96. 10.1016/j.autneu.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 150.Miller AJ, Stiles LE, Sheehan T, et al. Prevalence of hypermobile Ehlers-Danlos syndrome in postural orthostatic tachycardia syndrome. Auton Neurosci. 2020;224:102637. 10.1016/j.autneu.2020.102637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Deb A, Morgenshtern K, Culbertson CJ, Wang LB, Depold Hohler A. A survey-based analysis of symptoms in patients with postural orthostatic tachycardia syndrome. Proc (Bayl Univ Med Cent). 2015;28(2):157–159. 10.1080/08998280.2015.11929217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wallman D, Weinberg J, Hohler AD. Ehlers-Danlos syndrome and postural tachycardia syndrome: a relationship study. J Neurol Sci. 2014;340(1–2):99–102. 10.1016/j.jns.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 153.Boris JR, Bernadzikowski T. Demographics of a large paediatric postural orthostatic tachycardia syndrome program. Cardiol Young. 2018;28(5):668–674. 10.1017/S1047951117002888. [DOI] [PubMed] [Google Scholar]
- 154.Rowe PC, Barron DF, Calkins H, Maumenee IH, Tong PY, Geraghty MT. Orthostatic intolerance and chronic fatigue syndrome associated with Ehlers-Danlos syndrome. J Pediatr. 1999; 135(4):494–499. 10.1016/S0022-3476(99)70173-3. [DOI] [PubMed] [Google Scholar]
- 155.Leier CV, Call TD, Fulkerson PK, Wooley CF. The spectrum of cardiac defects in the Ehlers-Danlos syndrome, types I and III. Ann Intern Med. 1980;92(2 pt 1):171. 10.7326/0003-4819-92-2-171. [DOI] [PubMed] [Google Scholar]
- 156.McDonnell NB, Gorman BL, Mandel KW, et al. Echocardio-graphic findings in classical and hypermobile Ehlers-Danlos syndromes. Am J Med Genet Part A. 2006;140A(2):129–136. 10.1002/ajmg.a.31035. [DOI] [PubMed] [Google Scholar]
- 157.Wenstrup RJ, Meyer RA, Lyle JS, et al. Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genet Med. 2002; 4(3):112–117. 10.1097/00125817-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 158.Handler CE, Child A, Light ND, Dorrance DE. Mitral valve prolapse, aortic compliance, and skin collagen in joint hypermobility syndrome. Br Heart J. 1985;54(5):501–508. 10.1136/hrt.54.5.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Judge DP, Rouf R, Habashi J, Dietz HC. Mitral valve disease in Marfan syndrome and related disorders. J Cardiovasc Transl Res. 2011;4(6):741–747. 10.1007/s12265-011-9314-y. [DOI] [PubMed] [Google Scholar]
- 160.Asher SB, Chen R, Kallish S. Mitral valve prolapse and aortic root dilation in adults with hypermobile Ehlers-Danlos syndrome and related disorders. Am J Med Genet Part A. 2018; 176(9):1838–1844. 10.1002/ajmg.a.40364. [DOI] [PubMed] [Google Scholar]
- 161.Rauser-Foltz KK, Starr LJ, Yetman AT. Utilization of echocardiography in Ehlers-Danlos syndrome. Congenit Heart Dis. 2019;14(5):864–867. 10.1111/chd.12824. [DOI] [PubMed] [Google Scholar]
- 162.Seçkin Ü, Tur BS, Yilmaz Ö, Yagci I, Bodur H, Arasil T. The prevalence of joint hypermobility among high school students. Rheumatol Int. 2005;25(4):260–263. 10.1007/s00296-003-0434-9. [DOI] [PubMed] [Google Scholar]
- 163.Huang H, Deb A, Culbertson C, Morgenshtern K, Hohler AD. Dermatological manifestations of postural tachycardia syndrome are common and diverse. J Clin Neurol. 2016;12(1):75–78. 10.3988/jcn.2016.12.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dinc A, Keskin G. AB0866 presence of Acrocyanosis in patients with joint hypermobility. Ann Rheum Dis. 2014;73 (suppl 2):1088.3. 10.1136/annrheumdis-2014-eular.5595. [DOI] [Google Scholar]
- 165.Seneviratne SL, Maitland A, Afrin L. Mast cell disorders in Ehlers-Danlos syndrome. Am J Med Genet Part C Semin Med Genet. 2017;175(1):226–236. 10.1002/ajmg.c.31555. [DOI] [PubMed] [Google Scholar]
- 166.Daens S, Grossin D, Hermanns-Lê T, Peeters D, Manicourt D. Severe mast cell activation syndrome in a 15-year-old patient with an hypermobile Ehlers-Danlos syndrome. Rev Med Liege. 2018;73(2):61–64. [PubMed] [Google Scholar]
- 167.Molderings GJ, Brettner S, Homann J, Afrin LB. Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options. J Hematol Oncol. 2011;4:10. 10.1186/1756-8722-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Luzgina NG, Potapova OV, Shkurupiy VA. Structural and functional peculiarities of mast cells in undifferentiated connective tissue dysplasia. Bull Exp Biol Med. 2011;150(6):676–678. 10.1007/s10517-011-1220-4. [DOI] [PubMed] [Google Scholar]
- 169.Morgan AW, Pearson SB, Davies S, Gooi HC, Bird HA. Asthma and airways collapse in two heritable disorders of connective tissue. Ann Rheum Dis. 2007;66(10):1369–1373. 10.1136/ard.2006.062224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zeitoun J-D, Lefèvre JH, De Parades V, et al. Functional digestive symptoms and quality of life in patients with Ehlers-Danlos syndromes: results of a national cohort study on 134 patients. PLoS One. 2013;8:e80321. 10.1371/journal.pone.0080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zarate N, Farmer AD, Grahame R, et al. Unexplained gastrointestinal symptoms and joint hypermobility: is connective tissue the missing link? Neurogastroenterol Motil. 2010;22(3): 252–e78. 10.1111/j.1365-2982.2009.01421.x. [DOI] [PubMed] [Google Scholar]
- 172.Nelson AD, Mouchli MA, Valentin N, et al. Ehlers Danlos syndrome and gastrointestinal manifestations: a 20-year experience at Mayo Clinic. Neurogastroenterol Motil. 2015;27(11):1657–1666. 10.1111/nmo.12665. [DOI] [PubMed] [Google Scholar]
- 173.Hakim AJ, Grahame R. Letters to the Editor. Nonmusculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology. 2004;43: 1194–1195. 10.1093/rheumatology/keh279. [DOI] [PubMed] [Google Scholar]
- 174.Park K-J, Singer W, Sletten DM, Low PA, Bharucha AE. Gastric emptying in postural tachycardia syndrome: a preliminary report. Clin Auton Res. 2013;23:163–167. 10.1007/s10286-013-0193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Danese C, Castori M, Celletti C, et al. Screening for celiac disease in the joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type. Am J Med Genet Part A. 2011;155(9):2314–2316. 10.1002/ajmg.a.34134. [DOI] [PubMed] [Google Scholar]
- 176.Castori M, Sperduti I, Celletti C, Camerota F, Grammatico P. Symptom and joint mobility progression in the joint hypermobility syndrome. Clin Exp Rheumatol. 2011;29(6):998–1005. [PubMed] [Google Scholar]
- 177.Milhorat TH, Bolognese PA, Nishikawa M, McDonnell NB, Francomano CA. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and Chiari malformation Type I in patients with hereditary disorders of connective tissue. J Neurosurg. 2007;7(6):601–609. [DOI] [PubMed] [Google Scholar]
- 178.Pradini-Santos L, Craven CL, Sayal PP. Extradural compressive spinal cerebrospinal fluid leak in Ehlers-Danlos syndrome. World Neurosurg. 2019;132:67–68. 10.1016/j.wneu.2019.08.163. [DOI] [PubMed] [Google Scholar]
- 179.Henderson FC, Austin C, Benzel E, et al. Neurological and spinal manifestations of the Ehlers-Danlos syndromes. Am J Med Genet Part C Semin Med Genet. 2017;175(1):195–211. 10.1002/ajmg.c.31549. [DOI] [PubMed] [Google Scholar]
- 180.Voermans NC, Knoop H, van Engelen BG. High frequency of neuropathic pain in Ehlers-Danlos syndrome: an association with axonal polyneuropathy and compression neuropathy? J Pain Symptom Manage. 2011;41(5):e4–e6. 10.1016/j.jpainsymman.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 181.Cazzato D, Castori M, Lombardi R, et al. Small fiber neuropathy is a common feature of Ehlers-Danlos syndromes. Neurology. 2016;87(2):155–159. 10.1212/WNL.0000000000002847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Galan E, Kousseff BG. Peripheral neuropathy in Ehlers-Danlos syndrome. Pediatr Neurol. 1995;12(3):242–245. [DOI] [PubMed] [Google Scholar]
- 183.Kayed K, Kåss B. Acute multiple brachial neuropathy and Ehlers-Danlos syndrome. Neurology. 1979;29(12):1620–1621. [DOI] [PubMed] [Google Scholar]
- 184.Papapetropoulos T, Tsankanikas C, Spengos M. Brachial neuropathy and Ehlers-Danlos syndrome. Neurology. 1981;31(5): 642–643. 10.1212/wnl.31.5.642-a. [DOI] [PubMed] [Google Scholar]
- 185.Voermans NC, Drost G, Van Kampen A, et al. Recurrent neuropathy associated with Ehlers-Danlos syndrome. J Neurol. 2006;253(5):670–671. 10.1007/s00415-005-0056-0. [DOI] [PubMed] [Google Scholar]
- 186.Castori M, Voermans NC. Neurological manifestations of Ehlers-Danlos syndrome(s): a review. Iran J Neurol. 2014;13 (4):190–208. [PMC free article] [PubMed] [Google Scholar]
- 187.Puledda F, Viganò A, Celletti C, et al. A study of migraine characteristics in joint hypermobility syndrome a.k.a. Ehlers-Danlos syndrome, hypermobility type. Neurol Sci. 2015;36(8): 1417–1424. 10.1007/s10072-015-2173-6. [DOI] [PubMed] [Google Scholar]
- 188.Jacome D. Headache in Ehlers-Danlos syndrome. Cephalalgia. 1999;19(9):791–796. 10.1046/j.1468-2982.1999.1909791.x. [DOI] [PubMed] [Google Scholar]
- 189.Sacheti A, Szemere J, Bernstein B, Tafas T, Schechter N, Tsipouras P. Chronic pain is a manifestation of the Ehlers-Danlos syndrome. J Pain Symptom Manage. 1997;14(2):88–93. 10.1016/S0885-3924(97)00007-9. [DOI] [PubMed] [Google Scholar]
- 190.Rombaut L, Malfait F, Cools A, De Paepe A, Calders P. Musculoskeletal complaints, physical activity and health-related quality of life among patients with the Ehlers-Danlos syndrome hypermobility type. Disabil Rehabil. 2010;32(16):1339–1345. 10.3109/09638280903514739. [DOI] [PubMed] [Google Scholar]
- 191.Voermans NC, Knoop H. Both pain and fatigue are important possible determinants of disability in patients with the Ehlers-Danlos syndrome hypermobility type. Disabil Rehabil. 2011;33(8):706–707. 10.3109/09638288.2010.531373. [DOI] [PubMed] [Google Scholar]
- 192.Voermans NC, Knoop H, van de Kamp N, Hamel BC, Bleijenberg G, van Engelen BG. Fatigue is a frequent and clinically relevant problem in Ehlers-Danlos syndrome. Semin Arthritis Rheum. 2010;40(3):267–274. 10.1016/j.semarthrit.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 193.Castori M, Celletti C, Camerota F, Grammatico P. Chronic fatigue syndrome is commonly diagnosed in patients with Ehlers-Danlos syndrome hypermobility type/joint hypermobility syndrome. Clin Exp Rheumatol. 2011;29(3):597–598. [PubMed] [Google Scholar]
- 194.De Wandele I, Pocinki A, Rowe P, O’Callaghan C, Hakim A. Chronic fatigue in Ehlers-Danlos syndrome-hypermobile type. Am J Med Genet Part C Semin Med Genet. 2017;175(1):175–180. 10.1002/ajmg.c.31542. [DOI] [PubMed] [Google Scholar]
- 195.Voermans NC, Knoop H, Bleijenberg G, van Engelen BG. Fatigue is associated with muscle weakness in Ehlers-Danlos syndrome: an explorative study. Physiotherapy. 2011;97(2): 170–174. 10.1016/j.physio.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 196.Moore JR, Tolo VT, Weiland AJ. Painful subluxation of the carpometacarpal joint of the thumb in Ehlers-Danlos syndrome. J Hand Surg Am. 1985;10(5):661–663. 10.1016/S0363-5023(85)80203-3. [DOI] [PubMed] [Google Scholar]
- 197.Mulvey MR, MacFarlane GJ, Beasley M, et al. Modest association of joint hypermobility with disabling and limiting musculoskeletal pain: results from a large-scale general population-based survey. Arthritis Care Res. 2013;65(8):1325–1333. 10.1002/acr.21979. [DOI] [PubMed] [Google Scholar]
- 198.Berglund B, Nordström G, Harberg C, Mattiasson AC. Foot pain and disability in individuals with Ehlers-Danlos syndrome (EDS): impact on daily life activities. Disabil Rehabil. 2005;27(4): 164–169. 10.1080/09638280400009352. [DOI] [PubMed] [Google Scholar]
- 199.Sendur OF, Gurer G, Bozbas GT. The frequency of hypermobility and its relationship with clinical findings of fibromyalgia patients. Clin Rheumatol. 2007;26(4):485–487. 10.1007/s10067-006-0304-4. [DOI] [PubMed] [Google Scholar]
- 200.Ofluoglu D, Gunduz OH, Kul-Panza E, Guven Z. Hypermobility in women with fibromyalgia syndrome. Clin Rheumatol. 2006; 25(3):291–293. 10.1007/s10067-005-0040-1. [DOI] [PubMed] [Google Scholar]
- 201.Scheper MC, Pacey V, Rombaut L, et al. Generalized hyperalgesia in children and adults diagnosed with hypermobility syndrome and Ehlers-Danlos syndrome hypermobility type: a discriminative analysis. Arthritis Care Res. 2017;69(3):421–429. 10.1002/acr.22998. [DOI] [PubMed] [Google Scholar]
- 202.Leone CM, Celletti C, Gaudiano G, et al. Pain due to EhlersDanlos syndrome is associated with deficit of the endogenous pain inhibitory control. Pain Med. 2020. [DOI] [PubMed] [Google Scholar]
- 203.Domany KA, Hantragool S, Smith DF, Xu Y, Hossain M, Simakajornboon N. Sleep disorders and their management in children with Ehlers-Danlos syndrome referred to sleep clinics. J Clin Sleep Med. 2018;14(4):623–629. 10.5664/jcsm.7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Guilleminault C, Primeau M, Chiu HY, Yuen KM, Leger D, Metlaine A. Sleep-disordered breathing in Ehlers-Danlos syndrome: a genetic model of OSA. Chest. 2013;144(5):1503–1511. 10.1378/chest.13-0174. [DOI] [PubMed] [Google Scholar]
- 205.Mu W, Muriello M, Clemens JL, et al. Factors affecting quality of life in children and adolescents with hypermobile EhlersDanlos syndrome/hypermobility spectrum disorders. Am J Med Genet Part A. 2018;2019:1–9. 10.1002/ajmg.a.61055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Stöberl AS, Gaisl T, Giunta C, et al. Obstructive sleep apnoea in children and adolescents with Ehlers-Danlos syndrome. Respiration. 2019;97(4):284–291. 10.1159/000494328. [DOI] [PubMed] [Google Scholar]
- 207.Gaisl T, Giunta C, Bratton DJ, et al. Obstructive sleep apnoea and quality of life in Ehlers-Danlos syndrome: a parallel cohort study. Thorax. 2017;72(8):729–735. 10.1136/thoraxjnl-2016-209thinspace560. [DOI] [PubMed] [Google Scholar]
- 208.Bulbena A, Duró JC, Porta M, et al. Anxiety disorders in the joint hypermobility syndrome. Psychiatry Res. 1993;46(1):5968. 10.1016/0165-1781(93)90008-5. [DOI] [PubMed] [Google Scholar]
- 209.Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114–122. 10.1093/rheumatology/ket317. [DOI] [PubMed] [Google Scholar]
- 210.Hershenfeld SA, Wasim S, McNiven V, et al. Psychiatric disorders in Ehlers-Danlos syndrome are frequent, diverse and strongly associated with pain. Rheumatol Int. 2016;36(3):341–348. 10.1007/s00296-015-3375-1. [DOI] [PubMed] [Google Scholar]
- 211.Fatoye F, Palmer S, MacMillan F, Rowe P, Van Der Linden M. Pain intensity and quality of life perception in children with hypermobility syndrome. Rheumatol Int. 2012;32 (5):1277–1284. 10.1007/s00296-010-1729-2. [DOI] [PubMed] [Google Scholar]
- 212.Pacey V, Tofts L, Adams RD, Munns CF, Nicholson LL. Quality of life prediction in children with joint hypermobility syndrome. J Paediatr Child Health. 2015;51(7):689–695. 10.1111/jpc.12826. [DOI] [PubMed] [Google Scholar]
- 213.Shetreat-Klein M, Shinnar S, Rapin I. Abnormalities of joint mobility and gait in children with autism spectrum disorders. Brain Dev. 2014;36(2):91–96. 10.1016/j.braindev.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 214.Baeza-Velasco C, Pailhez G, Bulbena A, Baghdadli A. Joint hypermobility and the heritable disorders of connective tissue: clinical and empirical evidence of links with psychiatry. Gen Hosp Psychiatry. 2015;37(1):24–30. 10.1016/j.genhosppsych.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 215.Casanova EL, Sharp JL, Edelson SM, Kelly DP, Casanova MF. A cohort study comparing women with autism spectrum disorder with and without generalized joint hypermobility. Behav Sci (Basel). 2018;8(3):1–16. 10.3390/bs8030035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Banerjee G, Agarwal RK, Shembesh NM, Mauhoub ME. Ehlers Danlos syndrome—masquerading as primary muscle disease. Postgrad Med J. 1988;64(748):126–127. 10.1136/pgmj.64.748.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Adib N, Davies K, Grahame R, Woo P, Murray KJ. Joint hypermobility syndrome in childhood. A not so benign multisystem disorder? Rheumatology. 2005;44(6):744–750. 10.1093/rheumatology/keh557. [DOI] [PubMed] [Google Scholar]
- 218.Galli M, Rigoldi C, Celletti C, et al. Postural analysis in time and frequency domains in patients with Ehlers-Danlos syndrome. Res Dev Disabil. 2011;32(1):322–325. 10.1016/j.ridd.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 219.Galli M, Cimolin V, Vismara L, et al. The effects of muscle hypotonia and weakness on balance: a study on Prader-Willi and Ehlers-Danlos syndrome patients. Res Dev Disabil. 2011;32(3):1117–1121. 10.1016/j.ridd.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 220.Rombaut L, Malfait F, De Wandele I, et al. Balance, gait, falls, and fear of falling in women with the hypermobility type of Ehlers-Danlos syndrome. Arthritis Care Res (Hoboken). 2011; 63(10):1432–1439. 10.1002/acr.20557. [DOI] [PubMed] [Google Scholar]
- 221.Booshanam DS, Cherian B, Charles Joseph PAR, Mathew J, Thomas R. Evaluation of posture and pain in persons with benign joint hypermobility syndrome. Rheumatol Int. 2011;31: 1561–1565. 10.1007/s00296-010-1514-2. [DOI] [PubMed] [Google Scholar]
- 222.Sahin N, Baskent A, Cakmak A, Salli A, Ugurlu H, Berker E. Evaluation of knee proprioception and effects of proprioception exercise in patients with benign joint hypermobility syndrome. Rheumatol Int. 2008;28(10):995–1000. 10.1007/s00296-008-0566-z. [DOI] [PubMed] [Google Scholar]
- 223.Mallik AK, Ferrell WR, Mcdonaldtt AG, Sturrockt RD. Impaired proprioceptive acuity at the proximal interphalangeal joint in patients with the hypermobility syndrome. Br J Rheumatol. 1994;33(12):1192–1193. [DOI] [PubMed] [Google Scholar]
- 224.Hall MG, Ferrell WR, Sturrockj RD, Hamblenf DL, Baxendale RH. The Effect of the Hypermobility Syndrome on Knee Joint Proprioception. Br J Rheumatology. Vol 34; 1995(2):121–125. 10.1093/rheumatology/34.2.121. [DOI] [PubMed] [Google Scholar]
- 225.Clayton HA, Cressman EK, Henriques DYP. Proprioceptive sensitivity in Ehlers-Danlos syndrome patients. Exp Brain Res. 2013;230(3):311–321. 10.1007/s00221-013-3656-4. [DOI] [PubMed] [Google Scholar]
- 226.Clayton HA, Jones SAH, Henriques DYP. Proprioceptive precision is impaired in Ehlers-Danlos syndrome. Springerplus. 2015;4(1):1–8. 10.1186/s40064-015-1089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Galli M, Cimolin V, Rigoldi C, et al. Gait strategy in patients with Ehlers-Danlos syndrome hypermobility type: a kinematic and kinetic evaluation using 3D gait analysis. Res Dev Disabil. 2011;32(5):1663–1668. 10.1016/j.ridd.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 228.Lyons JJ, Yu X, Hughes JD, et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet. 2016;48(12):1564–1569. 10.1038/ng.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by Tenascin-X deficiency. N Engl J Med. 2002;345:1167–1175. 10.1056/nejmoa002939. [DOI] [PubMed] [Google Scholar]
- 230.Micale L, Guarnieri V, Augello B, et al. Novel TNXB variants in two Italian patients with classical-like Ehlers-Danlos syndrome. Genes (Basel). 2019;10(12):967. 10.3390/genes10120967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Rymen D, Ritelli M, Zoppi N, et al. Clinical and molecular characterization of classical-like Ehlers-Danlos syndrome due to a novel TNXB variant. Genes (Basel). 2019;10(11):843. 10.3390/genes10110843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kaufman CS, Butler MG. Mutation in TNXB gene causes moderate to severe Ehlers-Danlos syndrome HHS public access. World J Med Genet. 2016;6(2):17–21. 10.5496/wjmg.v6.i2.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Zweers MC, Dean WB, Van Kuppevelt TH, Bristow J, Schalkwijk J. Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with Tenascin-X mutations. Clin Genet. 2005;11:330–334. 10.1111/j.13990004.2004.00401.x. [DOI] [PubMed] [Google Scholar]
- 234.Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214–217. 10.1086/376564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Yamada K, Watanabe A, Takeshita H, et al. Measurement of serum Tenascin-X in joint hypermobility syndrome patients. Biol Pharm Bull. 2019;42(9):1596–1599. [DOI] [PubMed] [Google Scholar]
- 236.Burch GH, Gong Y, Liu W, et al. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet. 1997;17: 104–108. 10.1038/ng0997-104. [DOI] [PubMed] [Google Scholar]
- 237.Lao Q, Brookner B, Merke DP. High-throughput screening for CYP21A1P-TNXA/TNXB chimeric genes responsible for Ehlers Danlos syndrome in patients with congenital adrenal hyperplasia. J Mol Diagn. 2019;21:924–931. 10.1016/j.jmoldx.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Morissette R, Chen W, Perritt AF, et al. Broadening the spectrum of Ehlers Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2015;100(8): E1143–E1152. 10.1210/jc.2015-2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lethias C, Descollonges Y, Boutillon MM, Garrone R. Flexilin: a new extracellular matrix glycoprotein localized on collagen fibrils. Matrix Biol. 1996;15(1):11–19. 10.1016/S0945-053X(96)90122-5. [DOI] [PubMed] [Google Scholar]
- 240.Mao JR, Taylor G, Dean WB, et al. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat Genet. 2002;30(4):421–425. 10.1038/ng850. [DOI] [PubMed] [Google Scholar]
- 241.Chiquet M, Birk DE, Bönnemann CG, Koch M. Collagen XII: protecting bone and muscle integrity by organizing collagen fibrils. Int J Biochem Cell Biol. 2014;53:51–54. 10.1016/j.biocel.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bristow J, Carey W, Egging D, Schalkwijk J. Tenascin-X, collagen, elastin, and the Ehlers-Danlos syndrome. Am J Med Genet Part C Semin Med Genet. 2005;139C(1):24–30. 10.1002/ajmg.c.30071. [DOI] [PubMed] [Google Scholar]
- 243.Hausser I, Anton-Lamprecht I. Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum Genet. 1994; 93(4):394–407. 10.1007/BF00201664. [DOI] [PubMed] [Google Scholar]
- 244.Hermanns-Lê T, Piérard GE. Collagen fibril arabesques in connective tissue disorders. Am J Clin Dermatol. 2006; 7(5):323–326. 10.2165/00128071-200607050-00006. [DOI] [PubMed] [Google Scholar]
- 245.Holbrook KA, Byers PH. Structural abnormalities in the dermal collagen and elastic matrix from the skin of patients with inherited connective tissue disorders. J Invest Dermatol. 1982; 79(suppl 1):7–16. 10.1038/jid.1982.3. [DOI] [PubMed] [Google Scholar]
- 246.Hermanns-Lê T, Reginster MA, Piérard-Franchimont C, Delvenne P, Piérard GE, Manicourt D. Dermal ultrastructure in low beighton score members of 17 families with hypermobiletype Ehlers-Danlos syndrome. J Biomed Biotechnol. 2012;2012:1–3. 10.1155/2012/878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hermanns-Lê T, Piérard GE. Ultrastructural alterations of elastic fibers and other dermal components in Ehlers-Danlos syndrome of the hypermobile type. Am J Dermatopathol. 2007; 29(4):370–373. 10.1097/DAD.0b013e3180de3ec0. [DOI] [PubMed] [Google Scholar]
- 248.Kobayasi T. Abnormality of dermal collagen fibrils in Ehlers Danlos syndrome. Anticipation of the abnormality for the inherited hypermobile disorders. EurJDermatol. 2004;14:221–229. [PubMed] [Google Scholar]
- 249.Kobayasi T. Dermal elastic fibres in the inherited hypermobile disorders. J Dermatol Sci. 2006;41(3):175–185. 10.1016/j.jdermsci.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 250.Angwin C, Ghali N, Baker D, et al. Electron microscopy in the diagnosis of Ehlers-Danlos syndromes: correlation with clinical and genetic investigations. Br J Dermatol. 2019;182(4): 698–707. 10.1111/bjd.18165. [DOI] [PubMed] [Google Scholar]
- 251.Janecke AR, Li B, Boehm M, et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am J Med Genet Part A. 2016;170(1):103–115. 10.1002/ajmg.a.37383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Chiarelli N, Carini G, Zoppi N, et al. Transcriptome-wide expression profiling in skin fibroblasts of patients with joint hypermobility syndrome/ehlers-danlos syndrome hypermobility type. PLoS One. 2016;11:e0161347. 10.1371/journal.pone.0161347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Chiarelli N, Carini G, Zoppi N, Ritelli M, Colombi M. Molecular insights in the pathogenesis of classical Ehlers-Danlos syndrome from transcriptome-wide expression profiling of patients’ skin fibroblasts. PLoS One. 2019;14(2):e0211647. 10.1371/journal.pone.0211647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Zoppi N, Gardella R, De Paepe A, Barlati S, Colombi M. Human fibroblasts with mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate α2β1 integrin, and recruit α vβ3 instead of α5β1 integrin. J Biol Chem. 2004;279(18):18157–18168. 10.1074/jbc.M312609200. [DOI] [PubMed] [Google Scholar]
- 255.Zoppi N, Chiarelli N, Binetti S, Ritelli M, Colombi M. Dermal fibroblast-to-myofibroblast transition sustained by αvß3 integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4):1010–1023. 10.1016/j.bbadis.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 256.Wenstrup RJ, Florer JB, Davidson JM, et al. Murine model of the Ehlers-Danlos syndrome: col5a1 haploinsufficiency disrupts collagen fibril assembly at multiple stages. J Biol Chem. 2006; 281(18):12888–12895. 10.1074/jbc.M511528200. [DOI] [PubMed] [Google Scholar]
- 257.Johnston JM, Connizzo BK, Shetye SS, et al. Collagen V haploinsufficiency in a murine model of classic Ehlers-Danlos syndrome is associated with deficient structural and mechanical healing in tendons. J Orthop Res. 2017;35(12):2707–2715. 10.1002/jor.23571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Sun M, Connizzo BK, Adams SM, et al. Targeted deletion of collagen V in tendons and ligaments results in a classic Ehlers-Danlos syndrome joint phenotype. Am J Pathol. 2015;185(5):1436–1447. 10.1016/j.ajpath.2015.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kosho T, Mizumoto S, Watanabe T, Yoshizawa T, Miyake N, Yamada S. Recent advances in the pathophysiology of musculocontractural Ehlers-Danlos syndrome. Genes (Basel). 2020;11(43):1–14. 10.3390/genes11010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Bowen CJ, Giadrosic JFC, Burger Z, et al. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. J Clin Invest. 2020;130(2):686–698. 10.1172/JCI130730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Voermans NC, Verrijp K, Eshuis L, et al. Mild muscular features in Tenascin-X knockout mice, a model of Ehlers-Danlos syndrome. Connect Tissue Res. 2011;52(5):422–432. 10.3109/03008207.2010.551616. [DOI] [PubMed] [Google Scholar]
- 262.Hashimoto K, Kajitani N, Miyamoto Y, Matsumoto K. Ichi. Wound healing-related properties detected in an experimental model with a collagen gel contraction assay are affected in the absence of tenascin-X. Exp Cell Res. 2018;363(1):102–113. 10.1016/j.yexcr.2017.12.025. [DOI] [PubMed] [Google Scholar]
- 263.Grahame R. Joint hypermobility syndrome pain. Curr Pain Headache Rep. 2009;13(6):427–433. 10.1007/s11916-009-0070-5. [DOI] [PubMed] [Google Scholar]
- 264.Engelbert RHH, Juul-Kristensen B, Pacey V, et al. The evidence-based rationale for physical therapy treatment of children, adolescents, and adults diagnosed with joint hypermobility syndrome/hypermobile Ehlers Danlos syndrome. Am J Med Genet Part C Semin Med Genet. 2017;175(1):158–167. 10.1002/ajmg.c.31545. [DOI] [PubMed] [Google Scholar]
- 265.Simmonds JV, Keer RJ. Hypermobility and the hypermobility syndrome, part 2: assessment and management of hypermobility syndrome: illustrated via case studies. Man Ther. 2008;13(2):1–11. 10.1016/j.math.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 266.Simmonds JV, Keer RJ. Hypermobility and the hypermobility syndrome. Man Ther. 2007;12(4):298–309. 10.1016/j.math.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 267.World Health Organization. WHO Guidelines on Basic Training and Safety in Chiropractic Geneva 2005. Geneva, Switzerland: WHO Press; 2005. [Google Scholar]
- 268.Puentedura EJ, March J, Anders J, et al. Safety of cervical spine manipulation: are adverse events preventable and are manipulations being performed appropriately? A review of 134 case reports. J Man Manip Ther. 2012;20(2):66–74. 10.1179/2042618611Y.0000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Puentedura EJ, O’Grady WH. Safety of thrust joint manipulation in the thoracic spine: a systematic review. J Man Manip Ther. 2015;23(3):154–161. 10.1179/2042618615Y.0000000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Globe G, Farabaugh RJ, Hawk C, et al. Clinical practice guideline: chiropractic care for low back pain. J Manip Physiol Therap. 2016;39:1–22. 10.1016/j.jmpt.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 271.Weinberg J, Doering C, McFarland EG. Joint surgery in Ehlers-Danlos patients: results of a survey. Am J Orthop (Belle Mead NJ). 1999;28(7):406–409. [PubMed] [Google Scholar]
- 272.Beighton P, Horan FT. Surgical aspects of the Ehlers-Danlos syndrome a survey of 100 cases. Br J Surg. 1969;56(4):255–259. 10.1002/bjs.1800560404. [DOI] [PubMed] [Google Scholar]
- 273.Why the Zebra? | The Ehlers Danlos Society: The Ehlers Danlos Society. https://www.ehlers-danlos.com/why-the-zebra/. Accessed March 30, 2020.