

Abstract
Context:
Distinct from the muscle atrophy that develops from inactivity or disuse, atrophy that occurs after traumatic joint injury continues despite the patient being actively engaged in exercise. Recognizing the multitude of factors and cascade of events that are present and negatively influence the regulation of muscle mass after traumatic joint injury will likely enable clinicians to design more effective treatment strategies. To provide sports medicine practitioners with the best strategies to optimize muscle mass, the purpose of this clinical review is to discuss the predominant mechanisms that control muscle atrophy for disuse and posttraumatic scenarios, and to highlight how they differ.
Evidence Acquisition:
Articles that reported on disuse atrophy and muscle atrophy after traumatic joint injury were collected from peer-reviewed sources available on PubMed (2000 through December 2019). Search terms included the following: disuse muscle atrophy OR disuse muscle mass OR anterior cruciate ligament OR ACL AND mechanism OR muscle loss OR atrophy OR neurological disruption OR rehabilitation OR exercise.
Study Design:
Clinical review.
Level of Evidence:
Level 5.
Results:
We highlight that (1) muscle atrophy after traumatic joint injury is due to a broad range of atrophy-inducing factors that are resistant to standard resistance exercises and need to be effectively targeted with treatments and (2) neurological disruptions after traumatic joint injury uncouple the nervous system from muscle tissue, contributing to a more complex manifestation of muscle loss as well as degraded tissue quality.
Conclusion:
Atrophy occurring after traumatic joint injury is distinctly different from the muscle atrophy that develops from disuse and is likely due to the broad range of atrophy-inducing factors that are present after injury. Clinicians must challenge the standard prescriptive approach to combating muscle atrophy from simply prescribing physical activity to targeting the neurophysiological origins of muscle atrophy after traumatic joint injury.
Keywords: disuse, muscle wasting, neurophysiological, ACL
Physical inactivity poses a significant threat to the maintenance of skeletal muscle mass, strength, and power.55 In the absence of countermeasures such as exercise, nutritional, or pharmacological interventions, atrophy is inevitable. At the cellular level, disuse atrophy appears to be primarily driven by the deregulation of muscle protein synthesis rates to protein breakdown rates.38,98 Fortunately, this negative sequela can easily be rectified in healthy young patients. By mechanically engaging the muscle through exercise, individuals can positively influence pathways that regulate protein turnover, thereby increasing muscle strength and functional capacity.5,43 In this way, physical activity is an excellent prescriptive solution to maintaining skeletal muscle in healthy young patients.
Distinct from the skeletal muscle atrophy that develops from inactivity or disuse, atrophy that occurs after traumatic joint injury continues in spite of these patients being actively engaged in muscle-strengthening exercises (Figure 1).49,67 This presents clinicians with a major rehabilitation test, as traumatic joint injuries, such as anterior cruciate ligament (ACL) rupture, challenge the standard prescriptive approach to combating muscle atrophy. In a population that is already vulnerable to degenerative joint diseases,54,87 this unique atrophy profile can have devastating lifelong consequences. The inability of classic muscle-strengthening approaches to restore muscle mass after traumatic joint injury48,49,53,82,84 is likely due, in part, to the broad range of atrophy-inducing factors that are present and need to be effectively targeted with treatments (Figure 2).67 A rapidly growing body of evidence has begun to identify unique morphological, phenotypic, and signaling pathways that are altered after traumatic joint injury and the net results of these factors on muscle health.
Figure 1.

Simplified schematic demonstrating 2 routes of muscle atrophy: disuse or anterior cruciate ligament (ACL) injury. Inactivity or disuse can lead to muscle atrophy; however, mechanically engaging the muscle through exercise can lead to the reestablishment of healthy muscle tissue (left cycle of image). Conversely, muscle atrophy that occurs after ACL injury persists despite extensive strengthening exercises (right cycle of image). Resistance to traditional strengthening mechanisms is likely due, in part, to the various alterations in the morphological, phenotypic, and signaling pathways that are present after ACL injury and foster an environment where atrophy is more difficult to overcome. Unique to ACL injury, neurological disruptions uncouple the nervous system with muscle tissue, contributing to more complex manifestation of muscle loss, including fiber type transition, reduced satellite cells, and fatty tissue deposition, none of which are restored through traditional exercise.
Figure 2.
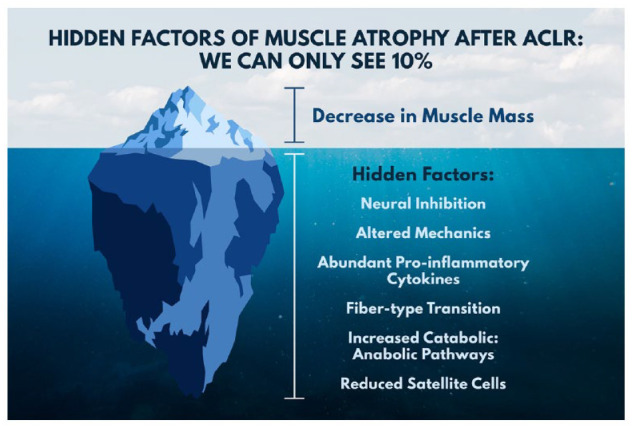
Muscle atrophy after anterior cruciate ligament injury clinically presents as reduced muscle mass; however, there are numerous underlying consequences that drive persistent muscle atrophy despite extensive strengthening exercises. Recognizing how these factors influence muscle atrophy can help clinicians develop targeted interventions aimed at mitigating muscle atrophy after traumatic joint injury. ACLR, anterior cruciate ligament reconstruction.
To more effectively treat muscle atrophy after traumatic joint injury, an understanding of the unique factors that underpin this condition are needed. Hence, the purpose of this clinical commentary was to describe the traditional model of disuse muscle atrophy, translate these mechanisms into clinical bottom lines, and then to contrast this to that of muscle atrophy after traumatic joint injury using ACL injury as a model.
Decoding the Mechanisms of Disuse Muscle Atrophy
Imbalance of Muscle Protein Synthesis to Breakdown Rates
Muscle mass is maintained via the regulated balance of muscle protein synthesis to muscle protein breakdown rates.4,8,11 Either side of the balance can be disturbed; as such, most atrophic conditions are thought to be a combination of decreased muscle protein synthesis and increased protein breakdown. In disuse conditions, blunted muscle protein synthesis seems to be the primary cause for the decline in muscle mass.4,17 Specifically, disuse leads to anabolic resistance, which reduces the ability of skeletal muscle to effectively increase protein synthesis in response to food intake.29 In scenarios where individuals become physically inactive but dietary intake is maintained, anabolic resistance is still present, such that skeletal muscle becomes less sensitive to the available protein and amino acids, and consequently muscle atrophy is observed.61 The mammalian target of rapamycin (mTOR) pathway is the major network involved in protein synthesis. In benchtop experiments, data suggest that this system senses important stimuli responsible for the regulation of muscle protein synthesis (though this pathway is not solely responsible for the maintenance of muscle mass31). In disuse conditions in humans, however, the role of mTOR in maintaining muscle mass is not as clear.17,29,31 Key markers of protein breakdown that have traditionally been explored in the literature as markers of mTOR deactivation are F-Box/atrogin-1 (MAFbx) and muscle-specific RING finger protein 1 (MuRF1). MAFbx and MuRF1 are two E3 ligases regulated by the Forkhead box O (FOXO) transcription factors that generally work to target proteins for turnover via the ubiquitin proteasome system. During periods of normal activity, FOXO transcription factors are inhibited, which allows for normal muscle growth through the activation of the mTOR pathway.11 In contrast, during periods of disuse, FOXO and MAFbx and MuRF1 are generally thought to be elevated, leading to muscle atrophy.76 The clinical bottom line—the net loss of skeletal muscle protein synthesis to muscle protein breakdown rates ultimately underpins muscle atrophy. In conditions of disuse, reduction in muscle protein synthesis rates is the main contributing factor to muscle atrophy.
Satellite Cells
Satellite cells are myogenic stem cells that lie along the sarcolemma of muscle fibers that can proliferate and differentiate into myoblasts. In adults, activated satellite cells (through damage or exercise) can proliferate and assist in the repair of damaged tissue or the formation of new muscle tissue by forming new myofibrils.25 Hence, augmentations in satellite cell function16 or the general abundance3,78 has been reported to accompany short-term disuse atrophy. Because of their location along the muscle fiber, satellite cells appear to be particularly sensitive to mechanical loading. During periods of unloading, preclinical models have shown a significant decline in the number of satellite cells.62 Similarly, in humans, after periods of disuse, significant declines in satellite cell count have been observed,3 in addition to moderate statistical relationships between augmentations in myofiber cross-sectional area and satellite cell content.74 However, it should be noted that not all studies reach the same conclusion. To this point, a 14-day unilateral leg immobilization protocol in young men yielded no change in satellite cell count,79 suggesting that relative level of disuse contributes to satellite cell maladaptation. The clinical bottom line—satellite cells help regulate skeletal muscle growth. If satellite cell function or number declines, the regenerative capacity of muscle can be impeded, contributing to muscle loss.
Myostatin
Myostatin is a well-established negative regulator of skeletal muscle growth.1,73 When myostatin is naturally mutated, reduced expression results in potent muscle growth, whereby significant muscle hypertrophy is observed in humans, sheep, dogs, and cattle.24 Experimentally, myostatin-deficient animals also exhibit marked muscle hypertrophy, a loss of fat mass, and greater contractile properties of muscle.40 Mechanistically, during disuse, overexpression of myostatin is thought to inhibit myoblast production via the mTOR pathway by upregulating the ubiquitin proteasome system through the E3 ligase, MuRF1. Myostatin has also been shown to negatively affect muscle tissue formation by inhibiting differentiation.44,85 Notably, others have also found that myostatin is highly colocalized with satellite cells57 and functionally plays a key role in regulating satellite cell function and promoting the overexpression of factors that regulate muscle growth.12 The clinical bottom line—myostatin is a protein that regulates muscle growth by limiting muscle mass. The absence of myostatin leads to dramatic increases in skeletal muscle, and conversely, overexpression of myostatin contributes to muscle atrophy.
Muscle Fiber Type Transitions
Human muscle contains 3 fiber types (type I, type IIa, and type IIx) that differ in size and function. Mechanistically, during periods of disuse, the lack of neurological influence or muscle loading is thought to suppress the activation of key factors that promote growth, such as insulin-like growth factor–1, or that help to maintain fiber-type composition, such as nuclear factor of activated T-cells (NFAT).23 Specific to the knee, the average percentage of muscle fiber types in the human vastus lateralis muscles are 40% to 70% slow-twitch oxidative (type I), 20% to 30% fast-twitch oxidative (type IIa), and 7% to 30% fast-twitch glycolytic (type IIa/IIx).73,81,88 In periods of disuse, the quadriceps appears to be particularly vulnerable to fiber-type transitions (more so than muscles dominated by fast-twitch fibers). Inactivity leads to a decrease in type I fibers relative to an increase in type IIa and IIa/IIx fibers, causing the quadriceps muscle to experience a slow-to-fast fiber transition.10,70,83 Preclinical models reinforce these observations, where scientists have observed that type I fibers show more sensitivity to disuse.18,86,89 Though the balance of data suggests a slow-to-fast type fiber transition,10,70,83 readers should be cautioned that others have also observed that atrophy of all fiber types occurs during periods of disuse.60 The clinical bottom line—muscle disuse can promote a slow-to-fast fiber-type transition.
Other Considerations for Traditional Mechanisms of Disuse Muscle Atrophy
Additional constructs involved in disuse muscle atrophy include reactive oxygen species, reduced insulin sensitivity, and alterations in transcription factors that regulate RNA and DNA production.38,98 These processes are not mutually exclusive and through an interplay of factors trigger a common program that stimulates the breakdown of proteins, ultimately leading to disuse muscle atrophy. To this point, nuclear factor–kappa B is a common factor that can link many of the aforementioned atrophying processes, as it can influence the ubiquitin proteasome system, interfere with myogenesis, and increase the expression of pro-inflammatory cytokines (eg, tumor necrosis factor–alpha, interleukin 1–beta, interleukin-6) that lead to muscle atrophy.51 Nutritional status may also play an important role in attenuating disuse muscle loss. Supplementing the diet with protein or amino acids has been found to help preserve muscle,92 although supplements do not always improve the protein synthesis response.61 Ultimately, disuse atrophy results in many undesirable complications. However, this negative sequela can easily be rectified in healthy young individuals by mechanically engaging the muscle through exercise (Figure 1).5,43
Muscle Atrophy after Traumatic Joint Injury: Different in Origin
In contrast to the obvious prescriptive solution commonly used to combat disuse muscle atrophy, the atrophy seen after traumatic joint injury continues despite these patients being actively engaged in exercise.41,52 Hence, simply exercising a muscle after traumatic joint injury is not enough to overcome the multitude of factors and cascade of events that are intensified and that negatively influence the regulation of muscle mass (Figure 2). Evidence in support of this comes from multiple systematic reviews reporting the general ineffectiveness of current rehabilitation programs to restore muscle after procedures such as ACL reconstruction (ACLR).48,49,53,67,82,84 In response, scientists have begun to utilize techniques that have improved sensitivity to uncover the intricacies of muscle atrophy factors that inhibit muscle growth after traumatic joint injuries. At the cellular level, it is becoming evident that the underlying mechanisms of atrophy after traumatic joint injury make for a more challenging environment to regrow/maintain muscle.9,59,64 Principally, deficiencies in the nervous system28,65 appear to be a major acute driver of reduced mass after traumatic joint injury (Figure 1). Recognition of the distinct difference between disuse atrophy and traumatic joint injury atrophy would enable clinicians to design more effective treatment strategies.
The following sections highlight the leading mechanisms of muscle atrophy after traumatic joint injury compared with those influencing disuse muscle atrophy. When available, emerging therapeutic countermeasures that can be used to combat the loss of muscle tissue, such as nontraditional exercises, nutritional supplements, and anti-inflammatory compounds, are referenced alongside the clinical bottom lines to help guide readers toward evidence-based solutions.
Neurophysiological Alterations
The underlying mechanisms that initiate and perpetuate disuse muscle atrophy appear to be triggered specifically by physical inactivity and the resultant failure to adequately stress muscle tissue. Although physical inactivity is also present after joint injury, and likely contributes to the overall atrophy observed, mounting evidence in the ACL literature points to the strong role that the wide neurological alterations after ACL injury have on the ability to grow and maintain muscle tissue.37,46,47,71,97 In particular, more so than any other muscle, the quadriceps experiences significant declines in muscle volume and muscle quality after ACL injury.2,65 Theoretically, this may be due to a protective inhibition role of the body in an attempt to dynamically stabilize the ACL-deficient knee against anterior tibial translation.93 Though the exact neural mechanism guiding this response is unknown, altered neural activity after ACL injury has been well documented at the spinal37,47 and cortical level,42,47,97 and it negatively influences the ability to generate a muscle contraction. Furthermore, even after the resumption of physical activity, neuroplasticity in cortical areas responsible for motor control32 are present. These central nervous system changes likely drive persistent alterations in the ability of the nervous system to generate muscle contractions, further hindering the reinstitution of healthy muscle.48 Together, these well-documented peripheral and central mechanisms limit the effectiveness of exercises targeting the involved muscle, directly affecting both muscle atrophy and the clinical weakness that is commonly observed.
Neural alterations have long been implicated as a resilient mechanism of atrophy in aging muscle and benchtop models of denervation.75 Animal studies have shown that by sectioning the dorsal root afferent neurons arising from arthritic joints, one can actually prevent muscle atrophy in the surrounding musculature, demonstrating the direct influence that afferent activity from the injured joint has on muscle atrophy.36 Altered neural activity also leads to progressive reductions in neuromuscular junctions and motoneurons in the periphery.94 In aged muscle, the lack of neural signaling is a primary event that can accelerate phases of muscle atrophy. Similar to individuals with aged muscle, those with a history of ACL injury also exhibit primary changes in neural health that influence muscle morphology.64 This direct neural-morphological interaction has recently been observed, where increased level of neural cell adhesion molecule (NCAM) in the quadriceps muscle of patients with ACLR has been found.28 NCAM is a glycoprotein molecule that uncouples nervous tissue with muscle tissue.14
Reductions in muscle fiber conduction velocity and spontaneous fiber discharge are other key indirect markers of denervation that have been observed after ACL injury20,39 and that are closely correlated with muscle atrophy.95 Notably, a key feature of quadriceps muscle atrophy after ACL injury is the accumulation of denervated fibers within the involved muscle(s). This directly distinguishes this type of muscle atrophy from disuse muscle atrophy and has a significant impact on prescriptive solutions. The change in afferent signaling, inability to excite motoneurons, and loss of neuromuscular connections can severely limit the ability to engage the muscle during exercise. Muscle denervation has also been linked to increased intermuscular fat deposits,21,55,77 which limits its functional capacity.91 Interventions that only incorporate exercise, without considering the underlying neural constructs of muscle atrophy, place a large burden on the remaining innervated fibers—rendering this approach ineffective. The clinical bottom line—neurological alterations after ACL injury have a direct impact on the morphological environment, as it uncouples the nervous system with muscle tissue. Countermeasures to combat neurophysiological alterations include electromagnetic modalities (transcutaneous electrical nerve stimulation, neuromuscular electrical stimulation, electromyographic biofeedback),13,35,63 eccentric exercise,50 and motor learning.30
Fiber-type Transitions
A reduction in neural activity reduces the muscle’s specificity for the maintenance of certain phenotypic profiles. Denervation experiments have routinely demonstrated that the absence of neural signaling causes a co-expression of fiber types, in which slow-twitch fibers begin to exhibit properties of fast-twitch fibers.70 In part, this is due to muscle phenotypes of one speed being re-innervated by a nerve from another phenotype.70 This phenomenon has been demonstrated in preclinical models, where denervation can lead to more hybrid fibers.75 To this point, similarly, after ACL injury the number of type IIa fibers has been found to be decreased, while the abundance of co-expressed type IIa/x fibers is increased (eg, change in composition of the muscle to faster fiber types).26,64 This form of change in fiber-type expression is similar to what occurs during periods of disuse atrophy; however, it should be considered that these changes appear after ACLR despite extensive strengthening.64 This supports the notion that posttraumatic fiber-type transitions are unlike disuse models of muscle atrophy that are responsive to unloading. In posttraumatic scenarios, it appears that the change in phenotypic expression is different in origin and that simply reloading the muscle with exercise is not enough to resist the change in fiber-type transition.15,56,80 The clinical bottom line—fiber type expression after traumatic joint injury is likely due to neurologic alterations. Countermeasures to combat fiber-type transitions include endurance-type stimuli,19 eccentric exercise,27 and the incorporation of interventions to blunt the common neurophysiological alterations that are observed in acute posttraumatic recovery.
Intrinsic Changes Within Muscle Fibers
Impairments in neural input have been cited as the primary cause of quadriceps muscle dysfunction after ACL injury; however, it is important to acknowledge that intrinsic changes in muscle architecture can also independently account for the loss of muscle functionality after injury. Evidence in support of this negative adaptation is beginning to emerge in the ACL literature, where the loss of myofibril density (the force generating organelles of muscle) appears to be a complication of the injury and surgical reconstruction.33 The acute activation of proteolytic processes within quadriceps muscle fibers may lead to the loss of myofibrils, which requires further investigation. The clinical bottom line—impairments in muscle functionality after injury may also be due to intrinsic changes in muscle fiber architecture. Exercises that mechanically tension the muscle for hypertrophy, such as eccentrics,27 are warranted.
Increased Circulating Factors of Atrophy
Molecular programing involved in the control of muscle mass is also dysregulated after ACLR. Specifically, MAFbx and MuRF1 (E3 ligases that signal to the ubiquitin proteasome system that protein is ready for degradation) are 2 molecular markers that are elevated after ACLR.22,59 Myostatin has also been shown to be upregulated after ACL injury58 and during acute key recovery points.59 This potent antigrowth factor can lead to profound levels of muscle atrophy. Myostatin also impairs satellite cells and is associated with chronically elevated levels of inflammatory cytokine systems that further underpin the net loss of muscle protein breakdown to muscle protein synthesis rates.78 Preliminary evidence also points to myostatin’s integral role in fibrotic pathways after ACL injury68 that degrade the quality of muscle tissue owing to the excessive accumulation of extracellular matrix components. The clinical bottom line—increased circulating factors of atrophy are upregulated after ACLR. Upregulation of these factors leads to a powerful reduction in muscle mass. Preclinical animal models96 and research in elderly patients6 suggests that postinjury myostatin-neutralizing antibodies may have a protective effect on maintaining muscle mass.
Cytokines
Several systemic inflammatory cytokines associated with tissue breakdown have been identified after traumatic joint injury. Although these cytokines have traditionally been investigated to examine the effects of cartilage and bone breakdown,66 they also represent key triggers that initiate muscle atrophy.9 Specific chemical factors that are present after traumatic injury, such as tumor necrosis factor–alpha, interleukin-1, and interleukin-6, can lead to increased protein breakdown. To this point, benchtop studies have shown that the local infusion of interleukin-6 in rodent muscle results in a loss of myofibrillar protein and decrease in growth factors that are critical for muscle growth, resulting in an overall catabolic profile causing muscle breakdown.34 A recent longitudinal study in ACLR patients has also found an increase in circulating cytokines that are influential in stimulating muscle atrophy.59 Importantly, these changes in cytokine levels were observed concurrently with changes in quadriceps muscle strength after ACLR, highlighting the importance of systemic cytokine factors in muscle atrophy after traumatic joint injury.59 The clinical bottom line—inflammatory cytokines that are present in posttraumatic scenarios have deleterious consequences on muscle breakdown and growth. Countermeasures to combat cytokines may include early joint aspiration and corticosteroid injection.45
Reduced Satellite Cells Abundance
Satellite cells play a critical role in coordinating muscle adaptation after injury, as the remodeling capacity of muscle is thought to be closely associated with satellite cell number (although the direct contribution of satellite cells to muscle remains controversial). Hence, it is concerning that early data indicate that satellite cell numbers are reduced prior to ACLR.64 This reduction in satellite cell content may impair muscle’s ability to adequately respond to rehabilitation.
Fibro-adipogenic progenitors (FAPs) are progenitor cells that support satellite cells and are employed to assist in repair by removing distressed tissue.7 Importantly, benchtop studies point to the central role that FAPs play in switching the regeneration environment from compensatory to pathologic.25 Conversion to a pathological environment promotes fibrotic tissue formation and fatty deposition, negatively influencing muscle quality.90 Direct data to support this mechanism are also available in ACL populations, where FAP content is known to be higher in the quadriceps after injury.28,68 This leads to an extracellular matrix content that is higher in collagen content28,68 and can potentially be a mechanism by which fat deposition occurs.53 Functionally, this fibrotic and fatty tissue deposition limits force generation, as less area of the quadriceps muscle is occupied by contractile components of muscle. The clinical bottom line—ACL injury is a condition that can lead to reduction in satellite cells and increased FAP content, promoting an environment that is geared toward muscle atrophy, fibrotic tissue formation, and fat infiltration, reducing muscle quality. Countermeasures to improve satellite cell abundance include eccentric exercise.58,69
Conclusion
Atrophy that occurs after traumatic joint injury continues in spite of being actively engaged in exercise (see Figure 1). This is distinctly different from the muscle atrophy that develops from disuse. The inability of classic muscle-strengthening approaches to restore muscle mass after traumatic joint injury, is likely due, in part, to the origin and the broad range of atrophy-inducing factors that are present as a result of the injury (see Figure 2).
Footnotes
The authors report no potential conflicts of interest in the development and publication of this article.
References
- 1. Allen DL, Unterman TG. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol. 2007;292:C188-C199. [DOI] [PubMed] [Google Scholar]
- 2. Arangio GA, Chen C, Kalady M, Reed JF., 3rd Thigh muscle size and strength after anterior cruciate ligament reconstruction and rehabilitation. J Orthop Sports Phys Ther. 1997;26:238-243. [DOI] [PubMed] [Google Scholar]
- 3. Arentson-Lantz EJ, English KL, Paddon-Jones D, Fry CS. Fourteen days of bed rest induces a decline in satellite cell content and robust atrophy of skeletal muscle fibers in middle-aged adults. J Appl Physiol (1985). 2016;120:965-975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Atherton PJ, Greenhaff PL, Phillips SM, Bodine SC, Adams CM, Lang CH, et al. Control of skeletal muscle atrophy in response to disuse: clinical/preclinical contentions and fallacies of evidence. Am J Physiol Endocrinol Metab. 2016;311:e594-e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Atherton PJ, Smith K. Muscle protein synthesis in response to nutrition and exercise. J Physiol. 2012;590:1049-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Becker C, Lord SR, Studenski SA, et al. Myostatin antibody (LY2495655) in older weak fallers: a proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015;3:948-957. [DOI] [PubMed] [Google Scholar]
- 7. Biferali B, Proietti D, Mozzetta C, Madaro L. Fibro-adipogenic progenitors cross-talk in skeletal muscle: the social network. Front Physiol. 2019;10:1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bodine SC. Disuse-induced muscle wasting. Int J Biochem Cell Biol. 2013;45:2200-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brocca L, Cannavino J, Coletto L, et al. The time course of the adaptations of human muscle proteome to bed rest and the underlying mechanisms. J Physiol. 2012;590:5211-5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brooks NE, Myburgh KH. Skeletal muscle wasting with disuse atrophy is multi-dimensional: the response and interaction of myonuclei, satellite cells and signaling pathways. Front Physiol. 2014;5:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carnac G, Vernus B, Bonnieu A. Myostatin in the pathophysiology of skeletal muscle. Curr Genomics. 2007;8:415-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carson RG, Buick AR. Neuromuscular electrical stimulation-promoted plasticity of the human brain [published online September 8, 2019]. J Physiol. doi: 10.1113/JP278298 [DOI] [PubMed] [Google Scholar]
- 14. Cashman NR, Covault J, Wollman RL, Sanes JR. Neural cell adhesion molecule in normal, denervated, and myopathic human muscle. Ann Neurol. 1987;21:481-489. [DOI] [PubMed] [Google Scholar]
- 15. Chang E, Kim K-M, Hertel J, Hart JM. Repeated bouts of exercise in patients with anterior cruciate ligament reconstruction. Med Sci Sports Exerc. 2014;46:769-775. [DOI] [PubMed] [Google Scholar]
- 16. Coker RH, Hays NP, Williams RH, Wolfe RR, Evans WJ. Bed rest promotes reductions in walking speed, functional parameters, and aerobic fitness in older, healthy adults. J Gerontol A Biol Sci Med Sci. 2014;70:91-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Boer MD, Selby A, Atherton P, et al. The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J Physiol. 2007;585(pt 1):241-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Desaphy J-F, Pierno S, Liantonio A, et al. Antioxidant treatment of hindlimb-unloaded mouse counteracts fiber type transition but not atrophy of disused muscles. Pharmacol Res. 2010;61:553-563. [DOI] [PubMed] [Google Scholar]
- 19. Desplanches D, Amami M, Dupré-Aucouturier S, et al. Hypoxia refines plasticity of mitochondrial respiration to repeated muscle work. Eur J Appl Physiol. 2014;114:405-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Drechsler WI, Cramp MC, Scott OM. Changes in muscle strength and EMG median frequency after anterior cruciate ligament reconstruction. Eur J Appl Physiol. 2006;98:613-623. [DOI] [PubMed] [Google Scholar]
- 21. Dulor JP, Cambon B, Vigneron P, et al. Expression of specific white adipose tissue genes in denervation-induced skeletal muscle fatty degeneration. FEBS Lett. 1998;439:89-92. [DOI] [PubMed] [Google Scholar]
- 22. Durigan JL, Delfino GB, Peviani SM, et al. Neuromuscular electrical stimulation alters gene expression and delays quadriceps muscle atrophy of rats after anterior cruciate ligament transection. Muscle Nerve. 2014;49:120-128. [DOI] [PubMed] [Google Scholar]
- 23. Ehlers ML, Celona B, Black BL. NFATc1 controls skeletal muscle fiber type and is a negative regulator of MyoD activity. Cell Rep. 2014;8:1639-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elkina Y, von Haehling S, Anker SD, Springer J. The role of myostatin in muscle wasting: an overview. J Cachexia Sarcopenia Muscle. 2011;2:143-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Farup J, Madaro L, Puri PL, Mikkelsen UR. Interactions between muscle stem cells, mesenchymal-derived cells and immune cells in muscle homeostasis, regeneration and disease. Cell Death Dis. 2015;6:e1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flück M, Viecelli C, Bapst AM, et al. Knee extensors muscle plasticity over a 5-years rehabilitation process after open knee surgery. Front Physiol. 2018;9:1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Franchi MV, Reeves ND, Narici MV. Skeletal muscle remodeling in response to eccentric vs. concentric loading: morphological, molecular, and metabolic adaptations. Front Physiol. 2017;8:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fry CS, Johnson DL, Ireland ML, Noehren B. ACL injury reduces satellite cell abundance and promotes fibrogenic cell expansion within skeletal muscle. J Orthop Res. 2017;35(9):1876-1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glover EI, Phillips SM, Oates BR, et al. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J Physiol. 2008;586:6049-6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gokeler A, Neuhaus D, Benjaminse A, Grooms DR, Baumeister J. Principles of motor learning to support neuroplasticity after ACL injury: implications for optimizing performance and reducing risk of second ACL injury. Sports Med. 2019;49:853-865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Greenhaff PL, Karagounis L, Peirce N, et al. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am J Physiol Endocrinol Metab. 2008;295:e595-e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grooms DR, Page SJ, Nichols-Larsen DS, Chaudhari AM, White SE, Onate JA. Neuroplasticity associated with anterior cruciate ligament reconstruction. J Orthop Sports Phys Ther. 2017;47:180-189. [DOI] [PubMed] [Google Scholar]
- 33. Gumucio JP, Sugg KB, Enselman ERS, et al. Anterior cruciate ligament tear induces a sustained loss of muscle fiber force production [published online January 18, 2018]. Muscle Nerve. doi:10.1002/mus.26075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haddad F, Zaldivar F, Cooper DM, Adams GR. IL-6-induced skeletal muscle atrophy. J Appl Physiol (1985). 2005;98:911-917. [DOI] [PubMed] [Google Scholar]
- 35. Harkey MS, Gribble PA, Pietrosimone BG. Disinhibitory interventions and voluntary quadriceps activation: a systematic review. J Athl Train. 2014;49:411-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hopkins JT, Ingersoll CD. Arthrogenic muscle inhibition: a limiting factor in joint rehabilitation. Journal of Sport Rehabilitation. 2000;9:135-159. [Google Scholar]
- 37. Ingersoll CD, Grindstaff TL, Pietrosimone BG, Hart JM. Neuromuscular consequences of anterior cruciate ligament injury. Clin Sports Med. 2008;27:383-404. [DOI] [PubMed] [Google Scholar]
- 38. Kandarian SC, Stevenson EJ. Molecular events in skeletal muscle during disuse atrophy. Exerc Sport Sci Rev. 2002;30:111-116. [DOI] [PubMed] [Google Scholar]
- 39. Kaneko F, Onari K, Kawaguchi K, Tsukisaka K, Roy SH. Electromechanical delay after ACL reconstruction: an innovative method for investigating central and peripheral contributions. J Orthop Sports Phys Ther. 2002;32:158-165. [DOI] [PubMed] [Google Scholar]
- 40. Kornegay JN, Bogan DJ, Bogan JR, et al. Dystrophin-deficient dogs with reduced myostatin have unequal muscle growth and greater joint contractures. Skeletal Muscle. 2016;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuenze CM, Blemker SS, Hart JM. Quadriceps function relates to muscle size following ACL reconstruction. J Orthop Res. 2016;34:1656-1662. [DOI] [PubMed] [Google Scholar]
- 42. Kuenze CM, Hertel J, Weltman A, Diduch D, Saliba SA, Hart JM. Persistent neuromuscular and corticomotor quadriceps asymmetry after anterior cruciate ligament reconstruction. J Athl Train. 2015;50:303-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumar V, Atherton P, Smith K, Rennie MJ. Human muscle protein synthesis and breakdown during and after exercise. J Appl Physiol (1985). 2009;106:2026-2039. [DOI] [PubMed] [Google Scholar]
- 44. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277:49831-49840. [DOI] [PubMed] [Google Scholar]
- 45. Lattermann C, Jacobs CA, Proffitt Bunnell M, et al. A multicenter study of early anti-inflammatory treatment in patients with acute anterior cruciate ligament tear. Am J Sports Med. 2017;45:325-333. [DOI] [PubMed] [Google Scholar]
- 46. Lepley AS, Ericksen HM, Sohn DH, Pietrosimone BG. Contributions of neural excitability and voluntary activation to quadriceps muscle strength following anterior cruciate ligament reconstruction. Knee. 2014;21:736-742. [DOI] [PubMed] [Google Scholar]
- 47. Lepley AS, Gribble PA, Thomas AC, Tevald MA, Sohn DH, Pietrosimone BG. Quadriceps neural alterations in anterior cruciate ligament reconstructed patients: a 6-month longitudinal investigation. Scand J Med Sci Sports. 2015;25:828-839. [DOI] [PubMed] [Google Scholar]
- 48. Lepley AS, Grooms DR, Burland JP, Davi SM, Kinsella-Shaw JM, Lepley LK. Quadriceps muscle function following anterior cruciate ligament reconstruction: systemic differences in neural and morphological characteristics. Exp Brain Res. 2019;237:1267-1278. [DOI] [PubMed] [Google Scholar]
- 49. Lepley LK. Deficits in quadriceps strength and patient-oriented outcomes at return to activity after ACL reconstruction: a review of the current literature. Sports Health. 2015;7:231-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lepley LK, Lepley AS, Onate JA, Grooms DR. Eccentric exercise to enhance neuromuscular control. Sports Health. 2017;9:333-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med (Berl). 2008;86:1113-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lindström M, Strandberg S, Wredmark T, Felländer-Tsai L, Henriksson M. Functional and muscle morphometric effects of ACL reconstruction. A prospective CT study with 1 year follow-up. Scand J Med Sci Sports. 2013;23:431-442. [DOI] [PubMed] [Google Scholar]
- 53. Liu X, Ning AY, Chang NC, et al. Investigating the cellular origin of rotator cuff muscle fatty infiltration and fibrosis after injury. Muscles Ligaments Tendons J. 2016;6:6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lohmander LS, Ostenberg A, Englund M, Roos H. High prevalence of knee osteoarthritis, pain, and functional limitations in female soccer players twelve years after anterior cruciate ligament injury. Arthritis Rheum. 2004;50:3145-3152. [DOI] [PubMed] [Google Scholar]
- 55. Marcus RL, Addison O, Kidde JP, Dibble LE, Lastayo PC. Skeletal muscle fat infiltration: impact of age, inactivity, and exercise. J Nutr Health Aging. 2010;14:362-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McHugh MP, Tyler TF, Nicholas SJ, Browne MG, Gleim GW. Electromyographic analysis of quadriceps fatigue after anterior cruciate ligament reconstruction.J Orthop Sports Phys Ther. 2001;31:25-32. [DOI] [PubMed] [Google Scholar]
- 57. McKay BR, Ogborn DI, Bellamy LM, Tarnopolsky MA, Parise G. Myostatin is associated with age-related human muscle stem cell dysfunction. FASEB J. 2012;26:2509-2521. [DOI] [PubMed] [Google Scholar]
- 58. McKay BR, Toth KG, Tarnopolsky MA, Parise G. Satellite cell number and cell cycle kinetics in response to acute myotrauma in humans: immunohistochemistry versus flow cytometry. J Physiol. 2010;588:3307-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mendias CL, Lynch EB, Davis ME, et al. Changes in circulating biomarkers of muscle atrophy, inflammation, and cartilage turnover in patients undergoing anterior cruciate ligament reconstruction and rehabilitation. Am J Sports Med. 2013;41:1819-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miokovic T, Armbrecht G, Felsenberg D, Belavý DL. Heterogeneous atrophy occurs within individual lower limb muscles during 60 days of bed rest. J Appl Physiol (1985). 2012;113:1545-1559. [DOI] [PubMed] [Google Scholar]
- 61. Moro T, Brightwell CR, Deer RR, et al. Muscle protein anabolic resistance to essential amino acids does not occur in healthy older adults before or after resistance exercise training. J Nutr. 2018;148:900-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nakanishi R, Hirayama Y, Tanaka M, et al. Nucleoprotein supplementation enhances the recovery of rat soleus mass with reloading after hindlimb unloading–induced atrophy via myonuclei accretion and increased protein synthesis. Nutr Res. 2016;36:1335-1344. [DOI] [PubMed] [Google Scholar]
- 63. Needle AR, Lepley AS, Grooms DR. Central nervous system adaptation after ligamentous injury: a summary of theories, evidence, and clinical interpretation. Sports Med. 2017;47:1271-1288. [DOI] [PubMed] [Google Scholar]
- 64. Noehren B, Andersen A, Hardy P, et al. Cellular and morphological alterations in the vastus lateralis muscle as the result of ACL injury and reconstruction. J Bone Joint Surg Am. 2016;98(18):1541-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Norte GE, Knaus KR, Kuenze C, et al. MRI-based assessment of lower-extremity muscle volumes in patients before and after ACL reconstruction. J Sport Rehabil. 2018;27:201-212. [DOI] [PubMed] [Google Scholar]
- 66. Olson SA, Horne P, Furman B, et al. The role of cytokines in posttraumatic arthritis. J Am Acad Orthop Surg. 2014;22:29-37. [DOI] [PubMed] [Google Scholar]
- 67. Palmieri-Smith RM, Thomas AC, Wojtys EM. Maximizing quadriceps strength after ACL reconstruction. Clin Sports Med. 2008;27:405-424. [DOI] [PubMed] [Google Scholar]
- 68. Peck BD, Brightwell CR, Johnson DL, Ireland ML, Noehren B, Fry CS. Anterior cruciate ligament tear promotes skeletal muscle myostatin expression, fibrogenic cell expansion, and a decline in muscle quality. Am J Sports Med. 2019;47:1385-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Petrella JK, Kim J-S, Mayhew DL, Cross JM, Bamman MM. Potent myofiber hypertrophy during resistance training in humans is associated with satellite cell-mediated myonuclear addition: a cluster analysis. J Appl Physiol. 2008;104:1736-1742. [DOI] [PubMed] [Google Scholar]
- 70. Pette D, Staron RS. Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech. 2000;50:500-509. [DOI] [PubMed] [Google Scholar]
- 71. Pietrosimone BG, McLeod MM, Lepley AS. A theoretical framework for understanding neuromuscular response to lower extremity joint injury. Sports Health. 2012;4:31-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Putman CT, Xu X, Gillies E, MacLean IM, Bell GJ. Effects of strength, endurance and combined training on myosin heavy chain content and fibre-type distribution in humans. Eur J Appl Physiol. 2004;92:376-384. [DOI] [PubMed] [Google Scholar]
- 73. Reardon KA, Davis J, Kapsa RM, Choong P, Byrne E. Myostatin, insulin-like growth factor-1, and leukemia inhibitory factor mRNAs are upregulated in chronic human disuse muscle atrophy. Muscle Nerve. 2001;24:893-899. [DOI] [PubMed] [Google Scholar]
- 74. Reidy PT, McKenzie AI, Brunker P, et al. Neuromuscular electrical stimulation combined with protein ingestion preserves thigh muscle mass but not muscle function in healthy older adults during 5 days of bed rest. Rejuvenation Res. 2017;20:449-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rowan SL, Rygiel K, Purves-Smith FM, Solbak NM, Turnbull DM, Hepple RT. Denervation causes fiber atrophy and myosin heavy chain co-expression in senescent skeletal muscle. PLoS One. 2012;7:e29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sandri M. Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol. 2013;45:2121-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sciorati C, Clementi E, Manfredi AA, Rovere-Querini P. Fat deposition and accumulation in the damaged and inflamed skeletal muscle: cellular and molecular players. Cell Mol Life Sci. 2015;72:2135-2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Silva JMS, Alabarse PVG, Teixeira VON, et al. Muscle wasting in osteoarthritis model induced by anterior cruciate ligament transection. PLoS One. 2018;13:e0196682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Snijders T, Wall BT, Dirks ML, et al. Muscle disuse atrophy is not accompanied by changes in skeletal muscle satellite cell content. Clin Sci (Lond). 2014;126:557-566. [DOI] [PubMed] [Google Scholar]
- 80. Snyder-Mackler L, Binder-Macleod SA, Williams PR. Fatigability of human quadriceps femoris muscle following anterior cruciate ligament reconstruction. Med Sci Sports Exerc. 1993;25:783-789. [DOI] [PubMed] [Google Scholar]
- 81. Staron RS, Hagerman FC, Hikida RS, et al. Fiber type composition of the vastus lateralis muscle of young men and women. J Histochem Cytochem. 2000;48:623-629. [DOI] [PubMed] [Google Scholar]
- 82. Strandberg S, Lindström M, Wretling M-L, Aspelin P, Shalabi A. Muscle morphometric effect of anterior cruciate ligament injury measured by computed tomography: aspects on using non-injured leg as control. BMC Musculoskelet Disord. 2013;14:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Talbot J, Maves L. Skeletal muscle fiber type: using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip Rev Dev Biol. 2016;5:518-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Thomas AC, Wojtys EM, Brandon C, Palmieri-Smith RM. Muscle atrophy contributes to quadriceps weakness after anterior cruciate ligament reconstruction. J Sci Med Sport. 2016;19:7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thomas M, Langley B, Berry C, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem. 2000;275:40235-40243. [DOI] [PubMed] [Google Scholar]
- 86. Thomason DB, Booth FW. Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol (1985). 1990;68:1-12. [DOI] [PubMed] [Google Scholar]
- 87. Tourville TW, Jarrell KM, Naud S, Slauterbeck JR, Johnson RJ, Beynnon BD. Relationship between isokinetic strength and tibiofemoral joint space width changes after anterior cruciate ligament reconstruction. Am J Sports Med. 2014;42:302-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Trappe S, Trappe T, Gallagher P, Harber M, Alkner B, Tesch P. Human single muscle fibre function with 84 day bed-rest and resistance exercise. J Physiol. 2004;557:501-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tsika R, Herrick R, Baldwin K. Effect of anabolic steroids on skeletal muscle mass during hindlimb suspension. J Appl Physiol (1985). 1987;63:2122-2127. [DOI] [PubMed] [Google Scholar]
- 90. Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010;12:143-152. [DOI] [PubMed] [Google Scholar]
- 91. Visser M, Goodpaster BH, Kritchevsky SB, et al. Muscle mass, muscle strength, and muscle fat infiltration as predictors of incident mobility limitations in well-functioning older persons. J Gerontol A Biol Sci Med Sci. 2005;60:324-333. [DOI] [PubMed] [Google Scholar]
- 92. Wall BT, Van Loon LJ. Nutritional strategies to attenuate muscle disuse atrophy. Nutr Rev. 2013;71:195-208. [DOI] [PubMed] [Google Scholar]
- 93. Wojtys EM, Huston LJ. Neuromuscular performance in normal and anterior cruciate ligament-deficient lower extremities. Am J Sports Med. 1994;22:89-104. [DOI] [PubMed] [Google Scholar]
- 94. Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wu P, Chawla A, Spinner RJ, et al. Key changes in denervated muscles and their impact on regeneration and reinnervation. Neural Regen Res. 2014;9(20):1796-1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wurtzel CN, Gumucio JP, Grekin JA, et al. Pharmacological inhibition of myostatin protects against skeletal muscle atrophy and weakness after anterior cruciate ligament tear. J Orthop Res. 2017;35:2499-2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zarzycki R, Morton SM, Charalambous CC, Marmon A, Snyder-Mackler L. Corticospinal and intracortical excitability differ between athletes early after ACLR and matched controls. J Orthop Res. 2018;36:2941-2948. [DOI] [PubMed] [Google Scholar]
- 98. Zhang P, Chen X, Fan M. Signaling mechanisms involved in disuse muscle atrophy. Med Hypotheses. 2007;69:310-321. [DOI] [PubMed] [Google Scholar]